

The nucleotide-binding domain (NBD) and leucine-rich repeat (LRR) pyrin-domain containing protein 1 (NLRP1) is an inflammasome sensor that mediates activation of caspase-1 to induce cytokine maturation and pyroptosis1–4. Gain-of-function NLRP1 mutations cause severe skin inflammatory diseases4–6. NLRP1 contains a function-to-find domain (FIIND) that autoproteolyzes into noncovalently associated subdomains7–9, and proteasomal degradation of the repressive N-terminal fragment (NT) releases the inflammatory C-terminal fragment (CT)10,11. The cytosolic dipeptidyl peptidases 8 and 9 (DPP8/9) interact with NLRP1, and small-molecule DPP8/9 inhibitors activate NLRP1 by unclear mechanisms10,12–14. Here, we report cryo-EM structures of the human NLRP1-DPP9 complex, alone and with the DPP8/9 inhibitor Val-boroPro (VbP). Surprisingly, the structures reveal a ternary complex comprising DPP9, full-length NLRP1 (NLRP1-FL), and NLRP1-CT. NLRP1-CT binding to DPP9 requires NLRP1-FL, suggesting regulation of NLRP1 activation by the critical NLRP1-CT to NLRP1-FL ratio. Consistently, inflammasome activation by ectopic NLRP1-CT expression is rescued by co-expressing autoproteolysis-deficient NLRP1-FL. The N-terminus of NLRP1-CT inserts into the DPP9 active site and VbP disrupts this interaction. Thus, VbP weakens the NLRP1-DPP9 interaction while also accelerating NT degradation10 to induce inflammasome activation. Overall, these data demonstrate that DPP9 quenches low levels of NLRP1-CT to function as a “bomb-defuser”, thereby serving as checkpoint for NLRP1 inflammasome activation.
NLRP1 senses danger-associated signals that cause protein destabilization and/or degradation, such as direct cleavage or ubiquitination by microbial proteases and ubiquitin ligases, respectively4,15. Recent studies demonstrated that functional NT degradation by the proteasome is a mechanism for NLRP1 activation10,11. Because FIIND autoproteolysis creates a break in the NLRP1 polypeptide chain between the ZU5 and UPA subdomains (Fig. 1a)7,8, degradation of the NLRP1-NT releases noncovalently associated NLRP1-CT (UPA-CARD) and enables CARD filament formation, recruitment of the apoptosis-associated speck-like protein containing a CARD (ASC) adaptor and pro-caspase-1, and caspase-1 activation10,11. Active caspase-1 processes interleukin-1 (IL-1) family pro-cytokines to their bioactive forms and cleaves gasdermin D (GSDMD) to liberate its pore-forming N-terminus, which oligomerizes and perforates cell membranes to mediate cytokine secretion and inflammatory cell death16.
Fig. 1. |. Structure of the NLRP1-DPP9 complex.
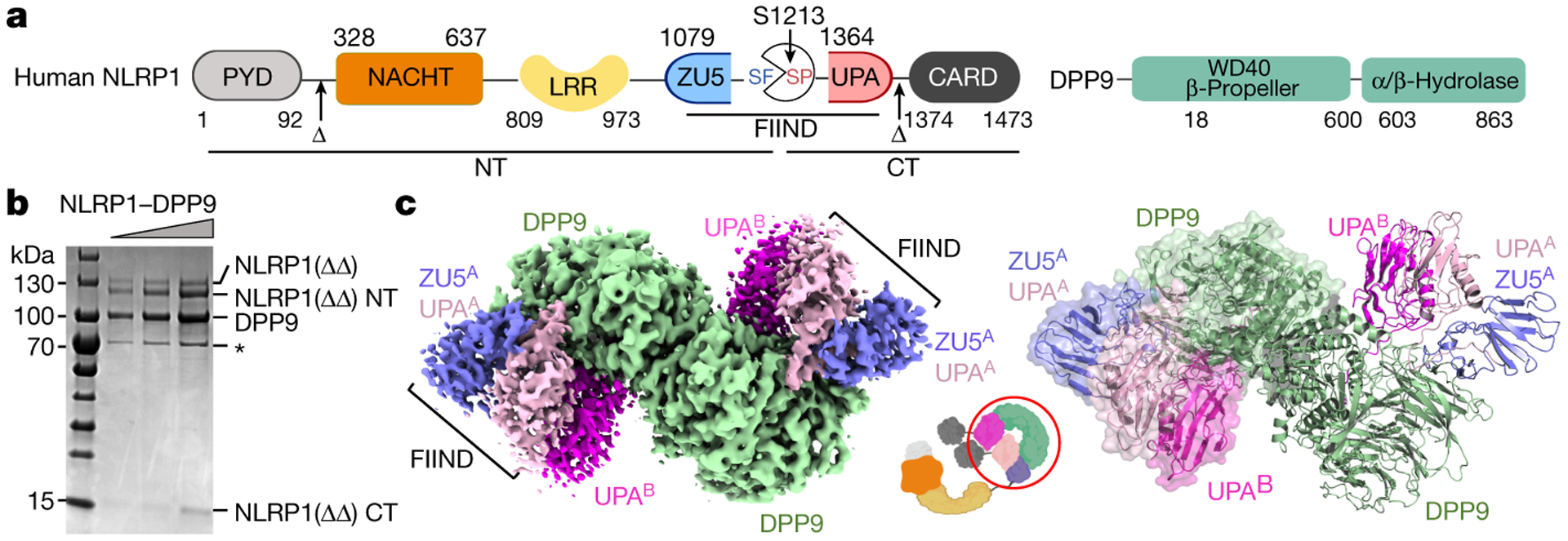
a, Domain organization. b, Representative (>3 independent experiments) SDS-PAGE of the purified NLRP1-DPP9 complex. HSP70 contamination is noted with an asterisk (*). c, Cryo-EM map (left) and the model (right) of the ternary NLRP1A-NLRP1B-DPP9 complex. The DPP9 dimer and the two copies of NLRP1 (A and B) are labelled with the colour scheme in (a). A schematic diagram (middle) denotes the entire NLRP1 and DPP9 molecules versus the ordered, resolved portions of the proteins (red circle).
In contrast to the selective activation of certain NLRP1 alleles by particular microbial effectors, small-molecule inhibitors of the dipeptidyl peptidases DPP8 and DPP9 (DPP8/9), such as VbP (also known as Talabostat or PT-100), activate all functional human and rodent NLRP1 homologs10,13,14,17,18. DPP8/9 cleave dipeptides following proline or alanine from the N-termini of polypeptides (NH2-X-P or NH2-X-A)19,20, and inhibition of this enzymatic activity accelerates NLRP1-NT ubiquitination and proteasomal degradation via an unknown mechanism10,12,21,22. In addition, DPP8/9 directly bind to NLRP112,21, but how DPP8/9 binding modulates the NLRP1 inflammasome activity remains unclear. Here, we sought to address this question through structural studies of the NLRP1-DPP9 complex.
Cryo-EM structure of the ternary complex
We co-expressed PYD and CARD-deleted NLRP1 (NLRP1ΔΔ) with DPP9 in mammalian cells and purified the NLRP1-DPP9 complex to homogeneity (Fig. 1b). We then determined the cryo-EM structure of the NLRP1-DPP9 complex at 3.6 Å resolution (Extended Data Fig. 1). The atomic model was validated independently by crosslinking mass spectrometry (Extended Data Fig. 2). The cryo-EM structure surprisingly revealed that two NLRP1 molecules bind each monomer of the DPP9 dimer, forming an NLRP1A-NLRP1B-DPP9 ternary complex (Fig. 1c). The first NLRP1 (molecule A) is composed of a complete FIIND with intimately associated ZU5 and UPA subdomains (Extended Data Fig. 3a–c). The β-sandwich folds in these subdomains are similar to previously defined ZU5 and UPA domains23,24, but with a number of differences, in particular the insertion of the first β-strand (β0) of UPA into the ZU5 fold as if it is the last β-strand of ZU5 (Extended Data Fig. 3b–c). Since our sample contained more autoprocessed than unprocessed NLRP1 (Fig. 1b), we built the model as processed FIIND. In this model, the catalytic triad residues (S1213 of UPA, H1186 and E1195 of ZU5) are nearly properly positioned for catalysis (Extended Data Fig. 3b), suggesting that there are likely limited conformational changes between unprocessed and processed forms.
The second NLRP1 (molecule B) contains only the UPA subdomain (Fig. 1c). If we place the ZU5 subdomain of the second NLRP1 using the ZU5-UPA structure of the first NLRP1, it clashes with both DPP9 and the first NLRP1 (Extended Data Fig. 3d), suggesting that the second NLRP1 cannot include the ZU5 subdomain or the NT fragment. Thus, the NLRP1A-NLRP1B-DPP9 complex can only be formed when FL-NLRP1 and NT degraded NLRP1 (NLRP1-CT) are both present. NACHT and LRR domains of NLRP1 are not visible in the density, likely due to a flexible linkage between the FIIND and the preceding LRR (Fig. 1a).
NLRP1-CT inserts into the DPP9 tunnel
A striking observation in the NLRP1-DPP9 complex is that the N-terminal segment of UPAB (β0, residue S1213-N1224) unfolds and inserts into the DPP9 substrate tunnel as an extended polypeptide chain (Fig. 2a). The inserted segment in UPAB shows dramatically different conformation from its UPAA counterpart, whereas the gross conformations of UPAA and UPAB are similar (Extended Data Fig. 3e–f). Notably, the UPAB N-terminus (NH2-S-P) is a potential DPP8/9 substrate, and the terminal amino group of the segment forms hydrogen-bond and salt-bridge interactions with E248 and E249 in the EE-helix of DPP9. The DPP9 active site is also known to undergo substantial rearrangement at a large loop segment, which partially folds into an α-helix known as the R-helix when an Arg residue on the helix engages a substrate25. In the NLRP1-DPP9 complex, R133 of DPP9 interacts with main chain carbonyl oxygen atoms in the N-terminal segment of UPAB, and the R-helix concomitantly undergoes a disorder to order transition (Fig. 2a), similar to substrate or inhibitor binding.
Fig. 2. |. Detailed interfaces in the NLRP1-DPP9 ternary complex and inhibition by the DPP9 inhibitor VbP.
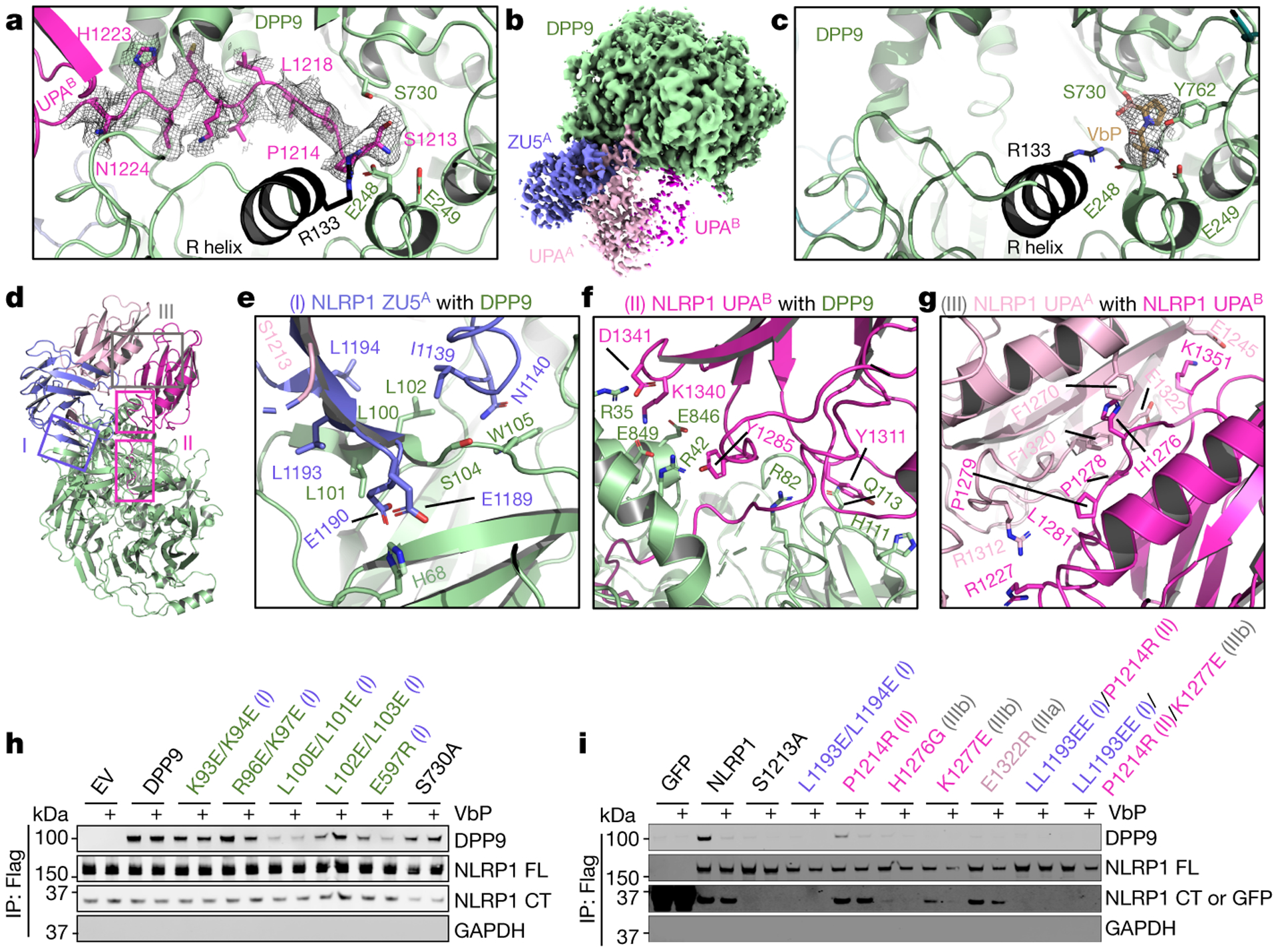
a, Insertion of the N-terminal peptide of UPAB into the DPP9 active site. b, Cryo-EM map of the NLRP1-DPP9 complex in the presence of VbP. VbP binding reduces UPAB occupancy. c, Displacement of the UPAB N-terminal peptide from the DPP9 active site by VbP. d, Overview of three interfaces important for NLRP1-DPP9 association. Regions blocked in rectangles are shown in detail in (a) and (e-g). e-g, Zoom-ins of each NLRP1-DPP9 binding interface. h, FLAG co-immunoprecipitation using FLAG-tagged WT NLRP1 and the indicated His-tagged DPP9 constructs expressed in DPP8/9 DKO 293T cells. EV: empty vector. i, FLAG co-immunoprecipitation using FLAG-tagged NLRP1 expressed in HEK293T cells. FLAG-tagged GFP was used as a negative control. The Roman numerals in parentheses (h-i) represent the three interfaces in the NLRP1-DPP9 ternary complex. Each immunoblot is representative of > 2 independent experiments.
We next explored the structural impact of DPP8/9 inhibitors on the NLRP1-DPP9 complex. We solved a 2.9 Å cryo-EM structure of the NLRP1-DPP9 complex purified in the presence of the small-molecule inhibitor VbP, a covalent peptidomimetic that modifies the catalytic S730 (Fig. 2b,c, Extended Data Fig. 4, 5a). The density of VbP facilitated its unambiguous placement, which showed that VbP interacts with several notable DPP9 residues in addition to the catalytic S730: the R-loop residue R133 and the EE motif composed of E248 and E249 (Fig. 2c, Extended Data Fig. 5b). VbP also induces the substrate-bound state of DPP9 as indicated by ordering of the R-helix. In fact, the VbP conformation closely resembles the pose of an isoleucine-proline substrate bound to bacterial DPP426 (Extended Data Fig. 5c).
Remarkably, the N-terminal peptide of UPAB was absent in the DPP9 substrate tunnel (Fig. 2c), and the entire UPAB molecule was weak in the cryo-EM density of the NLRP1-DPP9-VbP complex structure (Fig. 2b), suggesting that VbP can displace NLRP1-CT from DPP9. Superposition of VbP-bound and NLRP1-bound DPP9 structures showed that VbP displaces NLRP1-CT by direct competition for the DPP9 active site rather than by causing conformational changes (Extended Data Fig. 5d). Thus, these data rationalize prior reports that VbP weakened the NLRP1-DPP9 interaction as shown by co-immunoprecipitation and mass spectrometry12,21. Despite the binding of NLRP1-CT to the DPP9 active site and its displacement by VbP, DPP9 does not cleave NLRP1-CT (Extended Data Fig. 6) and likely does not repress the NLRP1-CT activity by direct dipeptide cleavage.
Three interfaces mediate complex assembly
There are three interaction sites in the NLRP1-FLA-DPP9-NLRP1-CTB complex: interface I (between ZU5A and DPP9), interface II (between UPAB and DPP9), and interface III (between UPAA and UPAB) (Fig. 2d). For interface I, only ZU5A, but not UPAA in the same FIIND, interacts with DPP9; however, UPAA helps recruit NLRP1B via interface III. Thus, site A selects for FL-NLRP1 via ZU5A at interface I, whereas site B selects for NLRP1-CT because a theoretical ZU5B cannot be accommodated (Extended Data Fig. 3d). All three interfaces are extensive, burying ~1,000 Å2, ~1,600 Å2 and ~800 Å2 surface areas per partner, respectively.
Interface I is formed by regions of β9 and α1-β5 loop in ZU5A and the WD40 domain in DPP9, with part of the interactions through hydrogen bonds between β-strands (Fig. 2e). Part of the blade I of DPP9 is disordered in the DPP9 crystal structure25 but becomes ordered upon NLRP1 interaction (Extended Data Fig. 7a). Interface II has two components, one formed by the N-terminal segment of UPAB (S1213-N1224) as described previously (Fig. 2a), and the other formed between one face of the UPAB globular domain and the α/β-hydrolase domain of DPP9 (Fig. 2f). In interface III, UPAA interacts intimately with UPAB using the sides of their β-sandwich folds (Fig. 2g, Extended Data Fig. 3e), comprising both hydrogen bonding interactions and hydrophobic packing. The relationship between these two UPA subdomains is mainly a translation with only a 9° rotation between them (Extended Data Fig. 3e), and this UPA-UPA interaction has not been observed in other ZU5 and UPA-containing structural homologs23,24. Given the translational relationship between these UPAs, the entire interaction surface differs between UPAA (IIIa) and UPAB (IIIb).
Structure-based NLRP1 and DPP9 mutations
We generated structure-based mutations on all three interfaces to determine their effects on NLRP1-DPP9 complex formation. Both LL100EE and E597R DPP9 mutants at interface I strongly reduced NLRP1 interaction without compromising DPP9’s post-proline cleavage activity or sensitivity to VbP (Fig. 2h, Extended Data Fig. 7b–d). We also managed to obtain the DPP9-binding defective NLRP1 interface I mutant LL1193EE, which, although partially defective in FIIND autoprocessing, was still active (Fig. 2i, 3a, Extended Data Fig. 7e). The interface II mutant P1214R at the UPAB N-terminus (Fig. 2a), a previously described autoactive patient mutation12,27, was fully autoprocessed with reduced DPP9 binding (Fig. 2i, Extended Data Fig. 7e). Mutations on interface III, the UPAA (IIIa)-UPAB (IIIb) dimerization surface, almost completely abolished DPP9 binding while retaining autoprocessing (Fig. 2i, Extended Data Fig. 7e). Similarly, the S1213A autoprocessing-defective mutation, or wild-type (WT) NLRP1 treated with VbP, also abolished DPP9 binding in the co-IP assay (Fig. 2h–i). These data suggest that there is cooperativity between the two NLRP1 molecules in binding DPP9, in which neither NLRP1A nor NLRP1B binds to DPP9 without the other. In vitro however, the high protein concentration could facilitate the binding of NLRP1-FLA to DPP9 even when NLRP1-CTB is largely absent (Fig. 2b).
Interface I/II mutations: autoactivation
Since DPP9 inhibition or its genetic ablation activates NLRP112,14,18, we hypothesized that loss of DPP9 interaction would result in NLRP1 autoactivation. We designed a HEK293T system that recapitulates key components of the inflammasome pathway to evaluate the functional consequences of specifically disrupting interactions of NLRP1-FLA, NLRP1-CTB, or both with DPP9. Specifically, we transiently reconstituted ASC and loss-of-DPP9 interaction NLRP1 mutants in HEK293T cells stably expressing caspase-1 and GSDMD, and assayed cell death by measuring the release of the intracellular enzyme lactate dehydrogenase (LDH). Indeed, mutations on interfaces I and II, including the previously reported patient mutation P1214R12,27, induced marked cell death and GSDMD processing in the absence of VbP, with no further increase upon VbP addition (Fig. 3a). Similarly, these NLRP1 mutants spontaneously formed ASC specks (Fig. 3b) in the absence of any activating signal. Thus, disruption of either NLRP1-DPP9 interface I or II leads to spontaneous inflammasome activation.
Fig. 3. |. Functional consequences of interfacial mutations in the NLRP1-DPP9 ternary complex.
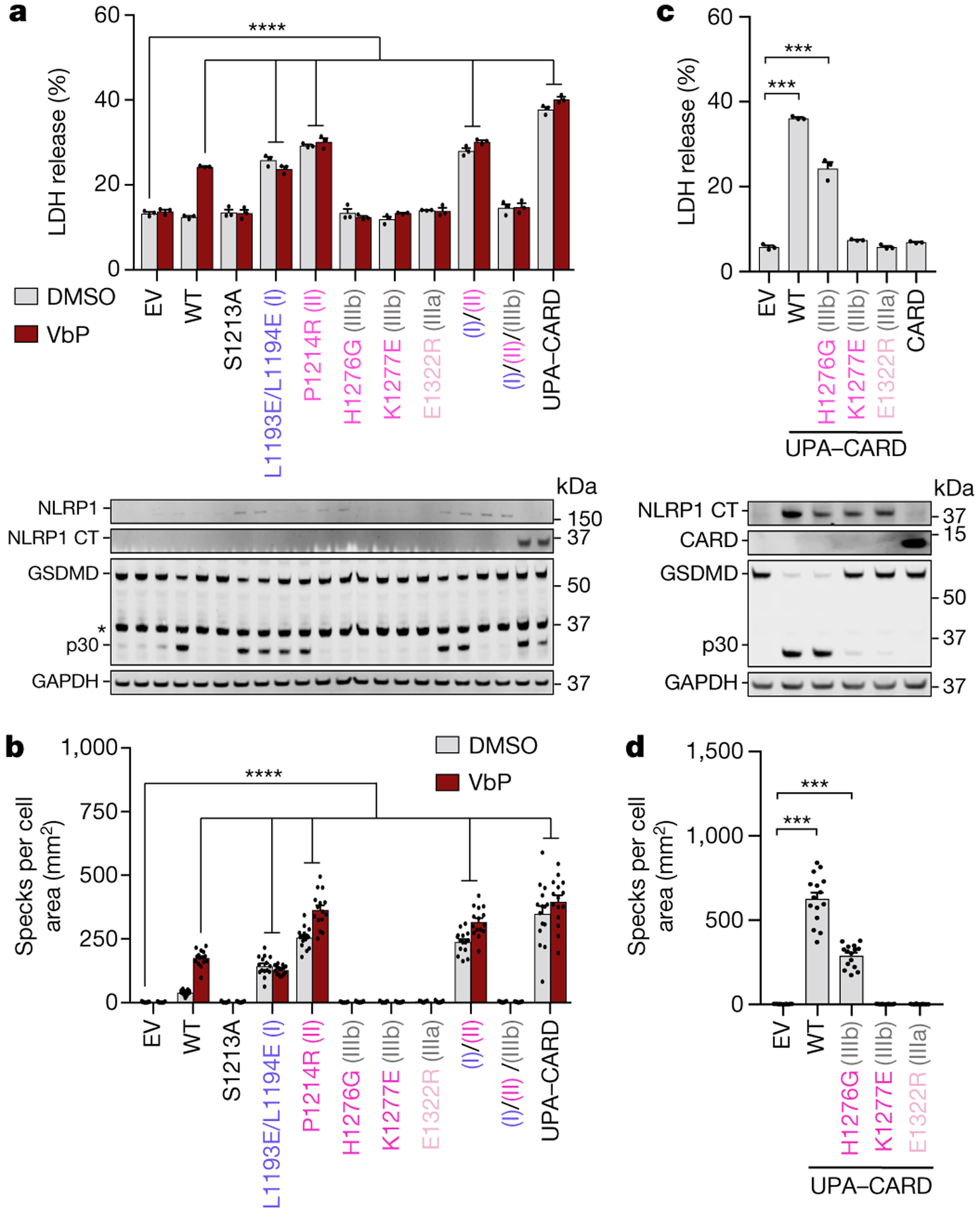
Interface I and II mutants cause NLRP1 autoactivation and UPA-UPA interactions are required for inflammasome activity. a, LDH release (top) and GSDMD processing (bottom) from transient expression of indicated constructs in a reconstituted HEK293T inflammasome system, with and without addition of VbP. n=3 independent biological replicates. *: non-specific bands; p30: GSDMD N-terminal fragment from caspase-1 cleavage. b, Quantification of speck formation induced by expression of indicated constructs in the presence and absence of VbP. n=15 quantified fields of view. c, LDH release (top) and GSDMD processing (bottom) by direct expression of WT or mutant NLRP1 UPA-CARDs. CARD alone was also included. n=3 independent biological replicates. d, Quantification of speck formation induced by expression of WT or mutant NLRP1 UPA-CARD. n=15 quantified fields of view. All data are representative of > 3 independent experiments. **** in (a) and (b): p < 0.0001 compared to EV by 2-way ANOVA with Tukey multiple comparison correction. *** in (c) and (d): p < 0.001 by unpaired two-sided t test, exact p-values are presented in the Source Data file. Data in (a-d) are mean ± SEM.
Interface III mutations abolish activity
Surprisingly, interface III UPAA-UPAB loss-of-interaction mutants were completely deficient in both spontaneous and VbP-mediated inflammasome signalling (Fig. 3a–b) despite retaining some autoprocessing (Fig. 2i). Furthermore, addition of an interface III mutant abolished the activity of an interface I/II double mutant (Fig. 3a–b). Since we and others recently demonstrated that the UPA subdomain promotes CARD filament formation and NLRP1 inflammasome signalling28,29 through self-oligomerization, we reasoned that this function is driven by interface III interactions. To test this hypothesis, we ectopically expressed NLRP1-CT in the same reconstituted HEK293T cell system, which as expected induced cell death and ASC speck formation (Fig. 3c–d). The interface III mutations abolished or reduced CT-mediated cell death, GSDMD cleavage, and ASC speck formation (Fig. 3c–d). An NLRP1-CARD construct was similarly deficient in inducing cell death (Fig. 3c). This requirement for the UPA domain and the UPA-UPA interface III contacts in NLRP1-CT inflammasome activation suggests that UPA plays an architectural role in inflammasome assembly.
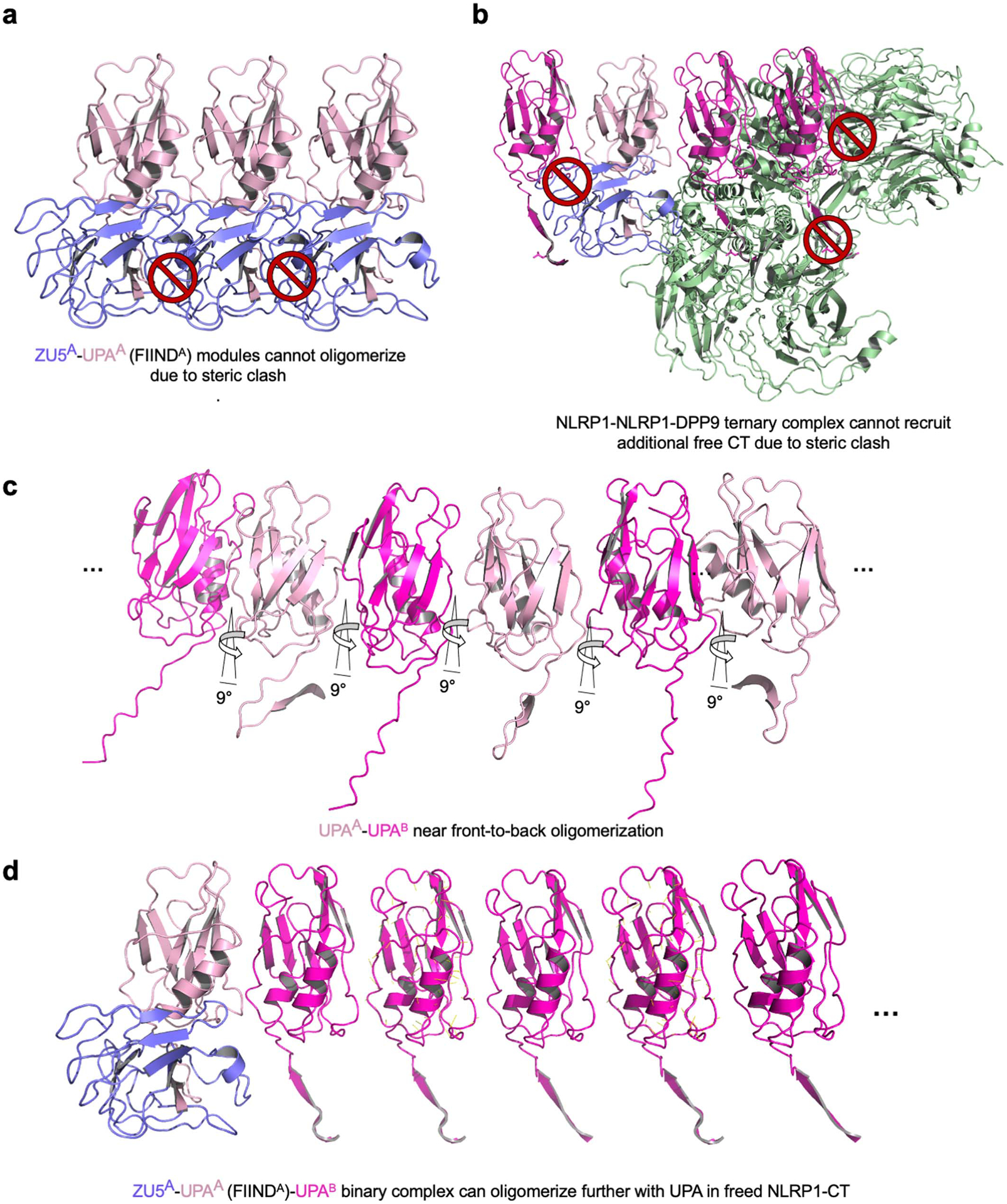
The UPA within an intact FIIND cannot self-oligomerize due to clashes with ZU5 (Extended Data Fig. 3d, 8a). In addition, freed NLRP1-CT cannot be further recruited to the ternary complex by an UPA-UPA interaction with either UPAA due to a clash with ZU5A or with UPAB due to clashes with both monomers of the DPP9 dimer (Extended Data Fig. 8b). However, the UPA domains in freed NLRP1-CTs could, in principle, oligomerize assuming that they maintain the same near front-to-back binding mode seen in the complex with DPP9 (Extended Data Fig. 8c–d). This UPA oligomerization would create a platform for CARD oligomerization, ASC recruitment, and inflammasome assembly. Thus, UPA oligomerization has two opposing functions: to mediate repression of the NLRP1-CT through DPP9 association and to facilitate inflammasome activation by promoting CARD oligomerization.
DPP9 sequesters NLRP1-CT with NLRP1-FL
Because mutations on either side of the UPAA-UPAB interface almost completely disrupted NLRP1 association with DPP9 while retaining autoprocessing (Fig. 2i), we reasoned that DPP9 binding by NLRP1-FL and NLRP1-CT is cooperative and that NLRP1-FL might dimerize with NLRP1-CT. We co-expressed FLAG-tagged NLRP1-FL S1213A that is autoproteolytically deficient and can only occupy site A with a construct for NLRP1-CT that can only occupy site B. For NLRP1-CT expression, we used an N-terminal ubiquitin (Ub) fusion (Ub-NLRP1-CT), which gets co-translationally processed30,31 to generate the native S1213 N-terminus (Fig. 4a). Indeed, NLRP1-S1213A-FL captured NLRP1-CT, and co-expression of these two components rescued DPP9 association (Fig. 4b), directly confirming that DPP9 engagement is cooperative. Addition of DPP8/9 inhibitors to this preformed ternary complex in lysates (Fig. 4b) or on beads (Extended data Fig. 9a,b) displaced DPP9, as expected. In contrast, DPP8/9 inhibitors did not disrupt the interaction between the NLRP1-CT and NLRP1-S1213A, indicating that dimerization (or oligomerization) can occur in the absence of DPP9 (Fig. 4b, Extended data Fig. 9a,b). It should be noted that, unlike the NLRP1-FLA-NLRP1-CTB-DPP9 ternary complex, the NLRP1-FLA-NLRP1-CTB binary complex should allow UPA-mediated oligomerization on one face of UPAB and thus not effectively repress NLRP1-CT inflammasome formation (Extended Data Fig. 8d).
Fig. 4. |. Repression of NLRP1-CT inflammasome activity by ternary complex formation.
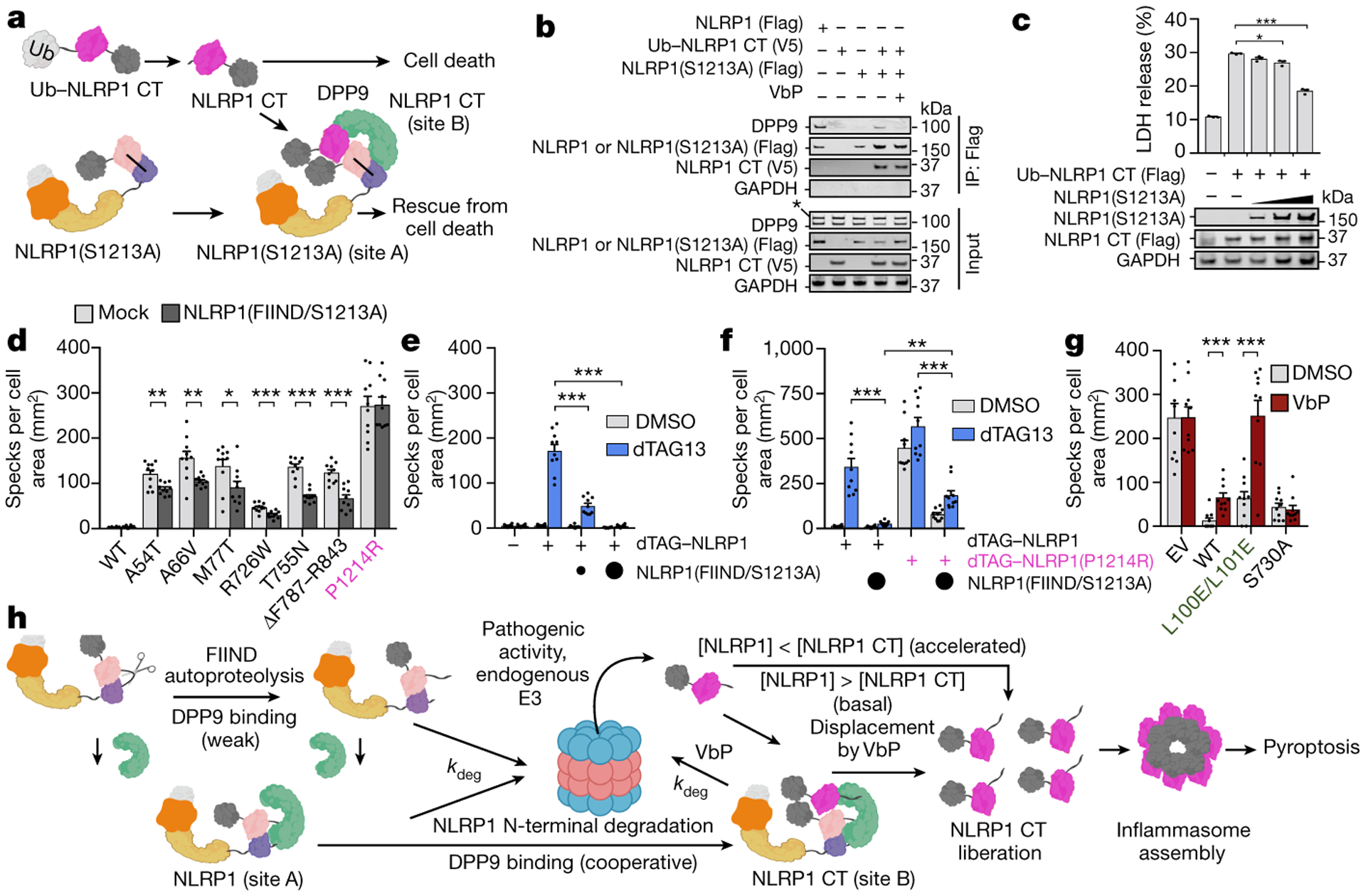
a, Schematic representation of ternary complex formation between the autocatalysis-deficient NLRP1-S1213A-FL, Ub-NLRP1-CT, and DPP9. Ub-NLRP1-CT is co-translationally processed by endogenous deubiquitinases, resulting in NLRP1-CT with the native S1213 N-terminus. b, FLAG co-immunoprecipitation using FLAG-tagged WT NLRP1-FL or S1213A-FL and V5-tagged Ub-NLRP1-CT. S1213A-FL (site A) alone did not pull down endogenous DPP9. In contrast, co-expression of the two constructs formed a ternary complex with DPP9. c, Increasing amounts of S1213A-FL suppress NLRP1-CT-induced cell death measured by LDH release in a reconstituted HEK293 inflammasome system. n=3 independent biological replicates. d, NLRP1 FIIND-SA rescues ASC specks resulting from patient mutations except for P1214R, which reduces DPP9 binding. e, NLRP1 FIIND-SA rescues ASC specks resulting from dTAG13-induced NLRP1 degradation. f, ASC speck formation and FIIND-SA rescue for the NLRP1 P1214R patient mutation. FIIND-SA partially rescues dTAG-P1214R, but rescue of dTAG-P1214R is significantly less than for wild-type (WT) dTAG-NLRP1. g, DPP9 binding and catalytic activity contribute to repression of the NLRP1 inflammasome. ASC specks from NLRP1 and the indicated DPP9 constructs expressed in DPP8/9 DKO 293T cells. (d-g) n=10 quantified fields of view. Data in (c-g) are mean ± SEM. All data are representative of > 2 independent experiments (c-d, g) *: p < 0.05, **: p < 0.01 and ***: p < 0.001 by unpaired two-sided t test, (e-f) **: p < 0.01 and ***: p < 0.001 by 2-way ANOVA with Tukey multiple comparison correction, exact p-values are presented in the Source Data file. h, Schematic of NLRP1 activation and repression by DPP9. NLRP1-FL, together with DPP9, represses some threshold of free NLRP1-CTs. Enhanced degradation of NLRP1-FL, or displacement of the NLRP1-CT, leads to inflammasome signalling.
Because sequestration of NLRP1-CT into the ternary complex should prevent UPA-UPA oligomerization (Extended Data Fig. 8b) and inhibit inflammasome formation, we posited that NLRP1-S1213A-FL co-expression would rescue inflammatory cell death mediated by the NLRP1-CT through DPP9 association. Indeed, titration of NLRP1-S1213A-FL reduced NLRP1-CT-mediated cell death in a dose-dependent manner (Fig. 4c). Similarly, NLRP1-FIIND-S1213A reduced speck formation resulting from all disease-associated NLRP1 mutations except for P1214R (Fig. 4d), a mutation that impairs the DPP9-NLRP1 interaction (Fig. 2i). We next leveraged the dTAG system to pharmacologically control the release of NLRP1-CT32. Briefly, we fused FKBP12F36V (dTAG) to the N-terminus of NLRP1 (dTAG-NLRP1), and used the small molecule dTAG13 to recruit the E3 ligase cereblon to the fusion protein and trigger NT degradation (Extended Data Fig. 9c). As expected, dTAG13 robustly induced the degradation and activation of dTAG-NLRP1, as measured by ASC speck formation and GSDMD cleavage (Fig. 4e, Extended Data Fig. 9d). Moreover, we observed dose-dependent rescue of dTAG13-induced inflammasome activation with NLRP1-FIIND-S1213A co-expression (Fig. 4e, Extended Data Fig. 9d). We next tested a dTAG-P1214R NLRP1 mutant in this system (Fig. 4f), which spontaneously formed ASC specks and was only partially rescued by NLRP1-FIIND-S1213A, consistent with disruption of its CT binding to the repressive ternary complex. Thus, DPP9 represses the inflammatory activity of NLRP1-CT in a manner that depends on the presence of NLRP1-FL. Finally, we confirmed that both DPP9’s enzymatic activity and binding repress NLRP1 activation (Fig. 4g).
Conclusion
In summary, our structural and cellular data show that NLRP1-FL and DPP9 act as a checkpoint for NLRP1 inflammasome activation by directly binding to and sequestering the inflammatory NLRP1-CT. We propose that free inflammatory NLRP1-CTs are not only generated by some as-yet-unknown danger-associated signal, but also during normal homeostatic protein turnover. Accordingly, ternary complex formation would be needed to avoid spurious inflammation from low, background levels of NLRP1-CT relative to NLRP1-FL. It is unclear what happens to these ternary complexes after they are formed, and in particular how the inflammatory NLRP1-CT fragment is turned over. Since we did not observe NLRP1-CT cleavage by DPP9 (Extended Data Fig. 6), we speculate that unknown cellular machinery might recognize the NLRP1-DPP9 ternary complex and destroy NLRP1-CT, preventing its accumulation.
How could cells overcome this important checkpoint for NLRP1 activation? In one scenario, accelerated degradation of NLRP1-FL induced by danger-associated signals (e.g., pathogen proteases or DPP8/9 inhibition) would lead to simultaneous increase of NLRP1-CT and decrease of NLRP1-FL to overwhelm this checkpoint and lead to inflammasome activation and pyroptotic cell death (Fig. 4h). Alternatively, any factor that impacts the interaction between NLRP1 and DPP9 (e.g., the P1214R mutation in NLRP1 or DPP8/9 inhibitors) would release NLRP1-CT to promote inflammasome activation (Fig. 4h). It is tempting to speculate that additional unknown endogenous- or pathogen-derived signals may act to accelerate NT degradation and/or alter ternary complex stability, and thereby regulate inflammasome formation. We expect that future studies based on our results will not only lead to greater mechanistic understanding of this long enigmatic inflammasome, but also eventually enable therapeutic modulation of NLRP1 to treat human disease.
Methods
Cell culture.
Expi293F cells were maintained in Expi293F Expression Medium (GIBCO, ThermoFisher) with constant shaking at 100 RPM, 37 °C, and 5% CO2. Sf9 insect cells were maintained in HyClone SFX-Insect Cell Media (Cytiva) supplemented with 1X antibiotic-antimycotic (ThermoFisher) at 27 °C with constant shaking at 100 RPM. HEK293T cell lines and their derivatives were maintained in DMEM (GIBCO, ThermoFisher) supplemented with 10% fetal bovine serum (GIBCO, ThermoFisher Scientific) at 37°C and 5% CO2. HEK293T (ATCC), Sf9 (ThermoFisher), and Expi293F (ThermoFisher) were authenticated by the manufacturer and frequently checked for their morphological features. HEK293T cells stably expressing GSDMD and caspase-1 were previously described33. DPP8/9 knockout HEK cells were generated in this study (below) using previously validated DPP9 KO cells21. Expi293F and Sf9 cells for protein production were not tested regularly for mycoplasma contamination. All other cell lines regularly tested negative for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza).
Constructs and cloning.
Mammalian codon-optimized full-length human NLRP1 cDNA (isoform 1, Uniprot ID Q9C000-1) was synthesized by Synbio Technologies (Source Data). Codon-optimized NLRP1 lacking the PYD and CARD (Residues W148-P1364, NLRP1ΔΔ) was subcloned into an in-house modified pcDNA3.1 LIC 6D (Addgene plasmid #30127) vector containing a C-terminal TEV linker, GFP tag, and FLAG tag (NLRP1ΔΔ-TEV-GFP-FLAG). NLRP1-FL was also cloned into pcDNA3.1 LIC 6A (Addgene plasmid #30124) with a C-terminal FLAG tag (NLRP1-FLAG). NLRP1 was also subcloned into pDONR221 (Thermo) and shuttled into pLEX 305-N-dTAG (Addgene plasmid #91797) with Gateway technology (Thermo). The short isoform of DPP9 (DPP9S, Uniprot ID Q86TI2-1) from the Harvard PlasmID Database was subcloned into pfastBac HTB (N-terminal His-TEV tag) for recombinant protein expression. DPP9 was then subcloned into pcDNA3.1 LIC 6A with either an N-terminal His- or FLAG-tag (His-TEV-DPP9 or FLAG-TEV-DPP9). Point mutations were introduced with Q5 site-directed mutagenesis (NEB) or QuikChange site-directed mutagenesis kit (Agilent). Ub-fused NLRP1 constructs were synthesized (GenScript) with an N-terminal synthetic ubiquitin sequence followed by NLRP1-CT (S1213-S1473, L1215-S1473 or L1218-S1473) and cloned into pcDNA3.1 and pLEX-307 (Addgene plasmid #41392) vectors. Study constructs are available on Addgene (https://www.addgene.org/Hao_Wu/ and https://www.addgene.org/Daniel_Bachovchin/).
Protein expression, purification, and Edman degradation.
Recombinant DPP9 was purified similarly to Ross et al., 201825. Baculoviruses containing DPP9 were prepared using the Bac-to-Bac system (Invitrogen), and used to generate baculovirus-infected insect cells. To express DPP9, 1 mL of these baculovirus-containing cells was used to infect each L of Sf9 cells. Cells were harvested 48 h after infection by centrifugation (1,682 × g, 20 min), washed once with phosphate buffered saline (PBS), flash-frozen in liquid nitrogen, and stored at −80 °C. The thawed pellet from 2 L of cells was resuspended in lysis buffer (80 mL, 25 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine abbreviated as TCEP, 5 mM imidazole), sonicated (3 s on 5 s off, 3.5 min total on, 45% power, Branson), and ultracentrifuged (186,000 × g, 1.5 h, 45 Ti fixed-angle rotor, Beckman). After centrifugation, the supernatant was incubated with 1 mL Ni-NTA resin at 4 °C for 1 h. The bound Ni-NTA beads washed once in batch and subsequently by gravity flow using 50–100 CV wash buffer (25 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM TCEP, 25 mM imidazole). The protein was eluted with buffer containing 500 mM imidazole (5 mL), spin concentrated to 0.5 mL (Amicon Ultra, 100 kDa MW cutoff), and further purified by size exclusion chromatography (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM TCEP) on a Superdex 200 increase 10/300 GL column (Cytiva). The yield was ~5 mg per L of insect cell culture.
To purify the NLRP1-DPP9 complex, expi293F cells (1 L, 2–3×106 cells/mL) were co-transfected with NLRP1ΔΔ-TEV-GFP-FLAG (0.7 mg) and the short isoform of DPP9 (His-TEV-DPP9, 0.3 mg) following incubation with polyethylenimine (3 mL, 1 mg/mL) in Opti-MEM (100 mL) for 30 min. 24 h later, cells were supplemented with glucose (9 mL, 45%) and valproic acid (10 mL, 300 mM). Cells were harvested 5 d after transfection by centrifugation (1,329 × g, 20 min), washed once with PBS, flash-frozen in liquid nitrogen, and stored at −80 °C. Later, the thawed pellet from 1 L of cells was resuspended in lysis buffer (50–100 mL, 25 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM TCEP), sonicated (2 s on 8 s off, 3.5 min total on, 40% power, Branson), and ultracentrifuged (186,000 × g, 1 h, 45 Ti fixed-angle rotor, Beckman). The soluble proteome was incubated with pre-equilibrated anti-FLAG M2 affinity gel (Sigma, 1.5 mL) for 4 h at 4 °C, washed in batch once with 5–10 column volume (CV) lysis buffer, and then washed by gravity flow with 25–50 CV lysis buffer. The NLRP1-DPP9 complex was eluted by on-column TEV protease cleavage at room temperature for 1 h using elution buffer (5 mL, 25 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.2 mM ADP, 1 mM TCEP, 0.2 mg TEV protease) and loaded onto a Mono Q 5/50 GL anion exchange column (Cytiva). Protein was eluted using a salt gradient from 150 mM to 1 M NaCl (25 mM Tris-HCl pH 8.0, 1 mM TCEP) over 15 CV. Subsequent purification steps employing size exclusion columns led to complete loss of protein, likely due to hydrophobic interactions between the protein and the column’s support. Nonetheless, the anion exchange eluent showed sufficiently homogeneous NLRP1-DPP9 complexes by negative stain electron microscopy. Mono Q eluent containing the complex was concentrated to 0.3–0.5 mg/mL (assuming ε = 1.25) using a 0.5 mL spin concentrator (Amicon Ultra, 50 kDa MW cutoff) and immediately supplemented with 0.2 mM ADP and 5 mM MgCl2. Concentrated eluent was dialyzed overnight into a HEPES buffer that is compatible with crosslinking for cryo-EM (25 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.2 mM ADP, 1 mM TCEP) using a 0.5 mL Slide-A-Lyzer (ThermoFisher). Total protein yield varied between 0.2–0.5 mg per L of mammalian culture. Purified protein was run on an SDS-PAGE gel, transferred to a PVDF membrane (iBLOT 2), and stained by Coomassie Brilliant Blue. The NLRP1-CT band was excised and sent to the Tufts University Core Facility for Edman Degradation.
Expression and purification of the NLRP1-DPP9-VbP complex was identical to NLRP1-DPP9 (described above) except for the addition of 10 μM VbP to all purification and dialysis buffers. Extended protocol including intermediary purification and quality control results are available on protocols.io (https://www.protocols.io/groups/hao-wu-lab).
Crosslinking mass spectrometry.
Amine-amine crosslinking of the purified NLRP1-DPP9 complex was performed at a final concentration of 0.24 mg/mL using either 0.5, 1, or 2 mM bis(sulfosuccinimidyl)suberate (BS3) in 50 mM HEPES pH 7.8 and 100 mM NaCl for 1 h at room temperature. The reaction was quenched with hydroxylamine to a final concentration of 100 mM. Urea was added to 5 M concentration and samples were reduced for 1 h in 10 mM TCEP and 50 mM HEPBS pH 8.3, followed by alkylation with 30 mM iodoacetamide in the dark for one hour and quenching with 50 mM β-mercaptoethanol. Samples were then diluted with 50 mM HEPBS pH 8.3 to reduce urea concentration down to 1 M and digested with trypsin (Promega) at 1:10 enzyme:substrate ratio overnight at 37°C. Digested peptides were acidified with 10% formic acid (FA) to pH ~2 and desalted using stage tips with Empore C18 SPE Extraction Disks (3M) and dried under vacuum.
Crosslinked samples were reconstituted in 5% FA/5% acetonitrile (ACN) and analysed in the Orbitrap Fusion Lumos Mass Spectrometer (ThermoFisher) coupled to an EASY-nLC 1200 (ThermoFisher) ultra-high pressure liquid chromatography (UHPLC) pump, as well as a high-field asymmetric waveform ion mobility spectrometry (FAIMS) FAIMSpro interface. Peptides were separated on an in-house column with 100 μM inner diameter and packed with 35 cm of Accucore™ C18 resin (2.6 μm, 150 Å, ThermoFisher), using a gradient consisting of 5–35% (ACN, 0.125% FA) over 135 min at ~500 nL/min. The instrument was operated in data-dependent mode. FTMS1 spectra were collected at a resolution of 120,000, with an automatic gain control (AGC) target of 5 × 105, and a max injection time of 50 ms. The most intense ions were selected for MS/MS for 1 s in top-speed mode, while switching among three FAIMS compensation voltages: −40, −60, and −80 V in the same method. Precursors were filtered according to charge state (allowed 3 <= z <= 7), and monoisotopic peak assignment was turned on. Previously interrogated precursors were excluded using a dynamic exclusion window (60 s ± 7 ppm). MS2 precursors were isolated with a quadrupole mass filter set to a width of 0.7 Th and analysed by FTMS2, with the Orbitrap operating at 30,000 resolution, an AGC target of 100,000, and a maximum injection time of 150 ms. Precursors were then fragmented by high-energy collision dissociation (HCD) at a 32% normalized collision energy.
Mass spectra were processed and searched using the PIXL search engine34. Precursor tolerance was set to 15 ppm and fragment ion tolerance to 10 ppm. Methionine oxidation was set as a variable modification in addition to mono-linked mass of +156.0786 for BS3. Crosslinker mass shift of +138.0681 was used for BS3 reagent. All crosslinked searches included 50 most abundant protein sequences to ensure sufficient statistics for false discovery rate (FDR) estimation. Matches were filtered to 1% FDR on the unique peptide level using linear discriminant features as previously described34.
Cryo-EM grid preparation and data collection.
The purified NLRP1-DPP9 complex (0.3–0.5 mg/mL, 25 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.2 mM ADP, 1 mM TCEP, ±10 μM VbP) was crosslinked with 0.02% glutaraldehyde on ice for 5 min and immediately loaded onto a glow-discharged Quantifoil grid (R1.2/1.3 400-mesh gold-supported holey carbon, Electron Microscopy Sciences), blotted for 3–5 s under 100% humidity at 4 °C, and plunged into liquid ethane using a Mark IV Vitrobot (ThermoFisher). These sample conditions were optimized extensively using insights gleaned from different small datasets collected at the Pacific Northwest Center for Cryo-EM (PNCC), the NCI’s National Cryo-EM Microscope Facility (NCEF), and the Harvard Cryo-EM Center (HMS). Final collection parameters are summarized in Extended Data Table 1.
Prior to data collection, all grids were screened for ice and particle quality. For data collection, movies were acquired at HMS using a Titan Krios microscope (ThermoFisher) at an acceleration voltage of 300 keV equipped with BioQuantum K3 Imaging Filter (Gatan, slit width 20 eV). Movies were recorded with a K3 Summit direct electron detector (Gatan) operating in counting mode at 105,000 × magnification (0.825 Å/pix).
For the NLRP1-DPP9 data set at 0° stage tilt: 7,840 movies were collected using SerialEM35 to vary the defocus range between −1 to −2.5 μm and to record two shots for each of the four holes per stage movement through image shift. All movies were exposed with a total dose of 67.5 e−/Å2 for 1.8 s fractionated over 45 frames. Orientation preference was apparent, and an ideal tilt angle of 37° was calculated from this processed dataset (below) using cryoEF36. For the NLRP1-DPP9 data set at a stage tilt of 37°: 1,916 movies were collected using SerialEM35 to vary the defocus range between −1.5 to −3.0 μm with two shots recorded for each hole per stage movement through image shift. All movies were exposed with a total dose of 67.6 e−/Å2 for 2.6 s fractionated over 45 frames.
For the NLRP1-DPP9-VbP data set at 0° stage tilt: 3,553 movies were collected using SerialEM35 to vary the defocus range between −0.8 to −2.2 μm and to record two shots for each of the four holes per stage movement through image shift. All movies were exposed with a total dose of 52.0 e−/Å2 for 2.0 s fractionated over 50 frames. Orientation preference was apparent, and an ideal tilt angle of 37° was calculated from this processed dataset (below) using cryoEF36. For NLRP1-DPP9-VbP at a stage tilt of 37°: 1,954 movies were collected using SerialEM35 to vary the defocus range between −1.5 to −3.0 μm with two shots recorded for each hole per stage movement through image shift. All movies were exposed with a total dose of 65.0 e−/Å2 for 2.4 s fractionated over 43 frames.
Cryo-EM data processing.
Data processing leveraged SBgrid Consortium37 and NSF XSEDE38 computing support (Extended Data Fig. 1, 4). Movies collected at HMS were pre-processed on-the-fly by the facility’s pipeline script. Movies were corrected by gain reference and for beam-induced motion, and summed into motion-corrected images using the Relion 3.08 implementation of the MotionCor2 algorithm39. CTFFIND440 or goCTF41 was used to determine the defocus of each micrograph (Extended Data Fig. 1, 4). These pre-processed micrographs were used for subsequent analysis.
For the NLRP1-DPP9 complex collected at 0° stage tilt, crYOLO42 (generalized training by the HMS facility) picked 1,1197,166 particles from the 7840 micrographs. Relion 3.143 was used for subsequent image processing. Initially, picked particles were subjected to multiple rounds of 2D classification until the classes appeared visually homogenous. A randomized set of 100,000 particles was selected for the de novo reconstruction of an initial model, which was low-pass-filtered to 40 Å to use as the input reference map for 3D classification. Multiple rounds of 3D classification were used to produce homogeneous particle stacks among the heterogeneous particle populations (unbound DPP9, 2:2 and 2:4 DPP9:NLRP1 complexes). One selected 3D class (81,705 particles) was further 3D refined. Subsequently, particles were symmetry expanded (C2) and 3D refined locally followed by per-particle CTFRefine and Bayesian polishing to reach an overall resolution of 3.7 Å for the NLRP1-DPP9 complex. However, the map suffered from anisotropic resolution, and cryoEF36 analysis estimated that data collected at a tilt angle of 37° degrees was ideal to fill the orientation gaps in Fourier space.
The overall processing scheme for the 37° tilt data of the NLRP1-DPP9 complex was derived from the workflow described in Relion 3.043. Template-free auto picking with crYOLO42 picked 412,419 particles from then pre-processed micrographs. After multiple rounds of 2D classification, 378,410 visually homogenous particles remained for further processing. A random subset comprising 100,000 of these particles was used for de novo initial model construction, which was low-passed filtered and used as the initial reference map for 3D classification. After multiple rounds of 3D classification, a 3D class with 89,601 particles remained. The first 3D refinement using these particles yielded an 8.9 Å resolution structure, and the Fourier shell correlation (FSC) curve showed strong fluctuations indicating imprecisions in CTF estimation. To rectify this, we utilized CTF refinement and Bayesian polishing implemented in Relion 3.1, including higher order aberrations correction, anisotropic magnification correction, and per-particle defocus estimation44. Iterative rounds of 3D refinement followed by CTF refinement and Bayesian polishing gradually improved the resolution and converged at a resolution plateau of 3.8 Å. Refined particle sets from 0° and 37° stage tilts were then merged and a final 3D refinement was performed. The merged data was reconstructed to an overall resolution of 3.6 Å with much improved FIIND density. Gold-standard FSC between half maps are shown (Extended Data Fig. 1).
A similar data processing scheme was applied to 0° and 37° stage-tilted data of the NLRP1-DPP9-VbP complex. Briefly, crYOLO picked particles were 2D and 3D classified leading to final sets composed of 118,113 and 124,781 particles at 0° and 37° stage-tilts, respectively. 3D refinement followed by one iteration of CTF refinement and Bayesian polishing led to a 3.1 Å resolution map for the non-tilt data. 37° stage-tilted particles initially resolved to 6.1 Å resolution owing to inaccuracies in CTF estimation. To overcome this issue, particles were further refined; iterative 3D refinement followed by CTF refinement (higher order aberrations correction, anisotropic magnification corrections, and per-particle defocus estimation) gradually improved the resolution. Finally, tilted particles were subjected to Bayesian polishing followed by a final round of 3D refinement, which yielded a structure at 3.3 Å resolution. These two refined and polished particle sets (0° and 37° data) were then joined and refined, yielding a final map at 2.9 Å resolution calculated with the gold-standard FSC between half maps (Extended Data Fig. 4). Local resolution estimation for all maps was calculated with ResMap45.
Atomic model building and structure representation.
The cryo-EM maps were first fit with the crystal structure of DPP9 dimer (PDB ID: 6EOQ)25. A homology model of hNLRP1-FIIND was generated with Schrodinger Prime46 using the crystal structure of the rat NLRP1-FIIND47 (Jijie Chai, personal communications) as a template. Initial model fitting in the map was performed with UCSF-Chimera48. Manual adjustment and de novo building of missing segments, rigid-body fitting, flexible fitting, and segment-based real-space refinement were performed in distinct parts of the initial model to fit in the density in Coot49 and real-space refinement in Phenix50. A few unstructured regions, including parts of the UPA subdomain, were omitted owing to poor density. The full model represents residues Asp18-Met1356 of DPP9, and ZU5 residues Phe1079-Phe1212, UPAA residues Ser1213-Val1350, and UPAB residues Ser1213-Met1356 of NLRP1.
For the VbP-bound structure, the full model represents residues Asp18-Met1356 of DPP9, and ZU5A residues Phe1079-Phe1212, UPAA residues Ser1213-Val1350. UPAB was not modelled owing to poor density. We modelled covalently linked DPP9 S730-VbP in the cryo-EM map density based on the DPP8-VbP complex structure (PDB ID: 6HP8)51 and independently validated fitting with the GemSpot pipeline52.
For both structures, interaction analysis was conducted visually and using PISA53. Structure representations were generated in UCSF-ChimeraX54 and Pymol55. Per-residue Cα-Cα distances between the NLRP1-bound and VbP-bound DPP9 structures (Extended Data Fig. 5e) were calculated with the matchmaker function in UCSF-Chimera48 and the ColorByRMSD script in Pymol55 script. Ligand interaction analysis was conducted with Maestro56. Pymol and ChimeraX session files are available on our Open Science Framework repository (https://osf.io/x7dv8/). Schematics were created with BioRender.
CRISPR/Cas9 gene editing.
5 × 105 DPP9 KO HEK293T cells stably expressing Cas921 were seeded in 6-well tissue culture dishes in 2 mL of media per well. The next day cells were transfected according to manufacturer’s instructions (FuGENE HD, Promega) with a mix of four single guide RNA (sgRNA) plasmids targeting DPP8. After 48 h of transfection, cells were transferred to a 10 cm tissue culture dish and selected with puromycin (2 μg/mL) until untransfected control cells were dead. Single cell clones were isolated by serial dilution and confirmed by genomic sequencing (Extended Data Fig. 7b).
Immunoblotting.
Samples were run on either NuPAGE™ 4 to 12%, Bis-Tris 1.0 mm, Mini Protein Gel (Invitrogen) for 30 min at 175 V or NuPAGE™ 4 to 12%, Bis-Tris, 1.0 mm, Midi Protein Gel (Invitrogen) for 45–60 min at 175 V. Gels were transferred to nitrocellulose with the Trans-Blot Turbo Transfer System (BIO-RAD). Membranes were blocked with Intercept™ (TBS) Blocking Buffer (LI-COR) for 30 min at ambient temperature, prior to incubating with primary antibody overnight at 4 °C. Blots were washed 3 times with TBST buffer prior to incubating with secondary antibody for 60 min at ambient temperature. Blots were washed 3 times, rinsed with water and imaged via Odyssey CLx (LI-COR). Primary antibodies used in this study include: DPP9 rabbit polyclonal Ab (1:1000, Abcam, Ab42080), FLAG® M2 mouse monoclonal Ab (1:1000, Sigma, F3165), GAPDH rabbit monoclonal Ab (1:1000, Cell Signaling Technology, 14C10), NLRP1-CT mouse monoclonal Ab (1:1000, R&D Systems, MAB6788), CASP1 rabbit polyclonal Ab (1:1000, Cell Signaling Technology, 2225), GSDMDC1 rabbit polyclonal Ab (1:1000, Novus, NBP2-33422), V5 rabbit polyclonal Ab (1:1000, Abcam, Ab9116), HA tag rabbit monoclonal Ab (1:1000, Cell Signaling Technology, 3724). Secondary antibodies used in this study include: IRDye 680 RD Streptavidin (1:1000, LI-COR, 926-68079), IRDye 800CW anti-rabbit (1:10000, LI-COR, 925-32211), IRDye 800CW anti-mouse (1:10000, LI-COR, 925-32210), IRDye 680CW anti-rabbit (1:10000, LI-COR, 925-68073), IRDye 680CW anti-mouse (1:10000, LI-COR, 925-68072).
Immunoprecipitation assays.
HEK293T cells were seeded at 5 × 105 cells/well in 6-well tissue culture dishes. The following day the cells were transfected with plasmids encoding the indicated FLAG-tagged protein (2 μg) with FuGENE HD according to manufacturer’s instructions (Promega). After 48 h cells were harvested and washed 3 times with PBS. Pellets were lysed in Tris-Buffered Saline (TBS) with 0.5% NP-40 using pulse sonication and centrifuged at 20,000 × g for 10 min at 4 °C. Protein concentration of the soluble proteome was determined using the DC Protein Assay kit (Bio-Rad) and adjusted to 2 mg/mL. Lysates were treated with DMSO or VbP (10 μM) for 1 h. Lysates were incubated with 20 μL of anti-FLAG-M2 agarose resin (Sigma) overnight at 4 °C. After washing 3 × 500 μL with cold PBS in microcentrifuge spin columns (Pierce), bound proteins were eluted by incubating resin with 40 μL of PBS with 150 ng/μL 3x-FLAG peptide for 1 h at 4 °C. An equal volume of 2x sample loading was added to the eluate and boiled. Immunoblots were developed with the Odyssey CLx imaging system (LI-COR).
For DPP9 mutants, DPP8/9 double knockout (DKO) HEK293T cells were seeded at 1 × 106 cells/well in 6-well tissue culture dishes. The following day cells were transfected with plasmids encoding for FLAG-tagged NLRP1 (2 μg) or the indicated DPP9 construct (2 μg) with FuGENE HD according to manufacturer’s instructions (Promega). Cells were harvested, lysed, and normalized as per above. Lysates were mixed in a one-to-one ratio of DPP9 and NLRP1 prior to treating with DMSO or VbP (10 μM) for 1 h. Assay was completed as above.
For on-bead displacement assay HEK293T cells were seeded at 3 × 106 cells in a 10 cm tissue culture dish. The following day the cells were transfected with plasmids encoding for FLAG-tagged NLRP1-S1213A (5 μg), V5-tagged DPP9 (3 μg), and MYC-tagged Ub-NLRP1-CT (2 μg) with FuGENE HD according to manufacturer’s instructions (Promega). Cells were harvested and washed 3x with PBS. Pellets were lysed in Tris-Buffered Saline (TBS) with 0.5% NP-40 using pulse sonication and centrifuged at 20,000 × g for 10 min at 4 °C. Lysates were incubated with 100 μL of anti-FLAG-M2 agarose resin (Sigma) for 2 h at 4 °C. The agarose was washed once with PBS, and subsequently split into 4 × 25 μL aliquots which were incubated with 50 μL of PBS containing DMSO, VbP (10 μM), 8J (50 μM), Bestatin methyl ester (MeBS, 10 μM) at RT for 1 h in microcentrifuge spin columns (Pierce). Displaced proteins were collected via centrifugation. The resin was washed 3x with cold PBS, followed by elution with 3x-FLAG peptide as above.
DPP9 activity assay.
A solution of substrate (1 mM Gly-Pro-AMC) was prepared in DMSO. 19 μL of PBS, recombinant DPP9 (1 nM), or the indicated cell lysate was added to a 384-well, black, clear-bottom plate (Corning) followed by 1 μL of substrate to initiate the reaction. Substrate cleavage was measured as increasing fluorescence signal recorded at ambient temperature every minute at 380-nm excitation and 460-nm emission wavelengths over a 30 min period. Activity was calculated by linear regression (Prism 7). For NLRP1 peptide assays, peptide or DMSO was added to the indicated final concentration and incubated for 30 min on ice prior to addition of GP-AMC substrate.
LDH cytotoxicity assay.
HEK293T cells stably expressing CASP1 and GSDMD were seeded at 1.25 × 105 cells/well in 12-well tissue culture dishes. The following day the cells were transfected with: (Fig. 3a,c, Extended Data Fig. 6g) plasmids encoding for ASC (0.01 μg), the indicated NLRP1 construct (0.02 μg) and RFP (to 2 μg); (Fig. 4c) plasmids encoding for ASC (0.01 μg), Ub-NLRP1-CT (0.02 μg), NLRP1-S1213A (0.25, 1, 2 μg) and RFP (to 2 μg); with FuGENE HD according to manufacturer’s instructions (Promega) per 125 μL of Opti-MEM. At 24 h cells were treated with DMSO or VbP (10 μM). 24 h later supernatants were analysed for LDH activity using the Pierce LDH Cytotoxicity Assay Kit (ThermoFisher) and lysate protein content was evaluated by immunoblotting. For NLRP1-CT experiments LDH was measured and lysates were collected 24 h post transfection.
dTAG-NLRP1 assay.
HEK293T cells stably expressing CASP1 and GSDMD were seeded at 1.25 × 105 cells/well in 12-well tissue culture dishes. The following day the cells were transfected with plasmids encoding for ASC (0.01 μg), dTAG NLRP1 (0.025 μg), NLRP1 FIIND-SA (indicated concentration) and RFP (to 1 μg) with FuGENE HD according to manufacturer’s instructions (Promega). At 24 h cells were treated with DMSO, dTAG13 (500 nM) and/or VbP (10 μM) for 6 h. Lysates were collected and protein content was evaluated by immunoblotting.
ASC puncta formation assay.
HEK293T cells were seeded at 2.5 × 105 cells/mL in 12-well tissue culture dishes. The following day the cells were transfected with: (Fig. 3b,d, Extended Data Fig. 6h) plasmids encoding for GFP-ASC (0.033 μg), the indicated NLRP1 construct (0.066 μg), and RFP (to 2 μg); (Fig. 4d) plasmids encoding for GFP-ASC (0.02 μg), the indicated NLRP1 construct (0.05 μg), NLRP1 FIIND-S1213A (0.5 μg) and RFP (to 2 μg) with FuGENE HD according to manufacturer’s instructions (Promega) per 125 μL of Opti-MEM. For the DPP9 experiment (Fig. 4g), DPP8/9 DKO HEK293T cells were seeded at 2.5 × 105 cells/mL in 12-well tissue culture dishes and transfected the following day with plasmids encoding for GFP-ASC (0.02 μg), NLRP1 (0.05 μg), NLRP1-FIIND-S1213A (0.25 μg), the indicated DPP9 (0.5 μg) and RFP (to 2 μg) with FuGENE HD according to manufacturer’s instructions (Promega) per 125 μL of Opti-MEM. After 24 h cells were treated with DMSO or VbP (10 μM) as indicated for 6 h. Nuclei were stained by treating cells with Hoechst dye (1 μL of 1 mg/ml solution). Live cell imaging was performed on a Zeiss Axio Observer.Z1 inverted wide-field microscope. For each chamber 5–15 positions were imaged on DAPI, red and green fluorescence channels at a single time point from a given experiment. Data were exported as raw .czi files and analysed using custom macro written in ImageJ/FIJI. Total cell area was estimated from RFP-positive signal, and the number of GFP-ASC specks was quantified using the “Analyze particles” function following threshold adjustment in the GFP positive images.
dTAG ASC puncta formation assay.
HEK293T cells were seeded at 2.5 × 105 cells/mL in 12-well tissue culture dishes. The following day the cells were transfected with constructs encoding for GFP-ASC (0.02 μg), dTAG-NLRP1 (0.05 μg), NLRP1-FIIND-S1213A (0.25 or 0.5 μg) and RFP (to 2 μg) with FuGENE HD according to manufacturer’s instructions (Promega) per 125 μL of Opti-MEM. After 24 h cells were treated with DMSO or dTAG13 (500 nM) as indicated for 24 h. For the dTAG-NLRP1-P1214R experiment (Fig. 4f), HEK293T cells were seeded at 2.5 × 105 cells/mL in 12-well tissue culture dishes. The following day the cells were transfected with constructs encoding for GFP-ASC (0.02 μg), the indicated dTAG construct (0.05 μg), NLRP1-FIIND-S1213A (0.5 μg) and RFP (to 2 μg) with FuGENE HD according to manufacturer’s instructions (Promega) per 125 μL of Opti-MEM. After 24 h cells were treated with DMSO or dTAG13 (500 nM) as indicated for 16 h. Cells were imaged and quantified as detailed above.
Chemical enrichment of protease substrates (CHOPS).
DPP8/9 DKO HEK293T cells were seeded at 1 × 106 cells/well in 6-well tissue culture dishes. The following day cells were transfected with plasmids encoding for FLAG-tagged NLRP1 (2 μg) with FuGENE HD according to manufacturer’s instructions (Promega). After 48 h, cells were harvested, washed 3x with PBS, lysed in PBS with pulse sonication, and centrifuged at 20,000 × g for 10 min at 4 °C. Protein concentration of the soluble proteome was determined using the DC Protein Assay kit (Bio-Rad) and adjusted to 1.5 mg/mL.
250 μL of lysate was incubated with either PBS or recombinant DPP9 (rDPP9, ~3 μg). The activity of rDPP9 was confirmed by GP-AMC assay prior to its addition. The mixtures were allowed to incubate for 16 h at 37 °C, after which samples were boiled for 10 min to deactivate rDPP9. 2PCA-biotin probe was added to lysates (10 mM final concentration) and incubated with shaking at 37 °C for an additional 16 h. Excess probe was removed by buffer exchange into fresh PBS with Amicon Ultra-0.5 mL Centrifugal Filters, 10k MW cutoff (3 × 500 μL). SDS was added to 1% and samples were boiled for 10 min, prior to diluting further with 5 mL of PBS. 100 μL of high capacity neutravidin agarose resin (Pierce) was added, before rotating tubes end-over-end for 1 h at room temperature. Samples were centrifuged at 1,000 × g for 2 min, supernatant was removed and beads were washed with 10 mL of PBS. This was step was repeated for a total of 3 times. Bound proteins were eluted by boiling the resulting neutravidin resin in 100 μL of 2x loading dye and evaluated by immunoblot.
Data availability
Extended protein purification protocols are available on Protocols.io (https://www.protocols.io/groups/hao-wu-lab). Raw cryo-EM data are available on EMPIAR under the accession numbers EMPIAR-10594 (NLRP1-DPP9) and EMPIAR-10595 (NLRP1-DPP9-VbP). The cryo-EM maps are available on the Electron Microscopy Data Bank under the accession numbers EMD-22074 (NLRP1-DPP9) and EMD-22075 (NLRP1-DPP9-VbP). The atomic coordinates are available on the Protein Data Bank under the accession numbers 6X6A (NLRP1-DPP9) and 6X6C (NLRP1-DPP9-VbP). Pymol session files and the image analysis macro are available on OSF [http://doi.org/10.17605/OSF.IO/X7DV8]. All other data can be obtained from the corresponding authors upon reasonable request.
Extended Data
Extended Data Fig. 1 |. Structure determination of the NLRP1-DPP9 complex.
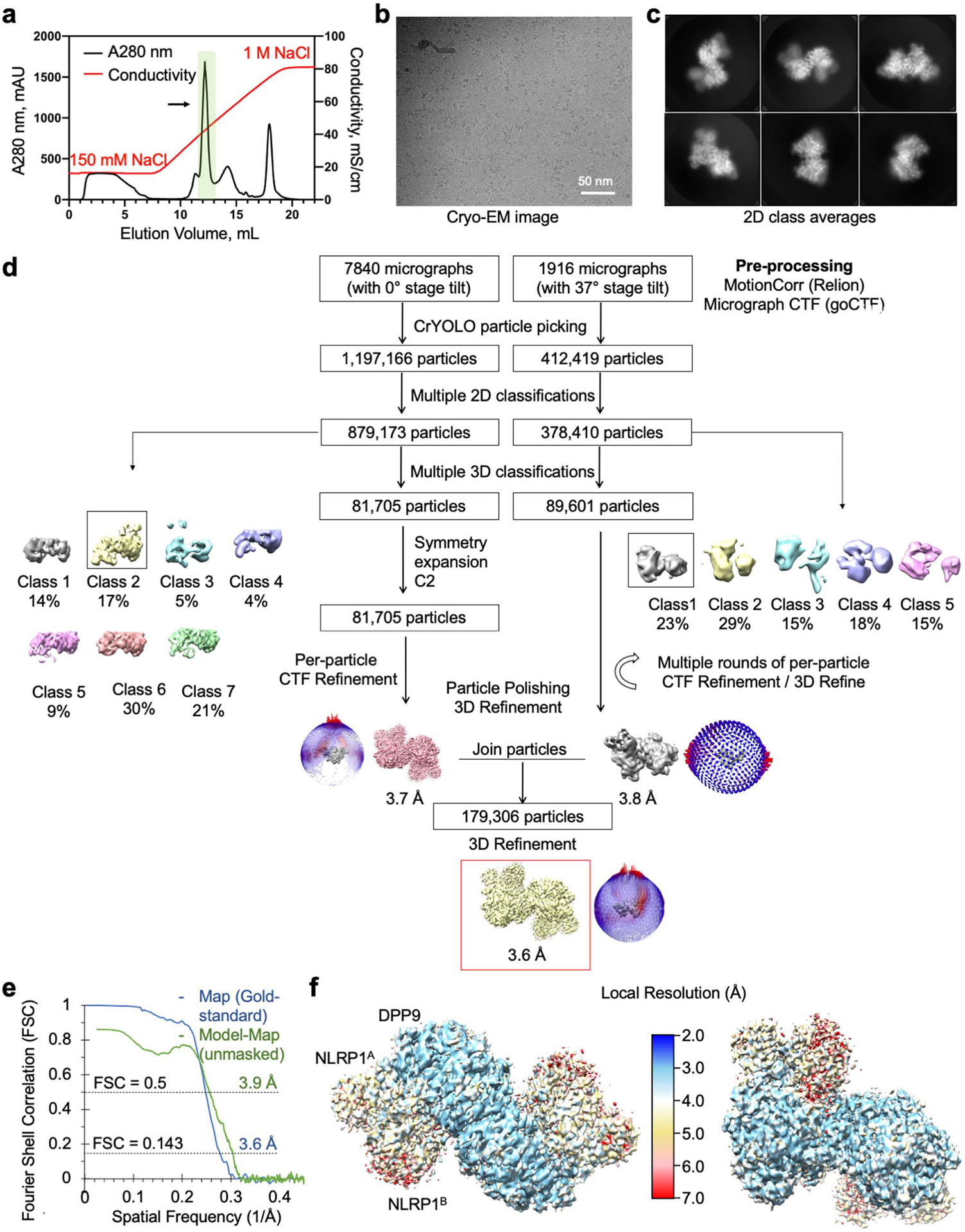
a, Purification of the NLRP1-DPP9 complex by ion exchange chromatography. The ternary complex peak is shaded in green and labelled with an arrow. b, A representative (>1000 images) cryo-EM micrograph. c, Representative 2D class averages. d, Workflow for the NLRP1-DPP9 complex structure determination. e, Map-map and map-model FSC curves. f, Local resolution distribution of the final map calculated with ResMap45 and coloured as indicated.
Extended Data Fig. 2 |. Crosslinking mass spectrometry analysis of the NLRP1-DPP9 complex.
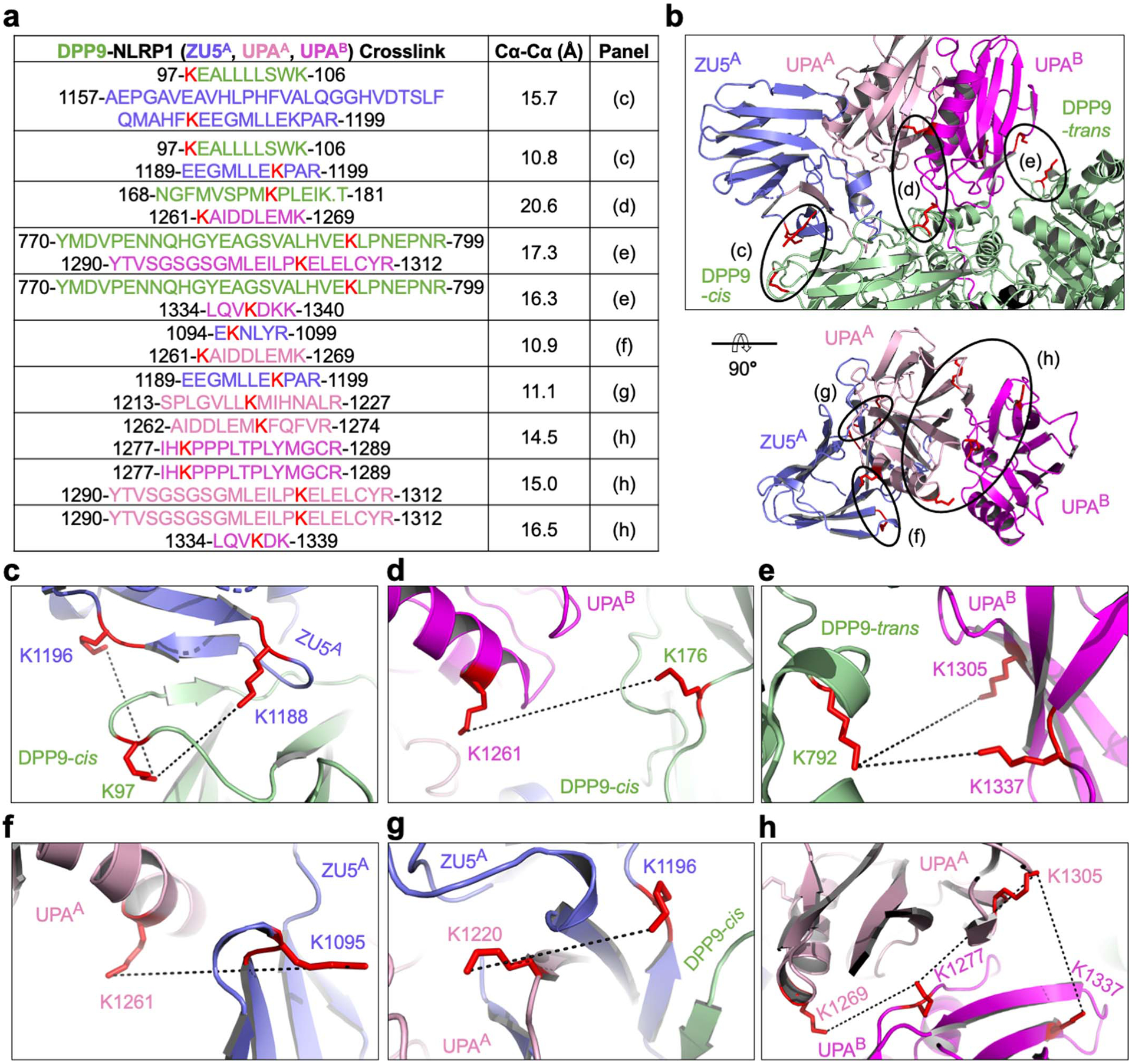
a, Summary of BS3 crosslinking between DPP9 and NLRP1. High confidence crosslinked peptides are displayed, with residue ranges labelled and colours coded by domains. Crosslinked lysine pairs are indicated in red. Cα-Cα distances between these lysine residues interpreted by the final NLRP1-DPP9 model are shown, along with the figure panel names for their detailed depictions. All detected peptide pairs are tabulated in the Source Data file. b, Overview of BS3-mediated crosslinks. c-h, Zoom-ins highlighting crosslinked lysine pairs (red) interpreted by the final NLRP1-DPP9 model.
Extended Data Fig. 3 |. Sequence and structural analysis of FIIND.
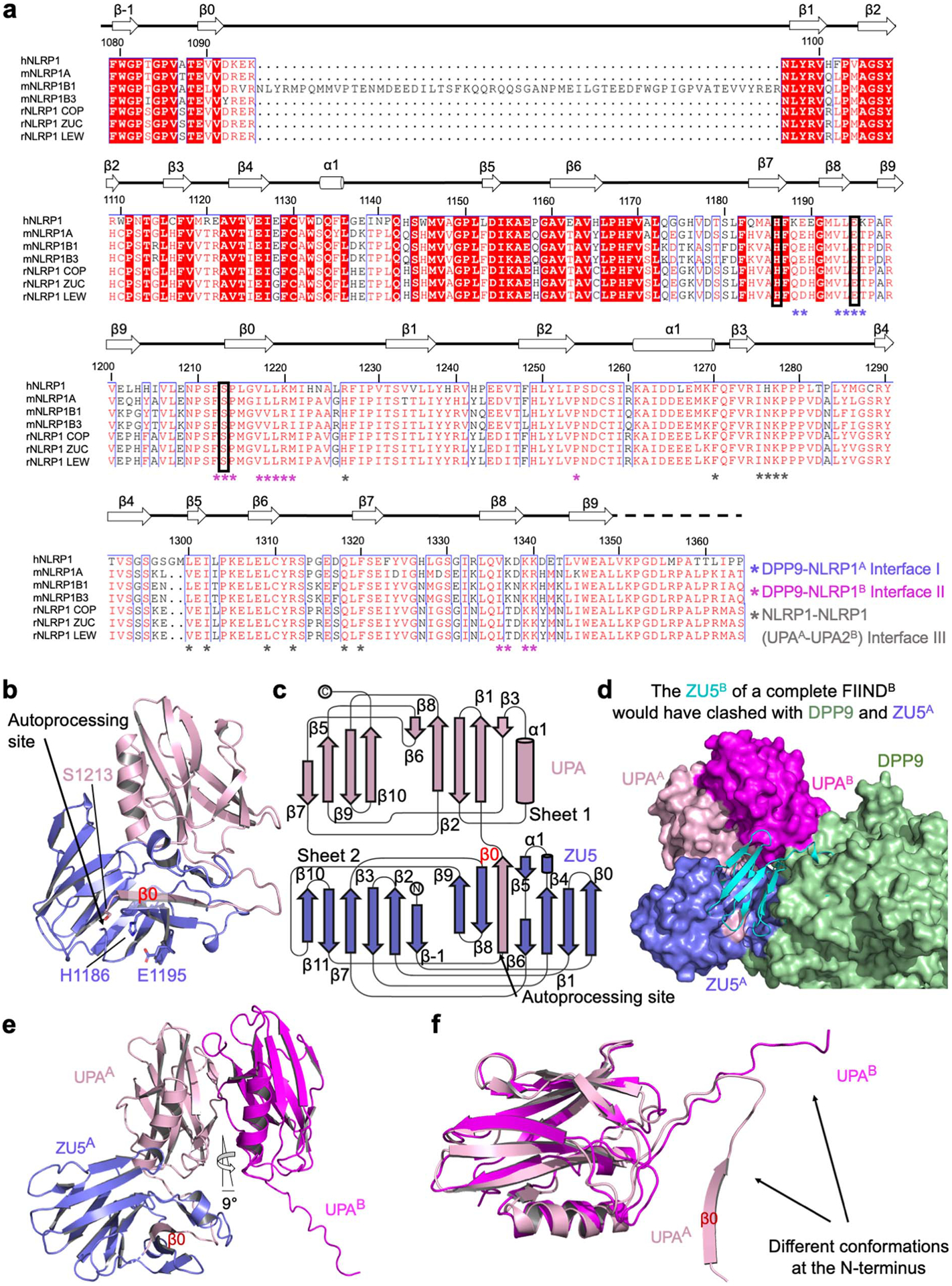
a, ClustalW multiple sequence alignment between human NLRP1 (hNLRP1), mouse NLRP1 (mNLRP1, different isoforms), and rat NLRP1 (rNLRP1, different isoforms). COP: Copenhagen; ZUC: Zucker; LEW: Lewis; SD: Sprague Dawley; and CDF: Fischer. Secondary structures and residue numbers are denoted based on the human FIINDA structure in the NLRP1-DPP9 ternary complex. Interfacial residues in the NLRP1-DPP9 complex are annotated with asterisks, and residues in the catalytic triad (H1186, E1195, S1213) are boxed in black. b, FIINDA overview with ZU5 (blue) and UPA (light pink) subdomains. The catalytic triad residues (H1186, E1195, and S1213) are shown in sticks. c, Topology of the FIIND with secondary structures labelled. d, Superimposition of FIINDA onto the UPAB. NLRP1B must be free NLRP1-CT because a ZU5 subdomain at site B would have clashed with ZU5 and UPA at site A and with DPP9. e, The ZU5A-UPAA-UPAB module that binds DPP9. UPAA and UPAB interact with each other in a front-to-back manner with only a 9° rotation between them. f, Altered conformation of the UPAB N-terminus that binds in the DPP9 active site tunnel in comparison to UPAA in a complete FIINDA.
Extended Data Fig. 4 |. Structure determination of the NLRP1-DPP9 complex with VbP.
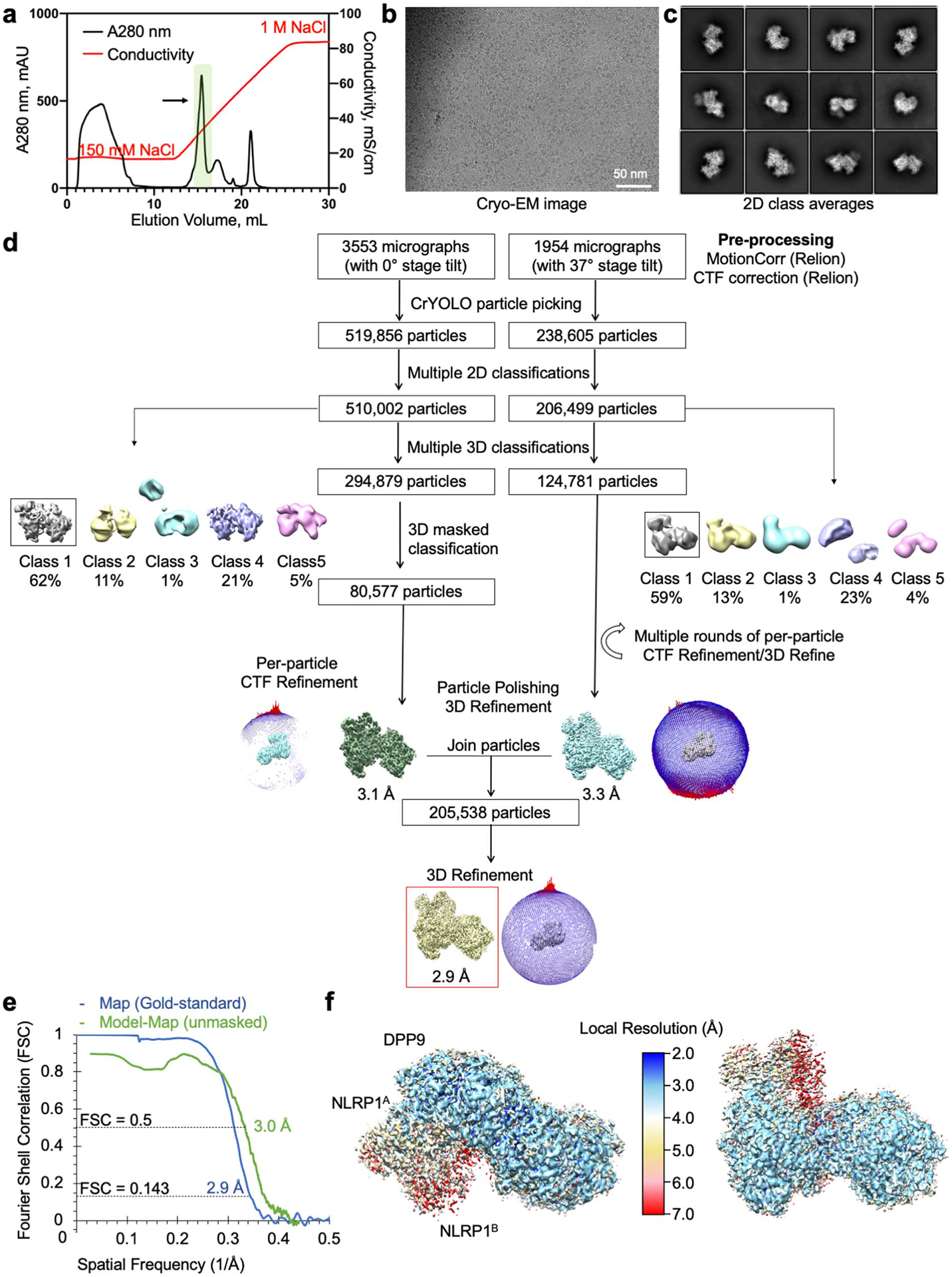
a, Purification of the NLRP1-DPP9 complex in the presence of VbP by ion exchange chromatography. The ternary complex peak is shaded in green and labelled with an arrow. b, A representative (>1000 images) cryo-EM micrograph. c, Representative 2D class averages. d, Workflow for the NLRP1-DPP9-VbP complex structure determination. e, Map-map and map-model FSC curves. f, Local resolution distribution of the final map calculated with ResMap45 and coloured as indicated.
Extended Data Fig. 5 |. VbP interactions in the DPP9 active site and comparison to a DPP substrate and NLRP1.
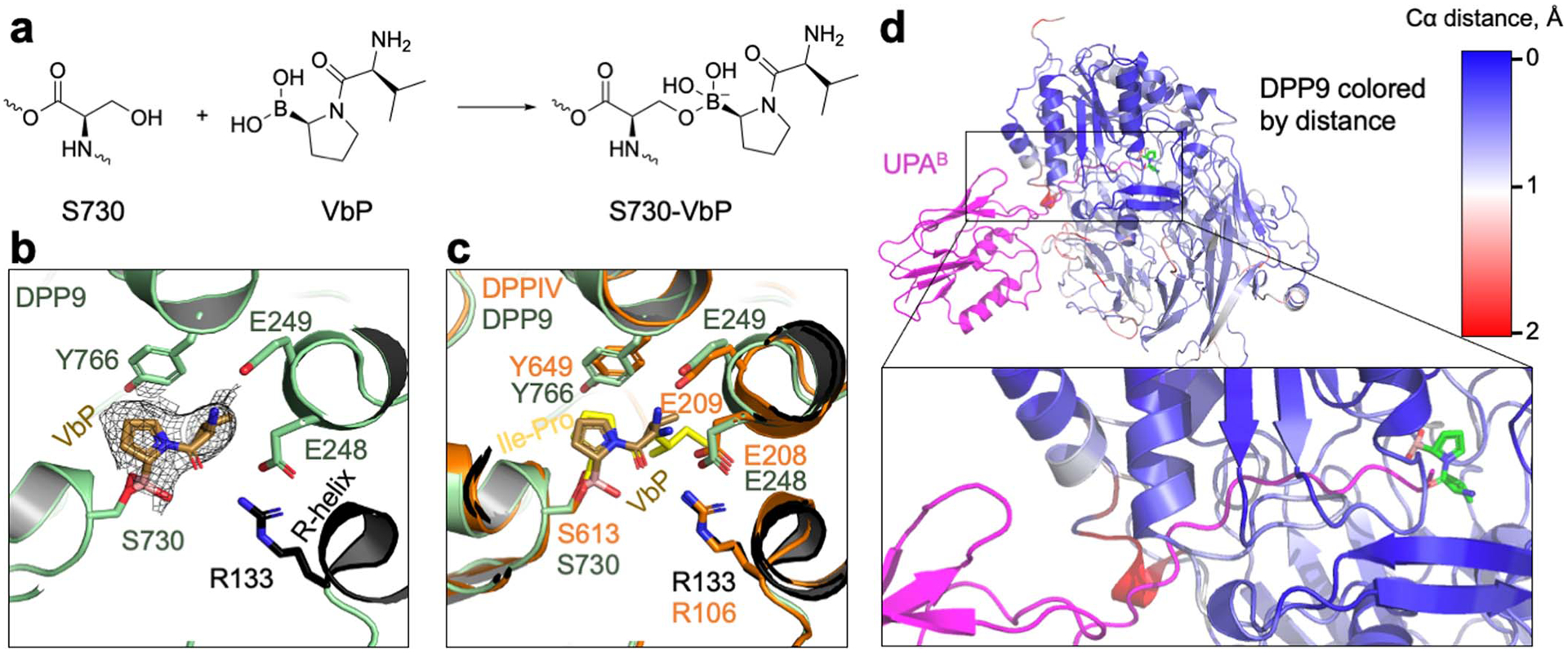
a, Schematic of covalent linkage between DPP9’s S730 and VbP. b, Fit of VbP into the cryo-EM density. VbP is shown in stick with carbon atoms in light brown. The charged amino group of VbP interacts with the DPP9 EE loop which also coordinates a substrate N-terminus, and the carbonyl oxygen of VbP interacts with R133 of the R-helix. The covalent linkage of VbP with S730, the catalytic serine, is displayed. c, Structural alignment of the VbP-bound DPP9 model (green) and the crystal structure of bacterial DPP4 bound to the substrate Ile-Pro (PDB ID: 5YP3, orange)26. VbP assumes a pose remarkably like a model substrate. d, NLRP1-CT-DPP9 complex in which DPP9 is coloured by Cα-Cα distances between NLRP1-bound and VbP-bound structures as indicated. A distance scale bar is shown, and VbP is displayed in sticks to mark the active site, with carbon atoms in green, oxygen atoms in red, nitrogen atoms in blue, and boron atoms in orange. NLRP1-CT (UPA) is shown in magenta.
Extended Data Fig. 6 |. Lack of cleavage of intact NLRP1-CT but the cleavage of its isolated N-terminal peptide by DPP9.
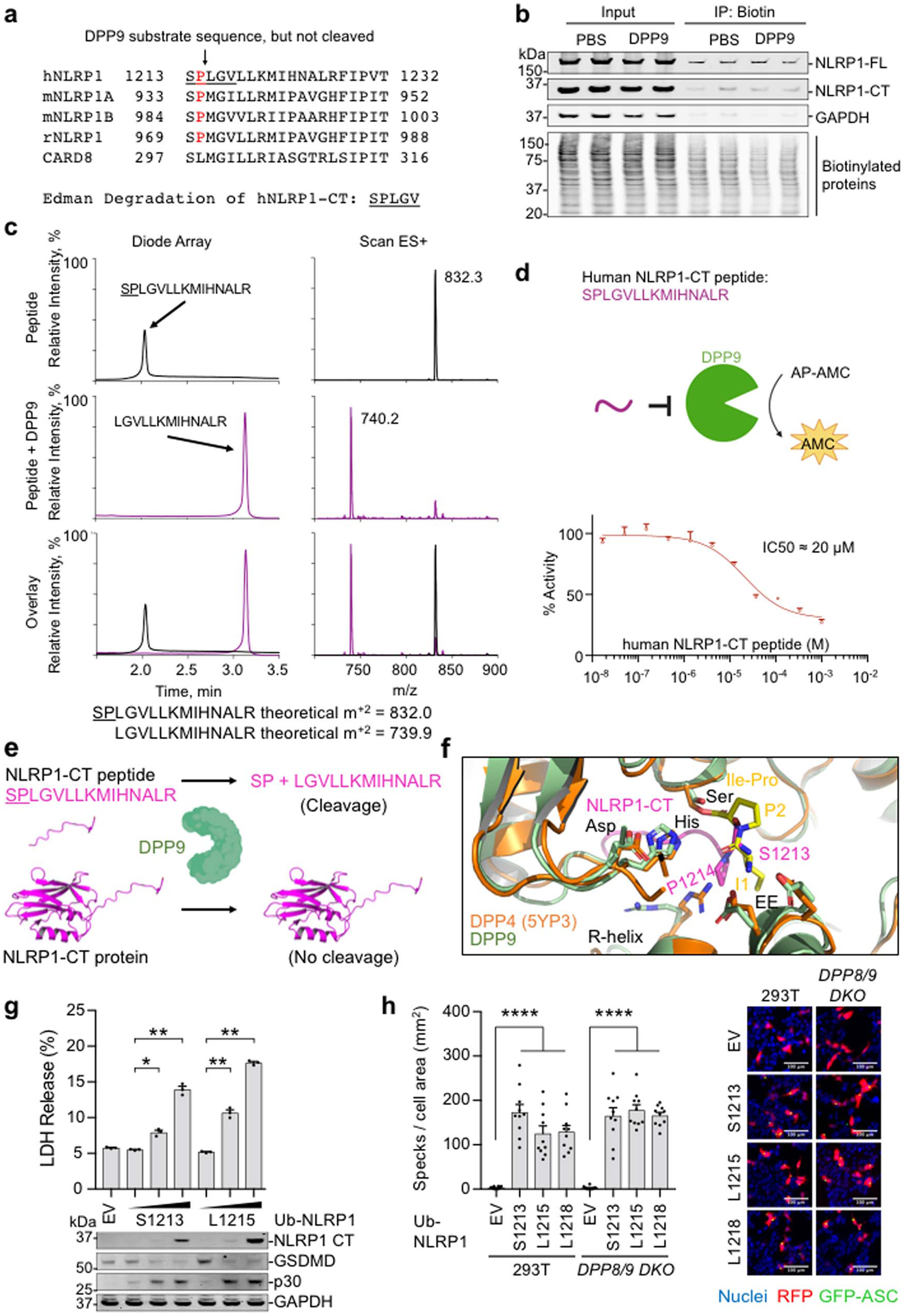
a, N-terminal sequencing of the purified NLRP1-DPP9 complex showing that the NLRP1-CT is not cleaved by the co-expressed DPP9. b, Chemical enrichment of protease substrates (CHOPS) assay showing that DPP9 does not cleave NLRP1-CT. Briefly, WT NLRP1 expressed in DPP8/9 DKO HEK293T cells was incubated with PBS or recombinant DPP9 prior to labelling with a 2-pyridinecarboxaldehyde (2PCA)-biotin probe which selectively biotinylates free N-termini except those with a proline in the second position, followed by capture of biotinylated proteins. The inputs and the eluents were analysed by immunoblots using anti-NLRP1-CT (NLRP1-FL and NLRP1-CT), anti-GAPDH, and anti-Streptavidin (biotinylated proteins) antibodies. DPP9 treatment did not increase the biotinylation of NLRP1-CT, as would be expected after the removal of the N-terminal Ser-Pro dipeptide. c, Evidence of cleavage of the isolated 15 residue N-terminal peptide in NLRP1-CT by recombinant DPP9 from mass spectrometry analysis. d, Inhibition of DPP9 catalytic activity against AP-AMC by the isolated NLRP1-CT peptide. e, Schematic illustrates the ability of DPP9 to cleave an isolated UPA N-terminal peptide, but not dipeptides from an intact NLRP1-CT. f, Comparison of the binding modes of the UPAB N-terminal peptide in the NLRP1-DPP9 complex and the Ile-Pro dipeptide in an acyl-enzyme intermediate26. g, Theoretical dipeptide cleavage does not dampen NLRP1 inflammasome activity by LDH release or inflammasome signalling. n=3 independent biological replicates. Data are mean ± SEM. Anti-FLAG (NLRP1-CT), anti-GSDMD, and anti-GAPDH antibodies were used in the immunoblots. p30: GSDMD N-terminal fragment from caspase-1 cleavage. h, Theoretical dipeptide cleavage does not dampen NLRP1 inflammasome activity by ASC speck formation. n = 10 quantified fields of view. Data are mean ± SEM. Right, representative superimposed images of nuclei (blue), RFP (red) and GFP-ASC (green). All data are representative of > 2 independent experiments. *: p < 0.05, **: p < 0.01, ***: p < 0.001 and ****: p < 0.0001 by unpaired two-sided t test, exact p-values are presented in the Source Data file.
Extended Data Fig. 7 |. Mutational analysis of the interactions in the NLRP1-DPP9 ternary complex.
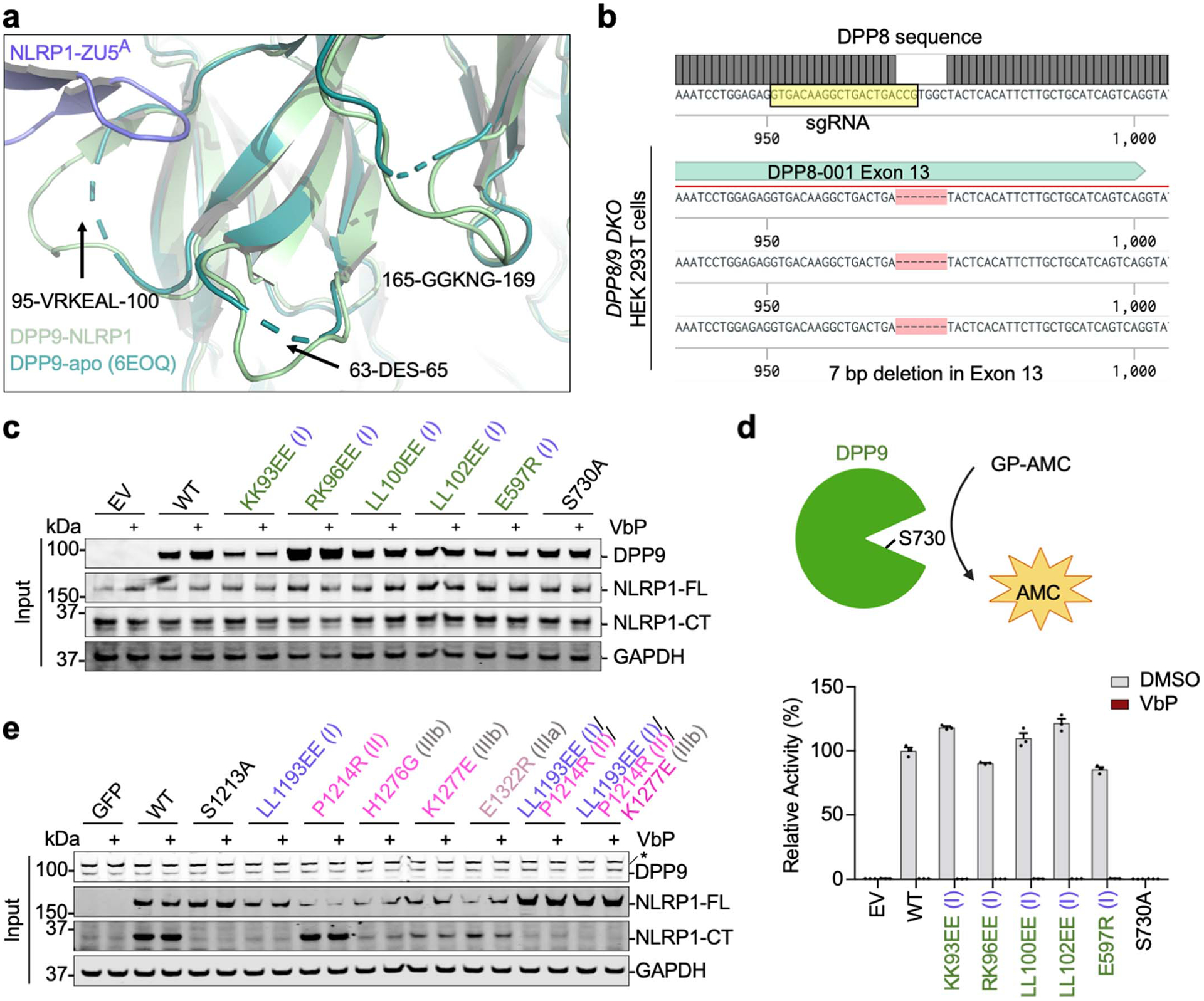
a, Disorder to order transition of several DPP9 surface loops from the isolated DPP9 crystal structure (PDB ID: 6EOQ)25 to the NLRP1-bound DPP9 cryo-EM structure. b, Genomic confirmation of DPP8 KO generated in DPP9 KO HEK293T cells stably expressing Cas921 to create DPP8/9 DKO HEK293T cells. Single guide RNA (sgRNA) sequence is highlighted. c, Immunoblots of the input lysates for the FLAG co-immunoprecipitation with WT or mutant DPP9 and WT NLRP1-FLAG, related to Fig. 2h. Anti-DPP9, anti-NLRP1 (NLRP1-FL, NLRP1-CT), and anti-GAPDH antibodies were used in the immunoblots. d, Cleavage rate of a model DPP9 substrate, GP-AMC, by WT DPP9 and its structure-guided mutants. Only the catalytically dead mutant S730A disrupts catalytic activity and sensitivity to the DPP9 inhibitor, VbP. n=3 technical replicates. Data are mean ± SEM. e, Immunoblots of the input lysates for the FLAG co-immunoprecipitation with WT or mutant NLRP1-FLAG and WT DPP9, related to Fig. 2i. Anti-DPP9, anti-NLRP1 (NLRP1-FL, NLRP1-CT), and anti-GAPDH antibodies were used in the immunoblots. All data are representative of > 2 independent experiments.
Extended Data Fig. 8 |. The ZU5 domain and DPP9 sterically hinder UPA polymerization.
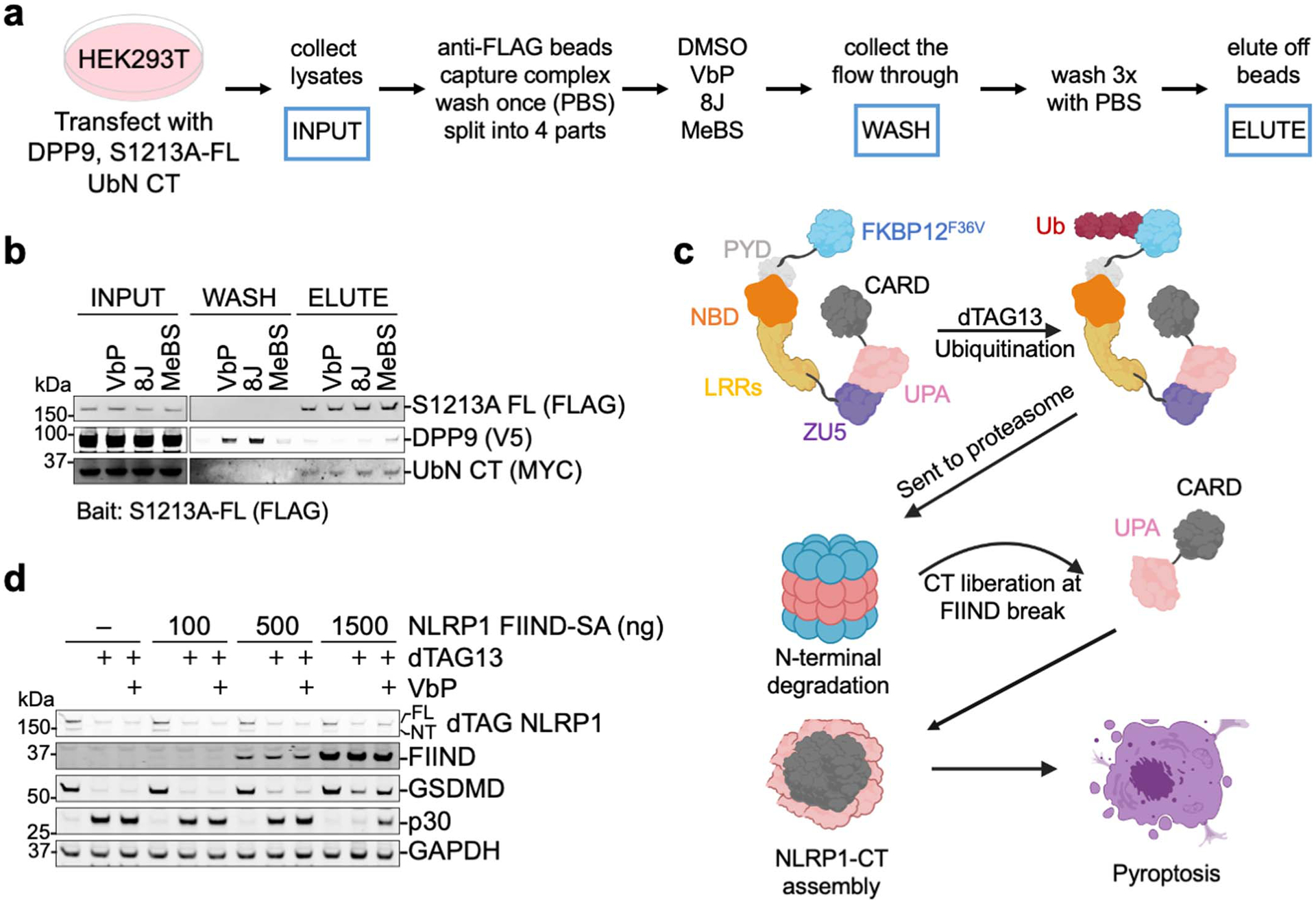
a, Modelling of a FIIND polymer using the observed UPAA-UPAB relationship. Adjacent ZU5 molecules would clash, suggesting that UPA polymerization cannot occur in complete FIIND. b, Modelled recruitment of free UPA adjacent to UPAA and UPAB in the ternary complex with DPP9. The additional UPA subdomain next to FINDA clashes with the ZU5 subdomain, and the additional UPA next to UPAB clashes with both DPP9 monomers in the complex, suggesting that DPP9 inhibits UPA oligomerization. c, A modelled UPA oligomer based on the near front-to-back interaction in the NLRP1-DPP9 ternary complex. In the model, the N-terminal tails of free UPAs are shown in either the UPAA (pink) or UPAB (magenta) conformation in complex with DPP9, but in reality, this conformation is likely to be different. d, Modelling of a UPA oligomer formed on one side of a FIINDA-CTB complex. The NLRP1-FIINDA-NLRP1-CTB binary complex can polymerize with freed NLRP1-CT. a-d, DPP9 is coloured in green, and NLRP1 domains are coloured as indicated.
Extended Data Fig. 9 |. VbP displaces NLRP1 from DPP9 in vitro and in cells.
a, Schematic of the on-bead displacement experiment. The DPP9-ternary complex is expressed in 293T cells, which are then lysed and incubated with FLAG beads. Once bound, beads are split equally and washed with compounds or DMSO. The remainder of the protein is eluted off of the beads. MeBS, Bestatin methyl ester. b, Two structurally distinct VbP inhibitors, VbP and 8J, displace DPP9 from NLRP1-S1213A by the on-bead displacement assay. Anti-FLAG (S1213A-FL), anti-MYC (NLRP1-CT), and anti-V5 (DPP9) antibodies were used in the immunoblots. Representative of 2 independent experiments. c, Schematic of the dTAG experiment. FKBP12 with the F36V mutation (dTAG) is fused to the N-terminus of NLRP1-FL. The dTAG13 ligand recruits an E3-ligase to FKBP12-F36V, leading to its ubiquitination and N-terminal degradation of the fusion protein. NLRP1-CTs (UPA-CARDs) resulting from FIIND autoprocessing are released to assemble the inflammasome. d, NLRP1 FIIND-SA expression in reconstituted HEK293T inflammasome system rescues GSDMD cleavage resulting from dTAG13-induced NLRP1 degradation. VbP prevents GSDMD rescue without inducing additional NLRP1 degradation. Anti-HA (dTAG-NLRP1-FL, dTAG-NLRP1-NT), anti-FLAG (FIIND-SA), anti-GSDMD, and anti-GAPDH antibodies were used in the immunoblots. p30: GSDMD N-terminal fragment from caspase-1 cleavage. Representative of 2 independent experiments.
Extended Data Table 1.
Cryo-EM data collection and refinement statistics of NLRP1-DPP9
| NLRP1-DPP9 | NLRP1-DPP9-VbP | |||
|---|---|---|---|---|
| Stage tilt (°) | 0° | 37° | 0° | 37° |
| Movies collected | 7,840 | 1,916 | 3,553 | 1,954 |
| Magnification | 105,000 | 105,000 | 105,000 | 105,000 |
| Voltage (kV) | 300 | 300 | 300 | 300 |
| Electron exposure (e−/Å2) | 67.54 | 67.6 | 52 | 65 |
| Defocus range (μm) | −1.0 to −2.5 | −1.5 to −3.0 | −0.8 to −2.2 | −1.5 to −3.0 |
| Exposure time (s) | 1.8 | 2.6 | 2.0 | 2.4 |
| Pixel size (Å) | 0.825 | 0.825 | 0.825 | 0.825 |
| Symmetry imposed | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 758,461 | 398,698 | 1,1609,585 | 239,416 |
| Merged Data | Merged Data | |
|---|---|---|
| Final particle images (no.) | 179,306 | 205,538 |
| Map resolution (Å) | 3.6 | 2.9 |
| FSC threshold | 0.143 | 0.143 |
| Refinement | ||
| Model resolution (Å) | 3.9 | 2.9 |
| FSC threshold | 0.5 | 0.5 |
| Map sharpening B factor (Å2) | −81.4 | −49.0 |
| Model composition | ||
| Non-hydrogen atoms | 20,269 | 15,806 |
| Protein residues | 2,524 | 1964 |
| Ligands | 2 | |
| B factors (Å2) | 110.1 | 82.7 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.011 | 0.008 |
| Bond angles (°) | 1.327 | 1.363 |
| Validation | ||
| MolProbity score | 1.93 | 1.88 |
| Clashscore | 8.55 | 7.21 |
| Poor rotamers (%) | 0.00 | 0.00 |
| Ramachandran plot | ||
| Favoured (%) | 92.1 | 93.3 |
| Allowed (%) | 7.7 | 6.5 |
| Disallowed (%) | 0.1 | 0.2 |
Supplementary Material
Acknowledgements.
We thank Dr. Tian-min Fu of the Wu lab for initiating an apo-NLRP1 project and all members of the Wu and Bachovchin labs for helpful discussions. We thank Dr. Jijie Chai for sharing their crystal structure of the rat NLRP1 FIIND prior to submission, Dr. Min Su for his advice on running goCTF and CTF estimation for tilt datasets, Dr. Maria Ericsson at the HMS EM facility for advice and training, and Drs. Richard Walsh, Sarah Sterling, and Shaun Rawson at the Harvard Cryo-EM Center for Structural Biology for cryo-EM training and productive consultation. We also thank Drs. Georgios Skiniotis and Mike Robertson for sharing their GemSpot script and for troubleshooting advice. We thank Dr. Thomas Edwards and the National Cancer Institute’s National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research under contract HSSN261200800001E. We also thank Dr. Janette Myers and the Pacific Northwest Center for Cryo-EM at Oregon Health & Science University for screening and preliminary dataset collection, under the NIH grant U24GM129547 and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. The pcDNA3.1 LIC 6A and pcDNA 3.1 LIC 6D plasmids were gifts from Scott Gradia (Addgene plasmids #30124 and #30127). The pLEX_305-N-dTAG plasmid was a gift from James Bradner & Behnam Nabet (Addgene plasmid #91797). The pLEX_307 plasmid was a gift from David Root (Addgene plasmid #41392). Our research used software and computing support at SBGrid and the Extreme Science and Engineering Discovery Environment (XSEDE) Bridges at the Pittsburgh Supercomputing Center through allocation TG-MCB190086. This work was supported by the National Institutes of Health and the Cancer Research Institute (DP1 HD087988 and R01 AI124491 to H.W., T32-GM007726 to L.R.H., the CRI Irvington Postdoctoral Fellowship to P.F., R01GM132129 to J.A.P, R01GM67945 to S.P.G., T32 GM007739-Andersen and F30 CA243444 to A.R.G., and the Memorial Sloan Kettering Cancer Center Core Grant P30 CA008748 and R01 AI137168 to D.A.B.). We apologize to authors whose work could not be cited because of space limitation.
Footnotes
Competing interests. H.W. is a co-founder of Ventus Therapeutics. The other authors declare no competing interests.
References
- 1.Martinon F, Burns K & Tschopp J The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol. Cell 10, 417–426 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Shen C, Sharif H, Xia S & Wu H Structural and mechanistic elucidation of inflammasome signaling by cryo-EM. Curr. Opin. Struct. Biol 58, 18–25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitchell PS, Sandstrom A & Vance RE The NLRP1 inflammasome: new mechanistic insights and unresolved mysteries. Curr. Opin. Immunol 60, 37–45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taabazuing CY, Griswold AR & Bachovchin DA The NLRP1 and CARD8 inflammasomes. Immunol. Rev, 297, 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soler VJ et al. Whole exome sequencing identifies a mutation for a novel form of corneal intraepithelial dyskeratosis. J. Med. Genet 50, 246–254 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong FL et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 167, 187–202.e117 (2016). [DOI] [PubMed] [Google Scholar]
- 7.D’Osualdo A et al. CARD8 and NLRP1 Undergo Autoproteolytic Processing through a ZU5-Like Domain. PLoS One 6, e27396 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finger JN et al. Autolytic Proteolysis within the Function to Find Domain (FIIND) Is Required for NLRP1 Inflammasome Activity. J. Biol. Chem 287, 25030–25037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frew BC, Joag VR & Mogridge J Proteolytic Processing of Nlrp1b Is Required for Inflammasome Activity. PLoS Pathog. 8, e1002659 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chui AJ et al. N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandstrom A et al. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364, eaau1330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhong FL et al. Human DPP9 represses NLRP1 inflammasome and protects against auto-inflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem 293, 18864–18878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vasconcelos NM et al. DPP8/DPP9 inhibition elicits canonical Nlrp1b inflammasome hallmarks in murine macrophages. Life Sci. Alliance 2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okondo MC et al. Inhibition of Dpp8/9 Activates the Nlrp1b Inflammasome. Cell Chem. Biol 25, 262–267.e265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell PS, Sandstrom A & Vance RE The NLRP1 inflammasome: new mechanistic insights and unresolved mysteries. Curr. Opin. Immunol 60, 37–45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia S, Hollingsworth LR IV & Wu H in Cell Survival and Cell Death (eds Newton K, Murphy JM, & Miao EA) (CSHL Press, Cold Spring Harb Perspect Biol, 2019). [Google Scholar]
- 17.Okondo MC et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol 13, 46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gai K et al. DPP8/9 inhibitors are universal activators of functional NLRP1 alleles. Cell Death Dis. 10, 587 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griswold AR et al. A Chemical Strategy for Protease Substrate Profiling. Cell Chem. Biol 26, 901–907.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geiss-Friedlander R et al. The cytoplasmic peptidase DPP9 is rate-limiting for degradation of proline-containing peptides. J. Biol. Chem 284, 27211–27219 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griswold AR et al. DPP9’s Enzymatic Activity and Not Its Binding to CARD8 Inhibits Inflammasome Activation. ACS Chem. Biol 14, 2424–2429 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chui AJ et al. Activation of the CARD8 Inflammasome Requires a Disordered Region. Cell Rep. 33, 108264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang R et al. Autoinhibition of UNC5b revealed by the cytoplasmic domain structure of the receptor. Mol. Cell 33, 692–703 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Yu C, Ye F, Wei Z & Zhang M Structure of the ZU5-ZU5-UPA-DD tandem of ankyrin-B reveals interaction surfaces necessary for ankyrin function. Proc. Natl. Acad. Sci. USA 109, 4822–4827 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross B et al. Structures and mechanism of dipeptidyl peptidases 8 and 9, important players in cellular homeostasis and cancer. Proc. Natl. Acad. Sci. USA 115, E1437–E1445 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roppongi S et al. Crystal structures of a bacterial dipeptidyl peptidase IV reveal a novel substrate recognition mechanism distinct from that of mammalian orthologues. Sci. Rep 8, 2714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grandemange S et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis 76, 1191–1198 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Hollingsworth L et al. Mechanism of filament formation in UPA-promoted CARD8 and NLRP1 inflammasomes. Nat. Commun 12, 189 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong Q et al. Structural basis for distinct inflammasome complex assembly by human NLRP1 and CARD8. Nat. Commun 12, 188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bachmair A, Finley D & Varshavsky A In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186 (1986). [DOI] [PubMed] [Google Scholar]
- 31.Baker RT & Varshavsky A Inhibition of the N-end rule pathway in living cells. Proc. Natl. Acad. Sci. USA 88, 1090–1094 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nabet B et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson DC et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med 24, 1151–1156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mintseris J & Gygi SP High-density chemical cross-linking for modeling protein interactions. Proc. Natl. Acad. Sci. USA 117, 93–102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Naydenova K & Russo CJ Measuring the effects of particle orientation to improve the efficiency of electron cryomicroscopy. Nat. Commun 8, 629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morin A et al. Collaboration gets the most out of software. eLife 2, e01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Towns J et al. XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng 16, 62–74 (2014). [Google Scholar]
- 39.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Meth 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su M goCTF: Geometrically optimized CTF determination for single-particle cryo-EM. J. Struct. Biol 205, 22–29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wagner T et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol 2, 218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zivanov J, Nakane T & Scheres SHW Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kucukelbir A, Sigworth FJ & Tagare HD Quantifying the local resolution of cryo-EM density maps. Nat. Meth 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacobson MP, Friesner RA, Xiang Z & Honig B On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol 320, 597–608 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Huang M et al. Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. BioRxiv, 2020.2008.2013.250241 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goddard TD, Huang CC & Ferrin TE Visualizing density maps with UCSF Chimera. J. Struct. Biol 157, 281–287 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta. Cryst. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klaholz B Deriving and refining atomic models in crystallography and cryo-EM: the latest Phenix tools to facilitate structure analysis. Acta. Cryst. D 75, 878–881 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Díaz BHR Improvement of Protein Crystal Diffraction Using Post-Crystallization Methods: Infrared Laser Radiation Controls Crystal Order, Universität Hamburg, (2018). [Google Scholar]
- 52.Robertson MJ, van Zundert GCP, Borrelli K & Skiniotis G GemSpot: A Pipeline for Robust Modeling of Ligands into Cryo-EM Maps. Structure 28, 707–716.e703 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krissinel E & Henrick K Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Goddard TD et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. [Google Scholar]
- 56.Schrödinger Release 2020–4: Maestro, Schrödinger, LLC, New York, NY, 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Extended protein purification protocols are available on Protocols.io (https://www.protocols.io/groups/hao-wu-lab). Raw cryo-EM data are available on EMPIAR under the accession numbers EMPIAR-10594 (NLRP1-DPP9) and EMPIAR-10595 (NLRP1-DPP9-VbP). The cryo-EM maps are available on the Electron Microscopy Data Bank under the accession numbers EMD-22074 (NLRP1-DPP9) and EMD-22075 (NLRP1-DPP9-VbP). The atomic coordinates are available on the Protein Data Bank under the accession numbers 6X6A (NLRP1-DPP9) and 6X6C (NLRP1-DPP9-VbP). Pymol session files and the image analysis macro are available on OSF [http://doi.org/10.17605/OSF.IO/X7DV8]. All other data can be obtained from the corresponding authors upon reasonable request.