

To the Editor:
Somatic mutations of the X-linked phosphatidylinositol N-acetylglucosaminyltransferase subunit A (PIGA) gene in hematopoietic stem cells are the key molecular events in the pathogenesis of paroxysmal nocturnal hemoglobinuria (PNH) [1]. PIGA mutations lead to impaired biosynthesis of glycosylphosphatidylinositol (GPI)-anchor [2]. Resultant lack of GPI-anchored proteins (GPI-AP), primarily CD55 and CD59, generates clonal selection of PNH clone(s) and increases sensitivity of PNH red cells (RBCs) to complement-mediated hemolysis [3]. Based on immuno-fluorescent staining, the grade of GPI-APs deficiency on the RBCs can distinguish: PNH type-1 (TI) with normal expression; PNH type-2 (TII) with partial reduction; and PNH type-3 (TIII) with total absence of two common GPI-APs (CD55 and CD59) [4]. Fractions of these RBC types are believed to be a result of the molecular spectrum of PIGA mutations, ranging from missense in various amino acids positions to truncations and locus deletions, which can be concurrent in some patients [5, 6]. Consequently, the presence of TII and TIII cells may indicate the overall severity of PIGA defect, and thus, individual phenotypic subtypes can be defined as predominantly TII or TIII according to the dominant clonal defect [7, 8].
We hypothesize that PIGA genotype may shape the PNH phenotype trajectory with regard to the severity of GPI-AP deficiency and clinical course. For instance, incomplete TII RBCs PNH phenotype should result in slower expansion due to incomplete selection advantage as compared to complete loss of PIGA function indicated by TIII RBCs, typical of hemolytic PNH. However, the process of “GPI-AP painting”, a passive transfer from normal to TIII RBC could result in the presence of pseudo-TII RBCs with an intermediate phenotype [9]. Here, we first used modern deep targeted DNA sequencing to characterize the molecular landscape of patients with PNH and then correlated the type of PIGA mutations with flow-cytometric phenotypes. For improved resolution, our study focused on patients with large clones and hemolytic PNH.
Among 295 diagnosed patients who had a PNH clone (>0.5% of PNH granulocytes), we identified 60 who fulfilled the criteria for fully blown hemolytic PNH (PNH clone size >20% and lactate dehydrogenase >2.5x upper limit of normal). Test for PNH was conducted before initiation of complement blockade therapy [10, 11]. Whole blood DNA was subjected to multi-amplicon deep next generation sequencing (NGS) using primers covering all coding exons of PIGA gene as previously shown [12]. Five-color flow cytometry strategy was conducted as previously described [13, 14] for detection of PNH granulocytes. In particular, fresh specimens were stained with CD15-V450, CD45-PC7, CD64-APC, CD157-PE, FLAER-Alexa 488, while CD59-PE and CD235a-FITC antibodies were used to determine PNH RBCs (see also Supplementary Appendix).
Of 60 patients (median age 37 years, range 5–84, see also Table 1 and Supplementary Table 1), 13/60 (22%) were classified as having TII and 47/60 (78%) TIII PNH. Overall, median PNH granulocyte clone size was 71.4% (21.5–99.4) with no differences between TII/TIII subgroups (p = 0.872). Median TII RBC clone was 40.9% (6.8–84.1) and 1.9% (0.54–30) in TII and TIII PNH patients while median TIII RBC clone was 3.2% (1.2–21.8) and 22.1% (11.4–93.4), respectively. No differences were found at baseline between the two subgroups for male:female ratio, blood counts, and previous history of aplastic anemia (AA) while TIII patients had higher lactate dehydrogenase (LDH) levels (571 vs. 346 U/L in TII, p = 0.015), probably expression of the increased complement sensitivity resulting from total loss of GPI-APs. Thrombotic events occurred in 17/60 (28%) of patients with no differences in TII vs. TIII cases (46% vs. 23%, p = 0.162). Majority of cases 15/17 (88%) were registered before anti-complement treatment start and typically involved venous districts (16/17, 94%) with no fatal events. (See also Supplementary Table 2).
Table 1.
Patients’ characteristics.
| ALL | Type-II dominant | Type-III dominant | p value | ||
|---|---|---|---|---|---|
| Overall | N | 60 | 13 (21%) | 47 (79%) | |
| Gender | F | 33 (55%) | 9 (69%) | 24 (51%) | |
| M | 27 (45%) | 4 (31%) | 23 (49%) | 0.348 | |
| Age | 37 [5–84] | 39 [15–80] | 36 [5–84] | 0.501 | |
| Diagnosis | pPNH | 28 (46%) | 4 (30%) | 24 (51%) | |
| sPNH | 32 (53%) | 9 (70%) | 23 (49%) | 0.348 | |
| Granulocytes clone size (%)a | 71.49 (21.5–99.9) | 69.5 (21.5–99.5) | 78.2 (22.1–99.9) | 0.872 | |
| Type II RBC clone size (%)a | 2.41 (0–84.6) | 40.9 (6.8–84.1) | 1.9 (0.54–30) | ||
| Type III RBC clone size (%)a | 14.75 (0–93.4) | 3.2 (1.2–21.8) | 22.1 (11.4–93.4) | ||
| Hb (gr/dl) at diagnosisa | 9.9 [8–11] | 9.3 (8.3–10.45) | 10(8.1–11.4) | 0.452 | |
| ANC (109/L) diagnosisa | 2 (1.19–3) | 2.4 (1.17–2.85) | 2 (1.27–3.05) | 0.98 | |
| PLT (109/L) at diagnosisa | 68 (5–241) | 37 (15–148) | 87 (5–241) | 0.085 | |
| LDH (U/L) at diagnosisa | 498 (170–3666) | 346 (170–939) | 571 (196–3666) | 0.015 | |
| Median Follow-up (months) | 102.7 (0.5–375) | 82.3 (11–328) | 109 (0.5–375) |
All values are expressed in median (ranges).
F female, M male, RBC red blood cells, Hb hemoglobin, ANC absolute neutrophils count, PLT platelets, pPNH primary paroxysmal nocturnal hemoglobinuria, sPNH secondary paroxysmal nocturnal hemoglobinuria, LDH lactate dehydrogenase.
A total of 137 PIGA mutations were found with 27 in TII and 110 in TIII dominant patients (Fig. 1a–c, Fig. S1). Overall, >1 and >2 mutations (clonal mosaicism) were found in 85% and 57% of patients, respectively. Frameshift (n = 54, 39%) and missense (n = 41, 30%) mutations were most common, followed by splice site (n = 22, 16%), nonsense (n = 15, 11%) and non-frameshift insertion/deletion (n = 5, 4%). Although PIGA mutations are distributed throughout the entire coding region, TIII patients harbouring non-frameshift hits more often had truncating mutations mapping within the first 200 bps across the PIGA domain. Median total variant allele frequency (VAF) adjusted for X-chromosome clonality burden was 17.9% (range, 1.2–63%) and correlated with number of mutation (p = 0.0007) but without differences between TII/TIII patients (p = 0.345). Overall, PIGA mutations VAF correlated with PNH granulocytes clone size in both the entire cohort (p < 0.0001) and in the two subgroups of patients (TII, p = 0.02; and TIII, p < 0.0001; Fig. S2).
Fig. 1. PIGA mutational landscape of hemolytic PNH.
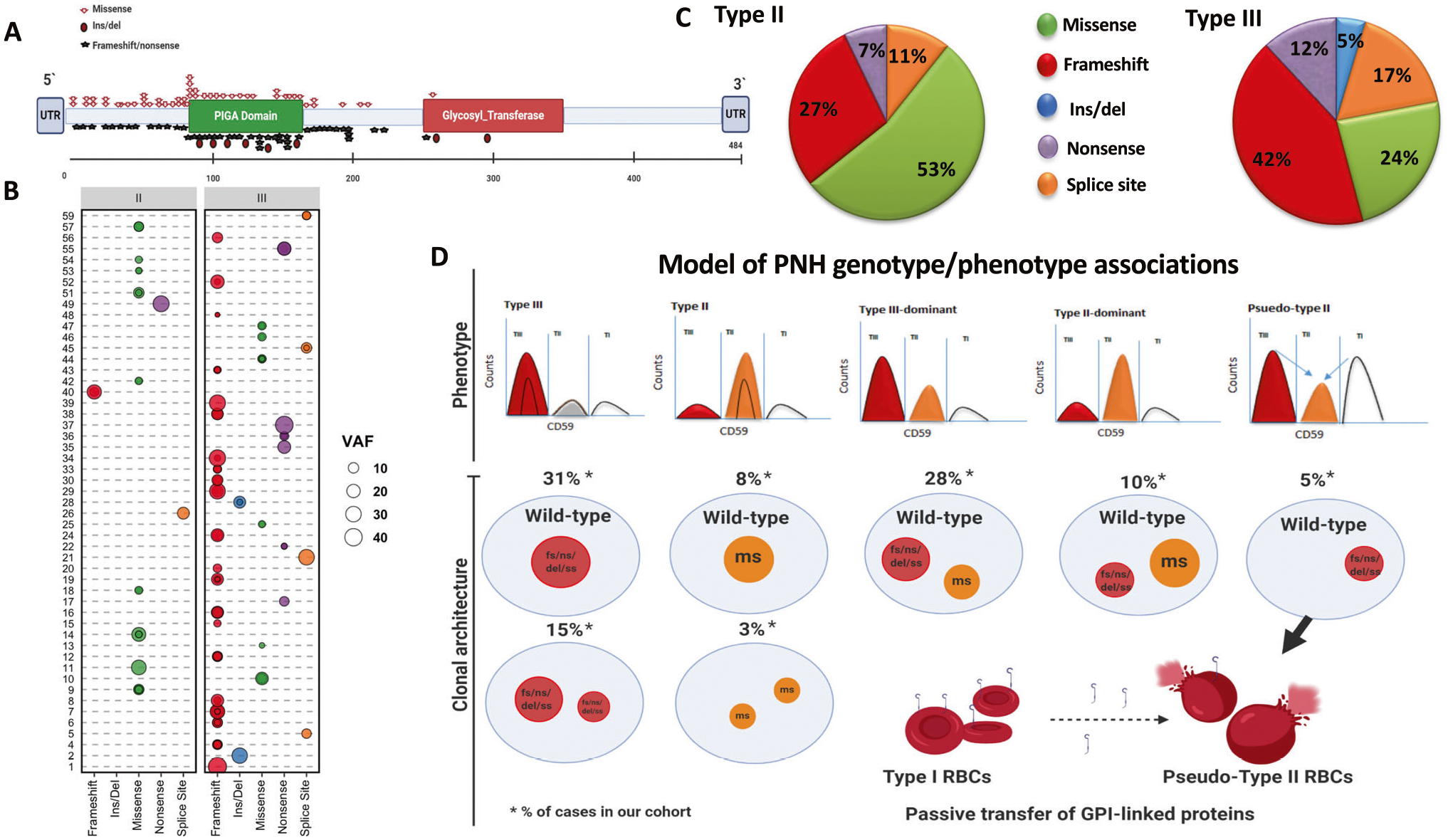
A Lollipop plot for amino acid changes resulting from PIGA mutations. B Bubble chart outlining PNH patients and their mutations according to the type and the size (VAF) of PIGA somatic hits (the area of the bubble is proportional to PIGA VAF). C Pie charts showing the differences in mutation types between patients with type II and type III RBCs. D Model depicting the various possibilities of PIGA mutations and the resultant phenotypes: TII RBCs phenotype in the presence of a dominant missense mutation (8% of cases in our cohort) or multiple missense mutations (3%); TIII RBCs phenotype in the presence of a truncating mutation (31%) or in the presence of multiple truncating mutations (15%); T-III dominant in case of a dominant truncating and a subclonal missense mutation (28%); TII-dominant in case of a dominant missense and a subclonal truncating mutation(10%); pseudo-TII RBCs phenotype in case of patients with TII RBCs phenotype harboring truncating mutations (5%). Pseudo-TII RBC may result from the passive transfer of GPI-linked proteins from normal RBCs (type I) to the GPI-deficient TIII RBCs generated by non-missense mutations in TII dominant patients (pseudo-TII), a phenomenon called “painting” process.Fs frameshift, del/ins deletion/insertion, ss splice site, ns nonsense, ms missense, VAF variant allelic frequency calculated as the sum of all mutations found in an individual patient (*adjusted for X-chromosome zigosity).
In cases negative for PIGA mutations by NGS (9/60, of which 8/9 presenting TIII-dominant PNH RBC clones), no mutations were found even after sorting to enrich for GPI (−) cells. This is likely due to inability to detect deletions larger than 250 bp by targeted sequencing of PIGA gene [6, 12]. To further understand the molecular basis of cases with wild type PIGA, we collected results of single-nucleotide polymorphism (SNP) array-based karyotyping to detect microdeletions of Xp22.2 encompassing the PIGA locus, an alternative mechanism that ultimately leads to the same selective clonal advantage as hypomorphic mutation of the PIGA gene as we previously showed [6]. Of the 9 negative cases, one of the patients (UPN 3) harboured a submicroscopic chromosomal Xp22.2 deletion. Furthermore, we investigated the presence of mutations in other genes involved in GPI biosynthetic pathway e.g., PIGT, previously shown to be responsible for atypical PNH cases without PIGA mutations and with a clinical picture characterized by the presence of inflammatory symptoms. Of note, our cases were negative for PIGT and did not show any of these symptoms [15].
Our results demonstrate that missense PIGA variants were enriched in TII PNH, while frameshift deletions in TIII dominant patients (p = 0.0021 and 0.0201, respectively). Moreover, when only the dominant clone (>2×the remaining VAF) was analysed, missense and frameshift mutations were strong predictors of PNH RBC phenotype, defining TII RBC (p = 0.0002) and TIII RBC patients (p = 0.02), respectively. Of note, we also found higher LDH levels in patients carrying truncating vs. missense mutations (median LDH 900 vs 378 U/L, p = 0.04), further emphasizing the higher hemolysis tendency of completely GPI-deficient TIII cases deriving from disruptive genomic lesions. When patients harbouring one unique somatic hit were studied (16/60, 27%), missense mutations accounted only for 19% of cases. Finally, analysing an internal cohort of 89 AA patients with small PNH granulocytes clones (<20%), we found that missense mutations accounted for 43% of cases indicating incomplete immune privilege.
PIGA missense and truncating mutations dictate the susceptibility of PNH RBCs to complement. In sum, we show that while missense mutations may still lead to a deficient but not totally absent GPI-APs synthesis, truncating mutations seem to be responsible for an entirely disruptive phenotype, causing the complete absence of GPI-APs on RBC membrane surface. Moreover, the non-missense TII dominant patients (pseudo-TII) may be an epiphenomenon of the “painting” process [9, 15] (Fig.1d). Our results suggest also that complete molecular screening and results of SNP array-based karyotyping to detect mutations or microdeletions of PIGA locus and other genes of the GPI synthetic pathway may help to rule out other causes explaining the PNH phenotype of cases lacking PIGA mutations. Finally, to overcome the limits of conventional targeted sequencing strategies, whole genome scanning approaches may also better magnify the diagnostic power in selected cases resulting negative for the aforementioned classical approaches.
Supplementary Material
Acknowledgements
We thank Edward P. Evans Foundation, the HENRY & MARILYN TAUB FOUNDATION, grants R01HL118281, R01HL123904, R01HL132071, R35HL135795 (all to J.P.M), AA&MDSIF, VeloSano Pilot Award, and Vera and Joseph Dresner Foundation–MDS (to VV), and Lilli Foundation (to CG). Supported by the American-Italian Cancer Foundation Post-Doctoral Research Fellowship (to CG).
Footnotes
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41375-020-01113-0.
Conflict of interest The authors declare that they have no conflict of interest.
Consent for publication Written informed consent was obtained from all patients.
Ethics approval and patients’ consent to participate Ethics approval and patients’ consent to participate to the study was approved by The Institutional Review Board of the Cleveland Clinic Foundation. All procedures were carried out in accordance with guidelines set forth by the Declaration of Helsinki.
References
- 1.Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–11. Epub 1993/05/21. eng [DOI] [PubMed] [Google Scholar]
- 2.Takahashi M, Takeda J, Hirose S, Hyman R, Inoue N, Miyata T, et al. Deficient biosynthesis of N-acetylglucosaminyl-phosphatidylinositol, the first intermediate of glycosyl phosphatidylinositol anchor biosynthesis, in cell lines established from patients with paroxysmal nocturnal hemoglobinuria. J Exp Med. 1993;177:517–21. Epub 1993/02/01. eng [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Risitano AM. Paroxysmal nocturnal hemoglobinuria and other complement-mediated hematological disorders. Immunobiology 2012;217:1080–7. Epub 2012/09/12. eng [DOI] [PubMed] [Google Scholar]
- 4.Sutherland DR, Keeney M, Illingworth A. Practical guidelines for the high-sensitivity detection and monitoring of paroxysmal nocturnal hemoglobinuria clones by flow cytometry. Cytom Part B, Clin Cytom. 2012;82:195–208. Epub 2012/04/14. eng [DOI] [PubMed] [Google Scholar]
- 5.Young NS, Maciejewski JP. Genetic and environmental effects in paroxysmal nocturnal hemoglobinuria: this little PIG-A goes “Why? Why? Why?”. J Clin Investig. 2000;106:637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Keefe CL, Sugimori C, Afable M, Clemente M, Shain K, Araten DJ, et al. Deletions of Xp22.2 including PIG-A locus lead to paroxysmal nocturnal hemoglobinuria. Leukemia. 2011;25:379–82. [DOI] [PubMed] [Google Scholar]
- 7.Endo M, Ware RE, Vreeke TM, Singh SP, Howard TA, Tomita A, et al. Molecular basis of the heterogeneity of expression of glycosyl phosphatidylinositol anchored proteins in paroxysmal nocturnal hemoglobinuria. Blood 1996;87:2546–57. Epub 1996/03/15. eng [PubMed] [Google Scholar]
- 8.Nishimura J, Inoue N, Wada H, Ueda E, Pramoonjago P, Hirota T, et al. A patient with paroxysmal nocturnal hemoglobinuria bearing four independent PIG-A mutant clones. Blood. 1997;89:3470–6. Epub 1997/05/01. eng [PubMed] [Google Scholar]
- 9.Sloand EM, Mainwaring L, Keyvanfar K, Chen J, Maciejewski J, Klein HG, et al. Transfer of glycosylphosphatidylinositol-anchored proteins to deficient cells after erythrocyte transfusion in paroxysmal nocturnal hemoglobinuria. Blood. 2004;104:3782–8. [DOI] [PubMed] [Google Scholar]
- 10.Parker C, Omine M, Richards S, Nishimura J-i, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112:3099–106. [DOI] [PubMed] [Google Scholar]
- 12.Clemente MJ, Przychodzen B, Hirsch CM, Nagata Y, Bat T, Wlodarski MW, et al. Clonal PIGA mosaicism and dynamics in paroxysmal nocturnal hemoglobinuria. Leukemia. 2018;32:2507–11. Epub 2018/05/12. eng [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutherland DR, Illingworth A, Keeney M, Richards SJ. High-sensitivity detection of PNH red blood cells, red cell precursors, and white blood cells. Curr Protoc Cytom. 2015;72:6.37.1–6.29. [DOI] [PubMed] [Google Scholar]
- 14.Illingworth A, Marinov I, Sutherland DR, Wagner-Ballon O, DelVecchio L. ICCS/ESCCA consensus guidelines to detect GPI-deficient cells in paroxysmal nocturnal hemoglobinuria (PNH) and related disorders part 3 – data analysis, reporting and case studies. Cytom Part B: Clin Cytom. 2018;94:49–66. [DOI] [PubMed] [Google Scholar]
- 15.Höchsmann B, Murakami Y, Osato M, Knaus A, Kawamoto M, Inoue N, et al. Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoin-flammation. J Clin Invest. 2019;129:5123–36. Epub 2019/08/21. eng [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.