

Abstract
Cells within the lung micro-environment are continuously subjected to dynamic mechanical stimuli which are converted into biochemical signaling events in a process known as mechanotransduction. In pulmonary diseases, the abrogated mechanical conditions modify the homeostatic signaling which influences cellular phenotype and disease progression. The use of in vitro models has significantly expanded our understanding of lung mechanotransduction mechanisms. However, our ability to match complex facets of the lung including three-dimensionality, multicellular interactions, and multiple simultaneous forces is limited and it has proven difficult to replicate and control these factors in vitro. The goal of this review is to (a) outline the anatomy of the pulmonary system and the mechanical stimuli that reside therein, (b) describe how disease impacts the mechanical micro-environment of the lung, and (c) summarize how existing in vitro models have contributed to our current understanding of pulmonary mechanotransduction. We also highlight critical needs in the pulmonary mechanotransduction field with an emphasis on next-generation devices that can simulate the complex mechanical and cellular environment of the lung. This review provides a comprehensive basis for understanding the current state of knowledge in pulmonary mechanotransduction and identifying the areas for future research.
1 Introduction
The lung is a complex mechanical organ and cells within the respiratory system experience a range of complex and dynamic mechanical forces. Although the heterogeneous mechanical forces that occur within the lung in vivo are well understood, the use of in vitro systems to recapitulate these complex physiological and pathophysiological mechanical stresses has only recently been appreciated. This review will provide a macro- and microscopic overview of lung anatomy, identify the magnitude of mechanical stimuli that cells experience within the pulmonary micro-environment, summarize current in vitro models and mechanotransduction findings, and discuss future directions for this field. We will focus on the need for novel in vitro systems that not only capture physiologically relevant mechanical conditions but also incorporate heterogenous cell-types to account for multicellular interactions. This review aims to introduce the complex micro-environment of the lung and the current mechanotransduction studies within this field to facilitate the development of new systems that more accurately recapitulate the pulmonary micro-environment.
1.1 Macroscopic Anatomy of the Lung.
The respiratory system extends from the nasal/oral passages, through the pharynx, down the larynx, and into the trachea which splits into right and left bronchi followed by five generations of branching. Airway branching continues for 10–12 generations in humans, leading into terminal bronchioles, which is the last unit of the lung's conducting zone where no gas exchange occurs (Fig. 1(a)). Air then travels into the respiratory zone, denoted by the presence of alveoli within the bronchioles (respiratory bronchioles), which terminates into the alveolar duct, alveolar sac, and alveoli (Figs. 1(b)–1(c)). The process of gas exchange occurs within the alveoli lined by a thin layer of epithelial cells forming a tight, single-celled membrane atop a basement membrane (1.66 μ ±0.128 μ in normal lungs [1]) with direct connections to the endothelial lining of the capillary network. The close positioning of the air sac and blood flow across a thin epithelial-endothelial barrier enables the diffusive exchange of carbon dioxide and oxygen. The space between epithelial and endothelial linings is known as the pulmonary interstitium and contains a variety of cells and extracellular matrix components that provide structure and support for the lung. The visceral pleura is a thin membrane that lines the entirety of the pleural cavity in one continuum and changes names based on the structure it lines (mediastinal, diaphragmatic, costal, cervical, and visceral pleura). The pleura maintains a negative intrapleural pressure of −4 mm Hg and reduces friction at the lung-wall interface during breathing.
Fig. 1.
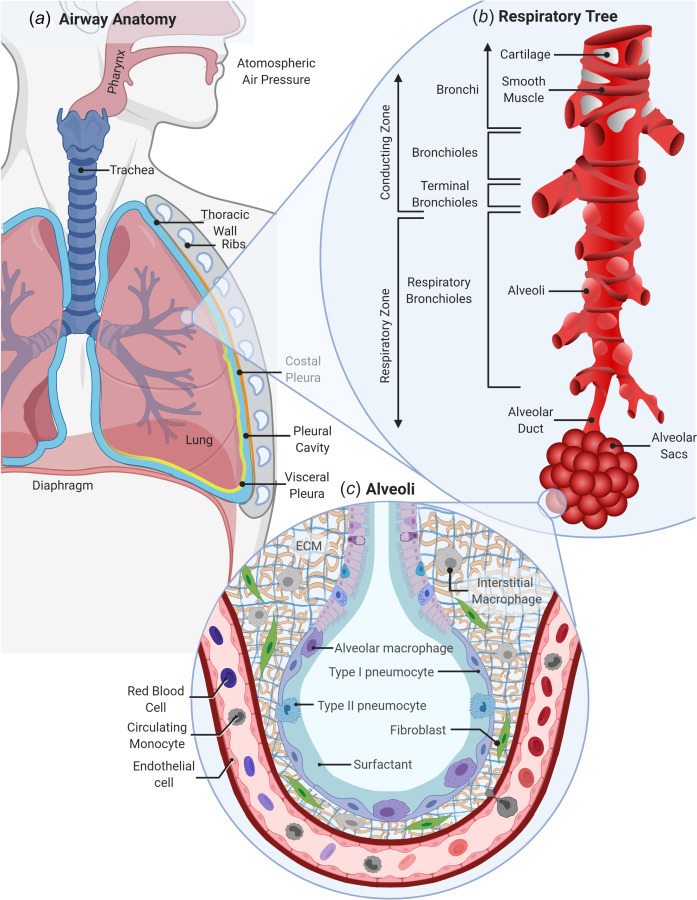
Anatomy of the respiratory system. (a) In humans, the respiratory system includes the mouth, nose, pharynx, and proximal trachea which branches into right and left lung lobes and a pleural cavity that borders the thoracic wall. (b) The macro-environment of the respiratory tree contains a conducting zone with bronchi located proximally leading into bronchioles, and terminal bronchioles and a respiratory zone with respiratory bronchioles, the alveolar duct, and alveolar sacs. (c) The micro-environment of the alveoli includes type 1 and type 2 pneumocytes/epithelial cells attached to a thin basement membrane, alveolar macrophages, endothelial cells and fibroblasts, and interstitial macrophages within a collagen and elastin ECM. Created with the link in a note.3
The main function of the lung is gas exchange. Inhalation is initiated through the contraction of the diaphragm and surrounding chest muscles, pulling the chest wall outward. The resulting reduction in intrapleural pressures draws the lung parenchyma outward, increasing the volume of the alveoli. This movement creates a negative or subambient pressure within the alveoli and draws air into the lung until equilibrium is reached. Once chest muscles and diaphragm contraction are relaxed, the elastic recoil of the lung releases the chest wall inward and decreases alveolar volumes. This decrease in volume compresses the air, increasing its pressure and forcing it out of the lung, i.e., exhalation.
1.2 Microscopic Anatomy of the Lung.
There are many different types of cells and structural supports within the respiratory tract [2]. The trachea and primary generations of branching bronchi are supported by a cartilage superstructure along with smooth muscle that dilates or constricts to regulate airflow [3,4]. Bronchi are lined with primarily ciliated columnar epithelial cells that act as physical barriers within the lung to remove inhaled debris [5] or trap the debris in a mucus layer secreted by goblet cells [6] via the mucociliary escalator. Basal cells residing beneath this epithelial layer can differentiate and replace damaged cells as needed [7]. As bronchi branching continues, the main cell type shifts from ciliated epithelial to nonciliated club cells [7–9] which line the respiratory bronchioles [5,10] before terminating in alveolar ducts. Smooth muscle cells continue to support the airway tree down to the respiratory bronchioles [4]. The alveoli are lined with type one and two pneumocytes (alveolar epithelial cells). Type one pneumocytes are thin squamous cells that stretch to cover large areas making up the majority (95%) of the alveolar surface and participating in immune host defense [5]. This thin membrane allows for diffusive gas exchange and prevents plasma fluid entry into the alveoli. Type two pneumocytes are granular, cuboidal in shape, and are responsible for surfactant secretion. Previous work has demonstrated that type two pneumocytes replicate to replace damaged or dying type one pneumocytes [5] and actively participate in immune defense via the expression of pathogen recognition receptors [11,12].
Although not a cellular component, pulmonary surfactant is a crucial part of the respiratory system which acts to reduce the surface tension at the air–liquid interface. Reduced surface tensions in the alveoli stabilize the lung by preventing alveolar collapse during exhalation, maintains low alveoli opening pressures, ensures uniform alveoli inflation [13], and establishes a barrier to prevent pathogen access to cell surfaces [14]. Pulmonary surfactant constituents include lipids (90%) and proteins (10%) [15] and an extensive breakdown of surfactant composition can be found in the reviews by Perez-Gil [14,16].
Since the lung is the largest internal organ with exposure to the outside environment, it contains a robust immunological system to distinguish pathogens from inert particles. It contains both innate and adaptive immune cells and can be considered an immunological organ [17]. Several macrophage subtypes exist within the lung micro-environment including alveolar macrophages and interstitial macrophages. Alveolar macrophages reside along the airway in the epithelial lining fluid and are responsible for identifying and phagocytosing inhaled microbes and/or particles and clearing pulmonary surfactant [5]. Interstitial macrophages are located within the lung tissue though their exact location is debated as their clear isolation from other macrophage types is complex [18]. They are able to phagocytose bacteria and particulate matter, express cytokines and act as immunoregulatory cells [18]. There are also a variety of other immune cells that are present in the lung during homeostasis such as lymphocytes, dendritic cells, and natural killer cells.
The lung also contains structural cells and proteins that play important roles within the pulmonary micro-environment. The lung parenchyma is composed of extracellular matrix (ECM) proteins, connective tissues, and cells. Collagen and elastin are two of the main proteins found within the ECM and this matrix composition provides important structural support for the lung micro-environment during the active process of inhalation and avoids collapse while maintaining prestress after exhalation [19]. Fibroblasts, long, spindle shaped cells that reside within the ECM, are responsible for maintaining and repairing the ECM [20]. When an injury occurs, fibroblasts can be activated and differentiated into myofibroblasts, which express alpha-smooth muscle actin and remodel or contract their surroundings to initiate the wound healing and repair response. A final component of the lung is the visceral pleura which are composed of five layers of cells and connective tissues [21]. A detailed account of each layer and function can be found in Sevin and Light's review [21]. The pleural membranes secrete fluid into the pleural cavity, reducing friction between the lungs and chest wall during respiration [22]. These cellular and protein components work together allowing the lungs to accomplish their essential task of gas exchange.
1.3 Mechanical Forces Within the Lung.
In addition to the matrix and cellular components of the lung, there are also a variety of mechanical forces which occur in the respiratory system. Mechanotransduction is the process by which cells sense and translate these mechanical forces into downstream signaling and phenotypic changes. Within the scientific community, the importance of mechanotransduction has been recognized in a variety of diseases and cell types such as cancer, where shear stress and compression increases proliferation, chemoresistance, and progression [23–25], lung fibrosis where stiffness promotes fibrotic buildup [26], and bone cell survival which is influenced by tension [27]. Through the study of physiological mechanical forces in specific micro-environments, the importance of a three-dimensional (3D) micro-environment, compressive stimulus, shear stress, and tension forces influence not only cellular phenotypes but also proliferation, cell death, morphology, differentiation, and response to drug treatments [25,28,29].
As evident from the large size change and continuous use of the respiratory system, cells within the lung experience an ongoing dynamic range of mechanical stresses. In the setting of normal breathing, the lung parenchyma is prestressed at end expiration and transmural pressure keeps alveoli open preventing collapse [19]. During inhalation, the alveolar-capillary barrier, including the epithelial cell layer, basement membrane, and endothelial cell layer, is stretched in tension creating internal strain affecting all cells within the micro-environment. During exhalation, the elasticity of the lung contracts the parenchyma and alveoli, initially increasing the localized pressure within the alveolar sacs and creating a momentary compressive force on the alveolar epithelial lining. The continuous expansion and contraction of the lung alveolar surfaces also create a shear force along the surface epithelium [30–32]. Additionally, the lung also has viscoelastic properties, meaning that it responds in a time-dependent manner to applied forces dissipating some applied energy. The mechanism by which this viscoelasticity impacts the cellular components and their response to a mechanical stimulus is largely unknown and will be discussed further in Sec. 3.5. Although these forces are normal within a healthy lung micro-environment, in the setting of disease or during mechanical ventilation these forces are significantly altered in both magnitude and distribution [31]. Where healthy lung tissue is primarily homogeneous in terms of mechanical properties/forces, diseases introduce significant heterogeneity and new or aberrant forces within the lung. A summary of physiological force magnitudes experienced within the lung is provided in Table 1.
Table 1.
Mechanical properties of the lung in normal and diseased states
| Parameter | Magnitude in healthy lung | Magnitude during pulmonary disease |
|---|---|---|
| Alveolar tissue strain (%) | 4% normal tidal breathing [33–35] 12% deep sigh (calculation from excised dog lungs) [36,37] | Inhomogeneity of the diseased lung creates focal stressors that increase alveolar strain [35,38,39] |
| 0–5% (Linear distension estimate for normal tidal breathing) [33] | ||
| 15–40% or higher (for functional residual capacity to TLC) [33] | ||
| Noncollagenous lung tissue stiffness (kPa) | 0.5 to >3 (mice saline treatment) [26] | 3 to >15 (mice bleo treatment) [26] |
| 1.96 ± 0.13 (human lung tissue) [40] | 16.52 ± 2.25 (IPF human lung tissue) [40] | |
| 1.9137 rat lung parenchyma [41] | 9-38.5 (IPF human lung tissue) [42] | |
| 3.7 ± 1.3 (human lung tissue) [42] | 2.9 ± 0.8 (COPD GOLD IV human lung tissue) [42] | |
| Collagenous Bronchi Stiffness (kPa) | Pseudo-elatic linear modulus in Pig [3] | |
| Axial 30.31 ± 3.1 | ||
| Circumferential | ||
| -Small bronchi 12.5 ± 1.9 | ||
| -Trachea 6.0 ± 0.6 | ||
| -Large bronchi 6.6 ± 0.9 | ||
| Shear stress in alveoli (dyn/cm2) | <15 [31] | Several magnitudes higher in alveolar reopening conditions [31] |
| Respiratory frequency (Hz) | 0.20 (normal tidal breathing) | 0.44 (cystic fibrosis (CF)) [43] |
| 0.55 (heavy exercise) [44] | ||
| 0.285 [43] | ||
| Tidal volume (mL) | 410 [43]; 500 [44] | 403 (CF) [43] |
| Surfactant based surface tension (mN/m) | Minimum surface tension: | |
| Inspiration ∼ 20 | ACB > 12 [45] | |
| Expiration <2 [14] | CF > 15 [46] | |
| (near zero [15]) | ARDS > 20 [15] |
2 Respiratory Diseases
In general, pulmonary diseases are classified into two categories: restrictive, which involves a reduced lung expansion capacity, or obstructive, which involves increased airway resistance and limited airflow. Classification of restrictive or obstructive disease is based on clinical measurements including the ratio of forced expired volume in one second to full forced vital capacity (FEV1/FVC%) and total lung capacity percentage (TLC%) [47]. The obstructive disease involves a low FEV1 (due to obstructed airways) and a high TLC%. The restrictive disease is characterized by a reduced TLC% and no change or an increase in FEV1/FVC ratio as both measures are reduced simultaneously or FEV1 is increased with reduced lung compliance. Restrictive diseases such as pulmonary fibrosis (PF), interstitial lung disease, and sarcoidosis often involve stiffening of the lung parenchyma due to the buildup of ECM and scar-like tissue around the alveoli, restricting the lung's expansion capacity. Obstructive diseases, such as asthma, bronchopulmonary dysplasia (BPD), bronchiolitis obliterans (BO), and chronic obstructive pulmonary disease (COPD), involve airway swelling and decreased matrix stiffness and elasticity due to the degradation of the lung connective tissue which reduces the lung retraction force and ability of the individual to fully exhale. Pulmonary diseases can also be classified as either acute or chronic. Chronic and/or progressive conditions such as asthma, COPD, and idiopathic pulmonary fibrosis (IPF) can last up to a lifetime, and interestingly some acute conditions, such as acute respiratory distress syndrome (ARDS) can lead to chronic lung damage (i.e., fibrosis). During acute/chronic lung disease, the complex mechanical environment of the lung is altered, changing the cellular forces experienced within the tissue. This section will focus on the mechanical stress changes associated with pulmonary diseases and the pathophysiological alterations of the lung micro-environment.
2.1 Acute Respiratory Distress Syndrome.
Acute respiratory distress syndrome is characterized by fluid buildup in the lungs preventing gas exchange in the alveoli resulting in hypoxemia. This acute disorder occurs rapidly and is thought to be a secondary response to an initial insult such as viral infection or injury [48]. Unfortunately, the primary treatment for ARDS, protective mechanical ventilation, provides essential oxygenation to the body but also alters mechanical stress within the lungs resulting in pathophysiological forces that further disrupt the alveolar-capillary barrier resulting in edema. Previous work has demonstrated that excess ECM deposition causes alveolar remodeling in ARDS patients [49] contributing to the resistance of lung expansion (Fig. 2(c)) [50]. Long-term effects for ARDS survivors include: mild fibrosis (majority ventilation-induced), exercise limitations, cognitive impairment, muscle weakness, diminished oxygen transfer, and reduced mental health [51]. Additional effects of ventilator-induced lung injury will be discussed in the ventilator-induced lung injury section below.
Fig. 2.
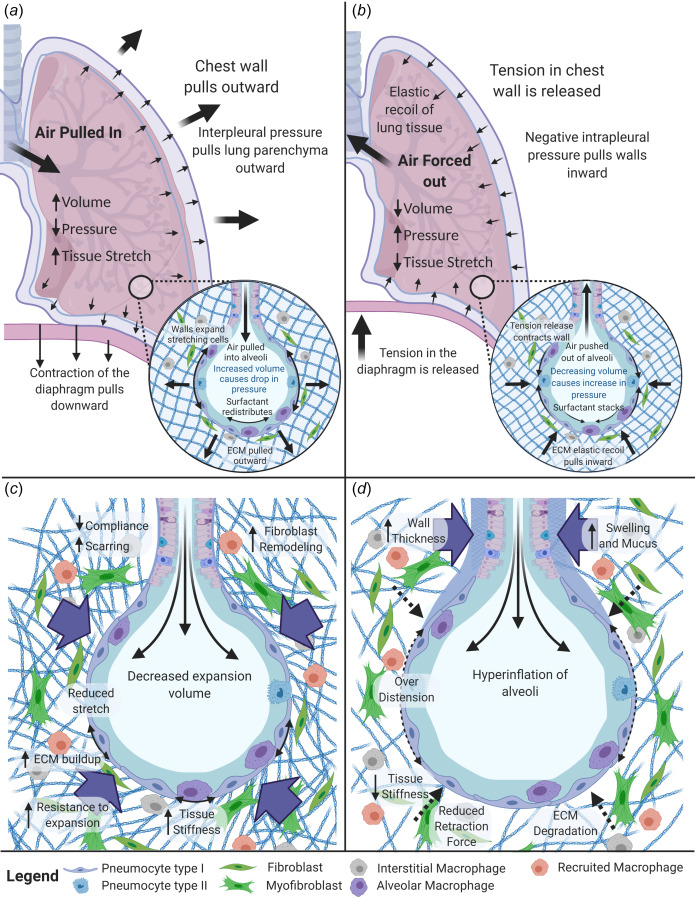
Mechanical Forces within the lung. (a) Contraction of the diaphragm and chest muscles during inhalation results in negative interpleural pressure that expands lung tissue, stretches the alveoli, and increases lung volume driving air inflow. (b) Relaxation of the diaphragm and chest muscles during expiration allows for elastic recoil that reduces lung volume and air compression that drives air outflow. (c) In restrictive diseases, fibroblast-mediated remodeling causes increased tissue stiffness which limits expansion capability during inspiration. (d) In obstructive pulmonary disease, ECM degradation leads to hyperinflation and reduced elastic recoil inhibiting full exhalation. Created with the link in a note.3
2.2 Asthma.
Asthma is a chronic disease characterized by hyper-responsiveness of the airway to a stimulant that would normally not cause airway narrowing in a healthy patient [52]. Restricted breathing, caused by the contraction of airway smooth muscle cells, is thought to be responsible for these hyperactive responses [52,53]. Asthma encompasses a wide range of pathophysiological mechanisms and subtypes including allergic and nonallergic (obesity, viruses, etc.) stimulants. Patients experience shortness of breath due to acute airway constriction, swelling, and mucus production. This intense constriction induces extreme compressive forces on the airway epithelium influencing cellular phenotypic responses [54,55]. Although airway constriction is an acute response, repeated exposure induces chronic inflammation leading to airway wall thickening, permanent loss of lung function [56], mucus production, airway remodeling [56], enlargement of the surrounding smooth muscles, and ECM structural changes [57]. In addition, a variety of immune cells alter the cellular makeup in the proximal airways [58] as well as the lung periphery [59,60] contributing to airway remodeling and perpetuation of the disease [61,62]. Asthmatic patients have increased and irregular collagen deposition in both large and small airways, and this effect on lung biomechanics requires future modeling investigations [61].
2.3 Bronchiolitis Obliterans.
Bronchiolitis obliterans is an obstructive respiratory disease where inflammation and luminal fibrosis are found in the respiratory bronchioles (Fig. 2(d)) [63,64] as a result of lung or bone marrow transplant, toxin exposure, or infections [65]. This scar tissue buildup blocks gas exchange in the small airways and patients present with dry cough, shortness of breath, and fatigue [66]. BO is a major complication of lung transplantation representing chronic organ rejection with an unknown cause [64] and is the leading cause of death after the first year of transplantation [63]. It is characterized by air trapping in the lungs during complete expiration, narrowing of the lumen without alteration in the parenchyma, along with collagen deposition in the submucosa constricting the small airways sometimes causing complete occlusion [65]. The alterations in lung biomechanics associated with this disease and how changes in force contribute to BO progression are currently understudied.
2.4 Bronchopulmonary Dysplasia.
Bronchopulmonary dysplasia is a chronic disease associated with premature infants that receive oxygen and mechanical ventilation for respiratory distress [67]. It is caused by injury to the developing lungs, either before or after birth resulting in an abnormal healing response [68]. Advances in preterm neonatal care have greatly improved the survival rate for preterm infants [69]. Infants that survive BPD often experience delayed lung development, reduced alveolarization, and diminished generation of alveoli units as they age [70]. The impact of altered mechanical forces on alveolarization remains understudied. BPD survivors can also experience chronic pulmonary issues in adulthood such as pulmonary dysfunction, altered lung structure, obstructive lung functions, and exercise intolerance alluding to a predisposition to pulmonary complications later in life. The mechanisms that regulate this phenomenon are currently unknown [70]. Additional research is needed to understand the long-term effects of this early lung injury and how the altered mechanical environment influences recovery and prolonged pulmonary complications.
2.5 Chronic Obstructive Pulmonary Disease.
Chronic obstructive pulmonary disease is a chronic inflammatory disease causing obstruction of the lower airways and is currently one of the leading causes of death worldwide [71,72]. There are a variety of causes for COPD with most patients having exposure to long-term irritants or particulates, such as smoking. The acute progression of the disease has been observed with infection [73], but patients diagnosed with COPD experience difficulty breathing due to narrowing of the airways and inflammation-induced destruction of the normal ECM [74]. The disruption of collagen IV [75] and elastin degradation [76] within the lung results in reduced elastic recoil during exhalation [76] and softening the lung tissue (∼30%). This parenchymal destruction leads to enlarged alveoli and possibly emphysema (Fig. 2(d)) [77].
2.6 Pulmonary Fibrosis.
Pulmonary fibrosis is a chronic, restrictive lung disease with excessive collagen deposition from the buildup of scar tissue in the lung caused by pulmonary injury, inflammation, and/or long-term exposure to toxins or particles. PF patients experience a range of symptoms including shortness of breath, cough, and fatigue. The most severe form of pulmonary fibrosis is IPF which has an undetermined cause and a median survival of fewer than three years after diagnosis [78,79]. Unresolved scar tissue in the lungs stiffens the lung parenchyma and restricts lung expansion. Excessive matrix deposition occurs in the distal airway structures, where fibroblastic foci are constructed from excess collagen, fibrin, and other ECM components that fail to be resolved and worsen over time [77]. The heterogeneous lung stiffness creates foci of intensified stress and strains (stress raisers) influencing cellular mechanotransduction and progression of the disease (Fig. 2(c)) [80].
2.7 Lymphangioleiomyomatosis.
Lymphangioleiomyomatosis (LAM) is a rare condition where patients exhibit cysts in the lungs and lymphatics as well as abdominal tumors [81]. It primarily affects females of childbearing age with initial symptoms of dyspnea, pneumothorax, pleural effusions, ascites, abdominal hemorrhage, airway hyperreactivity, or bloody cough [82]. There are two forms of LAM including the sporadic form, which has an unknown cause, and the familiar form which includes mutations of the tuberous sclerosis complex (TSC) [83]. LAM cells, abnormal smooth muscle-like cells with mutations in TSC1 or TSC2 genes, exhibit over activation of the mTOR pathway [81,83], proliferate to form the lung and/or lymphatic infiltrating cysts that destroying the lung parenchyma. The infiltration of the LAM cells cause vascular wall thickening, occlusions in airways and veins, hemorrhage, lung degradation, and hinder gas exchange and airflow in alveoli [83–86]. It is predicted that changes in matrix-degrading enzymes, such as matrix metalloproteinases (MMPs), alter the lung structure and enable cyst formation in the lungs [85,86]. Lung degradation corresponds to cyst score (proportion of lung occupied by cysts) and severity of the disease but can be mitigated with sirolimus treatment [84]. The obstructive behavior of this disease can cause air trapping, and the limited airflow restriction is believed to be mainly caused by increased airway resistance not loss of elastic lung recoil (Fig. 2(d)) [86].
2.8 Sarcoidosis.
Sarcoidosis is characterized by abnormal inflammatory cell aggregates known as granulomas that form in a variety of organs but most commonly in the lungs [87]. The cause of sarcoidosis is unknown although some predisposition genes have been identified [88]. In roughly two thirds of patients, sarcoidosis resolves on its own, but for the remaining patients, the disease results in a chronic condition with reduced pulmonary function [89]. Sarcoidosis is often accompanied by fibrosis and can be thought of as both a restrictive and obstructive disease (Figs. 2(c) and 2(d)) [88,90] with some individuals displaying a mixed ventilatory pattern (reduced FEV1/FVC and below normal TLC) [47]. As the disease progresses, granulomas become more fibrotic and form nodules with excessive collagen deposition, and histology of end-stage disease resembles the fibrotic buildup observed in IPF [88].
2.9 Ventilator Induced Lung Injury.
The use of a ventilator is critical for treatment in a variety of acute and chronic pulmonary conditions; however, it can also be a source of further damage to the lung tissue. During the application of positive pressure to inflate the lungs, excessive pressure and stretch to accommodate the volume change can damage the alveolar lining. This over-distention causes additional damage and inflammation initiating an acute response, which can also exacerbate a chronic condition [91,92]. Mechanical ventilation can exacerbate damage to lung tissue through several mechanisms. Volutrauma injures the lung when high tidal volumes result in over-distension/excessive stretch of the alveoli. Atelectrauma damages the lung by shear/interfacial forces generated from repetitive opening/closing of fluid occluded alveoli/airways. Finally, barotrauma causes damage via elevated transmural pressure through the accumulation of air in extra alveolar spaces often due to alveolar rupture [93–95]. In addition to physical damage, these mechanical forces can also lead to pro-inflammatory cytokine release that not only exacerbates lung injury but can also cause distal organ dysfunction. Some of the acute injury induced by mechanical ventilation can be avoided through the use of low tidal volumes [96], although it is not entirely mitigated because ventilation can also damage the lung through the repetitive opening and collapse of fluid-filled alveoli at low lung volumes (atelectrauma) [92]. This action induces damage through high-pressure gradients that are generated by moving air–liquid interfaces [95,97] which can be lessened by the use of positive end expiratory pressures to maintain an open lung and minimize recruitment/derecruitment of alveoli [91,97]. However, the effectiveness of positive end expiratory pressures modulation currently remains ambiguous [94,98].
2.10 Viral Induced Lung Injury (Influenza and COVID-19).
The lungs are subjected to constant exposure to the pathogens and irritants of the outside world in the air we breathe. To protect against harmful infection the immune system monitors the airway and alveolar spaces but in the setting of viral infection can become overwhelmed and unable to contain and clear the infection. Viral lung infections affect large portions of the population seasonally and with the COVID-19 pandemic, more people than ever are exposed to this form of lung injury. Viruses infiltrate the respiratory tract targeting the airway and alveolar epithelium resulting in destruction of the epithelial lining and leakage of plasma fluid and protein into the alveolar space. This resulting edema fluid within the airspace significantly diminishes gas exchange [99]. Secondarily, activation of the patient's innate and adaptive immune response, though necessary to clear the infection, can also further injure the lungs by damaging the epithelium and endothelium [100]. As a result, patients develop critical changes within the lung micro-environment and severe infections can lead to the development of ARDS and the necessity for mechanical ventilation.
The cellular destruction and resulting fluid within alveoli compartments create tissue heterogeneity augmenting force distribution throughout the lung and increasing the work required for normal tidal breathing. Due to the increase in work, patients can also self-inflict further injury within their lungs from respiratory contraction without mechanical ventilation [100]. For example, increased breathing effort concentrated in open as opposed to occluded lung regions can lead to enhanced localized strain even with normal overall tidal volume. In addition, increased diaphragm contraction force causes increased pendelluft, tidal recruitment, and regional strain [100]. Areas of enhanced strain in mouse models of over inflation have preliminary shown elevated cell death and markers of inflammation [100] which coincide with elevated strain models in the whole lung [101]. Further study of the mechanism of lung injury under viral load conditions is needed.
Although the lung has a robust repair mechanism, these repair mechanisms, which include a wound healing/re-epithelization response, can be extensively damaged by a viral infection and the effect of viral infection on lung repair is not completely understood [99]. Previous work has demonstrated that recovery from severe viral infections can result in fibrosis and intimal thickening [102] which would stiffen and restrict normal alveoli expansion. Further studies on how these fibrotic changes influence disease progression or resolution as well as the long-term functional impacts on patients are needed specifically for those recovering from COVID-19 [103].
3 In Vitro Lung Mechanotransduction Studies
Many investigators have developed in vitro systems to examine how lung cells respond to mechanical forces. These models typically investigate how one type of mechanical force influences one cell type, with little work combining multidimensional forces and multicellular models to accurately recapitulate the complex interactions that occur in vivo. In this section, we review the current literature on lung cell types and their responses to a mechanical stimulus in vitro.
3.1 Epithelial Cells.
Lung epithelial cells line pulmonary airways and alveoli and are exposed to cyclic tension due to alveolar expansion, low levels of shear stress from air flow in the proximal lung, and the stiffness of the basement membrane. Minor levels of tension occur from bronchodilation, shear stress via air, mucus, or surfactant fluid flow, and compression from air pressures within the lung. In the diseased state, epithelial cells are subjected to higher levels of shear, altered tensile and stiffness profiles, and in some conditions aberrant air–liquid interfacial flows in the deep lung. Additionally, alveolar collapse during some conditions can cause epithelial cells to undergo complex hydrodynamic stresses during airway reopening [95]. The air–liquid interfacial movement along with altered surfactant properties can influence the mechanics of the entire lung [95]. The range and impact of these mechanical stimuli can significantly modify epithelial cell phenotypes, cellular metabolism, and gene expression profiles.
Many studies have focused on the response of epithelial cells to tensile stresses. Investigations of normal physiological conditions include cyclical application of uniaxial or biaxial strain of ∼10% and show that within this strain range alveolar epithelial cells remain viable [104] with enhanced proliferation [105–107]. Some controversy exists regarding the role of focal adhesion kinase (FAK) in the mechanotransduction response to strain [105–107], and this disparity may be attributed to differences in strain magnitude, e.g., 10% versus 20% which mimics lung over-distension [105,106]. The involvement of FAK in regulating mechanotransduction in the lung is based on previous work demonstrating its a critical role in other diseases, such as cancer [108,109], and with other cell types, such as endothelial cells [110]. Application of 20% strain induced an increase in reactive oxygen species (ROS [111]) as well as generation and involvement of the p42/44 MAP kinase pathway leading to increased proliferation [107]. Interestingly, even at physiological cyclic stretch magnitudes of 10%, Felder et al. Demonstrated impaired wound healing response when compared to static culture [112]. The standard strain used for over-distension in vitro is typically 20% or greater [113] and this has repeatedly been shown to injure epithelial layers grown on elastic membranes [113–115] with static stretch application following these same trends in cell injury [119]. The recent work of Arora et al. investigated ventilated mice lung strains using synchrotron radiation microcomputed tomography. They found heterogeneous deformations throughout the respiratory cycle with strains ranging from 80 to 100% regionally and up to 150% locally within the alveoli [121]. The ex vivo studies performed on mouse and pig lungs using digital image correlation by Mariano et al. found heterogeneous and anisotropic strains in the parenchyma [122].
Work done by Tschumperlin et al. has demonstrated that cellular injury is proportional to the magnitude of deformation, seeding density, and culture time [113]. Cellular response to strain occurs quickly with 20% strain resulting in ROS induction within 15 min [107,123] while the duration, magnitude, and type of cell stimulated influences the specific response rate [124,125]. With heightened stretch (10–20%) or the addition of asbestos fibers concurrent with 5% strain, IL-8 production is amplified [104,126,127] via mitogen-activated protein kinase and p38 pathways [104]. The pro-inflammatory response to lipopolysaccharide (LPS) stimulation was shown by Rentzsch et al. to be reduced using variable stretch techniques (random peak stretch 1–15% average 7.5%) compared to constant peak stretch (7.5%) mediated through the extracellular signaling regulated kinase (ERK) 1/2 pathway [128]. Additional involvement of the ERK pathway [105,125,128], increases Na+-K+-ATPase activity [129], and enhanced intracellular adhesion molecules (ICAM)-1 expression [125] have been associated with cyclic mechanical loading of epithelial cells with duration, load magnitude, and cell type all influencing the extent of pathway activity.
Shear stress exposure is often investigated using a microfluidic device. When epithelial cells were exposed to low shear stresses in the alveoli (0.69 × 10−3 to 2.8 × 10−3 dynes/cm2), Nalayanda et al. observed that A549 cells exhibit decreased growth rates with increased shear stress magnitude [130]. Interestingly, Mahto et al. demonstrated that shear stress can differentially impact alveolar cells depending on their source. The human cell line A549 had no change in surfactant secretion below 8 dynes/cm2 and impaired secretion above this level; whereas murine type 2 alveolar epithelial cells (ATII) (murine lung epithelial (MLE)-12) cells showed enhanced secretion with increasing stimulus [117]. Shear stress also impacts mechanical properties by altering the keratin intermediate filament (KIF) network in alveolar epithelial cells. Shear stress of 30 dynes/cm2 induced KIF network disassembly (not under tensile stress) [131] while a moderate stimulation, 7–15 dynes/cm2, lead to the strengthening of the KIF networks [132,133].
Complex interfacial forces affect the lung when repetitive airway/alveolar opening and closing are present. This occurs when plasma fluid infiltration into the lungs alters the pulmonary surfactant's ability to maintain near zero surface tension in the alveoli. The epithelial layers are subjected to a combination of tangential shear and normal pressures that vary temporally and spatially during airway/alveolar reopening and these forces cause significant cell injury and barrier disruption [95]. Gaver and colleagues were the first to show that increased spatial gradients in pressure are responsible for elevated epithelial cell damage at slower reopening velocities and that repeated reopening events exacerbate cell damage [134,135]. The impact of pressure gradients on cell damage and death was confirmed by the work of Yalcin et al. who showed reduced velocity and airway diameter and the resulting elevated pressure gradient increased epithelial cell death [136]. These authors also demonstrated reduced cellular confluence and repeated reopening events induced cell detachment and death [136] and that “fluidization” of the actin cytoskeleton (decreased stiffness with increased viscosity) leads to significantly less injury during reopening [137]. Importantly, these authors also demonstrated that pharmacological modulation of the cytoskeleton with statins also reduces cell injury and inflammatory cytokine secretion (IL-6, IL-8) [138]. A thorough review of the recent models and studies investigating interfacial forces due to ARDS can be found in the work by Viola et al. [139]. The chronic long-term effects of interfacial forces are still unknown and better in vitro models are needed to investigate the physiological response of all cells in the lung micro-environment under extended force stimulus.
Compression is the most understudied stimulus for pulmonary epithelial cells as it is considered a minor component of the micro-environment compared to tensile forces. Huang et al. demonstrated that cyclic and static compression activates nuclear factor kappaB (NF-kB) in a magnitude-dependent fashion at low frequencies [116]. Tschumperlin et al. demonstrated that compressive stress activates phosphorylation of ERK and production of heparin binding epidermal growth factor (HB-EGF) in bronchial epithelial cells [140] and Chu et al. found that compression induces expression of epidermal growth factor receptor (EGFR) ligands HB-EGF, epiregulin, and amphiregulin for sustained periods after 1–2 h of 30 cm H2O (2.9 kPa) of pressure [141]. Finally, Savla and Waters demonstrated that compression slows wound repair in epithelial cells [142]. For a detailed review of the impact of mechanical compression on asthma airway response please see the review by Veerati et al. [55].
The role of substrate stiffness on epithelial cell function has some contradictory findings in the literature. Eisenberg et al. demonstrated that substrate stiffness influences cellular morphology, actin cytoskeleton, and focal adhesions but does not alter epithelial-mesenchymal transition (EMT) or the differentiation of ATII cells into ATI cell types in primary murine alveolar epithelial cells [143]. Conversely, Dysart et al. and Markowski et al. found that increased substrate stiffness induced EMT, contractile phenotypes, and activation of transforming growth factor beta (TGF)-β via Rho/Rho associated coiled-coil containing protein kinase (ROCK) signaling [144] and that these responses could be enhanced through exposure to particulate matter [145] in rat lung epithelial T antigen negative cells. When combining the reopening forces with varying substrate stiffness, Higuita-Castro et al. demonstrated greater stiffness induced increased cell death but prevented epithelial detachment and monolayer disruption along with increased expression of FAK and phosphorylated paxillin [146]. Taken together, these studies highlight the importance of further dissecting the role of mechanical forces in epithelial cells.
3.2 Macrophages.
Macrophages exist in all areas of the lung and can be derived from either proliferation of tissue-resident cells or via differentiation from circulating monocytes. Within different lung regions, macrophages experience a range of mechanical stimuli which can alter their function. Macrophages can be activated or polarized into an M1, pro-inflammatory, classically activated phenotype, or M2, alternatively activated phenotype, and these phenotypes are distinguished by the expression of a variety of cell markers and production of cytokines/chemokines. Activation states exist on a continuum and are not a result of terminal differentiation but rather in response to their micro-environment; therefore, understanding how macrophages respond to various mechanical forces and how these mechanotransduction events govern macrophage phenotype has been an active area of research.
Macrophages exposed to cyclic strain increase various factors in relation to the amount of strain and frequency of stretch. Pugin et al. showed increased production of pro-inflammatory cytokines, such as TNFα, IL-6, IL-8, MMP9, and NF-κB activation, in human alveolar macrophages with 12% elongation of the culture membrane [127]. A 20% strain in murine alveolar macrophages induced IL-1β and IL-18 release, the latter dependent on caspase 1 and TLR4 signaling, along with inflammasome activation which required ROS [147]. Using macrophages derived from primary human peripheral blood mononuclear cells (PBMCs), Ballotta et al. showed that 7% deformation polarized macrophages toward an alternatively activated, M2 phenotype [148]. Mathesona et al. validated the Flexcell cyclic strain system and demonstrated that 10% strain increased IL-6, esterase, and acid phosphatase activity in the macrophage-like U937 cells and also led to an aligned and lengthened morphology [149]. Pakshir et al. discovered that macrophage attraction to sites of tissue repair was dependent on the dynamic strain of fibular collagen produced by fibroblast remodeling of the ECM and not the ECM structure itself [150]. The tethered fibers transmitted movement over hundreds of micrometers away and the macrophage mechanosensing was attributed to α2β1 integrin and stretch activated ion channels [150].
Varying the substrate stiffness on which macrophages are cultured influences phenotypic and morphologic changes. Culturing murine bone marrow derived macrophages (BMDMs) on a soft matrix (2.55 kPa) was associated with the expression of M1 markers including increased CD86 expression, rounded shape, and production of ROS, IL-1β, and TNFα [151]. However, macrophages cultured on stiff matrices (∼34.8 kPa) exhibited an M2 or alternatively activated phenotype as measured by elevated CD206 expression, diminished ROS production, elevated IL-4 and TGF-β secretion, and spindle-shaped morphology [151]. The morphological change associated with stiffness was confirmed by Fereol et al. who demonstrated that transitions from rounded cells to flattened cells accompanied an F-actin independent increase in cell stiffness [152]. Interestingly, in THP-1 cells, Sridhara et al. found classical activation or M1 phenotypes were associated with stiff culture gels (323 kPa) and M2 characteristics were associated with softer substrates (11 and 88 kPa) with M2 properties being ROCK dependent [153]. Investigation of macrophage response to either shear or compressive stimulus has been mostly limited to vascular applications with little to no consideration for the lung micro-environment. For a comprehensive review of macrophage mechanical stimulus studies beyond the pulmonary micro-environment please see Adams et al. [154].
3.3 Fibroblasts.
Fibroblasts are located within the connective tissue between alveoli (i.e., the pulmonary interstitium) and this interstitial space is subjected to three-dimensional tension as well as compression during respiration. Fibroblasts also experience shear stress from the interstitial fluid flow that accompanies dynamic changes in lung volume and fibroblasts can sense and contribute to stiffness changes via secretion of ECM proteins.
Pulmonary fibroblasts subjected to strain have altered responses depending on the strain magnitude and cell type. 5% strain has been shown to inhibit proliferation and increase apoptosis in fetal rat fibroblasts [155]. Interestingly, 20% strain on fetal human fibroblasts, IMR-90 cells, increased type I collagen expression [156] while the cells align perpendicular to the force [156] application. However, 10% strain on adult human tracheal fibroblasts increased proliferation [157], and cyclic strain reduced α-smooth muscle actin and expression of tenascin C, collagen I, III, and V [158]. A 30% strain increased the expression of inflammatory markers, such as IL-8, MMPs, and collagens, in fibroblasts isolated from asthmatic patients or normal, donor controls [159].
The influence of shear stress on pulmonary fibroblasts function has been less thoroughly explored, specifically when accounting for the 3D micro-environment. Under two-dimensional (2D) stimulus in a parallel plate flow chamber, Lee et al. found the nuclear movement in 3T3 cells was enhanced under shear flows and that microtubule structure was responsible for nuclei rotation and translocation [160]. Ng and Swartz investigated interstitial fluid flow effects on 3D human fibroblast (CCD1079sk) and concluded that cells align perpendicular to the flow direction and become spindle-like in shape [120]. Cell morphology has been implicated in phenotypic responses of cells including proliferation, apoptosis, migration, and mechanotransduction signaling [161].
Fibroblast response to matrix stiffness has been thoroughly investigated and an increase in stiffness is associated with heightened proliferation, matrix synthesis, and contractile functions [26,162,163]. The cell source, either diseased or normal lung tissue, impacts fibroblast behavior although these tendencies can be overcome by culturing diseased cells on soft substrates [162]. Additionally, Booth et al. demonstrated that decellularized matrices of diseased lungs (∼7.34 kPa stiffness) increase myofibroblast differentiation from normal fibroblasts when compared to normal lung matrices (∼1.6 kPa stiffness) [40]. Taken together, these studies indicate stiffness as a key component dictating the fibroblast function in both healthy and diseased lungs. The recent work by Matera et al. found opposite trends in stiffness response in 2D versus 3D, with less α-smooth muscle actin (myofibroblast indicator) and Ki67 (proliferation marker) expression at higher stiffnesses in 3D [164]. They determined that it was fiber density that drove myofibroblast differentiation, proliferation, and yes-associated protein (YAP) nuclear localization in 3D (functionalized dextran) culture not the stiffness of the environment [164]. Across studies, the key mechanotransduction pathways identified in orchestrating substrate stiffness responses in fibroblasts include YAP [164]/transcriptional co-activator with PDZ-binding motif (TAZ) [165], Rho/ROCK via actin, and MKL1 [163], and PGE2 [26,162]. However, PGE2 signaling may not be required for fibroblast stiffness response [162]. Finally, amplified substrate stiffness increases myofibroblast invasion of the basement membrane via the mechanosensing molecule α6-integrin [166]. By contrast, although fibroblasts experience compressive forces within the lung micro-environment little is known about the response of lung fibroblasts to compressive stimulus and is thus an opportunity for future investigations [167].
3.4 Pulmonary Endothelial Cells.
Pulmonary endothelial cells line blood vessel networks within the lung and are continuously exposed to fluid shear forces, ECM stiffness, cyclic strain, and fluid pressure [168,169]. Many studies have investigated the effect of physiologic and pathologic strains on pulmonary endothelial cells and their contribution to ventilator-induced lung injury. Physiologic strains (∼5% cyclic strain) do not induce a pro-inflammatory response [170,171] but can cause some reduction in endothelial barrier function [172] along with reduced fibronectin secretion at 4.9% and 12.5% strains [173]. Higher strains (∼10%) activate ERK, c-JUN N-terminal kinase, and p38 pathways [168] and can induce endothelial cell alignment and actin cytoskeletal remodeling with responses dependent on the magnitude of strain and type of mechanical stretch. The actin cytoskeleton and cell morphology reorienting in the direction of minimum substrate deformation and is more reliant on magnitude than deformation rate [174]. In multicellular models of physiologic breathing, 10% strain enhanced response to particulate exposure via ICAM-1 expression that was sufficient to attract circulating neutrophils [118]. Interestingly, 10% stretch did not activate Ca2+ channels but 20–30% strain resulted in activation of the actin cytoskeleton and Ca2+ influx [175]. At 15% strain, Russo et al. found reduced cellular migration and diminished capillary like tube formation but increased expression of a variety of proteoglycans, adhesive proteins, and growth factors that contributed to ECM remodeling and cell-matrix interactions [176]. Iwaki et al. demonstrated that 20% strain induced IL-8, IL-6, monocyte chemoattractant protein-1 production, and caused cellular alignment perpendicular to the stretch orientation with actin polymerization [170]. Tian et al. confirmed these inflammatory findings at 18% cyclic strain along with increased ICAM1 and monolayer barrier disruption via LPS [171]. Birukov et al. Confirmed endothelial barrier disruption with 18% elongation but in the absence of concurrent LPS stimulation [172]. Overall, it is evident that endothelial responses fluctuate with the magnitude of stretch becoming more inflammatory and disruptive as strain increases.
The effects of endothelial cells under shear stress stimulus have been thoroughly investigated by a variety of sources and we will only provide a brief overview of shear stress stimulated mechanotransduction responses in the pulmonary system. Shear stress is a known influencer of endothelial cell morphology (elongation and alignment in the direction of flow) [177], cytoskeletal remodeling [178], and cytosolic Ca2+ levels [179–181] which is a major signaling cascade in endothelial function [182]. Nitric oxide release is one of the most important and well documented effects of shear stress stimulation [183] contributing to the maintenance of vasomotor action, anti-inflammatory mechanisms, and anti-oxidant capacity of plasma [184]. With the application of pulsatile shear stress, endothelial cells upregulate their ACE2 expression which contributes to NO production and a reduction of proliferation and inflammation [185]. The recent work of Song et al. Demonstrated increased ACE2 expression when endothelial cells were stimulated in a Flexcell streamer [185]. As all mechanotransduction shear stress, endothelial studies are beyond the scope of this review, readers are referred to recent reviews for further details [186–188].
Compressive forces on endothelial cells are most commonly applied in vitro using hydrostatic fluid pressure which results in cell elongation and altered morphology via rearranged actin cytoskeleton [189,190], increased proliferation, and the formation of multilayer cell stacks [189–193]. The results of these hydrostatic pressure studies are remarkably consistent between research groups and the mechanisms of these observed changes may include the release of bFGF [191], αv integrins [193], vascular endothelial growth factor (VEGF)-C [194], and VE-cadherin [190]. Using pure uniaxial compression, Wille et al. found that endothelial cells are more responsive to stimulus magnitude not compression rate, and that actin stress fibers are disrupted and reoriented along with the cells long axes in response to the compression [195].
The majority of endothelial stiffness studies have been carried out with pulmonary hypertension as the primary target for investigation; however, many of these findings are relevant to the aberrant stiffness that pulmonary endothelial cells experience within a diseased lung. At higher stiffnesses (∼50 kPa), endothelial cells increased proliferation and migration through mechanotransduction pathways YAP/TAZ [196,197] and activate TGF-B, Toll-like receptors, transient receptor potential channels, and NF-kB [197]. Focal adhesions are increased on stiff substrates (10.3 kPa) [198] and heterogeneous matrix stiffness disrupts cellular connections [198]. Barrier disruption and recovery following thrombin stimulus were optimally regulated through the Rac/Rho signaling cascade when cultured on substrates of physiological stiffness (8.6 kPa) as oppose to stiffer (42 kPa) and softer (0.55 kPa) ranges [199]. Mambetsariev et al. confirmed these stiffness responses on a 40 kPa substrate which enhanced inflammatory response to LPS stimulus in the form of ICAM-1, vascular cell adhesion molecule (VCAM)-1, fibronectin, and guanine nucleotide factor (GEF)-H1 a Rho activator when compared to a soft (1.5 or 2.8 kPa) culture substrate [200].
The mechanotransduction studies discussed here are summarized in Table 2 for easy reference based on both cell type and mechanical stimulus.
Table 2.
Current findings of pulmonary mechanical stimulation studies in vitro
| Epithelial cells | Macrophages | Fibroblasts | Endothelial cells | |
|---|---|---|---|---|
| Strain | Tsuda et al. [126] | Pugin et al. [127] | Breen [156] | Gorfien et al. [173] |
| Savla and Waters [142] | Sanchez-Esteban et al. [155] | Kito et al. [168] | ||
| Tschumperlin et al. [113] | Matheson et al. [149] | Webb et al. [157] | Wang et al. [174] | |
| Waters et al. [129] | Wu et al. [147] | Blaauboerab et al. [158] | Birukov et al. [172] | |
| Chess et al. [106] | Ballotta et al. [148] | Manuyakorn et al. [159] | Iwaki et al. [170] | |
| Oswari et al. [114] | Pakshir et al. [150] | Ito et al. [175] | ||
| Oudin and Pugin [104] | Huh et al. [118] | |||
| Chess et al. [123] | Vion et al. [201] | |||
| Chapman et al. [124] | Tian et al. [171] | |||
| Chess et al. [107] | Russo et al. [176] | |||
| Chaturvedi et al. [105] | ||||
| Hu et al. [125] | ||||
| Felder et al. [119] | ||||
| Rapalo et al. [115] | ||||
| Rentzsch et al. [128] | ||||
| Felder et al. [112] | ||||
| Interfacial forces | Bilek et al. [134] | Area of need | N/A | N/A |
| Kay et al. [135] | ||||
| Huh et al. [202] | ||||
| Yalcin et al. [136] | ||||
| Ghadiali and Gaver [95] | ||||
| Ha et al. [203] | ||||
| Yalcin et al. [137] | ||||
| Tavana et al. [204] | ||||
| Higuita-Castro et al. [146] | ||||
| Higuita-Castro et al. [138] | ||||
| Viola et al. [139] | ||||
| Shear | Ridge et al. [131] | Area of need | Ng and Swartz [120] | Birukov et al. [178] |
| Nalayanda et al. [130] | Lee et al. [160] | Fisher et al. [188] | ||
| Sivaramakrishnan et al. [133] | Yamamoto et al. [180] | |||
| Flitney et al. [132] | Kumar et al. [183] | |||
| Mahto et al. [117] | Tousoulis et al. [184] | |||
| Szulcek et al. [177] | ||||
| Yamamoto et al. [181] | ||||
| Charbonier et al. [186] | ||||
| Wu and Birukov [187] | ||||
| Song et al. [185] | ||||
| Compression | Savla and Waters [142] | Area of need | Area of need | Acevedo et al. [191] |
| Tschumperlin et al. [140] | Sumpio et al. [192] | |||
| Chu et al. [141] | Salwen et al. [189] | |||
| Huang et al. [116] | Schwartz et al. [193] | |||
| Veerati et al. [55] | Shin et al. [194] | |||
| Wille et al. [195] | ||||
| Ohashi et al. [190] | ||||
| Stiffness | Eisenberg et al. [143] | Chen et al. [151] | Liu et al. [26] | Birukova et al. [199] |
| Markowski et al. [144] | Fereol et al. [152] | Huang et al. [163] | Mambetsariev et al. [200] | |
| Dysart et al. [145] | Sridharan et al. [153] | Booth et al. [40] | Bertero et al. [196] | |
| Marinkovic et al. [162] | Lampi et al. [198] | |||
| Chen et al. [166] | Thenappan et al. [197] | |||
| Noguchi et al. [165] | ||||
| Matera et al. [164] |
4 Discussion and Future Directions
The lung is a dynamic organ with a complex mechanical environment at the microscale. As a result, mechanotransduction plays a crucial role in both pulmonary health and disease. Over many years of investigation, researchers have confirmed the influence of mechanical forces on cellular proliferation, morphology, ECM production and restructuring, gene regulation, and inflammatory responses. While specific aspects of the lung mechanical environment have been investigated in isolation through various in vitro systems, the compound effects of cellular diversity, concurrent forces, and immune regulation have not been fully understood. For example, in the setting of IPF, although it is known that stiffening of the ECM regulates the functionality of fibroblasts, endothelial cells, and epithelial cells, we do not have information on how cell–cell interactions and communication alter the response to a stiffening environment. Importantly, we also do not understand how immune cells interact with other cell types and if these interactions alter the mechanotransduction responses that perpetuate disease conditions. Additionally, IPF alters the temporal and spatial strain/stiffness gradients within the lung while the dynamic remodeling of the ECM is ongoing, and these facets have yet to be recapitulated using in vitro bioreactor systems.
Despite the wide array of mechanotransduction studies performed in the field of pulmonary medicine, there remain large gaps in our understanding of the influence and response of macrophages and fibroblasts to the lung micro-environment collective. Where some work has explored the importance of tensile stress and substrate stiffness on polarization and morphology of macrophages, little effort has been devoted to understanding the role of shear stress, interfacial forces, and compressive stimulus in these innate immune cells. Additionally, current literature has not taken into account the various subtypes of macrophages present in the lung micro-environment and therefore the effects of mechanotransduction in regulating activation and functionality of interstitial macrophages, alveolar macrophages, and recruited macrophages remain unknown. The response of fibroblasts to compressive forces is also neglected despite its clear relevance in the contracting airways of asthmatics and shifting parenchyma throughout the breathing cycle. Once the impact of these individual force and cell pairings are investigated, a more in-depth understanding of mechanotransduction in the lung can be developed.
The mechanical profile of the lung needs to be better described throughout the progression of the disease. With novel sensing and imaging tools to characterize in vivo mechanics an accurate and immediate readout would both assist researchers in accurately recreating a physiological in vitro system and quantifying patient conditions for improved diagnosis. Techniques such as magnetic resonance elastography [205,206] and ultrasound elastography (US-E) [207] are particularly promising for the ability to map stiffnesses of body tissue. Combining these noninvasive imaging techniques with computational simulations would further quantify and personalize fibrosis assessment ideally predicting and preventing the progression of the disease. Additionally, the heterogeneous composition of the lung could be measured in vivo and personalized medicine could be applied based on our understanding of cell response to the degree of aberrant forces.
To achieve in vitro systems that recapitulate the complex physiology of the lung, we need next generation bioreactors that account for the innate complexity of the lung including cell–cell interactions, 3D geometry, cell-based matrix remodeling, long term cultures, and multiforce stimulants. No cell exists in isolation and intercellular communications across cell types are ongoing and continuously influence phenotypic responses. Better in vitro models will capture the multicellular interactions between macrophages, fibroblasts, epithelial, and endothelial cells and are necessary for a holistic view on the cooperative cellular responses. In addition, these cells in vivo do not exist in 2D, and since dimensionality has shown to drastically alter the phenotypic response, careful consideration for culture conditions should be made in the next generation devices. The 3D matrix in vivo experiences ongoing modifications from its cell inhabitants and allowing for cell mediated remodeling over long-term culture could thus recapitulate disease progression in vitro. Incorporating primary cells into these systems would also improve accuracy in culture response compared to immortalized cell lines often used as representative systems. Finally, the use of multiforce systems will be a challenging but necessary development for the progression of lung mechanotransduction in vitro research. To accomplish this, the current bioreactor designs will grow in complexity and it will be advantageous to independently control each force and confirm cell response to known stimulus magnitudes. Therefore, the ability to distinctly characterize the force fields within culture conditions needs to be maintained, and a strong association between in vitro modeling and computational models should be incorporated to confirm mechanical conditions. Though the complete integration of all these aspects may not be feasible initially, continual advances in the development of in vitro systems that recapitulate the complex physiology of the lung micro-environment will greatly expand our understanding of the mechanotransduction mechanisms responsible for lung diseases. These next-generation devices would also be an enabling technology that could be used for advanced drug screening and may help reduce the need for costly animal studies.
As cellular interactions and experimental results become more convoluted with the increasing complexity of in vitro systems, it will be necessary to incorporate cross discipline expertise. Integrating the work of molecular biologists, immunologists, physicians, and engineers will allow for faster and more accurate progress in this field. Identification of the signaling transduction pathways initiated by the different mechanical forces may provide specific targets to inhibit and possibly reverse changes that occur in the lung micro-environment during chronic disease. Drawing from these and other areas of research will give us a better avenue to develop drugs and treatment options as well as physiological in vitro high throughput screening mechanisms.
5 Conclusion
In conclusion, in the past several decades, scientists and engineers have developed numerous in vitro systems that expose pulmonary cells to specific mechanical conditions and these devices have greatly expanded our knowledge of how these mechanical factors contribute to disease pathogenesis. However, most of these systems investigate how a single force influenced one specific cell type and do not capture the complex multicellular, multiforce, and multidimensional conditions that exist in the lung. Therefore, the new goal within the pulmonary mechanotransduction field should be the development and incorporation of next generation in vitro models that more accurately simulate the lung micro-environment and its complex physiology. Studying the combined effects of these areas will build upon the established mechanotransduction findings and provide a clearer picture of lung physiology and disease progression. With the help of these advanced in vitro systems and collaboration across disciplines, researchers will be better equipped to develop improved treatment options and ultimately improved patient outcomes.
Conflict of Interest
The authors have no conflicts of interest to disclose.
Footnotes
References
- [1]. Divertie, M. B. , Cassan, S. M. , and Brown, A. L. , 1976, “ Ultrastructural Morphometry of the Blood-Air Barrier in Pulmonary Sarcoidosis,” Chest, 69(2), pp. 154–157. 10.1378/chest.69.2.154 [DOI] [PubMed] [Google Scholar]
- [2]. Bai, A. , Eidelman, D. H. , Hogg, J. C. , James, A. L. , Lambert, R. K. , Ludwig, M. S. , Martin, J. , McDonald, D. M. , Mitzner, W. A. , Okazawa, M. , and Et, A. , 1994, “ Proposed Nomenclature for Quantifying Subdivisions of the Bronchial Wall,” J. Appl. Physiol., 77(2), pp. 1011–1014. 10.1152/jappl.1994.77.2.1011 [DOI] [PubMed] [Google Scholar]
- [3]. Eskandari, M. , Nordgren, T. M. , and O'Connell, G. D. , 2019, “ Mechanics of Pulmonary Airways: Linking Structure to Function Through Constitutive Modeling, Biochemistry, and Histology,” Acta Biomater., 97, pp. 513–523. 10.1016/j.actbio.2019.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Gunst, S. J. , 2012, Chapter 104 - Airway Smooth Muscle and Asthma, Hill J. A. and Olson E. N., eds., Academic Press, Boston/Waltham, MA, pp. 1359–1369. [Google Scholar]
- [5]. Alper, S. , and Janssen, W. J. , eds., 2018, Lung Innate Immunity and Inflammation: Methods and Protocols, Springer, New York. [Google Scholar]
- [6]. Rogers, D. F. , 2003, “ The Airway Goblet Cell,” Int. J. Biochem. Cell Biol., 35(1), pp. 1–6. 10.1016/S1357-2725(02)00083-3 [DOI] [PubMed] [Google Scholar]
- [7]. Whitsett, J. A. , 2018, “ Airway Epithelial Differentiation and Mucociliary Clearance,” Ann. Am. Thorac. Soc., 15(Suppl_3), pp. S143–S148. 10.1513/AnnalsATS.201802-128AW [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8]. Zuo, W.-L. , Shenoy, S. A. , Li, S. , O'Beirne, S. L. , Strulovici-Barel, Y. , Leopold, P. L. , Wang, G. , Staudt, M. R. , Walters, M. S. , Mason, C. , Kaner, R. J. , Mezey, J. G. , and Crystal, R. G. , 2018, “ Ontogeny and Biology of Human Small Airway Epithelial Club Cells,” Am. J. Respir. Crit. Care Med., 198(11), pp. 1375–1388. 10.1164/rccm.201710-2107OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9]. Rokicki, W. , Rokicki, M. , Wojtacha, J. , and Dżeljijli, A. , 2016, “ The Role and Importance of Club Cells (Clara Cells) in the Pathogenesis of Some Respiratory Diseases,” Kardiochirurgia Torakochirurgia Pol. Pol. J. Cardio-Thorac. Surg., 1(1), pp. 26–30. 10.5114/kitp.2016.58961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Plopper, C. G. , and Hyde, D. M. , 2015, “ Chapter 7—Epithelial Cells of the Bronchiole,” Comparative Biology of the Normal Lung, Parent R. A., ed., 2nd ed., Academic Press, San Diego, CA, pp. 83–92. [Google Scholar]
- [11]. Juarez, E. , Nuñez, C. , Sada, E. , Ellner, J. J. , Schwander, S. K. , and Torres, M. , 2010, “ Differential Expression of Toll-Like Receptors on Human Alveolar Macrophages and Autologous Peripheral Monocytes,” Respir. Res., 11(1), p. 2. 10.1186/1465-9921-11-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]. Wissel, H. , Schulz, C. , Koehne, P. , Richter, E. , Maass, M. , and Rüdiger, M. , 2005, “ Chlamydophila Pneumoniae Induces Expression of Toll-Like Receptor 4 and Release of TNF-α and MIP-2 Via an NF-ΚB Pathway in Rat Type II Pneumocytes,” Respir. Res., 6(1), p. 51. 10.1186/1465-9921-6-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Barman, S. , Davidson, M. L. , Walker, L. M. , Anna, S. L. , and Zasadzinski, J. A. , 2020, “ Inflammation Product Effects on Dilatational Mechanics Can Trigger the Laplace Instability and Acute Respiratory Distress Syndrome,” Soft Matter, 16(29), pp. 6890–6901. 10.1039/D0SM00415D [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Autilio, C. , and Pérez-Gil, J. , 2019, “ Understanding the Principle Biophysics Concepts of Pulmonary Surfactant in Health and Disease,” Arch. Dis. Child. Fetal Neonatal Ed., 104(4), pp. F443–F451. 10.1136/archdischild-2018-315413 [DOI] [PubMed] [Google Scholar]
- [15]. Markart, P. , Ruppert, C. , Wygrecka, M. , Colaris, T. , Dahal, B. , Walmrath, D. , Harbach, H. , Wilhelm, J. , Seeger, W. , Schmidt, R. , and Guenther, A. , 2007, “ Patients With ARDS Show Improvement But Not Normalisation of Alveolar Surface Activity With Surfactant Treatment: Putative Role of Neutral Lipids,” Thorax, 62(7), pp. 588–594. 10.1136/thx.2006.062398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Parra, E. , and Pérez-Gil, J. , 2015, “ Composition, Structure and Mechanical Properties Define Performance of Pulmonary Surfactant Membranes and Films,” Chem. Phys. Lipids, 185, pp. 153–175. 10.1016/j.chemphyslip.2014.09.002 [DOI] [PubMed] [Google Scholar]
- [17]. Bienenstock, J. , 1984, “ The Lung as an Immunologic Organ,” Annu. Rev. Med., 35(1), pp. 49–62. 10.1146/annurev.me.35.020184.000405 [DOI] [PubMed] [Google Scholar]
- [18]. Liegeois, M. , Legrand, C. , Desmet, C. J. , Marichal, T. , and Bureau, F. , 2018, “ The Interstitial Macrophage: A Long-Neglected Piece in the Puzzle of Lung Immunity,” Cell. Immunol., 330, pp. 91–96. 10.1016/j.cellimm.2018.02.001 [DOI] [PubMed] [Google Scholar]
- [19]. Suki, B. , Stamenović, D. , and Hubmayr, R. , 2011, “Lung Parenchymal Mechanics,” Comprehensive Physiology, American Cancer Society, Boston, MA, pp. 1317–1351. 10.1002/cphy.c100033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. White, E. S. , 2015, “ Lung Extracellular Matrix and Fibroblast Function,” Ann. Am. Thorac. Soc., 12(Suppl. 1), pp. S30–S33. 10.1513/AnnalsATS.201406-240MG [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Sevin, C. M. , and Light, R. W. , 2011, “ Microscopic Anatomy of the Pleura,” Thorac. Surg. Clin., 21(2), pp. 173–175. 10.1016/j.thorsurg.2010.12.003 [DOI] [PubMed] [Google Scholar]
- [22]. El-Hashash, A. , 2018, “ Brief Overview of the Human Respiratory System Structure and Development,” Lung Stem Cell Behavior, El-Hashash A., ed., Springer International Publishing, Cham, Switzerland, pp. 1–3. [Google Scholar]
- [23]. Novak, C. M. , Horst, E. N. , Taylor, C. C. , Liu, C. Z. , and Mehta, G. , “Fluid Shear Stress Stimulates Breast Cancer Cells to Display Invasive and Chemoresistant Phenotypes While Upregulating PLAU in a 3D Bioreactor,” Biotechnol. Bioeng., 116(11), pp. 3084–3097. 10.1002/bit.27119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Novak, C. M. , Horst, E. N. , Lin, E. , and Mehta, G. , 2020, “ Compressive Stimulation Enhances Ovarian Cancer Proliferation, Invasion, Chemoresistance, and Mechanotransduction Via CDC42 in a 3D Bioreactor,” Cancers, 12(6), p. 1521. 10.3390/cancers12061521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Novak, C. , Horst, E. , and Mehta, G. , 2018, “ Mechanotransduction in Ovarian Cancer: Shearing Into the Unknown,” APL Bioeng., 2(3), p. 031701. 10.1063/1.5024386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]. Liu, F. , Mih, J. D. , Shea, B. S. , Kho, A. T. , Sharif, A. S. , Tager, A. M. , and Tschumperlin, D. J. , 2010, “ Feedback Amplification of Fibrosis Through Matrix Stiffening and COX-2 Suppression,” J. Cell Biol., 190(4), pp. 693–706. 10.1083/jcb.201004082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Li, F. , Sun, X. , Zhao, B. , Ma, J. , Zhang, Y. , Li, S. , Li, Y. , and Ma, X. , 2015, “ Effects of Cyclic Tension Stress on the Apoptosis of Osteoclasts In Vitro,” Exp. Ther. Med., 9(5), pp. 1955–1961. 10.3892/etm.2015.2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. Bregenzer, M. E. , Horst, E. N. , Mehta, P. , Novak, C. M. , Repetto, T. , and Mehta, G. , 2019, “ The Role of Cancer Stem Cells and Mechanical Forces in Ovarian Cancer Metastasis,” Cancers, 11(7), p. 1008. 10.3390/cancers11071008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. Jain, R. K. , Martin, J. D. , and Stylianopoulos, T. , 2014, “ The Role of Mechanical Forces in Tumor Growth and Therapy,” Annu. Rev. Biomed. Eng., 16(1), pp. 321–346. 10.1146/annurev-bioeng-071813-105259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. Ravasio, A. , Hobi, N. , Bertocchi, C. , Jesacher, A. , Dietl, P. , and Haller, T. , 2011, “ Interfacial Sensing by Alveolar Type II Cells: A New Concept in Lung Physiology?,” Am. J. Physiol.-Cell Physiol., 300(6), pp. C1456–C1465. 10.1152/ajpcell.00427.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Chen, Z. , Song, Y. , Hu, Z. , Zhang, S. , and Chen, Y. , 2015, “ An Estimation of Mechanical Stress on Alveolar Walls During Repetitive Alveolar Reopening and Closure,” J. Appl. Physiol., 119(3), pp. 190–201. 10.1152/japplphysiol.00112.2015 [DOI] [PubMed] [Google Scholar]
- [32]. Tarran, R. , Button, B. , and Boucher, R. C. , 2006, “ Regulation of Normal and Cystic Fibrosis Airway Surface Liquid Volume by Phasic Shear Stress,” Annu. Rev. Physiol., 68(1), pp. 543–561. 10.1146/annurev.physiol.68.072304.112754 [DOI] [PubMed] [Google Scholar]
- [33]. Roan, E. , and Waters, C. M. , 2011, “ What Do We Know About Mechanical Strain in Lung Alveoli?,” Am. J. Physiol. Lung Cell. Mol. Physiol., 301(5), pp. L625–L635. 10.1152/ajplung.00105.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Knudsen, L. , and Ochs, M. , 2018, “ The Micromechanics of Lung Alveoli: Structure and Function of Surfactant and Tissue Components,” Histochem. Cell Biol., 150(6), pp. 661–676. 10.1007/s00418-018-1747-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35]. Waters, C. M. , Roan, E. , and Navajas, D. , 2012, “ Mechanobiology in Lung Epithelial Cells: Measurements, Perturbations, and Responses,” Compr. Physiol., 2(1), pp. 1–29. 10.1002/cphy.c100090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Fredberg, J. J. , Inouye, D. , Miller, B. , Nathan, M. , Jafari, S. , Helioui Raboudi, S. , Butler, J. P. , and Shore, S. A. , 1997, “ Airway Smooth Muscle, Tidal Stretches, and Dynamically Determined Contractile States,” Am. J. Respir. Crit. Care Med., 156(6), pp. 1752–1759. 10.1164/ajrccm.156.6.9611016 [DOI] [PubMed] [Google Scholar]
- [37]. Hughes, J. M. , Hoppin, F. G. , and Mead, J. , 1972, “ Effect of Lung Inflation on Bronchial Length and Diameter in Excised Lungs,” J. Appl. Physiol., 32(1), pp. 25–35. 10.1152/jappl.1972.32.1.25 [DOI] [PubMed] [Google Scholar]
- [38]. Nieman, G. F. , Satalin, J. , Andrews, P. , Habashi, N. M. , and Gatto, L. A. , 2016, “ Lung Stress, Strain, and Energy Load: Engineering Concepts to Understand the Mechanism of Ventilator-Induced Lung Injury (VILI),” Inten. Care Med. Exp., 4(1), p. 16. 10.1186/s40635-016-0090-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]. Albert, R. K. , Smith, B. , Perlman, C. E. , and Schwartz, D. A. , 2019, “ Is Progression of Pulmonary Fibrosis Due to Ventilation-Induced Lung Injury?,” Am. J. Respir. Crit. Care Med., 200(2), pp. 140–151. 10.1164/rccm.201903-0497PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40]. Booth, A. J. , Hadley, R. , Cornett, A. M. , Dreffs, A. A. , Matthes, S. A. , Tsui, J. L. , Weiss, K. , Horowitz, J. C. , Fiore, V. F. , Barker, T. H. , Moore, B. B. , Martinez, F. J. , Niklason, L. E. , and White, E. S. , 2012, “ Acellular Normal and Fibrotic Human Lung Matrices as a Culture System for In Vitro Investigation,” Am. J. Respir. Crit. Care Med., 186(9), pp. 866–876. 10.1164/rccm.201204-0754OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41]. Birzle, A. M. , Martin, C. , Uhlig, S. , and Wall, W. A. , 2019, “ A Coupled Approach for Identification of Nonlinear and Compressible Material Models for Soft Tissue Based on Different Experimental Setups – Exemplified and Detailed for Lung Parenchyma,” J. Mech. Behav. Biomed. Mater., 94, pp. 126–143. 10.1016/j.jmbbm.2019.02.019 [DOI] [PubMed] [Google Scholar]
- [42]. de Hilster, R. H. J. , Sharma, P. K. , Jonker, M. R. , White, E. S. , Gercama, E. A. , Roobeek, M. , Timens, W. , Harmsen, M. C. , Hylkema, M. N. , and Burgess, J. K. , 2020, “ Human Lung Extracellular Matrix Hydrogels Resemble the Stiffness and Viscoelasticity of Native Lung Tissue,” Am. J. Physiol. Lung Cell. Mol. Physiol., 318(4), pp. L698–L704. 10.1152/ajplung.00451.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43]. Browning, I. B. , D'Alonzo, G. E. , and Tobin, M. J. , 1990, “ Importance of Respiratory Rate as an Indicator of Respiratory Dysfunction in Patients With Cystic Fibrosis,” Chest, 97(6), pp. 1317–1321. 10.1378/chest.97.6.1317 [DOI] [PubMed] [Google Scholar]
- [44]. Doryab, A. , Tas, S. , Taskin, M. B. , Yang, L. , Hilgendorff, A. , Groll, J. , Wagner, D. E. , and Schmid, O. , 2019, “ Evolution of Bioengineered Lung Models: Recent Advances and Challenges in Tissue Mimicry for Studying the Role of Mechanical Forces in Cell Biology,” Adv. Funct. Mater., 29(39), p. 1903114. 10.1002/adfm.201903114 [DOI] [Google Scholar]
- [45]. Al-Saiedy, M. , Gunasekara, L. , Green, F. , Pratt, R. , Chiu, A. , Yang, A. , Dennis, J. , Pieron, C. , Bjornson, C. , Winston, B. , and Amrein, M. , 2018, “ Surfactant Dysfunction in ARDS and Bronchiolitis is Repaired With Cyclodextrins,” Mil. Med., 183(suppl_1), pp. 207–215. 10.1093/milmed/usx204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46]. Griese, M. , Essl, R. , Schmidt, R. , Ballmann, M. , Paul, K. , Rietschel, E. , and Ratjen, F. , and the Beat Study Group, 2005, “ Sequential Analysis of Surfactant, Lung Function and Inflammation in Cystic Fibrosis Patients,” Respir. Res., 6(1), p. 133. 10.1186/1465-9921-6-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47]. Kouranos, V. , Ward, S. , Kokosi, M. A. , Castillo, D. , Chua, F. , Judge, E. P. , Thomas, S. , Van Tonder, F. , Devaraj, A. , Nicholson, A. G. , Maher, T. M. , Renzoni, E. A. , and Wells, A. U. , 2020, “ Mixed Ventilatory Defects in Pulmonary Sarcoidosis: Prevalence and Clinical Features,” Chest, 158(5), pp. 2007–2014. 10.1016/j.chest.2020.04.074 [DOI] [PubMed] [Google Scholar]
- [48]. Umbrello, M. , Formenti, P. , Bolgiaghi, L. , and Chiumello, D. , 2016, “ Current Concepts of ARDS: A Narrative Review,” Int. J. Mol. Sci., 18(1), p. 64. 10.3390/ijms18010064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49]. Em, N. , C, H. , Cs, B. , Gs, M. , Ph, S. , and Vl, C. , 2002, “ Acute Remodeling of Parenchyma in Pulmonary and Extrapulmonary ARDS. An Autopsy Study of Collagen-Elastic System Fibers,” Pathol. Res. Pract., 198(5), pp. 355–361. 10.1078/0344-0338-00266 [DOI] [PubMed] [Google Scholar]
- [50]. Du, K. , 2019, “ Noninvasive Ventilation in Patients With Acute Respiratory Distress Syndrome,” Crit. Care, 23(1), p. 358. 10.1186/s13054-019-2666-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51]. Mart, M. F. , and Ware, L. B. , 2020, “ The Long-Lasting Effects of the Acute Respiratory Distress Syndrome,” Expert Rev. Respir. Med., 14(6), pp. 577–586. 10.1080/17476348.2020.1743182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52]. Chapman, D. G. , and Irvin, C. G. , 2015, “ Mechanisms of Airway Hyperresponsiveness in Asthma: The Past, Present and Yet to Come,” Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol., 45(4), pp. 706–719. 10.1111/cea.12506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53]. Lauzon, A.-M. , and Martin, J. G. , 2016, “ Airway Hyperresponsiveness; Smooth Muscle as the Principal Actor,” F1000Research, 5, p. 306. 10.12688/f1000research.7422.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54]. Kılıç, A. , Ameli, A. , Park, J.-A. , Kho, A. T. , Tantisira, K. , Santolini, M. , Cheng, F. , Mitchel, J. A. , McGill, M. , O'Sullivan, M. J. , De Marzio, M. , Sharma, A. , Randell, S. H. , Drazen, J. M. , Fredberg, J. J. , and Weiss, S. T. , 2020, “ Mechanical Forces Induce an Asthma Gene Signature in Healthy Airway Epithelial Cells,” Sci. Rep., 10(1), p. 966. 10.1038/s41598-020-57755-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55]. Veerati, P. C. , Mitchel, J. A. , Reid, A. T. , Knight, D. A. , Bartlett, N. W. , Park, J.-A. , and Grainge, C. L. , 2020, “ Airway Mechanical Compression: Its Role in Asthma Pathogenesis and Progression,” Eur. Respir. Rev., 29(157), p. 190123. 10.1183/16000617.0123-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56]. Boonpiyathad, T. , Sözener, Z. C. , Satitsuksanoa, P. , and Akdis, C. A. , 2019, “ Immunologic Mechanisms in Asthma,” Semin. Immunol., 46, p. 101333. 10.1016/j.smim.2019.101333 [DOI] [PubMed] [Google Scholar]
- [57]. Wright, D. , Sharma, P. , Ryu, M.-H. , Rissé, P.-A. , Ngo, M. , Maarsingh, H. , Koziol-White, C. , Jha, A. , Halayko, A. J. , and West, A. R. , 2013, “ Models to Study Airway Smooth Muscle Contraction In Vivo, Ex Vivo and In Vitro: Implications in Understanding Asthma,” Pulm. Pharmacol. Ther., 26(1), pp. 24–36. 10.1016/j.pupt.2012.08.006 [DOI] [PubMed] [Google Scholar]
- [58]. Barnes, P. J. , 2008, “ Immunology of Asthma and Chronic Obstructive Pulmonary Disease,” Nat. Rev. Immunol., 8(3), pp. 183–192. 10.1038/nri2254 [DOI] [PubMed] [Google Scholar]
- [59]. Kaminsky, D. A. , 2011, “ Peripheral Lung Mechanics in Asthma: Exploring the Outer Limits,” Pulm. Pharmacol. Ther., 24(2), pp. 199–202. 10.1016/j.pupt.2010.12.001 [DOI] [PubMed] [Google Scholar]
- [60]. Hamid, Q. , 2012, “ Pathogenesis of Small Airways in Asthma,” Respir. Basel, 84(1), pp. 4–11. 10.1159/000339550 [DOI] [PubMed] [Google Scholar]
- [61]. Mostaço-Guidolin, L. B. , Osei, E. T. , Ullah, J. , Hajimohammadi, S. , Fouadi, M. , Li, X. , Li, V. , Shaheen, F. , Yang, C. X. , Chu, F. , Cole, D. J. , Brandsma, C.-A. , Heijink, I. H. , Maksym, G. N. , Walker, D. , and Hackett, T.-L. , 2019, “ Defective Fibrillar Collagen Organization by Fibroblasts Contributes to Airway Remodeling in Asthma,” Am. J. Respir. Crit. Care Med., 200(4), pp. 431–443. 10.1164/rccm.201810-1855OC [DOI] [PubMed] [Google Scholar]
- [62]. Elias, J. A. , Zhu, Z. , Chupp, G. , and Homer, R. J. , 1999, “ Airway Remodeling in Asthma,” J. Clin. Invest., 104(8), pp. 1001–1006. 10.1172/JCI8124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63]. Hayes, D. , 2011, “ A Review of Bronchiolitis Obliterans Syndrome and Therapeutic Strategies,” J. Cardiothorac. Surg., 6(1), p. 92. 10.1186/1749-8090-6-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64]. Sharples, L. D. , McNeil, K. , Stewart, S. , and Wallwork, J. , 2002, “ Risk Factors for Bronchiolitis Obliterans: A Systematic Review of Recent Publications,” J. Heart Lung Transplant., 21(2), pp. 271–281. 10.1016/S1053-2498(01)00360-6 [DOI] [PubMed] [Google Scholar]
- [65]. Laohaburanakit, P. , Chan, A. , and Allen, R. P. , 2003, “ Bronchiolitis Obliterans,” Clin. Rev. Allergy Immunol., 25(3), pp. 259–274. 10.1385/CRIAI:25:3:259 [DOI] [PubMed] [Google Scholar]
- [66].National Institutes of Health, National Center for Advancing Translational Sciences, 2016, “ Bronchiolitis Obliterans | Genetic and Rare Diseases Information Center (GARD) – An NCATS Program,” Genetic and Rare Diseases Information Center, Gaithersburg, MD, accessed Aug. 14, 2020, https://rarediseases.info.nih.gov/diseases/9551/bronchiolitis-obliterans
- [67]. Kinsella, J. P. , Greenough, A. , and Abman, S. H. , 2006, “ Bronchopulmonary Dysplasia,” Lancet, 367(9520), pp. 1421–1431. 10.1016/S0140-6736(06)68615-7 [DOI] [PubMed] [Google Scholar]
- [68]. Thébaud, B. , Goss, K. N. , Laughon, M. , Whitsett, J. A. , Abman, S. H. , Steinhorn, R. H. , Aschner, J. L. , Davis, P. G. , McGrath-Morrow, S. A. , Soll, R. F. , and Jobe, A. H. , 2019, “ Bronchopulmonary Dysplasia,” Nat. Rev. Dis. Primer, 5(1), p. 78. 10.1038/s41572-019-0127-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69]. Abman, S. H. , Bancalari, E. , and Jobe, A. , 2017, “ The Evolution of Bronchopulmonary Dysplasia After 50 Years,” Am. J. Respir. Crit. Care Med., 195(4), pp. 421–424. 10.1164/rccm.201611-2386ED [DOI] [PubMed] [Google Scholar]
- [70]. Lignelli, E. , Palumbo, F. , Myti, D. , and Morty, R. E. , 2019, “ Recent Advances in Our Understanding of the Mechanisms of Lung Alveolarization and Bronchopulmonary Dysplasia,” Am. J. Physiol. Lung Cell. Mol. Physiol., 317(6), pp. L832–L887. 10.1152/ajplung.00369.2019 [DOI] [PubMed] [Google Scholar]
- [71]. Halbert, R. J. , Natoli, J. L. , Gano, A. , Badamgarav, E. , Buist, A. S. , and Mannino, D. M. , 2006, “ Global Burden of COPD: Systematic Review and Meta-Analysis,” Eur. Respir. J., 28(3), pp. 523–532. 10.1183/09031936.06.00124605 [DOI] [PubMed] [Google Scholar]
- [72].World Health Organization, 2020, “ The Top 10 Causes of Death,” World Health Organization, Geneva, Switzerland, accessed Aug. 14, 2020, https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death
- [73]. Alqahtani, J. S. , Oyelade, T. , Aldhahir, A. M. , Alghamdi, S. M. , Almehmadi, M. , Alqahtani, A. S. , Quaderi, S. , Mandal, S. , and Hurst, J. R. , 2020, “ Prevalence, Severity and Mortality Associated With COPD and Smoking in Patients With COVID-19: A Rapid Systematic Review and Meta-Analysis,” PLoS One, 15(5), p. e0233147. 10.1371/journal.pone.0233147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74]. Tan, W. S. D. , Shen, H.-M. , and Wong, W. S. F. , 2019, “ Dysregulated Autophagy in COPD: A Pathogenic Process to Be Deciphered,” Pharmacol. Res., 144, pp. 1–7. 10.1016/j.phrs.2019.04.005 [DOI] [PubMed] [Google Scholar]
- [75]. Uriarte, J. J. , Uhl, F. E. , Pouliot, R. A. , Bou Jawde, S. A M. , Rolandsson Enes, S. , Suki, B. , and Weiss, D. J. , 2019, “Nanomechanical Assessment of Decellularized COPD Lung Scaffolds,” A107. Engineered and Remodelled Matrix Compartments, American Thoracic Society, New York, pp. A2557–A2557. 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A2557 [DOI] [Google Scholar]
- [76]. Rønnow, S. R. , Langholm, L. L. , Sand, J. M. B. , Thorlacius-Ussing, J. , Leeming, D. J. , Manon-Jensen, T. , Tal-Singer, R. , Miller, B. E. , Karsdal, M. A. , and Vestbo, J. , 2019, “ Specific Elastin Degradation Products Are Associated With Poor Outcome in the ECLIPSE COPD Cohort,” Sci. Rep., 9(1), p. 4064. 10.1038/s41598-019-40785-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77]. Burgess, J. K. , Mauad, T. , Tjin, G. , Karlsson, J. C. , and Westergren‐Thorsson, G. , 2016, “ The Extracellular Matrix – The Under‐Recognized Element in Lung Disease?,” J. Pathol., 240(4), pp. 397–409. 10.1002/path.4808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78]. Günther, A. , Korfei, M. , Mahavadi, P. , Beck, D. von der , Ruppert, C. , and Markart, P. , 2012, “ Unravelling the Progressive Pathophysiology of Idiopathic Pulmonary Fibrosis,” Eur. Respir. Rev., 21(124), pp. 152–160. 10.1183/09059180.00001012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79]. Ley, B. , Collard, H. R. , and King, T. E. , 2011, “ Clinical Course and Prediction of Survival in Idiopathic Pulmonary Fibrosis,” Am. J. Respir. Crit. Care Med., 183(4), pp. 431–440. 10.1164/rccm.201006-0894CI [DOI] [PubMed] [Google Scholar]
- [80]. Deng, Z. , Fear, M. W. , Suk Choi, Y. , Wood, F. M. , Allahham, A. , Mutsaers, S. E. , and Prêle, C. M. , 2020, “ The Extracellular Matrix and Mechanotransduction in Pulmonary Fibrosis,” Int. J. Biochem. Cell Biol., 126, p. 105802. 10.1016/j.biocel.2020.105802 [DOI] [PubMed] [Google Scholar]
- [81]. Taveira-Dasilva, A. M. , Steagall, W. K. , and Moss, J. , 2006, “ Lymphangioleiomyomatosis,” Cancer Control, 13(4), pp. 276–285. 10.1177/107327480601300405 [DOI] [PubMed] [Google Scholar]
- [82]. Glasgow, C. G. , Taveira–DaSilva, A. , Pacheco–Rodriguez, G. , Steagall, W. K. , Tsukada, K. , Cai, X. , El–Chemaly, S. , and Moss, J. , 2009, “ Involvement of Lymphatics in Lymphangioleiomyomatosis,” Lymphat. Res. Biol., 7(4), pp. 221–228. 10.1089/lrb.2009.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83]. Xu, K.-F. , Xu, W. , Liu, S. , Yu, J. , Tian, X. , Yang, Y. , Wang, S.-T. , Zhang, W. , Feng, R. , and Zhang, T. , 2020, “ Lymphangioleiomyomatosis,” Semin. Respir. Crit. Care Med., 41(02), pp. 256–268. 10.1055/s-0040-1702195 [DOI] [PubMed] [Google Scholar]
- [84]. Gopalakrishnan, V. , Yao, J. , Steagall, W. K. , Avila, N. A. , Taveira-DaSilva, A. M. , Stylianou, M. , Chen, M. Y. , and Moss, J. , 2019, “ Use of CT Imaging to Quantify Progression and Response to Treatment in Lymphangioleiomyomatosis,” Chest, 155(5), pp. 962–971. 10.1016/j.chest.2019.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85]. Weber, E. , Sozio, F. , Borghini, A. , Sestini, P. , and Renzoni, E. , 2018, “ Pulmonary Lymphatic Vessel Morphology: A Review,” Ann. Anat. Anat. Anz., 218, pp. 110–117. 10.1016/j.aanat.2018.02.011 [DOI] [PubMed] [Google Scholar]
- [86]. Taveira-Dasilva, A. M. , and Moss, J. , 2016, “ Epidemiology, Pathogenesis and Diagnosis of Lymphangioleiomyomatosis,” Expert Opin. Orphan Drugs, 4(4), pp. 369–378. 10.1517/21678707.2016.1148597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87]. James, W. E. , and Judson, M. A. , 2020, “ Therapeutic Strategies for Pulmonary Sarcoidosis,” Expert Rev. Respir. Med., 14(4), pp. 391–403. 10.1080/17476348.2020.1721284 [DOI] [PubMed] [Google Scholar]
- [88]. Spagnolo, P. , Rossi, G. , Trisolini, R. , Sverzellati, N. , Baughman, R. P. , and Wells, A. U. , 2018, “ Pulmonary Sarcoidosis,” Lancet Respir. Med., 6(5), pp. 389–402. 10.1016/S2213-2600(18)30064-X [DOI] [PubMed] [Google Scholar]
- [89]. Lynch, J. P. , Kazerooni, E. A. , and Gay, S. E. , 1997, “ Pulmonary Sarcoidosis,” Clin. Chest Med., 18(4), pp. 755–785. 10.1016/S0272-5231(05)70417-2 [DOI] [PubMed] [Google Scholar]
- [90]. Laohaburanakit, P. , and Chan, A. , 2003, “ Obstructive Sarcoidosis,” Clin. Rev. Allergy Immunol., 25(2), pp. 115–129. 10.1385/CRIAI:25:2:115 [DOI] [PubMed] [Google Scholar]
- [91]. Nieman, G. F. , Satalin, J. , Andrews, P. , Aiash, H. , Habashi, N. M. , and Gatto, L. A. , 2017, “ Personalizing Mechanical Ventilation According to Physiologic Parameters to Stabilize Alveoli and MinimizeVentilator Induced Lung Injury (VILI),” Inten. Care Med. Exp., 5(1), pp. 1–21. 10.1186/s40635-017-0121-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92]. Gaver, D. P. , Nieman, G. F. , Gatto, L. A. , Cereda, M. , Habashi, N. M. , and Bates, J. H. T. , 2020, “ The POOR Get POORer: A Hypothesis for the Pathogenesis of Ventilator-Induced Lung Injury,” Am. J. Respir. Crit. Care Med., 202(8), pp. 1081–1087. 10.1164/rccm.202002-0453CP [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93]. McGuinness, G. , Zhan, C. , Rosenberg, N. , Azour, L. , Wickstrom, M. , Mason, D. M. , Thomas, K. M. , and Moore, W. H. , 2020, “ High Incidence of Barotrauma in Patients With COVID-19 Infection on Invasive Mechanical Ventilation,” Radiology, 297(2), pp. E252–E262. 10.1148/radiol.2020202352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94]. Madahar, P. , and Beitler, J. R. , 2020, “ Emerging Concepts in Ventilation-Induced Lung Injury,” F1000Research, 9, p. 222. 10.12688/f1000research.20576.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95]. Ghadiali, S. N. , and Gaver, D. P. , 2008, “ Biomechanics of Liquid–Epithelium Interactions in Pulmonary Airways,” Respir. Physiol. Neurobiol., 163(1–3), pp. 232–243. 10.1016/j.resp.2008.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96]. Hager, D. N. , Krishnan, J. A. , Hayden, D. L. , and Brower, R. G. , 2005, “ Tidal Volume Reduction in Patients With Acute Lung Injury When Plateau Pressures Are Not High,” Am. J. Respir. Crit. Care Med., 172(10), pp. 1241–1245. 10.1164/rccm.200501-048CP [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97]. Ghadiali, S. N. , and Huang, Y. , 2011, “ Role of Airway Recruitment and Derecruitment in Lung Injury,” Crit. Rev. Biomed. Eng., 39(4), pp. 297–318. 10.1615/CritRevBiomedEng.v39.i4.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98]. Walkey, A. J. , Del Sorbo, L. , Hodgson, C. L. , Adhikari, N. K. J. , Wunsch, H. , Meade, M. O. , Uleryk, E. , Hess, D. , Talmor, D. S. , Thompson, B. T. , Brower, R. G. , and Fan, E. , 2017, “ Higher PEEP Versus Lower PEEP Strategies for Patients With Acute Respiratory Distress Syndrome. A Systematic Review and Meta-Analysis,” Ann. Am. Thorac. Soc., 14(Suppl_4), pp. S297–S303. 10.1513/AnnalsATS.201704-338OT [DOI] [PubMed] [Google Scholar]
- [99]. Herold, S. , Becker, C. , Ridge, K. M. , and Budinger, G. R. S. , 2015, “ Influenza Virus-Induced Lung Injury: Pathogenesis and Implications for Treatment,” Eur. Respir. J., 45(5), pp. 1463–1478. 10.1183/09031936.00186214 [DOI] [PubMed] [Google Scholar]
- [100]. Cruces, P. , Retamal, J. , Hurtado, D. E. , Erranz, B. , Iturrieta, P. , González, C. , and Díaz, F. , 2020, “ A Physiological Approach to Understand the Role of Respiratory Effort in the Progression of Lung Injury in SARS-CoV-2 Infection,” Crit. Care, 24(1), p. 494. 10.1186/s13054-020-03197-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101]. Yen, S. , Preissner, M. , Bennett, E. , Dubsky, S. , Carnibella, R. , O'Toole, R. , Roddam, L. , Jones, H. , Dargaville, P. A. , Fouras, A. , and Zosky, G. R. , 2019, “ The Link Between Regional Tidal Stretch and Lung Injury During Mechanical Ventilation,” Am. J. Respir. Cell Mol. Biol., 60(5), pp. 569–577. 10.1165/rcmb.2018-0143OC [DOI] [PubMed] [Google Scholar]
- [102]. Pociask, D. A. , Robinson, K. M. , Chen, K. , McHugh, K. J. , Clay, M. E. , Huang, G. T. , Benos, P. V. , Janssen-Heininger, Y. M. W. , Kolls, J. K. , Anathy, V. , and Alcorn, J. F. , 2017, “ Epigenetic and Transcriptomic Regulation of Lung Repair During Recovery From Influenza Infection,” Am. J. Pathol., 187(4), pp. 851–863. 10.1016/j.ajpath.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103]. Grillo, F. , Barisione, E. , Ball, L. , Mastracci, L. , and Fiocca, R. , 2020, “ Lung Fibrosis: An Undervalued Finding in COVID-19 Pathological Series,” Lancet Infect. Dis., 21(4), p. e72. 10.1016/S1473-3099(20)30582-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104]. Oudin, S. , and Pugin, J. , 2002, “ Role of MAP Kinase Activation in Interleukin-8 Production by Human BEAS-2B Bronchial Epithelial Cells Submitted to Cyclic Stretch,” Am. J. Respir. Cell Mol. Biol., 27(1), pp. 107–114. 10.1165/ajrcmb.27.1.4766 [DOI] [PubMed] [Google Scholar]
- [105]. Chaturvedi, L. S. , Marsh, H. M. , and Basson, M. D. , 2007, “ SRC and Focal Adhesion Kinase Mediate Mechanical Strain-Induced Proliferation and ERK1/2 Phosphorylation in Human H441 Pulmonary Epithelial Cells,” Am. J. Physiol. Cell Physiol., 292(5), pp. C1701–C1713. 10.1152/ajpcell.00529.2006 [DOI] [PubMed] [Google Scholar]
- [106]. Chess, P. R. , Toia, L. , and Finkelstein, J. N. , 2000, “ Mechanical Strain-Induced Proliferation and Signaling in Pulmonary Epithelial H441 Cells,” Am. J. Physiol. Lung Cell. Mol. Physiol., 279(1), pp. L43–L51. 10.1152/ajplung.2000.279.1.L43 [DOI] [PubMed] [Google Scholar]
- [107]. Chess, P. R. , O'Reilly, M. A. , Sachs, F. , and Finkelstein, J. N. , 2005, “ Reactive Oxidant and P42/44 MAP Kinase Signaling is Necessary for Mechanical Strain-Induced Proliferation in Pulmonary Epithelial Cells,” J. Appl. Physiol., 99(3), pp. 1226–1232. 10.1152/japplphysiol.01105.2004 [DOI] [PubMed] [Google Scholar]
- [108]. Ward, K. K. , Tancioni, I. , Lawson, C. , Miller, N. L. G. , Jean, C. , Chen, X. L. , Uryu, S. , Kim, J. , Tarin, D. , Stupack, D. G. , Plaxe, S. C. , and Schlaepfer, D. D. , 2013, “ Inhibition of Focal Adhesion Kinase (FAK) Activity Prevents Anchorage-Independent Ovarian Carcinoma Cell Growth and Tumor Progression,” Clin. Exp. Metastasis, 30(5), pp. 579–594. 10.1007/s10585-012-9562-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109]. Xiong, N. , Li, S. , Tang, K. , Bai, H. , Peng, Y. , Yang, H. , Wu, C. , and Liu, Y. , 2017, “ Involvement of Caveolin-1 in Low Shear Stress-Induced Breast Cancer Cell Motility and Adhesion: Roles of FAK/Src and ROCK/p-MLC Pathways,” Biochim. Biophys. Acta BBA Mol. Cell Res., 1864(1), pp. 12–22. 10.1016/j.bbamcr.2016.10.013 [DOI] [PubMed] [Google Scholar]
- [110]. Zebda, N. , Dubrovskyi, O. , and Birukov, K. G. , 2012, “ Focal Adhesion Kinase Regulation of Mechanotransduction and Its Impact on Endothelial Cell Functions,” Microvasc. Res., 83(1), pp. 71–81. 10.1016/j.mvr.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111]. Finkel, T. , 2011, “ Signal Transduction by Reactive Oxygen Species,” J. Cell Biol., 194(1), pp. 7–15. 10.1083/jcb.201102095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112]. Felder, M. , Trueeb, B. , Stucki, A. O. , Borcard, S. , Stucki, J. D. , Schnyder, B. , Geiser, T. , and Guenat, O. T. , 2019, “ Impaired Wound Healing of Alveolar Lung Epithelial Cells in a Breathing Lung-On-A-Chip,” Front. Bioeng. Biotechnol., 7, p. 3. 10.3389/fbioe.2019.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113]. Tschumperlin, D. J. , and Margulies, S. S. , 1998, “ Equibiaxial Deformation-Induced Injury of Alveolar Epithelial Cells In Vitro,” Am. J. Physiol. Lung Cell. Mol. Physiol., 275(6), pp. L1173–L1183. 10.1152/ajplung.1998.275.6.L1173 [DOI] [PubMed] [Google Scholar]
- [114]. Oswari, J. , Matthay, M. A. , and Margulies, S. S. , 2001, “ Keratinocyte Growth Factor Reduces Alveolar Epithelial Susceptibility to In Vitro Mechanical Deformation,” Am. J. Physiol. Lung Cell. Mol. Physiol., 281(5), pp. L1068–L1077. 10.1152/ajplung.2001.281.5.L1068 [DOI] [PubMed] [Google Scholar]
- [115]. Rápalo, G. , Herwig, J. D. , Hewitt, R. , Wilhelm, K. R. , Waters, C. M. , and Roan, E. , 2015, “ Live Cell Imaging During Mechanical Stretch,” J. Vis. Exp. JoVE, (102), p. 52737. 10.3791/52737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116]. Huang, Y. , Haas, C. , and Ghadiali, S. N. , 2010, “ Influence of Transmural Pressure and Cytoskeletal Structure on NF-ΚB Activation in Respiratory Epithelial Cells,” Cell. Mol. Bioeng., 3(4), pp. 415–427. 10.1007/s12195-010-0138-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117]. Mahto, S. K. , Tenenbaum-Katan, J. , Greenblum, A. , Rothen-Rutishauser, B. , and Sznitman, J. , 2014, “ Microfluidic Shear Stress-Regulated Surfactant Secretion in Alveolar Epithelial Type II Cells In Vitro,” Am. J. Physiol. Lung Cell. Mol. Physiol., 306(7), pp. L672–L683. 10.1152/ajplung.00106.2013 [DOI] [PubMed] [Google Scholar]
- [118]. Huh, D. , Matthews, B. D. , Mammoto, A. , Montoya-Zavala, M. , Hsin, H. Y. , and Ingber, D. E. , 2010, “ Reconstituting Organ-Level Lung Functions on a Chip,” Science, 328(5986), pp. 1662–1668. 10.1126/science.1188302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119]. Felder, E. , Siebenbrunner, M. , Busch, T. , Fois, G. , Miklavc, P. , Walther, P. , and Dietl, P. , 2008, “ Mechanical Strain of Alveolar Type II Cells in Culture: Changes in the Transcellular Cytokeratin Network and Adaptations,” Am. J. Physiol. Lung Cell. Mol. Physiol., 295(5), pp. L849–L857. 10.1152/ajplung.00503.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120]. Ng, C. P. , and Swartz, M. A. , 2003, “ Fibroblast Alignment Under Interstitial Fluid Flow Using a Novel 3-D Tissue Culture Model,” Am. J. Physiol. Heart Circ. Physiol., 284(5), pp. H1771–H1777. 10.1152/ajpheart.01008.2002 [DOI] [PubMed] [Google Scholar]
- [121]. Arora, H. , Mitchell, R. L. , Johnston, R. , Manolesos, M. , Howells, D. , Sherwood, J. M. , Bodey, A. J. , and Wanelik, K. , 2021, “ Correlating Local Volumetric Tissue Strains With Global Lung Mechanics Measurements,” Materials, 14(2), p. 439. 10.3390/ma14020439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122]. Mariano, C. A. , Sattari, S. , Maghsoudi-Ganjeh, M. , Tartibi, M. , Lo, D. D. , and Eskandari, M. , 2020, “ Novel Mechanical Strain Characterization of Ventilated Ex Vivo Porcine and Murine Lung Using Digital Image Correlation,” Front. Physiol., 11, p. 600492. 10.3389/fphys.2020.600492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123]. Chess, P. R. , O'Reilly, M. A. , and Toia, L. , 2004, “ Macroarray Analysis Reveals a Strain-Induced Oxidant Response in Pulmonary Epithelial Cells,” Exp. Lung Res., 30(8), pp. 739–753. 10.1080/01902140490517782 [DOI] [PubMed] [Google Scholar]
- [124]. Chapman, K. E. , Sinclair, S. E. , Zhuang, D. , Hassid, A. , Desai, L. P. , and Waters, C. M. , 2005, “ Cyclic Mechanical Strain Increases Reactive Oxygen Species Production in Pulmonary Epithelial Cells,” Am. J. Physiol. Lung Cell. Mol. Physiol., 289(5), pp. L834–L841. 10.1152/ajplung.00069.2005 [DOI] [PubMed] [Google Scholar]
- [125]. Hu, X. , Zhang, Y. , Cheng, D. , Ding, Y. , Yang, D. , Jiang, F. , Zhou, C. , Ying, B. , and Wen, F. , 2008, “ Mechanical Stress Upregulates Intercellular Adhesion Molecule-1 in Pulmonary Epithelial Cells,” Respiration, 76(3), pp. 344–350. 10.1159/000137509 [DOI] [PubMed] [Google Scholar]
- [126]. Tsuda, A. , Stringer, B. K. , Mijailovich, S. M. , Rogers, R. A. , Hamada, K. , and Gray, M. L. , 1999, “ Alveolar Cell Stretching in the Presence of Fibrous Particles Induces Interleukin-8 Responses,” Am. J. Respir. Cell Mol. Biol., 21(4), pp. 455–462. 10.1165/ajrcmb.21.4.3351 [DOI] [PubMed] [Google Scholar]
- [127]. Pugin, J. , Dunn, I. , Jolliet, P. , Tassaux, D. , Magnenat, J.-L. , Nicod, L. P. , and Chevrolet, J.-C. , 1998, “ Activation of Human Macrophages by Mechanical Ventilation In Vitro,” Am. J. Physiol. Lung Cell. Mol. Physiol., 275(6), pp. L1040–L1050. 10.1152/ajplung.1998.275.6.L1040 [DOI] [PubMed] [Google Scholar]
- [128]. Rentzsch, I. , Santos, C. L. , Huhle, R. , Ferreira, J. M. C. , Koch, T. , Schnabel, C. , Koch, E. , Pelosi, P. , Rocco, P. R. M. , and Abreu, M. G. de , 2017, “ Variable Stretch Reduces the Pro-Inflammatory Response of Alveolar Epithelial Cells,” PLoS One, 12(8), p. e0182369. 10.1371/journal.pone.0182369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129]. Waters, C. M. , Ridge, K. M. , Sunio, G. , Venetsanou, K. , and Sznajder, J. I. , 1999, “ Mechanical Stretching of Alveolar Epithelial Cells Increases Na+-K+-ATPase Activity,” J. Appl. Physiol., 87(2), pp. 715–721. 10.1152/jappl.1999.87.2.715 [DOI] [PubMed] [Google Scholar]
- [130]. Nalayanda, D. D. , Puleo, C. M. , Fulton, W. B. , Wang, T.-H. , and Abdullah, F. , 2007, “ Characterization of Pulmonary Cell Growth Parameters in a Continuous Perfusion Microfluidic Environment,” Exp. Lung Res., 33(6), pp. 321–335. 10.1080/01902140701557754 [DOI] [PubMed] [Google Scholar]
- [131]. Ridge, K. M. , Linz, L. , Flitney, F. W. , Kuczmarski, E. R. , Chou, Y.-H. , Omary, M. B. , Sznajder, J. I. , and Goldman, R. D. , 2005, “ Keratin 8 Phosphorylation by Protein Kinase C δ Regulates Shear Stress-Mediated Disassembly of Keratin Intermediate Filaments in Alveolar Epithelial Cells,” J. Biol. Chem., 280(34), pp. 30400–30405. 10.1074/jbc.M504239200 [DOI] [PubMed] [Google Scholar]
- [132]. Flitney, E. W. , Kuczmarski, E. R. , Adam, S. A. , and Goldman, R. D. , 2009, “ Insights Into the Mechanical Properties of Epithelial Cells: The Effects of Shear Stress on the Assembly and Remodeling of Keratin Intermediate Filaments,” Faseb J., 23(7), pp. 2110–2119. 10.1096/fj.08-124453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133]. Sivaramakrishnan, S. , Schneider, J. L. , Sitikov, A. , Goldman, R. D. , and Ridge, K. M. , 2009, “ Shear Stress Induced Reorganization of the Keratin Intermediate Filament Network Requires Phosphorylation by Protein Kinase C ζ,” Mol. Biol. Cell, 20(11), pp. 2755–2765. 10.1091/mbc.e08-10-1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134]. Bilek, A. M. , Dee, K. C. , and Gaver, D. P. , 2003, “ Mechanisms of Surface-Tension-Induced Epithelial Cell Damage in a Model of Pulmonary Airway Reopening,” J. Appl. Physiol., 94(2), pp. 770–783. 10.1152/japplphysiol.00764.2002 [DOI] [PubMed] [Google Scholar]
- [135]. Kay, S. S. , Bilek, A. M. , Dee, K. C. , and Gaver, D. P. , 2004, “ Pressure Gradient, Not Exposure Duration, Determines the Extent of Epithelial Cell Damage in a Model of Pulmonary Airway Reopening,” J. Appl. Physiol., 97(1), pp. 269–276. 10.1152/japplphysiol.01288.2003 [DOI] [PubMed] [Google Scholar]
- [136]. Yalcin, H. C. , Perry, S. F. , and Ghadiali, S. N. , 2007, “ Influence of Airway Diameter and Cell Confluence on Epithelial Cell Injury in an In Vitro Model of Airway Reopening,” J. Appl. Physiol., 103(5), pp. 1796–1807. 10.1152/japplphysiol.00164.2007 [DOI] [PubMed] [Google Scholar]
- [137]. Yalcin, H. C. , Hallow, K. M. , Wang, J. , Wei, M. T. , Ou-Yang, H. D. , and Ghadiali, S. N. , 2009, “ Influence of Cytoskeletal Structure and Mechanics on Epithelial Cell Injury During Cyclic Airway Reopening,” Am. J. Physiol. Lung Cell. Mol. Physiol., 297(5), pp. L881–L891. 10.1152/ajplung.90562.2008 [DOI] [PubMed] [Google Scholar]
- [138]. Higuita-Castro, N. , Shukla, V. C. , Mihai, C. , and Ghadiali, S. N. , 2016, “ Simvastatin Treatment Modulates Mechanically-Induced Injury and Inflammation in Respiratory Epithelial Cells,” Ann. Biomed. Eng., 44(12), pp. 3632–3644. 10.1007/s10439-016-1693-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139]. Viola, H. , Chang, J. , Grunwell, J. R. , Hecker, L. , Tirouvanziam, R. , Grotberg, J. B. , and Takayama, S. , 2019, “ Microphysiological Systems Modeling Acute Respiratory Distress Syndrome That Capture Mechanical Force-Induced Injury-Inflammation-Repair,” APL Bioeng., 3(4), p. 041503. 10.1063/1.5111549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140]. Tschumperlin, D. J. , Shively, J. D. , Swartz, M. A. , Silverman, E. S. , Haley, K. J. , Raab, G. , and Drazen, J. M. , 2002, “ Bronchial Epithelial Compression Regulates MAP Kinase Signaling and HB-EGF-Like Growth Factor Expression,” Am. J. Physiol. Lung Cell. Mol. Physiol., 282(5), pp. L904–L911. 10.1152/ajplung.00270.2001 [DOI] [PubMed] [Google Scholar]
- [141]. Chu, E. K. , Foley, J. S. , Cheng, J. , Patel, A. S. , Drazen, J. M. , and Tschumperlin, D. J. , 2005, “ Bronchial Epithelial Compression Regulates Epidermal Growth Factor Receptor Family Ligand Expression in an Autocrine Manner,” Am. J. Respir. Cell Mol. Biol., 32(5), pp. 373–380. 10.1165/rcmb.2004-0266OC [DOI] [PubMed] [Google Scholar]
- [142]. Savla, U. , and Waters, C. M. , 1998, “ Mechanical Strain Inhibits Repair of Airway Epithelium In Vitro,” Am. J. Physiol. Lung Cell. Mol. Physiol., 274(6), pp. L883–L892. 10.1152/ajplung.1998.274.6.L883 [DOI] [PubMed] [Google Scholar]
- [143]. Eisenberg, J. L. , Safi, A. , Wei, X. , Espinosa, H. D. , Budinger, G. S. , Takawira, D. , Hopkinson, S. B. , and Jones, J. C. , 2011, “ Substrate Stiffness Regulates Extracellular Matrix Deposition by Alveolar Epithelial Cells,” Res. Rep. Biol., 2011(2), pp. 1–12. 10.2147/RRB.S13178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144]. Markowski, M. C. , Brown, A. C. , and Barker, T. H. , 2012, “ Directing Epithelial to Mesenchymal Transition Through Engineered Microenvironments Displaying Orthogonal Adhesive and Mechanical Cues,” J. Biomed. Mater. Res. A, 100A(8), pp. 2119–2127. 10.1002/jbm.a.34068 [DOI] [PubMed] [Google Scholar]
- [145]. Dysart, M. M. , Galvis, B. R. , Russell, A. G. , and Barker, T. H. , 2014, “ Environmental Particulate (PM2.5) Augments Stiffness-Induced Alveolar Epithelial Cell Mechanoactivation of Transforming Growth Factor Beta,” PLoS One, 9(9), p. e106821. 10.1371/journal.pone.0106821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146]. Higuita-Castro, N. , Mihai, C. , Hansford, D. J. , and Ghadiali, S. N. , 2014, “ Influence of Airway Wall Compliance on Epithelial Cell Injury and Adhesion During Interfacial Flows,” J. Appl. Physiol., 117(11), pp. 1231–1242. 10.1152/japplphysiol.00752.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147]. Wu, J. , Yan, Z. , Schwartz, D. E. , Yu, J. , Malik, A. B. , and Hu, G. , 2013, “ Activation of NLRP3 Inflammasome in Alveolar Macrophages Contributes to Mechanical Stretch-Induced Lung Inflammationand Injury,” J. Immunol. Baltim. Md. 1950, 190(7), pp. 3590–3599. 10.4049/jimmunol.1200860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148]. Ballotta, V. , Driessen-Mol, A. , Bouten, C. V. C. , and Baaijens, F. P. T. , 2014, “ Strain-Dependent Modulation of Macrophage Polarization Within Scaffolds,” Biomaterials, 35(18), pp. 4919–4928. 10.1016/j.biomaterials.2014.03.002 [DOI] [PubMed] [Google Scholar]
- [149]. Matheson, L. A. , Jack Fairbank, N. , Maksym, G. N. , Paul Santerre, J. , and Labow, R. S. , 2006, “ Characterization of the FlexcellTM UniflexTM Cyclic Strain Culture System With U937 Macrophage-Like Cells,” Biomaterials, 27(2), pp. 226–233. 10.1016/j.biomaterials.2005.05.070 [DOI] [PubMed] [Google Scholar]
- [150]. Pakshir, P. , Alizadehgiashi, M. , Wong, B. , Coelho, N. M. , Chen, X. , Gong, Z. , Shenoy, V. B. , McCulloch, C. A. , and Hinz, B. , 2019, “ Dynamic Fibroblast Contractions Attract Remote Macrophages in Fibrillar Collagen Matrix,” Nat. Commun., 10(1), pp. 1–17. 10.1038/s41467-019-09709-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151]. Chen, M. , Zhang, Y. , Zhou, P. , Liu, X. , Zhao, H. , Zhou, X. , Gu, Q. , Li, B. , Zhu, X. , and Shi, Q. , 2020, “ Substrate Stiffness Modulates Bone Marrow-Derived Macrophage Polarization Through NF-ΚB Signaling Pathway,” Bioact. Mater., 5(4), pp. 880–890. 10.1016/j.bioactmat.2020.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152]. Féréol, S. , Fodil, R. , Labat, B. , Galiacy, S. , Laurent, V. M. , Louis, B. , Isabey, D. , and Planus, E. , 2006, “ Sensitivity of Alveolar Macrophages to Substrate Mechanical and Adhesive Properties,” Cell Motil., 63(6), pp. 321–340. 10.1002/cm.20130 [DOI] [PubMed] [Google Scholar]
- [153]. Sridharan, R. , Cavanagh, B. , Cameron, A. R. , Kelly, D. J. , and O'Brien, F. J. , 2019, “ Material Stiffness Influences the Polarization State, Function and Migration Mode of Macrophages,” Acta Biomater., 89, pp. 47–59. 10.1016/j.actbio.2019.02.048 [DOI] [PubMed] [Google Scholar]
- [154]. Adams, S. , Wuescher, L. M. , Worth, R. , and Yildirim-Ayan, E. , 2019, “ Mechano-Immunomodulation: Mechanoresponsive Changes in Macrophage Activity and Polarization,” Ann. Biomed. Eng., 47(11), pp. 2213–2231. 10.1007/s10439-019-02302-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155]. Sanchez-Esteban, J. , Wang, Y. , Cicchiello, L. A. , and Rubin, L. P. , 2002, “ Cyclic Mechanical Stretch Inhibits Cell Proliferation and Induces Apoptosis in Fetal Rat Lung Fibroblasts,” Am. J. Physiol. Lung Cell. Mol. Physiol., 282(3), pp. L448–L456. 10.1152/ajplung.00399.2000 [DOI] [PubMed] [Google Scholar]
- [156]. Breen, E. C. , 2000, “ Mechanical Strain Increases Type I Collagen Expression in Pulmonary Fibroblasts In Vitro,” J. Appl. Physiol., 88(1), pp. 203–209. 10.1152/jappl.2000.88.1.203 [DOI] [PubMed] [Google Scholar]
- [157]. Webb, K. , Hitchcock, R. W. , Smeal, R. M. , Li, W. , Gray, S. D. , and Tresco, P. A. , 2006, “ Cyclic Strain Increases Fibroblast Proliferation, Matrix Accumulation, and Elastic Modulus of Fibroblast-Seeded Polyurethane Constructs,” J. Biomech., 39(6), pp. 1136–1144. 10.1016/j.jbiomech.2004.08.026 [DOI] [PubMed] [Google Scholar]
- [158]. Blaauboer, M. E. , Smit, T. H. , Hanemaaijer, R. , Stoop, R. , and Everts, V. , 2011, “ Cyclic Mechanical Stretch Reduces Myofibroblast Differentiation of Primary Lung Fibroblasts,” Biochem. Biophys. Res. Commun., 404(1), pp. 23–27. 10.1016/j.bbrc.2010.11.033 [DOI] [PubMed] [Google Scholar]
- [159]. Manuyakorn, W. , Smart, D. E. , Noto, A. , Bucchieri, F. , Haitchi, H. M. , Holgate, S. T. , Howarth, P. H. , and Davies, D. E. , 2016, “ Mechanical Strain Causes Adaptive Change in Bronchial Fibroblasts Enhancing Profibrotic and Inflammatory Responses,” PLoS One, 11(4), p. e0153926. 10.1371/journal.pone.0153926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160]. Lee, J. S. H. , Chang, M. I. , Tseng, Y. , and Wirtz, D. , 2005, “ Cdc42 Mediates Nucleus Movement and MTOC Polarization in Swiss 3T3 Fibroblasts Under Mechanical Shear Stress,” Mol. Biol. Cell, 16(2), pp. 871–880. 10.1091/mbc.e03-12-0910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161]. Ingber, D. E. , 2008, “ Tensegrity-Based Mechanosensing From Macro to Micro,” Prog. Biophys. Mol. Biol., 97(2–3), pp. 163–179. 10.1016/j.pbiomolbio.2008.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162]. Marinković, A. , Liu, F. , and Tschumperlin, D. J. , 2013, “ Matrices of Physiologic Stiffness Potently Inactivate Idiopathic Pulmonary Fibrosis Fibroblasts,” Am. J. Respir. Cell Mol. Biol., 48(4), pp. 422–430. 10.1165/rcmb.2012-0335OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163]. Huang, X. , Yang, N. , Fiore, V. F. , Barker, T. H. , Sun, Y. , Morris, S. W. , Ding, Q. , Thannickal, V. J. , and Zhou, Y. , 2012, “ Matrix Stiffness–Induced Myofibroblast Differentiation is Mediated by Intrinsic Mechanotransduction,” Am. J. Respir. Cell Mol. Biol., 47(3), pp. 340–348. 10.1165/rcmb.2012-0050OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164]. Matera, D. L. , DiLillo, K. M. , Smith, M. R. , Davidson, C. D. , Parikh, R. , Said, M. , Wilke, C. A. , Lombaert, I. M. , Arnold, K. B. , Moore, B. B. , and Baker, B. M. , 2020, “ Microengineered 3D Pulmonary Interstitial Mimetics Highlight a Critical Role for Matrix Degradation in Myofibroblast Differentiation,” Sci. Adv., 6(37), p. eabb5069. 10.1126/sciadv.abb5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165]. Noguchi, S. , Saito, A. , Mikami, Y. , Urushiyama, H. , Horie, M. , Matsuzaki, H. , Takeshima, H. , Makita, K. , Miyashita, N. , Mitani, A. , Jo, T. , Yamauchi, Y. , Terasaki, Y. , and Nagase, T. , 2017, “ TAZ Contributes to Pulmonary Fibrosis by Activating Profibrotic Functions of Lung Fibroblasts,” Sci. Rep., 7(1), p. 42595. 10.1038/srep42595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166]. Chen, H. , Qu, J. , Huang, X. , Kurundkar, A. , Zhu, L. , Yang, N. , Venado, A. , Ding, Q. , Liu, G. , Antony, V. B. , Thannickal, V. J. , and Zhou, Y. , 2016, “ Mechanosensing by the α 6 -Integrin Confers an Invasive Fibroblast Phenotype and Mediates Lung Fibrosis,” Nat. Commun., 7(1), p. 12564. 10.1038/ncomms12564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167]. Tschumperlin, D. J. , and Drazen, J. M. , 2001, “ Mechanical Stimuli to Airway Remodeling,” Am. J. Respir. Crit. Care Med., 164(Suppl_2), pp. S90–S94. 10.1164/ajrccm.164.supplement_2.2106060 [DOI] [PubMed] [Google Scholar]
- [168]. Kito, H. , Chen, E. L. , Wang, X. , Ikeda, M. , Azuma, N. , Nakajima, N. , Gahtan, V. , and Sumpio, B. E. , 2000, “ Role of Mitogen-Activated Protein Kinases in Pulmonary Endothelial Cells Exposed to Cyclic Strain,” J. Appl. Physiol., 89(6), pp. 2391–2400. 10.1152/jappl.2000.89.6.2391 [DOI] [PubMed] [Google Scholar]
- [169]. Ochoa, C. D. , Baker, H. , Hasak, S. , Matyal, R. , Salam, A. , Hales, C. A. , Hancock, W. , and Quinn, D. A. , 2008, “ Cyclic Stretch Affects Pulmonary Endothelial Cell Control of Pulmonary Smooth Muscle Cell Growth,” Am. J. Respir. Cell Mol. Biol., 39(1), pp. 105–112. 10.1165/rcmb.2007-0283OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170]. Iwaki, M. , Ito, S. , Morioka, M. , Iwata, S. , Numaguchi, Y. , Ishii, M. , Kondo, M. , Kume, H. , Naruse, K. , Sokabe, M. , and Hasegawa, Y. , 2009, “ Mechanical Stretch Enhances IL-8 Production in Pulmonary Microvascular Endothelial Cells,” Biochem. Biophys. Res. Commun., 389(3), pp. 531–536. 10.1016/j.bbrc.2009.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171]. Tian, Y. , Gawlak, G. , O'Donnell, J. J. , Mambetsariev, I. , and Birukova, A. A. , 2016, “ Modulation of Endothelial Inflammation by Low and High Magnitude Cyclic Stretch,” PLoS One, 11(4), p. e0153387. 10.1371/journal.pone.0153387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172]. Birukov, K. G. , Jacobson, J. R. , Flores, A. A. , Ye, S. Q. , Birukova, A. A. , Verin, A. D. , and Garcia, J. G. N. , 2003, “ Magnitude-Dependent Regulation of Pulmonary Endothelial Cell Barrier Function by Cyclic Stretch,” Am. J. Physiol. Lung Cell. Mol. Physiol., 285(4), pp. L785–L797. 10.1152/ajplung.00336.2002 [DOI] [PubMed] [Google Scholar]
- [173]. Gorfien, S. F. , Winston, F. K. , Thibault, L. E. , and Macarak, E. J. , 1989, “ Effects of Biaxial Deformation on Pulmonary Artery Endothelial Cells,” J. Cell. Physiol., 139(3), pp. 492–500. 10.1002/jcp.1041390307 [DOI] [PubMed] [Google Scholar]
- [174]. Wang, J. H.-C. , Goldschmidt-Clermont, P. , Wille, J. , and Yin, F. C.-P. , 2001, “ Specificity of Endothelial Cell Reorientation in Response to Cyclic Mechanical Stretching,” J. Biomech., 34(12), pp. 1563–1572. 10.1016/S0021-9290(01)00150-6 [DOI] [PubMed] [Google Scholar]
- [175]. Ito, S. , Suki, B. , Kume, H. , Numaguchi, Y. , Ishii, M. , Iwaki, M. , Kondo, M. , Naruse, K. , Hasegawa, Y. , and Sokabe, M. , 2010, “ Actin Cytoskeleton Regulates Stretch-Activated Ca2+ Influx in Human Pulmonary Microvascular Endothelial Cells,” Am. J. Respir. Cell Mol. Biol., 43(1), pp. 26–34. 10.1165/rcmb.2009-0073OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176]. Russo, T. A. , Stoll, D. , Nader, H. B. , and Dreyfuss, J. L. , 2018, “ Mechanical Stretch Implications for Vascular Endothelial Cells: Altered Extracellular Matrix Synthesis and Remodeling in Pathological Conditions,” Life Sci., 213, pp. 214–225. 10.1016/j.lfs.2018.10.030 [DOI] [PubMed] [Google Scholar]
- [177]. Szulcek, R. , Happé, C. M. , Rol, N. , Fontijn, R. D. , Dickhoff, C. , Hartemink, K. J. , Grünberg, K. , Tu, L. , Timens, W. , Nossent, G. D. , Paul, M. A. , Leyen, T. A. , Horrevoets, A. J. , de Man, F. S. , Guignabert, C. , Yu, P. B. , Vonk-Noordegraaf, A. , Amerongen, G. P. V N. , and Bogaard, H. J. , 2016, “ Delayed Microvascular Shear Adaptation in Pulmonary Arterial Hypertension. Role of Platelet Endothelial Cell Adhesion Molecule-1 Cleavage,” Am. J. Respir. Crit. Care Med., 193(12), pp. 1410–1420. 10.1164/rccm.201506-1231OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178]. Birukov, K. G. , Birukova, A. A. , Dudek, S. M. , Verin, A. D. , Crow, M. T. , Zhan, X. , DePaola, N. , and Garcia, J. G. N. , 2002, “ Shear Stress-Mediated Cytoskeletal Remodeling and Cortactin Translocation in Pulmonary Endothelial Cells,” Am. J. Respir. Cell Mol. Biol., 26(4), pp. 453–464. 10.1165/ajrcmb.26.4.4725 [DOI] [PubMed] [Google Scholar]
- [179]. Schilling, W. P. , Mo, M. , and Eskin, S. G. , 1992, “ Effect of Shear Stress on Cytosolic Ca2+ of Calf Pulmonary Artery Endothelial Cells,” Exp. Cell Res., 198(1), pp. 31–35. 10.1016/0014-4827(92)90145-X [DOI] [PubMed] [Google Scholar]
- [180]. Yamamoto, K. , Sokabe, T. , Ohura, N. , Nakatsuka, H. , Kamiya, A. , and Ando, J. , 2003, “ Endogenously Released ATP Mediates Shear Stress-Induced Ca2+ Influx Into Pulmonary Artery Endothelial Cells,” Am. J. Physiol. Heart Circ. Physiol., 285(2), pp. H793–H803. 10.1152/ajpheart.01155.2002 [DOI] [PubMed] [Google Scholar]
- [181]. Yamamoto, K. , Imamura, H. , and Ando, J. , 2018, “ Shear Stress Augments Mitochondrial ATP Generation That Triggers ATP Release and Ca2+ Signaling in Vascular Endothelial Cells,” Am. J. Physiol. Heart Circ. Physiol., 315(5), pp. H1477–H1485. 10.1152/ajpheart.00204.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182]. Tran, Q.-K. , and Watanabe, H. , 2006, “ Calcium Signalling in the Endothelium,” The Vascular Endothelium, Moncada I. S., and Higgs A., eds., Springer, Berlin, Heidelberg, pp. 145–187. [DOI] [PubMed] [Google Scholar]
- [183]. Kumar, S. , Sud, N. , Fonseca, F. V. , Hou, Y. , and Black, S. M. , 2010, “ Shear Stress Stimulates Nitric Oxide Signaling in Pulmonary Arterial Endothelial Cells Via a Reduction in Catalase Activity: Role of Protein Kinase Cδ,” Am. J. Physiol. Lung Cell. Mol. Physiol., 298(1), pp. L105–L116. 10.1152/ajplung.00290.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184]. Tousoulis, D. , Kampoli, A.-M. , Tentolouris, C. , Papageorgiou, N. , and Stefanadis, C. , 2012, “ The Role of Nitric Oxide on Endothelial Function,” Curr. Vasc. Pharmacol., 10(1), pp. 4–18. 10.2174/157016112798829760 [DOI] [PubMed] [Google Scholar]
- [185]. Song, J. , Hu, B. , Qu, H. , Wang, L. , Huang, X. , Li, M. , and Zhang, M. , 2020, “ Upregulation of Angiotensin Converting Enzyme 2 by Shear Stress Reduced Inflammation and Proliferation in Vascular Endothelial Cells,” Biochem. Biophys. Res. Commun., 525(3), pp. 812–818. 10.1016/j.bbrc.2020.02.151 [DOI] [PubMed] [Google Scholar]
- [186]. Charbonier, F. W. , Zamani, M. , and Huang, N. F. , 2019, “ Endothelial Cell Mechanotransduction in the Dynamic Vascular Environment,” Adv. Biosyst., 3(2), p. 1800252. 10.1002/adbi.201800252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187]. Wu, D. , and Birukov, K. , 2019, “ Endothelial Cell Mechano-Metabolomic Coupling to Disease States in the Lung Microvasculature,” Front. Bioeng. Biotechnol., 7, p. 172. 10.3389/fbioe.2019.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188]. Fisher, A. B. , Al-Mehdi, A. B. , and Manevich, Y. , 2002, “ Shear Stress and Endothelial Cell Activation,” Crit. Care Med., 30(5 Suppl), pp. S192–S197. 10.1097/00003246-200205001-00004 [DOI] [PubMed] [Google Scholar]
- [189]. Salwen, S. A. , Szarowski, D. H. , Turner, J. N. , and Bizios, R. , 1998, “ Three-Dimensional Changes of the Cytoskeleton of Vascular Endothelial Cells Exposed to Sustained Hydrostatic Pressure,” Med. Biol. Eng. Comput., 36(4), pp. 520–527. 10.1007/BF02523225 [DOI] [PubMed] [Google Scholar]
- [190]. Ohashi, T. , Sugaya, Y. , Sakamoto, N. , and Sato, M. , 2007, “ Hydrostatic Pressure Influences Morphology and Expression of VE-Cadherin of Vascular Endothelial Cells,” J. Biomech., 40(11), pp. 2399–2405. 10.1016/j.jbiomech.2006.11.023 [DOI] [PubMed] [Google Scholar]
- [191]. Acevedo, A. D. , Bowser, S. S. , Gerritsen, M. E. , and Bizios, R. , 1993, “ Morphological and Proliferative Responses of Endothelial Cells to Hydrostatic Pressure: Role of Fibroblast Growth Factor,” J. Cell. Physiol., 157(3), pp. 603–614. 10.1002/jcp.1041570321 [DOI] [PubMed] [Google Scholar]
- [192]. Sumpio, B. E. , Widmann, M. D. , Ricotta, J. , Awolesi, M. A. , and Watase, M. , 1994, “ Increased Ambient Pressure Stimulates Proliferation and Morphologic Changes in Cultured Endothelial Cells,” J. Cell. Physiol., 158(1), pp. 133–139. 10.1002/jcp.1041580117 [DOI] [PubMed] [Google Scholar]
- [193]. Schwartz, E. A. , Bizios, R. , Medow, M. S. , and Gerritsen, M. E. , 1999, “ Exposure of Human Vascular Endothelial Cells to Sustained Hydrostatic Pressure Stimulates Proliferation,” Circ. Res., 84(3), pp. 315–322. 10.1161/01.RES.84.3.315 [DOI] [PubMed] [Google Scholar]
- [194]. Shin, H. Y. , Smith, M. L. , Toy, K. J. , Williams, P. M. , Bizios, R. , and Gerritsen, M. E. , 2002, “ VEGF-C Mediates Cyclic Pressure-Induced Endothelial Cell Proliferation,” Physiol. Genomics, 11(3), pp. 245–251. 10.1152/physiolgenomics.00068.2002 [DOI] [PubMed] [Google Scholar]
- [195]. Wille, J. J. , Ambrosi, C. M. , and Yin, F. C.-P. , 2004, “ Comparison of the Effects of Cyclic Stretching and Compression on Endothelial Cell Morphological Responses,” ASME J. Biomech. Eng., 126(5), pp. 545–551. 10.1115/1.1798053 [DOI] [PubMed] [Google Scholar]
- [196]. Bertero, T. , Oldham, W. M. , Cottrill, K. A. , Pisano, S. , Vanderpool, R. R. , Yu, Q. , Zhao, J. , Tai, Y. , Tang, Y. , Zhang, Y.-Y. , Rehman, S. , Sugahara, M. , Qi, Z. , Gorcsan, J. , Vargas, S. O. , Saggar, R. , Saggar, R. , Wallace, W. D. , Ross, D. J. , Haley, K. J. , Waxman, A. B. , Parikh, V. N. , Marco, T. D. , Hsue, P. Y. , Morris, A. , Simon, M. A. , Norris, K. A. , Gaggioli, C. , Loscalzo, J. , Fessel, J. , and Chan, S. Y. , 2016, “ Vascular Stiffness Mechanoactivates Yap/TAZ-Dependent Glutaminolysis to Drive Pulmonary Hypertension,” J. Clin. Invest., 126(9), pp. 3313–3335. 10.1172/JCI86387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197]. Thenappan, T. , Chan, S. Y. , and Weir, E. K. , 2018, “ Role of Extracellular Matrix in the Pathogenesis of Pulmonary Arterial Hypertension,” Am. J. Physiol. Heart Circ. Physiol., 315(5), pp. H1322–H1331. 10.1152/ajpheart.00136.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198]. Lampi, M. C. , Guvendiren, M. , Burdick, J. A. , and Reinhart-King, C. A. , 2017, “ Photopatterned Hydrogels to Investigate the Endothelial Cell Response to Matrix Stiffness Heterogeneity,” ACS Biomater. Sci. Eng., 3(11), pp. 3007–3016. 10.1021/acsbiomaterials.6b00633 [DOI] [PubMed] [Google Scholar]
- [199]. Birukova, A. A. , Tian, X. , Cokic, I. , Beckham, Y. , Gardel, M. L. , and Birukov, K. G. , 2013, “ Endothelial Barrier Disruption and Recovery is Controlled by Substrate Stiffness,” Microvasc. Res., 87, pp. 50–57. 10.1016/j.mvr.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200]. Mambetsariev, I. , Tian, Y. , Wu, T. , Lavoie, T. , Solway, J. , Birukov, K. G. , and Birukova, A. A. , 2014, “ Stiffness-Activated GEF-H1 Expression Exacerbates LPS-Induced Lung Inflammation,” PLoS One, 9(4), p. e92670. 10.1371/journal.pone.0092670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201]. Vion, A. C. , Birukova, A. A. , Boulanger, C. M. , and Birukov, K. G. , 2013, “ Mechanical Forces Stimulate Endothelial Microparticle Generation Via Caspase-Dependent Apoptosis-Independent Mechanism,” Pulm. Circ., 3(1), pp. 95–99. 10.4103/2045-8932.109921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202]. Huh, D. , Fujioka, H. , Tung, Y.-C. , Futai, N. , Paine, R. , Grotberg, J. B. , and Takayama, S. , 2007, “ Acoustically Detectable Cellular-Level Lung Injury Induced by Fluid Mechanical Stresses in Microfluidic Airway Systems,” Proc. Natl. Acad. Sci. U. S. A., 104(48), pp. 18886–18891. 10.1073/pnas.0610868104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Belete, H. A., Godin, L. M., Stroetz, R. W., and Hubmayr, R. D., 2010, “Experimental Models to Study Cell Wounding and Repair,” Cell Physiol. Biochem., 25(1), pp. 71–80. 10.1159/000272052 [DOI] [PubMed] [Google Scholar]
- [204]. Tavana, H. , Zamankhan, P. , Christensen, P. J. , Grotberg, J. B. , and Takayama, S. , 2011, “ Epithelium Damage and Protection During Reopening of Occluded Airways in a Physiologic Microfluidic Pulmonary Airway Model,” Biomed. Microdev., 13(4), pp. 731–742. 10.1007/s10544-011-9543-5 [DOI] [PubMed] [Google Scholar]
- [205]. Fakhouri, F. , Dong, H. , and Kolipaka, A. , 2019, “ Magnetic Resonance Elastography of the Lungs: A Repeatability and Reproducibility Study,” NMR Biomed., 32(7), p. e4102. 10.1002/nbm.4102 [DOI] [PubMed] [Google Scholar]
- [206]. Marinelli, J. P. , Levin, D. L. , Vassallo, R. , Carter, R. E. , Hubmayr, R. D. , Ehman, R. L. , and McGee, K. P. , 2017, “ Quantitative Assessment of Lung Stiffness in Patients With Interstitial Lung Disease Using MR Elastography,” J. Magn. Reson. Imaging, 46(2), pp. 365–374. 10.1002/jmri.25579 [DOI] [PubMed] [Google Scholar]
- [207]. Zhou, B. , Yang, X. , Zhang, X. , Curran, W. J. , and Liu, T. , 2020, “ Ultrasound Elastography for Lung Disease Assessment,” IEEE Trans. Ultrason. Ferroelectr. Freq. Control, 67(11), pp. 2249–2257. 10.1109/TUFFC.2020.3026536 [DOI] [PMC free article] [PubMed] [Google Scholar]