

Abstract
[FeFe]-hydrogenases use a unique organometallic complex, termed the H cluster, to reversibly convert H2 into protons and low-potential electrons. It can be best described as a [Fe4S4] cluster coupled to a unique [2Fe]H center where the reaction actually takes place. The latter corresponds to two iron atoms, each of which is bound by one CN− ligand and one CO ligand. The two iron atoms are connected by a unique azadithiolate molecule (−S−CH2−NH−CH2−S−) and an additional bridging CO. This [2Fe]H center is built stepwise thanks to the well-orchestrated action of maturating enzymes that belong to the Hyd machinery. Among them, HydG converts l-tyrosine into CO and CN− to produce a unique l-cysteine-Fe(CO)2CN species termed complex-B. Very recently, HydE was shown to perform radical-based chemistry using synthetic complex-B as a substrate. Here we report the high-resolution crystal structure that establishes the identity of the complex-B-bound HydE. By triggering the reaction prior to crystallization, we trapped a new five-coordinate Fe species, supporting the proposal that HydE performs complex modifications of complex-B to produce a monomeric “SFe(CO)2CN” precursor to the [2Fe]H center. Substrate access, product release, and intermediate transfer are also discussed.
Graphical Abstract
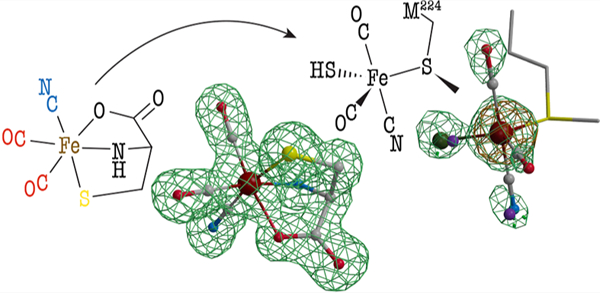
INTRODUCTION
Hydrogenases are metalloenzymes that efficiently catalyze the reversible oxidation of molecular hydrogen.1 Over the last 40 years, they have generated a constant interest in the context of using H2 as a renewable energy fuel. Depending on the metal content at their active site, three evolutionarily unrelated classes were defined, although all share a common feature: the presence of cyanide or carbon monoxide ligands bound to an iron atom. [FeFe]-hydrogenases are the most active in H2 generation. Their active site, the H cluster (see Figure 1), is deeply buried in the protein and consists of a [2Fe]H center linked to a [Fe4S4] cluster via a conserved cysteine residue.2 This [2Fe]H center consists of two iron atoms bound to a cyanide ligand and a carbon monoxide ligand. In addition, they are bridged by an additional CO ligand and a unique azadithiolate molecule. Whereas the [Fe4S4] cluster is synthesized and inserted into the apo-hydrogenase by generic house-keeping FeS cluster assembly machineries, the biosynthesis of the [2Fe]H center entails completely new biochemistry. Specifically, three enzymes—HydF, HydG, and HydE—orchestrate the formation of these components, which complement the diiron site.3,4 HydF is an [Fe4S4] cluster-containing GTPase and acts as a scaffold or insertase.5–7 Indeed, when incubated with either HydG or HydE or with a synthetic precursor of the [2Fe]H center, HydF can produce an active hydrogenase.6,8 HydG and HydE both belong to the radical (S)-adenosyl-l-methionine (SAM) superfamily.9 Most radical SAM enzymes use the one-electron reduction of an [Fe4S4] cluster to cleave the SAM into methionine and a highly reactive 5′-deoxyadenosyl radical (5′-dA●) species. The latter will, in turn, activate a broad range of substrates, usually through hydrogen-atom abstraction. HydG is a multifunctional enzyme responsible for the synthesis of the l-cysteine−Fe(CO)2(CN) complex, termed complex-B, as the precursor of the [2Fe]H subcluster (see Figure 1).10–14 It first uses 5′-dA● to abstract a hydrogen atom, likely from the amino group of its substrate l-tyrosine, which leads to the production of a 4-hydroxybenzyl radical and dehydroglycine (DHG).12,15 The former is subsequently converted into p-cresol, a byproduct of the reaction, while DHG is further processed to produce cyanide and carbon monoxide.10,11 The latter reaction involves a unique FeS cluster that in its resting state consists of a regular [Fe4S4] cluster combined with a dangling fifth iron that is coordinated to a l-cysteine, a conserved histidine residue, and either two water molecules or two hydroxyl groups (see Figure 1).13,16 Upon successive turnovers, this cluster is first converted into complex-A then complex-B (see Figure 1).17 Using a synthetic complex-B-containing compound termed syn-B, it was shown that cyanide, carbon monoxide, and the iron and sulfur atoms of complex-B are ultimately incorporated into the [2Fe]H subcluster of the [FeFe]-hydrogenase.14
Figure 1.
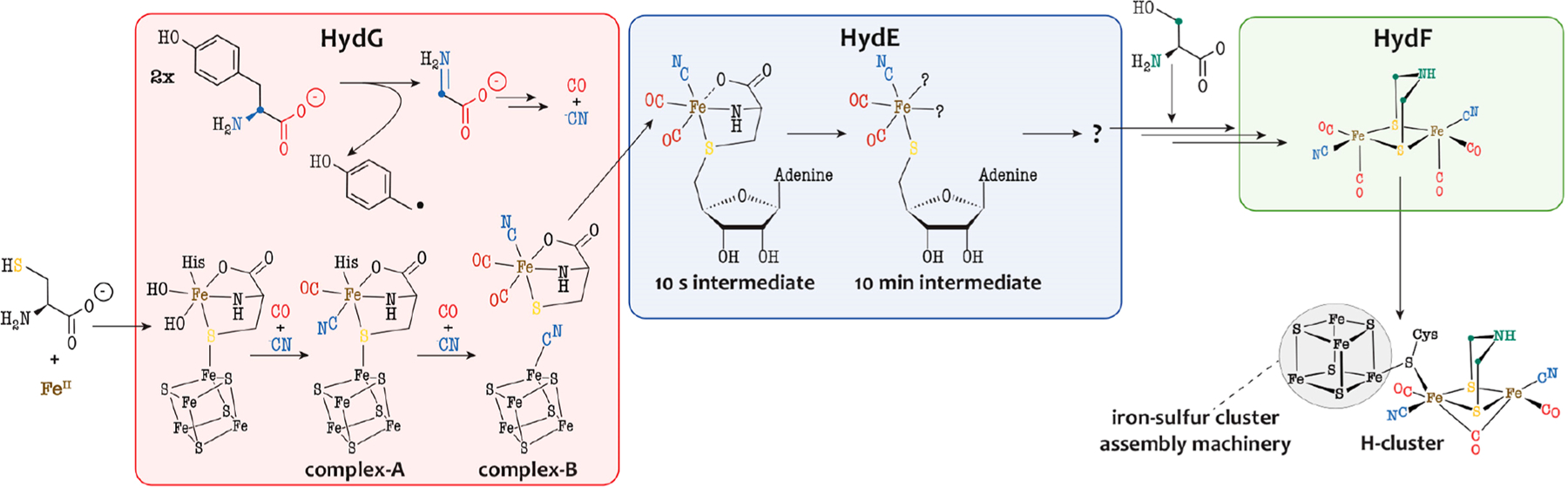
Biosynthesis of the [2Fe]H subcluster of the [FeFe]-hydrogenase active site. The Fe−CN−CO species depicted in the three Hyd proteins have been characterized by advanced spectroscopic techniques.6,17,20 In HydF, a synthetic [2Fe]H precursor was proposed to establish a [2Fe]Fe−NC−Fe[Fe4S4] link.
HydE was initially proposed to be involved in the synthesis of the azadithiolate ligand.18 Despite extensive studies, its substrate remained unidentified until very recently. HydE was shown to be able to process various thiazolidine compounds as nonphysiological substrates, catalyzing the radical-based addition of a 5′-dA● radical onto a sulfur atom to produce (S)-adenosyl-l-cysteine (SAC).19 Recently, it has been shown that, upon incubation with syn-B, HydE can successively produce two paramagnetic species (see Figure 1).20 The first one, which can be obtained 10 s after triggering the radical-based reaction, is a low-spin FeI complex coordinated by two carbon monoxide ligands, one cyanide ligand, and one SAC ligand (see Figure 1). Over the course of 10 min, this intermediate is converted into a second EPR-active species, which is another low-spin FeI complex coordinated by two carbon monoxide ligands, one cyanide ligand, and one 5′-thioadenosine ligand (see Figure 1).20 The formation of this second intermediate is coupled to the release of pyruvate as a byproduct from the l-cysteine C/N-fragment. This second intermediate was proposed to subsequently dimerize with release of a deoxyadenosyl fragment prior to its transfer to HydF, where the −CH2−NH−CH2− bridgehead issued from l-serine would be inserted.21
Here we report on the crystal structure of HydE from Thermotoga maritima (TmHydE) in complex with complex-B. We have identified the precise stereochemistry of the hexacoordinated octahedral complex-B. We also triggered the reaction prior to crystallization and trapped a new organometallic species, supporting the existence of a new mononuclear iron intermediate in the time course of the synthesis of the [2Fe]H subcluster of the [FeFe]-hydrogenase. Taken together, our findings support a mechanism that involves extensive ligand rearrangements and iron coordination changes to pass from complex-B to a unique HSFeI(CO)2CN intermediate. This work provides mechanistic insights into the role HydE plays in the enzymatic assembly of the H cluster.
RESULTS
Complex-B Bound to HydE and the Complex-B Structure.
HydE from T. maritima contains two [Fe4S4] clusters. The first one is strictly conserved among HydE enzymes and corresponds to the radical SAM cluster ([Fe4S4]RS) responsible for SAM binding and cleavage. The second cluster, termed the auxiliary cluster ([Fe4S4]Aux) is not conserved and can be removed without affecting the hydrogenase maturation process.22 During this study, we used two HydE constructs: the wild-type enzyme (TmHydE) and TmHydEΔFeS, where [Fe4S4]Aux cysteine residues are replaced by serine residues and therefore lack this auxiliary cluster (see the Supporting Information). Syn-B, which likely corresponds to a FeIII2(complex-B)4 complex,14 is unstable in an aqueous solution. When this material was solubilized in water at room temperature, we observed release of CO within seconds. In order to minimize this effect, we dissolved the syn-B powder in water a few seconds prior to adding the solution to a concentrated HydE sample. Incubating TmHydEΔFeS with 5 mM syn-B and (S)-adenosyl-l-homocysteine (SAH) led to hollow rod-shaped brownish crystals that diffracted to a 1.3 Å resolution (crystal-1; see Tables S1 and S2). SAH is often used as an inactive form of SAM in order to avoid the cleavage reaction of any uncoupled SAM. Despite being obtained in the same conditions as those previously reported,22 these crystals belong to a different space group (P21212 instead of P212121). The resulting crystal structure is, however, comparable to the previously reported ones, with a root means square deviation of 0.4 Å over 342 superimposed Cα atoms (see PDB 3IIX for the reference structure). The main difference is localized in a flexible loop (residues 309−315; see Figure S1), which does not affect the integrity of the active site cavity. Inspection of the Fo − Fc difference Fourier map clearly shows features that can best be modeled as complex-B bound in the “upper” part of the TmHydE β-barrel cavity (see Figures 2a and S2). Within these crystals, complex-B displays a 100% occupancy and is stable for weeks. The cyanide and carbon monoxide cannot be unambiguously distinguished based only on the diffraction data despite the high resolution. However, as observed in both [NiFe]- and [FeFe]-hydrogenases, cyanide is expected to establish hydrogen bonds, whereas carbon monoxide would occupy hydrophobic pockets (see Figures 2b and S3).23,24 Thus, considering the surrounding environment of the three diatomic ligands, the precise stereochemistry of the octahedral ferrous complex can be precisely determined. l-Cysteine is a facial, tridentate, and dianionic ligand. The carbon monoxide ligands are bound trans to the carboxylate and amine, while the cyanide ligand is trans to the thiolate. When positioning complex-B in the HydG structure,16,25 the two carbon monoxide ligands sit at the same positions as those of the two hydroxyl or water molecules. The cyanide ligand falls at the position of the conserved H265 residue (see Figure S4).
Figure 2.
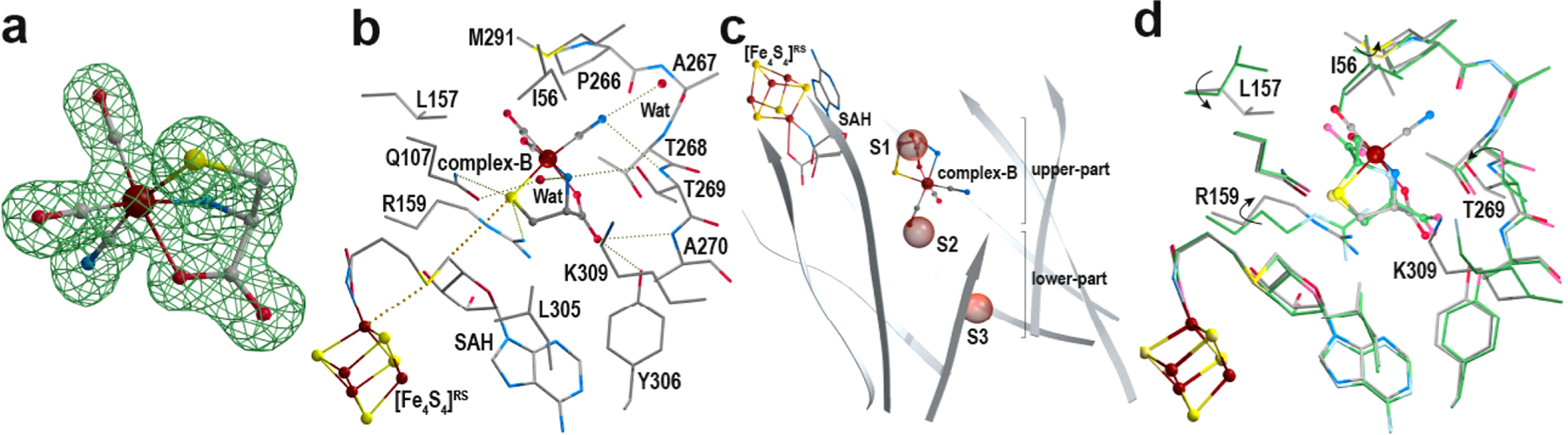
Complex-B crystal structure. (a) Structure of complex-B surrounded by its corresponding Fo − Fc “omit” difference Fourier electron density map, which was calculated using a model without complex-B. The difference Fourier map is depicted as a green mesh contoured at 4σ. The complex-B model is depicted as ball and sticks with the iron in brown and the N, C, O, and S atoms in blue, gray, red, and yellow, respectively. (b) Active site cavity (upper part of the β-barrel) of the complex-B-bound HydE crystal structure (crystal-1). Complex-B and the [Fe4S4]RS cluster are depicted in the ball-and-stick form. The surrounding residues and (S)-adenosyl-l-homocysteine (SAH) are depicted as sticks. The color code is the same as that in panel a. (c) View of the β-barrel cavity defined by the β-strands in gray. Complex-B and the [Fe4S4]RS cluster are depicted in the ball-and-stick form, whereas SAH is depicted as sticks. The color code is the same as that in panels a and b. Anion-binding sites S1, S2, and S3 are indicated as red semitransparent spheres.22 (d) Superposition of the crystal structures of HydE complexed with either complex-B or (2R,4R)-MeTDA (PDB 5FEP)19 in the same orientation as that in panel b. We want to point out here that the ligand-free and (2R,4R)-MeTDA-bound structures display identical conformations for the side-chain residues that define the active site cavity. Therefore, for simplicity, we only present the overlay between the complex-B- and (2R,4R)-MeTDA-bound structures. The latter is depicted in faded colors with the carbon atoms in green. Side chain rotations induced by complex-B binding are indicated with arrows. Stereoviews of each panel are available in Figures S2, S3, S5, and S6, respectively.
Conformational Responses to Binding Complex-B.
Previous structural studies on TmHydE showed that the β-barrel delineates a very large cavity consisting of two connected pockets.22 The radical-based reaction occurs in the first pocket near the [Fe4S4]RS cluster and the SAM binding site. The second pocket, which sits at the bottom of the β-barrel, was proposed to serve as a binding site for the product of the reaction in the upper pocket while it awaits a transfer to partner proteins such as HydF.22 Three anion binding sites, termed S1, S2, and S3, decorate the cavity and were proposed to play a role in HydE function, which has yet to be determined (see Figures 2c and S5). the binding of complex-B to the upper cavity induces the motions of side-chain residues (residues I56, L157, R159, T269, and K309; see Figures 2d and S6). Although several structural studies have been conducted on HydE, such side-chain shifts were not observed previously.26 For instance, the above-mentioned side-chain residues occupy the same position in all the previously reported HydE crystal structures, including the ligand-free and thiazolidine-bound ones. As can be seen in Figures 2d and S6, the rotation of residues R159 and L157 and the translation of T269 compress the active site cavity, clamping complex B. To check whether such an embrace is induced by complex-B itself or comes from changes in the crystal packing, we cocrystallized wild-type TmHydE with syn-B and SAH. The presence of the semiconserved [Fe4S4]Aux cluster, while it does not play any identified role in the hydrogenase maturation process,22 always induces crystallization in the original P212121 space group crystal form by locking the flexible loop into a P21212-incompatible conformation. Indeed, rod-shaped brownish crystals that belong to the P212121 space group and those that diffract to a 1.4 Å resolution were obtained (crystal-2; see Tables S1 and S2). Excluding both the above-mentioned motion of the loop containing residues 309−315 and the presence of a partially damaged [Fe4S4]Aux cluster,27 the two structures are identical (see Figure S1). This similarity includes the complex-B binding mode and the above-mentioned R159, L157, and T269 side-chain motions. Overall, these findings support a specific induced fit of the active site cavity upon binding with complex-B. In the ligand-free structure, site S1 bound a disordered chloride ion and was thus proposed as a binding site for the carboxylate moiety. This was further supported by crystal structures of HydE in complex with different ligands, notably (2R,4R)-2-methyl-1,3-thiazolidine-2,4-dicarboxylic acid ((2R,4R)-MeTDA, PDB 5FEP; see Figure S7).19 In addition, the (2R,4R)-MeTDA and complex-B sulfur atoms both sit at exactly the same position (see Figure 2d), further supporting a sulfur binding site that is sandwiched between the conserved residues R159 and Q107.19 It is known that substitution of these residues strongly affected the hydrogenase maturation process, supporting an impairment of the HydE activity.22 Complex-B binding also modifies the ratio of residue M291 side-chain alternative conformations. This residue has been proposed to be part of a valve that would separate the upper and lower parts of the β-barrel cavity.22 An iodide ion was also observed at site S3 in the lower part of the β-barrel. This iodide ion comes from the decomposition of syn-B.28 Iodide only accounts for about 50% occupancy, and the other 50% likely correspond to a chloride ion at the same position.22
In Vitro Reaction before Crystallization.
To investigate the conversion of enzyme-bound complex-B to the 10 s and 10 min EPR-active intermediates,20 we incubated TmHydEΔFeS with 1 equiv of SAM and a fivefold excess of syn-B. Dithionite was added to reductively trigger the reaction just prior to crystallization trials. The as-obtained brownish rod-shaped crystals belong to the P21212 space group and led to X-ray diffraction data at a 1.5 Å resolution (crystal-3 and crystal-4; see Tables S1 and S2). The intact complex-B was observed in the upper pocket of the β-barrel, as described above. In addition, features in the Fo − Fc difference Fourier electron density map (see Figures 3a and S8) clearly indicate that SAC is bound to the [Fe4S4]RScluster instead of SAM, SAH, or 5′-dA + methionine. The presence of SAC supports an attack on the sulfur atom of complex-B by the 5′-dA● radical. A similar experiment was conducted using the selenocysteine analogue of syn-B (syn-B-Se).28 X-ray diffraction data were recorded at energies near those of the selenium K-absorption edge (12.5 and 12.8 keV, Se K-edge at 12.666 keV; crystal-5, see Tables S1 and S2). The resulting anomalous difference Fourier electron density map clearly indicates the presence of a selenium atom and thus confirms the formation of either SAC or (S)-adenosyl-l-selenocysteine bound to the [Fe4S4]RS cluster (see Figures 3a and S8). We conclude that SAC results from the decay of the 10 s intermediate. The occupancy of the SAC molecule is nearly 100%, supporting a complete reaction between 5′-dA● and complex-B. The observed complex-B-bound state results from the displacement of SAC toward the [Fe4S4]RS cluster before the complete degradation of syn-B in solution. At this stage, we do not have any accurate kinetic data for the reaction. However, at odds with the almost 2 h necessary to observe a complete turnover when using (2R,4R)-MeTDA, it is likely that all these events, i.e., complex-B binding, SAM cleavage,10 s intermediate formation, and decay, occur within few minutes or at least before a complete decay of the remaining syn-B in solution.
Figure 3.
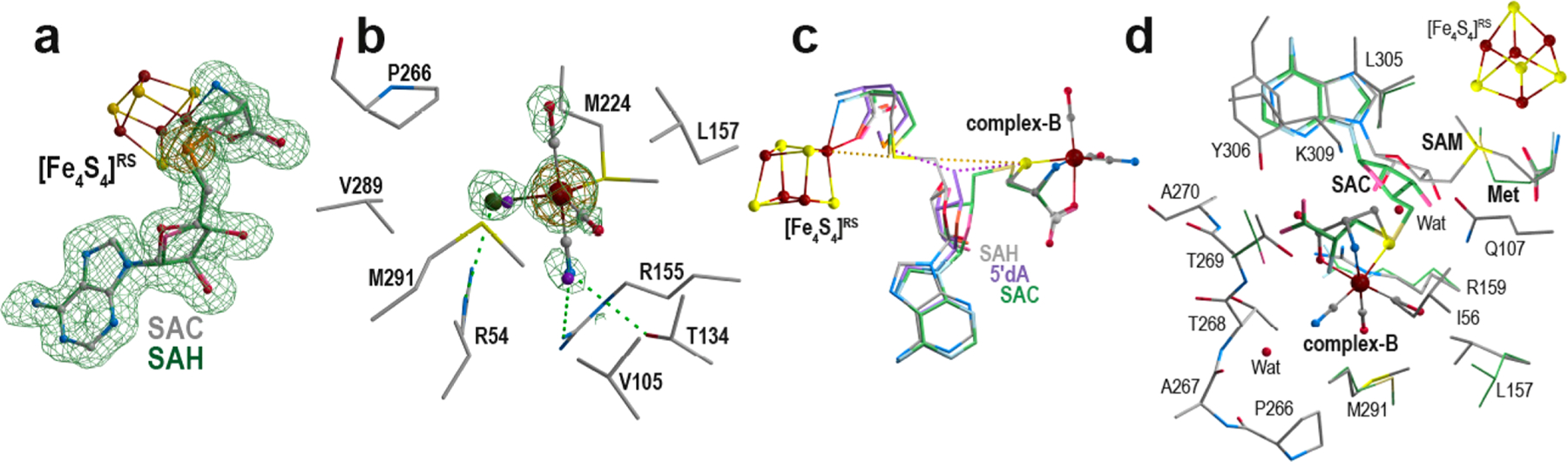
Complex-B conversion by HydE. (a) (S)-Adenosyl-l-selenocysteine molecule bound to the [Fe4S4]RS cluster in crystal RRU18. The corresponding Fo − Fc “omit” difference Fourier electron density map is contoured at 3σ and depicted as green mesh. The recorded anomalous difference Fourier map (energy of 12.8 keV) is contoured at 6σ and depicted as orange mesh. (S)-Adenosyl-l-selenocysteine and the [Fe4S4]RS cluster are depicted as ball and sticks with the same color code as that in in Figure 2 (the selenium is in orange). As a comparison, SAH is superimposed (faded colors and carbon atoms in green) to highlight the one-carbon difference between the two molecules. (b) Putative Fe(CO)2CNCl species bound to M224 at the lower part of the β-barrel (crystal-3). The surrounding residues are depicted as sticks. The new Fe-containing species is depicted as ball and sticks, and the multiple-conformation iodide ion (overall 70% occupancy) is depicted as purple spheres. The residual difference Fourier electron density map is contoured at 3σ and represented as green mesh. The recorded anomalous difference Fourier map (energy of 7.15 keV) is contoured at 4σ and represented as orange mesh. The anomalous difference Fourier map recorded at 7.0 keV does not exhibit any significant peak in that region and is not presented. (c) Superposition of the crystal structures of crystal-1 and crystal-6 and the SAC manual model. For clarity, only the [Fe4S4]RS cluster and complex-B from crystal-1 are presented (ball-and-stick form). SAH, 5′-dA + methionine, and SAC are depicted as sticks with carbon atoms colored in gray, purple, and green, respectively. The [Fe4S4]−Fe‧‧‧Sd‧‧‧C5′‧‧‧S‧‧‧Fe−complex-B alignment is highlighted as yellow dashed lines. The 5′-dA C5′ atom is equidistant to both the methionine and complex-B sulfur atoms. (d) In crystallo SAC formation. The mixture of states observed after triggering the reaction in HydE crystals (crystal-7) is depicted in sticks surrounding residues, and SAM and SAC + methionine are depicted as sticks. Complex-B and the [Fe4S4]RS cluster are in ball-and-stick form. The t = 0 and 60 min structures are depicted with carbon atoms colored in gray and green, respectively. Stereoviews of each panel are available in Figures S8, S9, S12, and S13, respectively.
Strikingly, when looking down the β-barrel, a strong peak with additional features can be seen in the Fo − Fc difference Fourier electron density map in addition to the iodide ion bound at site S3. This peak is located at 2.4 Å of the Sδ atom of residue M224 with Cγ−Sδ−X and Cε−Sγ−X angles of 110°, supporting a direct interaction with this residue (see Figures 3b and S9). Our previous ligand screenings only highlighted thiocyanate as capable of binding to site S3. With this single exception and the substitution of the usually observed chloride ion to either iodide or bromide, no other change was ever observed in that region of the β-barrel.22 The pattern of the water molecules filling the lower pocket was strictly conserved.26 In addition, residue M224 always displays two alternative conformations with a conserved ratio of 3:7. In the present structure, this ratio is 1:1, corresponding to an enrichment of the conformation closer to this additional electron density peak. The shape of the observed residual electron density suggests X would correspond to pentacoordinated iron with a partial occupancy of about 30%. To confirm the nature of X as an iron center, we recorded X-ray diffraction data at energies surrounding those of the iron K-absorption edge (7.0 and 7.15 keV, Fe K-edge of 7.112 keV; crystal-4, see Tables S1 and S2). To avoid contamination from both the normal and anomalous signals coming from the observed iodide ion that results from the decomposition of FeI2, the crystal was back-soaked in a cryoprotection solution that did not contain syn-B prior to flash-cooling. The resulting anomalous difference electron density maps show a peak at the position of X in only the anomalous difference electron density map calculated with the data set recorded at 7.15 keV, hence unambiguously confirming that X corresponds to iron (see Figures 3b and S9). Our many past TmHydE crystal soaking experiments using ferrous or ferric ions or a sulfide ion never led to any iron bound at that position. Meanwhile, such experiments afforded significant [Fe4S4]Aux cluster reconstitution, confirming that these ions can easily diffuse into the crystals.27 Interestingly, when incubating TmHydE with either syn-B or syn-B-Se and sodium dithionite, we also observed similar [Fe4S4]Aux cluster reconstitution and sulfide and selenide incorporation (see Figure S10); however, we observed no additional iron bound near site S3. To test whether the presence of this iron atom near site S3 comes from an artifactual reaction between syn-B and dithionite, we incubated TmHydE with SAH, syn-B, and dithionite as a control. The corresponding crystal structures displayed complex-B bound in the active site cavity. However, neither SAC nor the iron atom was present, demonstrating that this new species originates from the transformation of complex-B catalyzed by HydE.
The surrounding features in the difference Fourier electron density map are attributed to ligands of a trigonal bipyramid iron center (see Figures 3b and S9). Two ligands have an elongated shape, which is consistent with carbon monoxide or cyanide ligands. Because they both point toward hydrophobic pockets, they were assigned as carbon monoxide. A third ligand corresponds to the thioether from residue M224. The two remaining ligands overlap with the iodide or chloride ion and water molecule positions usually present at site S3. Previous isotopic labeling experiments support that the HydE product would contain at least one iron atom, two carbon monoxides, one cyanide ion, and a sulfur atom from the complex-B l-cysteine moiety.14,20 Yet, when using syn-B-Se as substrate (crystal-5, see Tables S1 and S2), X-ray diffraction data around the selenium K-absorption edge exclude a selenide ion and therefore sulfide bound to the iron atom. Subtracting the contribution of the empty structure, i.e., the residual water molecule and chloride ion, to the electron density led us to propose that these ligands are cyanide and a chloride ions. The former would establish hydrogen bonds with residues R155 and T134, and the latter would interact with residue R54. The refinement of this model led to a flat residual Fo – Fc difference Fourier map, supporting the good agreement between the model and the X-ray data. Interestingly, each fragment was refined as an independent moiety, and they all displayed the same partial 30% occupancy with very similar temperature factors, further supporting that they all are parts of the same R2SM224-Fe(CO)2CNCl species. Because the experiment was performed using a sample containing SAM and HydE in a 1:1 ratio, the almost 100% occupancy observed for the resulting SAC molecule (crystal-3, see Tables S1 and S2) strongly suggests that this Fe(CO)2CNCl species exhibiting about a 30% occupancy would result from the decay of the t = 10 s intermediate species previously detected by EPR.20 Another, yet less probable, hypothesis would be to consider SAM regenerating at the end of the reaction and HydE performing multiple turnover. Most of them would be abortive ones that lead to SAC, and a few successful ones would lead to this new species. Our many subsequent experimental attempts to enrich the occupancy of this species, including the use of excess SAM and syn-B, always led to the same shape in the electron density, accounting for about 30% of the model in that region.
5′-Deoxyadenosyl Radical Addition to Complex-B.
To further investigate how this new species is formed, we decided to track the radical-based reaction directly in crystals.19 A direct attack of the sulfur atom by the 5′-dA● species is supported by several lines of evidence as follows: (i) the <4 Å distance separating SAH C5′ and complex-B S atoms (see Figure 2b), (ii) the detection of SAC both in vitro and in the crystals, and (iii) the ENDOR characterization of t = 10 s and t = 10 min EPR-active intermediates.19,20 We thus cocrystallized TmHydE with syn-B, 5′-deoxyadenosine (5′-dA), and l-methionine. These species are good structural mimics of the SAM cleavage products. The Fo − Fc difference Fourier electron density map of the 1.5 Å resolution crystal structures (crystal-6 see Tables S1 and S2) displayed features that corresponded to complex-B, l-methionine bound to the [Fe4S4]RS cluster, and 5′-dA (Figure S11) in the same conformation as was previously observed. 5′-dA has exactly the same conformation as was previously reported, and C5′ is equidistant (3.25 Å) to the methionine Sδ and the complex-B sulfur atoms.29,19 All three atoms are roughly aligned with the unique iron of the [Fe4S4]RS cluster and the ferrous ion of complex-B (see Figures 3c and S12 and Table S3). This three-dimensional layout is reminiscent to that previously reported when using (2R,4R)-MeTDA as non-natural substrate.19 Overall, the Fe‧‧‧S‧‧‧C5′‧‧‧Sδ‧ ‧[Fe4S4]RS arrangement further supports a direct attack of the sulfur atom by the 5′-dA● radical.19,20 Following the approach described above, we probed the radical-based reaction directly in the crystals. For this experiment, we used the protein batch described in ref 19, which was copurified with SAM.19 The protein was incubated with 6 mM syn-B or syn-B-Se prior to crystallization. Crystals were treated with 6 mM dithionite and flash-frozen at time lapses ranging from 30 s to 60 min. The shorter time lapses indicated a very fast reaction, and no significant differences could be observed between 30 s and 60 min. In striking contrast, the dithionite-triggered reaction of (2R,4R)-MeTDA required hours to complete.19 Here, we only report the t = 60 min time-lapse structure (crystal-7 in Tables S1 and S2) because it corresponds to the best-diffracting crystal among the series. Despite our many trials, no more than 50% of the molecules reacted in the crystals, always leading to the same mixture of states observed in the corresponding electron density maps. Therefore, the first half of the molecules contain SAM bound to the [Fe4S4]RS cluster in addition to unreacted complex-B (complex-B-Se). The other half contain a SAC molecule in the same conformation as that was previously reported (PDB 5FF0; see Figures 3d and S13),19 which is in agreement with the presence of alternative conformations for residues R159, L157, and T269. The carboxyl moiety sits in the S1 site, while the amino nitrogen now roughly occupies the position of the oxygen ligand in complex-B (see Figure 3d). In contrast, the residual Fo − Fc difference Fourier electron density map does not contain any residual features that would correspond to an Fe(CO)2CN fragment bound to the SAC molecule, excluding the presence of the 10 s EPR-active intermediate, and we did not observe any residual peak corresponding to thet = 10 min EPR-active intermediate either. Likewise the residual electron density map does not indicate any peak corresponding to the Fe(CO)2CNCl species bound to the M224 Sδ atom. Yet despite the high-resolution data it is very difficult to unambiguously characterize very-low-occupancy species (lower than 10%) by X-ray crystallography without having any reference model, in contrast to advanced EPR spectroscopy.
DISCUSSION
syn-B Structure and Binding Mode.
OUR results establish several fundamental facts. First, the identity of complex-B as a (κ3-cysteinate)Fe(CN)(CO)2 center is confirmed. Previously, it was shown to be a complex of l-cysteine with Fe(CN)(CO)2, but the coordination mode of the cysteine was uncertain. However, plasticity cannot be excluded, and it is not certain that the structure of complex-B does not change upon binding to HydE.
The second major finding is that syn-B, a synthetic complex-B-containing compound, delivers complex-B to HydE (crystal structures crystal-1 and crystal-2; see Tables S1 and S2).
Of mechanistic significance, our third major finding is that complex-B binds in the vicinity of the SAM C5′ atom. Such binding induces a few adaptative residue side-chain rotations, which seem to stabilize complex-B. In contrast, syn-B, and presumably complex-B itself, are unstable in an aqueous solution. Complex-B is tightly embedded in the active site because the carbon monoxide and cyanide ligands are deeply anchored in hydrophobic and hydrophilic pockets, respectively. The cyanide ion is bound trans to the thiolate ligand. The crystallographic analysis revealed that a broad water-filled channel connects the complex-B binding site to the surface where the [Fe4S4]Aux cluster binds (Figure 4a and S14). In this region, the surface is covered by detergent molecules from the crystallization condition, suggesting that it may correspond to a contact zone between HydE and one of its upstream partners.22 It is tempting to propose that complex-B would be transferred from HydG to HydE through this channel. Furthermore, and in agreement with the EPR analysis, 5′-dA● would attack the complex-B sulfur atom upon SAM cleavage, producing a C5′−S bond and leading to the 10 s EPR-active FeI(CO)2CN-l-SAC intermediate.20 In our soaking experiments, we confirmed that SAC is produced when the radical-based reaction is triggered with dithionite. Thus, the mechanistic insights obtained when using (2R,4R)-MeTDA also apply to complex-B.19,20 Furthermore, a close comparison of the three following crystal structures (crystal-1, crystal-6, and crystal-7; see Tables S1 and S2) corresponding to HydE + syn-B + SAH, HydE + syn-B + 5′-dA + methionine, and HydE + methionine + SAC (post in crystal reaction), respectively, highlight that the 5′-dA moiety is trapped in its binding pocket with a highly limited freedom of movement upon SAM cleavage. Indeed, it can only afford a slight rotation around the N6 nitrogen atom along the adenine ring plane, thus maximizing the C5′atom displacement from a SAM-bound position to a SAC-bound position without perturbing its surrounding environment.
Figure 4.
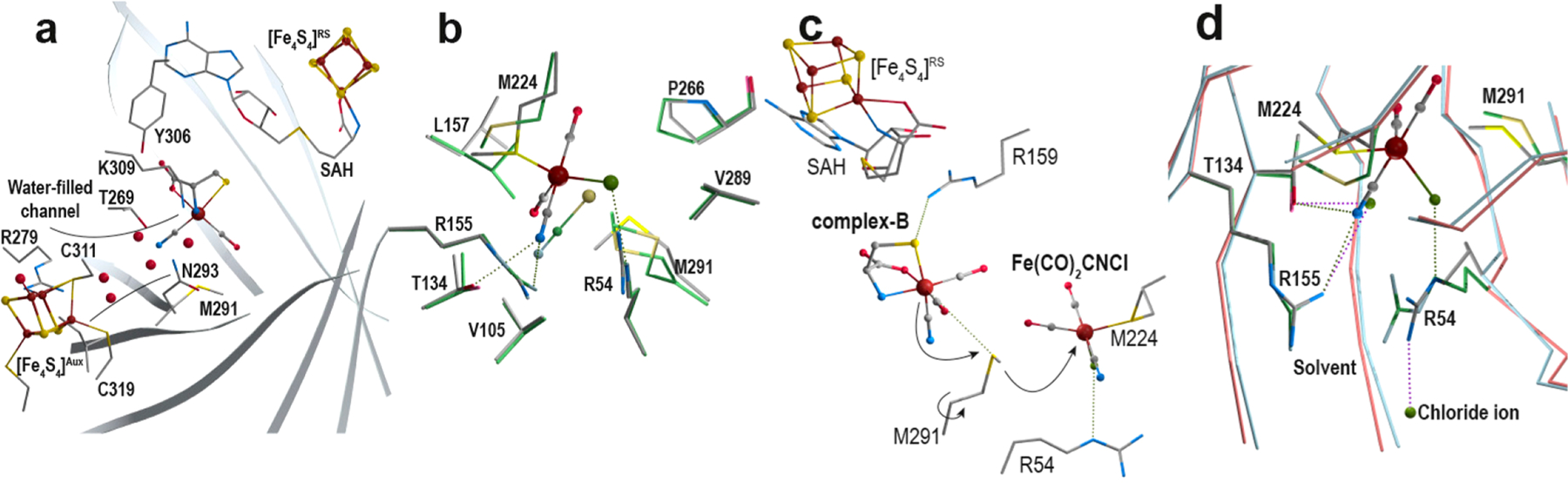
Transfers in HydE. (a) A water-filled channel connects the outside and the complex-B binding site. This channel reaches the protein surface near the [Fe4S4]Aux cluster binding site. Residues are depicted in sticks, clusters and complex-B as ball and stick,s and water molecules as red spheres. The barrel cavity is defined by the eight β-strands depicted as gray ribbons. (b) Superposition of the Fe(CO)2CNCl- and thiocyanate-bound HydE structures (crystal-3 and PDB 3CIX)22 with carbon colored in gray and green, respectively. (c) M291 as a putative relay to transfer the Fe(CO)2CN-containing species from top to bottom. M291 separates the upper and lower parts of the β-barrel. The rotation of the side-chain may help transfer the produced intermediate toward the lower pocket. Both sites are surrounded by conserved arginine and methionine residues. (d) Overlay of the crystal-3 and crystal-8 crystal structures, highlighting the opening of the cavity at the bottom of the β-barrel. Upon a C-terminal stretch displacement, direct access to the outside allows for product transfer. β-Strands corresponding to crystal-3 and crystal-8 are depicted in blue and red, respectively. The corresponding surrounding residues are depicted as sticks with carbons in gray and green, respectively. Stereoviews of each panel are available in Figures S14–S16 and S19, respectively.
Complex-B Reactivity in Crystals.
Starting with syn-B, two distinct HydE intermediates were previously characterized in solution by EPR/ENDOR spectroscopy (see Figure 1).20 None of these intermediates were observed by crystallography after triggering the reaction in solution either before crystallization or directly in a crystal. The observation of SAC in the active site in both cases confirms attack of the 5′-dA● radical on the complex-B sulfur. The previous EPR/ENDOR spectroscopic characterization of the 10 s intermediate confirmed the presence of the l-cysteine fragment bound to the FeI species.20 The hyperfine couplings of 15N, 13C2, and 13C3 were reported, but no 13C hyperfine signal of the carboxyl ligand was detected, suggesting that the carboxylate dissociates upon the reduction of the Fe(II) center. This scenario is supported by DFT calculations and agrees with the observation that low-spin FeI complexes favor pentacoordination.20,30 Manually modeling the FeI(CO)2CN-l-SAC 10 s intermediate in an octahedral fashion by combining5′-dA and the l-cysteine moiety of complex-B led to an acute C5′‧‧‧Sγ‧‧‧Cβ angle, highlighting the structural and conformational rearrangements and coordination changes necessary to relieve this strain. Therefore, the rearrangement of the coordination sphere of iron cannot be excluded upon the reaction with 5′-dA●. The SAC conformation in HydE crystals shows the carboxyl moiety located in site S1, whereas the amino group is found at a position similar to that of the oxygen ligand in complex-B. Such an “unstrained” binding mode agrees with a different coordination geometry for the FeI species. These modifications may be assisted by the surrounding residues (see Figures 3d and S13). When we monitored the conversion of l-cysteine to SAC within the crystals, neither 5′-thioadenosine nor the 10 min EPR-active intermediate were detected, which is at odds with the previous EPR/ENDOR and LC/MS characterizations.20 Perhaps crystal packing prevents further reactions of the FeI(CO)2CN-l-SAC. Instead the FeI(CO)2CN-l-SAC intermediate decays through the loss of the Fe(CO)2CN fragment. In fact, we observed that residues R159, L157, and T269 adopt nonrelaxed conformations when complex-B binds to HydE, which is again in agreement with possible conformational changes required during subsequent steps. Yet, the same conformations and same decay of the FeI(CO)2CN-l-SAC intermediate were observed in two different space groups exhibiting different crystal packing contacts. It is thus unclear what motions are necessary to convert the FeI(CO)2CN-l-SAC species into the 10 min intermediate and ultimately to the product. Yet, the strain applied by the surrounding residues to FeI(CO)2CN-l-SAC may weaken the S−Fe bond, which would explain why this intermediate is unstable in the crystal and might not accumulate sufficiently to be seen by X-ray crystallography. Attempts to manually model the 10 min EPR-active intermediate (see Figure 1) in the active site suggested that the FeI ion coordination may be a penta- or tetracoordinated ion.30 A Fe(CO)2CN(5′-thioadenosine) moiety can be modeled in a position similar to that of complex-B. Yet, the crowded cavity would preclude the previously proposed coordination of the ribose O4′ atom.20 Overall, the detailed structures of these low-spin FeI 10 s and 10 min species remain to be determined.
Fragmentation of the SAC Ligand.
Conversion of the 10 s intermediate to the 10 min intermediate entails the breaking of one C−S bond, one Fe−N bond, hydrolysis, and the release of pyruvate.20 Transition metals such as ferrous ions can act as Lewis acids when bound to l-cysteine, thus potentiating a base-catalyzed β-elimination through Cα deprotonation. The breakage of the resulting C−S bond ultimately leads to the release of a sulfide ion and aminoacrylate.31,32 The strictly conserved residue K309 is observed in the vicinity of the complex-B Cα atom (see Figures 2b and S3). Lysine residues often act as general bases in protein active sites. However, the K309 Nζ atom does not point toward the SAC Cα atom and rather establishes a salt bridge with the conserved residue E58. In this conformation, K309 cannot abstract the H−Cα proton. Indeed, the stability of complex-B is enhanced upon binding to HydE. In our experience, complex-B is stable for days when bound to HydE even though a rotation limited to the tip of the side chain (around Cδ-Cε bond) would ideally place the Nζ atom in a position to carry out such a deprotonation. Likewise, the deprotonation of the 10s intermediate entails the same reorganization. Yet, the reduction of the ferrous ion to FeI upon the addition of the 5′-dA● species and possible changes in the iron coordination, including the release of the carboxyl moiety, may break the salt-bridge between residues K309 and E58 and induce deprotonation, the ensuing sulfur atom β-elimination, and the release of the aminoacrylate molecule. The latter would subsequently be hydrolyzed into ammonia and pyruvate upon release, as was previously reported.20
Detection of a New Monoiron Species.
Isotopic labeling experiments shows that complex-B provides irons, cyanides, carbon monoxides, and sulfides of the [2Fe]H subcluster.14,33 The timing of the formation of the inorganic sulfur remains uncertain. For example, the (S)-adenosylated Fe center could conceivably be transferred to HydF for further processing. A key question is how to break the intermediate C5′−S bond. Although we did not observe any Fe(CO)2CN-S intermediates, a stable and unique Fe(CO)2CNCl species was bound to the thioether moiety of M224 at site S3. This species is only observed when the reaction is triggered in solution prior to crystallization and specifically requires syn-B, SAM, and a reductant. We conclude that Fe(CO)2CNCl results from HydE catalysis in solution. However, starting with one SAM molecule per HydE, the as-obtained crystals invariably contain 100% SAC bound to the RS cluster and 30% Fe(CO)2CNCl at site S3. Aside from considering SAM to be regenerated during catalysis, this newly observed species could originate from a rapid decay of the 10 s intermediate in solution prior to crystallization. As discussed above, its nondetection when triggering the reaction in a crystal may be either due to a reduced flexibility in the crystal that would impair its transfer to site S3 or because its abundance would be too low to be detected by X-ray crystallography. However, like the non-natural substrate thiazolidine was valuable in terms of the HydE reactivity, the unique observation of this Fe(CO)2CNCl species bound to site S3 might also be informative in regard to the enzyme function. This site most probably harbors the organometallic Fe(CO)2CNScys precursor to the H cluster in [FeFe]-hydrogenase.19,20 Furthermore, despite being far from the complex-B-binding site where the radical-based reaction occurs, site S3 is located at the lower part of the β-barrel in a pocket surrounded by conserved residues important to the HydE function.22 Such a tridimensional organization is reminiscent of that of HydG. Indeed, l-tyrosine binds in the upper part of the β-barrel, and the dehydroglycine intermediate crosses the broad cavity to a second site down the barrel where complex-B is produced.25 Strikingly, when superimposing the crystal-3 structure with that of a thiocyanate-bound HydE (PDB 3CIX), the N and S atoms of the thiocyanate ion occupy the positions of the NCFe(CO)2Cl species (see Figures 4b and S15).22 We now propose that like (2R,4R)-MeTDA mimics the true substrate complex-B bound to the active site, thiocyanate and the Fe(CO)2CNCl species may both mimic parts of a HydE intermediate or product bound to site S3. In view of both thiocyanate- and Fe(CO)2CN(Cl)-bound structures, we propose S3 as a binding site for the unstable HScys-Fe(CO)2CN species, the “monomeric” precursor to [2Fe]H (see Figure 5).
Figure 5.
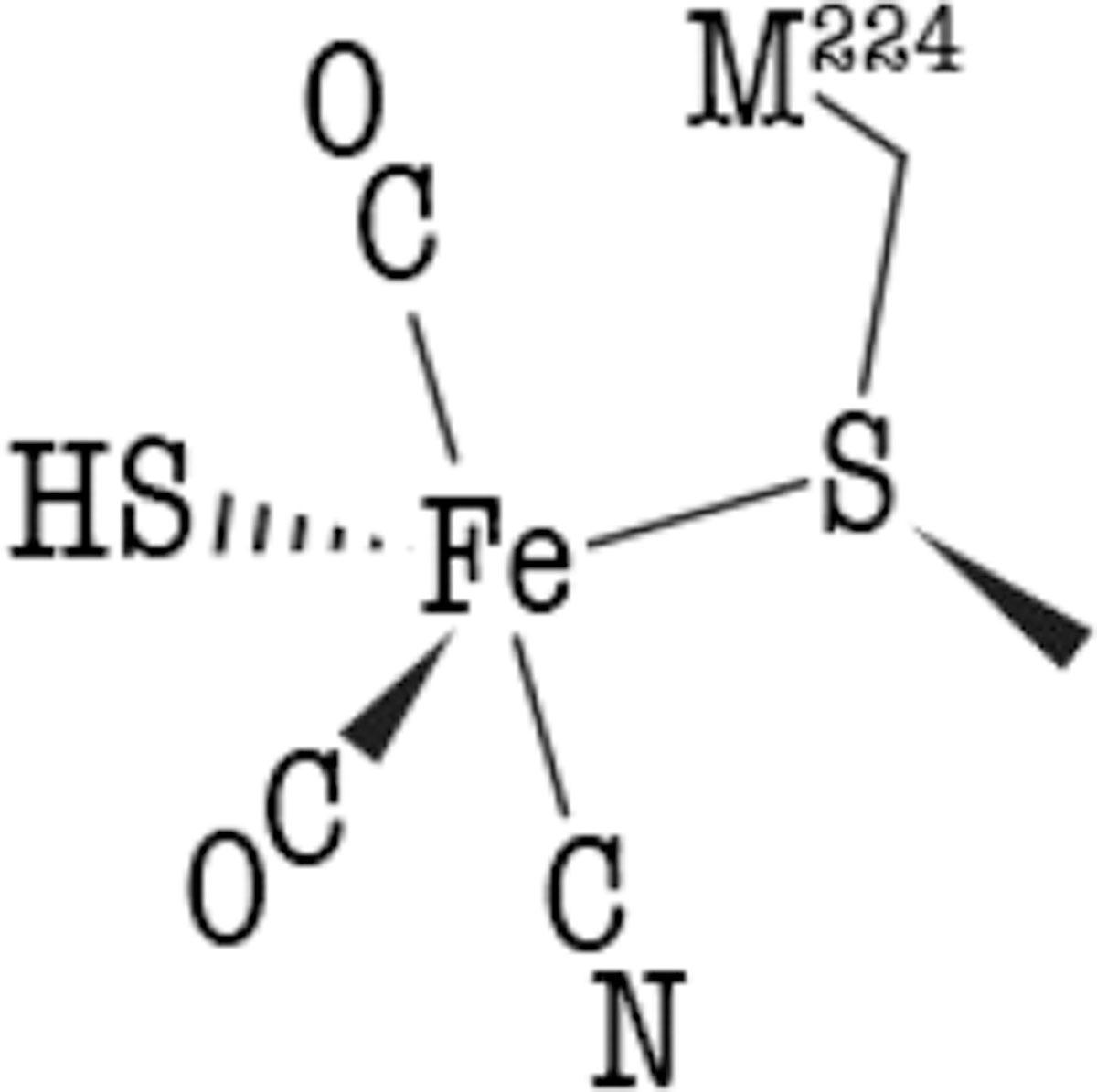
Scheme of the HScys-Fe(CO)2CN species as a possible intermediate or product bound to the lower pocket.
Dedicated Sulfur Atom Binding Sites.
The crystal structures of HydE complexed with complex-B and diverse thiazolidine or thiazinanes points out the existence of a sulfur-specific binding site located at the upper part of the β-barrel. This hypothesis is supported by the observation that thiol-containing molecules can also act as non-natural substrates to induce SAM cleavage despite their very low affinities.26,34 In this region, the strictly conserved residues R159 and Q109 both establish hydrogen bonds with the sulfur atom and may jointly act as a clamp to hold this atom in place during the reaction. Previous site-directed mutagenesis experiments targeting these two residues showed they indeed play a key role in the hydrogenase maturation process.22 When looking at the lower part of the β-barrel, the thiocyanate sulfur atom does not bind site S3 but instead interacts with the strictly conserved residue R54 that may define another dedicated sulfur binding site helpful to convey the HScys-Fe(CO)2CN precursor to [2Fe]H, which is at odds with what is intuitively expected.22
from the Active Site to Site S3 and Beyond.
HydE, like HydG, would function as an assembly line, affecting elaborate organosulfur reactions at the top of a β-barrel that are followed by transferal of the Fe(CO)2CN-containing product to the bottom of the barrel. The strictly conserved residue M291 is located between these two compartments, suggesting that it may function to relay the iron complex from the top worksite to the bottom worksite. M291 was initially proposed to merely separate the two pockets of the broad β-barrel cavity.22 Like M224, M291 displays multiple conformations, suggesting a dynamic role for this residue. Furthermore, an approximate twofold axis links residues M291 and R159 to residues M224 and R54 (see Figures 4c and S16). We thus propose that residue M291 transiently binds the HScys-Fe(CO)2CN species produced at the top compartment. A simple rotation would convey the highly reactive HScys-Fe(CO)2CN entity to site S3, where it would be further processed or transferred to HydF. Interestingly, it is also possible to manually model a Fe2S2 species between residue M291 and the M224 sulfur atoms with a reasonable geometry without changing the conformation of the surrounding residues (Figure S17). One cannot exclude that HydE, like HydG, which processes two l-tyrosines to produce one complex-B, would proceed twice to produce a Fe2S2(CO)4(CN)2 species. However, further functional characterizations are required to determine whether HydE would assemble a two-iron species before its transfer to HydF.
A Flexible Lid for Product Transfer.
HydE belongs to a subclass of radical SAM enzymes, such as NosL, PylB, or BioB,35–37 whose fold corresponds to a complete (βα)8-barrel.38 In these enzymes, the last α-helix ends with a semiconserved GxxP motif, followed by a short variable C-terminal stretch. In HydE, this C-terminal stretch covers the lower part of the β-barrel, impairing solvent access (see Figure S18). During our past search for HydE activity, we once obtained a crystal structure that lacked electron density corresponding to this C-terminal stretch (crystal-8, see Tables S1 and S2). The absence of this stretch induces major structural modifications at site S3. For instance, residue R54 now points toward the solvent (see Figures 4d and S19). A channel connects the β-barrel to the solvent, and a chloride ion is now visible outside the cavity. All these changes suggest that the C-terminal stretch would be displaced upon interaction with its partner, presumably HydF, allowing a direct transfer of the product while at the same time avoiding any hydrolysis from solvent. HydE would therefore act as a key player between HydG and HydF to process complex-B toward the [2Fe]H synthesis. Preliminary work supported the existence of several complexes between the three Hyd proteins.39 Further structural investigation are ongoing to determine how these proteins pass the baton, starting with l-tyrosine and an l-cysteine-bound ferrous ion to produce an active [FeFe]-hydrogenase.
CONCLUSION
From this work, we have established the structure of complex-B as a (κ3-cysteinate)FeII(CN)(CO)2 organometallic complex. Upon incubation with HydE, SAM, and a reductant, we confirmed that 5′-dA● attacks the complex-B sulfur atom, leading to SAC formation. Iron coordination changes are expected upon the reduction to an FeI species. The detection of a five-coordinate iron bound to residue M224 supports that HydE would produce an unstable HScys-FeI(CO)2CN monomeric precursor of the [2Fe]H subcluster of the [FeFe]-hydrogenase. However, further investigations are required to understand how the sulfur atom would be eventually “extracted” from the previously detected 10 min EPR-active intermediate. Finally, our structural analysis highlights channels for substrate access and product release through the broad cavity defined by the β-barrel. Taken together, all these results suggest that HydE acts as a nanofactory where the substrate is loaded by HydG on one side, a thorough radical-based chemistry process occurs inside, and product is unloaded to HydF on the other side. Subtle structural rearrangements are envisioned to control this whole process. Structural characterizations of complexes between the different Hyd proteins are eagerly awaited to complete our understanding of the [FeFe]-hydrogenase maturation.
Supplementary Material
ACKNOWLEDGMENTS
R.R. and Y.N. thank the ESRF staff for their constant support during remote data collection despite the COVID-19 pandemic. R.R. and Y.N. also thank Pierre Legrand from SOLEIL for his help with data collection. This work was supported by the French National Research Agency in the framework of the Investissements d’Avenir program (ANR-15-IDEX-02) through the funding of the “Origin of Life” project of the Université Grenoble Alpes. This work used the platforms of the Grenoble Instruct-ERIC center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology (PSB), which is supported by FRISBI (ANR-10-INBS-05-02) and GRAL and financed within the Université Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). This work was also supported by the National Institutes of Health Grant 1R35GM126961-01 to R.D.B and Grant GM61153 to T.B.R.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c03367.
Experimental materials and methods, X-ray data collection and refinement statistics, and supplementary figures and tables (PDF)
The authors declare no competing financial interest.
Contributor Information
Roman Rohac, Univ. Grenoble Alpes, CEA, CNRS, IBS, Metalloproteins Unit, F-38000 Grenoble, France.
Lydie Martin, Univ. Grenoble Alpes, CEA, CNRS, IBS, Metalloproteins Unit, F-38000 Grenoble, France.
Liang Liu, School of Chemical Sciences, University of Illinois at Urbana—Champaign, Urbana, Illinois 61801, United States.
Debashis Basu, School of Chemical Sciences, University of Illinois at Urbana—Champaign, Urbana, Illinois 61801, United States.
Lizhi Tao, Department of Chemistry, University of California—Davis, Davis, California 95616, United States.
R. David Britt, Department of Chemistry, University of California—Davis, Davis, California 95616, United States.
Thomas B. Rauchfuss, School of Chemical Sciences, University of Illinois at Urbana—Champaign, Urbana, Illinois 61801, United States.
Yvain Nicolet, Univ. Grenoble Alpes, CEA, CNRS, IBS, Metalloproteins Unit, F-38000 Grenoble, France.
REFERENCES
- (1).Lubitz W; Ogata H; Rüdiger O; Reijerse E Hydrogenases. Chem. Rev 2014, 114 (8), 4081–4148. [DOI] [PubMed] [Google Scholar]
- (2).Fontecilla-Camps JC; Volbeda A; Cavazza C; Nicolet Y Structure/Function Relationships of [NiFe]- and [FeFe]-Hydrogenases. Chem. Rev 2007, 107 (10), 4273–4303. [DOI] [PubMed] [Google Scholar]
- (3).Posewitz MC; King PW; Smolinski SL; Zhang L; Seibert M; Ghirardi ML Discovery of Two Novel Radical S-Adenosylmethionine Proteins Required for the Assembly of an Active [Fe] Hydrogenase. J. Biol. Chem 2004, 279 (24), 25711–25720. [DOI] [PubMed] [Google Scholar]
- (4).McGlynn SE; Ruebush SS; Naumov A; Nagy LE; Dubini A; King PW; Broderick JB; Posewitz MC; Peters JW In Vitro Activation of [FeFe] Hydrogenase: New Insights into Hydrogenase Maturation. JBIC, J. Biol. Inorg. Chem 2007, 12 (4), 443–447. [DOI] [PubMed] [Google Scholar]
- (5).Brazzolotto X; Rubach JK; Gaillard J; Gambarelli S; Atta M; Fontecave M The [Fe-Fe]-Hydrogenase Maturation Protein HydF from Thermotoga Maritima Is a GTPase with an Iron-Sulfur Cluster. J. Biol. Chem 2006, 281 (2), 769–774. [DOI] [PubMed] [Google Scholar]
- (6).Berggren G; Adamska A; Lambertz C; Simmons TR; Esselborn J; Atta M; Gambarelli S; Mouesca JM; Reijerse E; Lubitz W; Happe T; Artero V; Fontecave M Biomimetic Assembly and Activation of [FeFe]-Hydrogenases. Nature 2013, 499 (7456), 66–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Caserta G; Pecqueur L; Adamska-Venkatesh A; Papini C; Roy S; Artero V; Atta M; Reijerse E; Lubitz W; Fontecave M Structural and Functional Characterization of the Hydrogenase-Maturation HydF Protein. Nat. Chem. Biol 2017, 13 (7), 779–784. [DOI] [PubMed] [Google Scholar]
- (8).Byer AS; Shepard EM; Ratzloff MW; Betz JN; King PW; Broderick WE; Broderick JB H-Cluster Assembly Intermediates Built on HydF by the Radical SAM Enzymes HydE and HydG. JBIC, J. Biol. Inorg. Chem 2019, 24 (6), 783–792. [DOI] [PubMed] [Google Scholar]
- (9).Nicolet Y Structure-Function Relationships of Radical SAM Enzymes. Nat. Catal 2020, 3 (4), 337–350. [Google Scholar]
- (10).Driesener RC; Challand MR; McGlynn SE; Shepard EM; Boyd ES; Broderick JB; Peters JW; Roach PL [FeFe]-Hydrogenase Cyanide Ligands Derived from S-Adenosylmethionine-Dependent Cleavage of Tyrosine. Angew. Chem., Int. Ed 2010, 49 (9), 1687–1690. [DOI] [PubMed] [Google Scholar]
- (11).Shepard EM; Duffus BR; George SJ; McGlynn SE; Challand MR; Swanson KD; Roach PL; Cramer SP; Peters JW; Broderick JB [FeFe]-Hydrogenase Maturation: HydG-Catalyzed Synthesis of Carbon Monoxide. J. Am. Chem. Soc 2010, 132 (27), 9247–9249. [DOI] [PubMed] [Google Scholar]
- (12).Kuchenreuther JM; Myers WK; Stich TA; George SJ; Nejatyjahromy Y; Swartz JR; Britt RD A Radical Intermediate in Tyrosine Scission to the CO and CN-Ligands of FeFe Hydrogenase. Science 2013, 342 (6157), 472–475. [DOI] [PubMed] [Google Scholar]
- (13).Suess DLM; Bürstel I; De La Paz L; Kuchenreuther JM; Pham CC; Cramer SP; Swartz JR; Britt RD Cysteine as a Ligand Platform in the Biosynthesis of the FeFe Hydrogenase H Cluster. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (37), 11455–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rao G; Pattenaude SA; Alwan K; Blackburn NJ; Britt RD; Rauchfuss TB The Binuclear Cluster of [FeFe] Hydrogenase Is Formed with Sulfur Donated by Cysteine of an [Fe(Cys)(CO) (2)(CN)] Organometallic Precursor. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (42), 20850–20855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pilet E; Nicolet Y; Mathevon C; Douki T; Fontecilla-Camps JC; Fontecave M The Role of the Maturase HydG in [FeFe]-Hydrogenase Active Site Synthesis and Assembly. FEBS Lett 2009, 583 (3), 506–511. [DOI] [PubMed] [Google Scholar]
- (16).Dinis P; Suess DLM; Fox SJ; Harmer JE; Driesener RC; De La Paz L; Swartz JR; Essex JW; Britt RD; Roach PL X-Ray Crystallographic and EPR Spectroscopic Analysis of HydG, a Maturase in [FeFe]-Hydrogenase H-Cluster Assembly. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (5), 1362–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rao G; Tao L; Suess DLM; Britt RDA [4Fe-4S]-Fe(CO)(CN)-L-Cysteine Intermediate Is the First Organometallic Precursor in [FeFe] Hydrogenase H-Cluster Bioassembly. Nat. Chem 2018, 10 (5), 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Peters JW; Broderick JB Emerging Paradigms for Complex Iron-Sulfur Cofactor Assembly and Insertion. Annu. Rev. Biochem 2012, 81, 429–450. [DOI] [PubMed] [Google Scholar]
- (19).Rohac R; Amara P; Benjdia A; Martin L; Ruffie P; Favier A; Berteau O; Mouesca J-M; Fontecilla-Camps JC; Nicolet Y Carbon-Sulfur Bond-Forming Reaction Catalysed by the Radical SAM Enzyme HydE. Nat. Chem 2016, 8 (5), 491–500. [DOI] [PubMed] [Google Scholar]
- (20).Tao L; Pattenaude SA; Joshi S; Begley TP; Rauchfuss TB; Britt RD Radical SAM Enzyme HydE Generates Adenosylated Fe(I) Intermediates En Route to the [FeFe]-Hydrogenase Catalytic H-Cluster. J. Am. Chem. Soc 2020, 142 (24), 10841–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rao G; Tao L; Britt RD Serine Is the Molecular Source of the NH(CH2)2 Bridgehead Moiety of the in Vitro Assembled [FeFe] Hydrogenase H-Cluster. Chem. Sci 2020, 11 (5), 1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Nicolet Y; Rubach JK; Posewitz MC; Amara P; Mathevon C; Atta M; Fontecave M; Fontecilla-Camps JC X-Ray Structure of the [FeFe]-Hydrogenase Maturase HydE from Thermotoga Maritima. J. Biol. Chem 2008, 283 (27), 18861–18872. [DOI] [PubMed] [Google Scholar]
- (23).Volbeda A; Garcin E; Piras C; de Lacey AL; Fernandez VM; Hatchikian EC; Frey M; Fontecilla-Camps JC Structure of the [NiFe] Hydrogenase Active Site: Evidence for Biologically Uncommon Fe Ligands. J. Am. Chem. Soc 1996, 118 (51), 12989–12996. [Google Scholar]
- (24).Nicolet Y; Piras C; Legrand P; Hatchikian CE; Fontecilla-Camps JC Desulfovibrio Desulfuricans Iron Hydrogenase: The Structure Shows Unusual Coordination to an Active Site Fe Binuclear Center. Struct. London Engl 1999, 7 (1), 13–23. [DOI] [PubMed] [Google Scholar]
- (25).Pagnier A; Martin L; Zeppieri L; Nicolet Y; Fontecilla-Camps JC CO and CN-Syntheses by [FeFe]-Hydrogenase Maturase HydG Are Catalytically Differentiated Events. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (1), 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rohac R Etude Structurale et Fonctionnelle de La Proteíne à Radical SAM Hyde Ph.D. Thesis, Institut de Biologie Structurale, Grenoble, France, 2016 [Google Scholar]
- (27).Nicolet Y; Rohac R; Martin L; Fontecilla-Camps JC X-Ray Snapshots of Possible Intermediates in the Time Course of Synthesis and Degradation of Protein-Bound Fe4S4 Clusters. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (18), 7188–7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rao G; Pattenaude SA; Alwan K; Blackburn NJ; Britt RD; Rauchfuss TB The Binuclear Cluster of [FeFe] Hydrogenase Is Formed with Sulfur Donated by Cysteine of an [Fe(Cys)(CO)2-(CN)] Organometallic Precursor. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (42), 20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nicolet Y; Amara P; Mouesca J-M; Fontecilla-Camps JC Unexpected Electron Transfer Mechanism upon AdoMet Cleavage in Radical SAM Proteins. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (35), 14867–14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Therien MJ; Trogler WC Tricarbonylbis(Phosphine)Iron-(I) Cation Radicals. A Spectroscopic and Theoretical Study. J. Am. Chem. Soc 1986, 108 (13), 3697–3702. [Google Scholar]
- (31).Tchong S-I; Xu H; White RH L-Cysteine Desulfidase: An [4Fe-4S] Enzyme Isolated from Methanocaldococcus Jannaschii That Catalyzes the Breakdown of L-Cysteine into Pyruvate, Ammonia, and Sulfide. Biochemistry 2005, 44 (5), 1659–1670. [DOI] [PubMed] [Google Scholar]
- (32).Pagnier A; Nicolet Y; Fontecilla-Camps JC IscS from Archaeoglobus Fulgidus Has No Desulfurase Activity but May Provide a Cysteine Ligand for [Fe2S2] Cluster Assembly. Biochim. Biophys. Acta, Mol. Cell Res 2015, 1853 (6), 1457–1463. [DOI] [PubMed] [Google Scholar]
- (33).Suess DLM; Kuchenreuther JM; De La Paz L; Swartz JR; Britt RD Biosynthesis of the [FeFe] Hydrogenase H Cluster: A Central Role for the Radical SAM Enzyme HydG. Inorg. Chem 2016, 55 (2), 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Betz JN; Boswell NW; Fugate CJ; Holliday GL; Akiva E; Scott AG; Babbitt PC; Peters JW; Shepard EM; Broderick JB [FeFe]-Hydrogenase Maturation: Insights into the Role HydE Plays in Dithiomethylamine Biosynthesis. Biochemistry 2015, 54 (9), 1807–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nicolet Y; Zeppieri L; Amara P; Fontecilla-Camps JC Crystal Structure of Tryptophan Lyase (NosL): Evidence for Radical Formation at the Amino Group of Tryptophan. Angew. Chem., Int. Ed 2014, 53 (44), 11840–11844. [DOI] [PubMed] [Google Scholar]
- (36).Quitterer F; List A; Eisenreich W; Bacher A; Groll M Crystal Structure of Methylornithine Synthase (PylB): Insights into the Pyrrolysine Biosynthesis. Angew. Chem., Int. Ed 2012, 51 (6), 1339–1342. [DOI] [PubMed] [Google Scholar]
- (37).Berkovitch F; Nicolet Y; Wan JT; Jarrett JT; Drennan CL Crystal Structure of Biotin Synthase, an S-Adenosylmethionine-Dependent Radical Enzyme. Science 2004, 303 (5654), 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Nicolet Y; Drennan CL AdoMet Radical Proteins–from Structure to Evolution–Alignment of Divergent Protein Sequences Reveals Strong Secondary Structure Element Conservation. Nucleic Acids Res 2004, 32 (13), 4015–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).McGlynn SE; Shepard EM; Winslow MA; Naumov AV; Duschene KS; Posewitz MC; Broderick WE; Broderick JB; Peters JW HydF as a Scaffold Protein in [FeFe] Hydrogenase H-Cluster Biosynthesis. FEBS Lett 2008, 582 (15), 2183–2187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.