

Abstract
Structural, spectroscopic, and reactivity studies are presented for an electron transfer series of copper hydroxide complexes supported by a tridentate redox-active ligand. Single crystal X-ray crystallography shows that the mononuclear [CuOH]1+ core is stabilized via intramolecular H-bonds between the H-donors of the ligand and the hydroxide anion when the ligand is in its trianionic form. This complex undergoes two reversible oxidation processes that produce two metastable “high-valent” CuOH species, which can be generated by addition of stoichiometric amounts of 1e− oxidants. These CuOH species are characterized by an array of spectroscopic techniques including UV–vis absorption, electron paramagnetic resonance (EPR), and X-ray absorption spectroscopies (XAS), which together indicate that all redox couples are ligand-localized. The reactivity of the complexes in their higher oxidation states toward substrates with modest O─H bond dissociation energies (e.g., 4-substitued-2,6-di-tert-butylphenols) indicates that these complexes act as 2H+/2e− oxidants, differing from the 1H+/1e− reactivity of well-studied [CuOH]2+ systems.
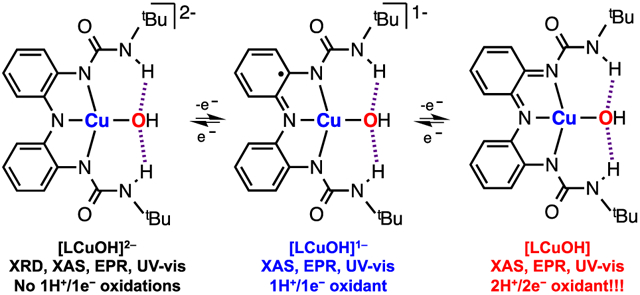
Graphical Abstract
INTRODUCTION
High-valent copper complexes have been invoked as active intermediates in the hydroxylation of C─H bonds performed by Cu-dependent metalloenzymes.1,2 For example, lytic polysaccharide monooxygenases (LPMOs) catalyze the oxidative degradation of cellulose using a mononuclear Cu center bound by two histidines, one acting as a bidentate ligand (Figure 1A).3,4 It has been proposed that the active species responsible for the oxidation of the cellulose substrate is a [CuO]1+ intermediate, which could be in equilibrium with a [CuOH]2+ analogue.5 In most of the members of this family of enzymes, a tyrosine residue is found in the vicinity of the active center. Recent reports have found that some LPMOs react with H2O2 (an alternative oxidant6,7) to generate a metastable Cu-tyrosyl species,8 which could be the active 2e− oxidant (Figure 1B).9 This species is similar to the active 2H+/2e− oxidant found in galactose-oxidase in which the dehydrogenation of alcohols to aldehydes is observed.10
Figure 1.

First step of cellulose degradation catalyzed by Cu-dependent LPMOs (A) and putative 2e− oxidants capable of performing C─H hydroxylation (B).
Inspired by our recent work on Cu complexes bearing bidentate redox-active ligands,11,12 we describe the synthesis and characterization of a mononuclear copper hydroxide complex bound by a tridentate redox-active ligand scaffold that stabilizes the CuOH core via intramolecular H-bonding interactions (see Figure 2). This [LCuOH]2− complex can be chemically oxidized using ferrocenium (Fc+) to generate two “high-valent” CuOH species, [LCuOH]1− and [LCuOH], that are characterized by multiple spectroscopies. The [LCuOH] species (generated by adding 2 equiv of Fc+ to the [LCuOH]2− complex) is capable of performing two sequential H-atom abstractions from phenols. This reactivity differs from the well-established 1H+/1e− oxidations promoted by mononuclear [CuOH]2+ cores.13-15
Figure 2.

Synthesis of the mononuclear complex [tBuCuOH]2− and displacement ellipsoid plot (50% probability level) at 110(2) K (see SI for details).
RESULTS AND DISCUSSION
Synthesis and Characterization of the Copper Hydroxide Complex.
Inspired by the work of Heyduk16 and Borovik,17 we designed tBuLH3, which contains a redox-active tridentate amine and two ureanyl H-bond donors (see Figure 2). tBuLH3 was synthesized by addition of 2 equiv of tert-butyl isocyanate (tBuNCO) to a THF solution of bis(2-aminophenyl)amine (see further details in the Supporting Information, SI).
In the glovebox, the addition of 3 equiv of KH to a solution of tBuLH3 led to the formation of the deprotonated scaffold (tBuL3−)K3, which, upon treatment with 1 equiv of Cu(OAc)2 and 2 equiv of NMe4(OH)·5H2O, formed the mononuclear copper hydroxide complex [Cu(tBuL3−)(OH)](NMe4)2, [tBuCuOH]2− (see SI).
Complex [tBuCuOH]2− was characterized by single crystal X-ray diffraction analysis (Figure 2). The Cu ion exhibits a distorted square-planar geometry around the metal ion (τ4 = 0.2. Note: for D4h structures, a τ4 value of 0.0 is expected18). Moreover, there is an intramolecular interaction between the hydroxide ligand and the ureanyl H-bond donors of the ligand scaffold (O…Hα’ distances ca. 1.9 Å). The average Cu─O distance is ca. 1.92 Å, which is comparable to the Cu─O distances found for the few mononuclear [CuOH]1+ complexes reported to date (ca. 1.90 Å).14,19-22
Electrochemistry.
Metal complexes bearing ligands derived from the bis(2-aminophenyl)amine scaffold are able to reach multiple oxidation states by oxidation/reduction of the ligand scaffold. For example, Heyduk reported that TaV complexes bound by the reduced form of a similar ligand scaffold generated TaV-semiquinone and TaV-quinone species via ligand oxidation.16 Similarly, we envisioned that [tBuCuOH]2− ([tBuCuOH]2− = tBuCuOHO.S.2) could be reduced to generate tBuCuOHO.S.1 or oxidized to form “high-valent” tBuCuOHO.S.3, tBuCuOHO.S.4, and tBuCuOHO.S.5 via ligand and/or metal oxidation (see Figure 3A in which tBuCuOHO.S.3 and tBuCuOHO.S.4 are depicted as [CuOH]+ complexes bound by the ligand scaffold oxidized to the semiquinone and quinone form, respectively). Cyclic voltammetry experiments revealed that tBuCuOHO.S.2 was reversibly oxidized to tBuCuOHO.S.3 and tBuCuOHO.S.4 at −1.05 and −0.28 V, respectively (V vs Fc0/+; see Figure 3B).
Figure 3.

(A) Possible oxidation states reached via reduction/oxidation of tBuCuOHO.S.2. (B) Electrochemical and spectroscopic characterization of tBuCuOH in three oxidation states (i.e., tBuCuOHO.S.2, tBuCuOHO.S.3, and tBuCuOHO.S.4).
The oxidation of tBuCuOHO.S.2 was also accomplished using stoichiometric amounts of ferrocenium hexafluorophosphate (FcPF6) (Figure 3B). Addition of 1 equiv of Fc+ to tBuCuOHO.S.2 in DMF at −40 °C (880 nm with ε = 400 M−1 cm−1) led to the formation of a new species with intense UV–vis absorption features at 625 nm (ε = 3600 M−1 cm−1) and 950 nm (ε = 4000 M−1 cm−1), which we assigned as tBuCuOHO.S.3 Addition of 1 equiv of Fc+ to tBuCuOHO.S.3 produced strong UV–vis absorptions centered at 442 nm (ε = 6200 M−1 cm−1) and 965 nm (ε = 6200 M−1 cm−1) which we assign as tBuCuOHO.S.4. Both isosbestic transformations required only 1 equiv of Fc+ (see titrations in Figure S6 and Figure S8 in the SI). The UV–vis absorption features of tBuCuOHO.S.3 are similar to those exhibited by metal complexes bound to the semiquinone form of the tridentate ligand, suggesting that tBuCuOHO.S.3 is a [CuOH]1+ complex bound by the radical ligand tBuL.2− (see Figure 3A).16 The UV–vis absorption spectrum of tBuCuOHO.S.4 strongly resembles the spectra of some metal complexes containing the quinone form of similar triamine ligands, which implies that tBuCuOHO.S.4 is a [CuOH]1+ core bound by the doubly oxidized form of the ligand (tBuL−).16,23
After the formation of tBuCuOHO.S.4 via addition of 2 equiv of Fc+ to tBuCuOHO.S.2, 1 equiv of cobaltocene (CoCp2) was added to generate tBuCuOHO.S.3, which was further reduced to tBuCuOHO.S.2 by adding a second equivalent of CoCp2 (see Figure S10 in SI). The reversibility of these redox processes agrees with the cyclic voltammetry experiments described above in which the tBuCuOHO.S.2 complex was oxidized to two “high-valent” species at low potentials (lower than Fc) and in which these species are sequentially reduced to tBuCuOHO.S.2 at higher potentials than CoCp2 (E1/2(CoCp2) = −1.29 V vs Fc0/+ in DMF).
The tBuCuOH platform in the different oxidation states was also characterized by perpendicular-mode continuous wave X-band EPR (Figure 3B). We observed an axial S = ½ EPR signal for tBuCuOHO.S.2 characteristic of square planar copper(II) complexes (g⊥ = 2.05, g∥ = 2.20, A∥ = 542 MHz).24 Upon addition of 1 equiv of Fc+, the EPR features of tBuCuOHO.S.2 disappeared to produce an EPR silent species, which could be due to the formation of an S = 0 [CuOH]2+ core or the formation of a Cu-semiquinone-like species in which the unpaired electron of the [CuOH]+ center is ferromagnetically coupled to the semiquinone ligand (S = 1) or antiferromagnetically coupled (S = 0).25 An addition of 1 equiv of Fc+ to the silent species tBuCuOHO.S.3 produced an EPR signal similar to tBuCuOHO.S.2 but distinct (g⊥ = 2.05, g∥ = 2.24, A∥ = 508 MHz), which suggests that tBuCuOHO.S.4 is a [CuOH]+ complex bound by the quinone form of the ligand. Magnetic susceptibilities were measured at room temperature using the Evans method for tBuCuOHO.S.2 (μeff = 1.50 μB), tBuCuOHO.S.3 (μeff = 0.00 μB), and tBuCuOHO.S.4 (μeff = 1.81 μB).26 This magnetic data supports the formulation of tBuCuOHO.S.2 and tBuCuOHO.S.4 as S = ½ complexes and indicates that the spin state of tBuCuOHO.S.3 is S = 0.
X-ray Absorption Spectroscopy.
Cu K-edge X-ray absorption spectra (XAS) were obtained for tBuCuOHO.S.2, tBuCuOHO.S.3, and tBuCuOHO.S.4 (Figure 4). Each spectrum exhibits a weak pre-edge absorption feature assigned by convention to Cu 1s → Cu 3d excitations. The maxima of these 1s → 3d bands occur at 8979.5 eV (tBuCuOHO.S.2), 8979.4 eV (tBuCuOHO.S.3), and 8979.2 eV (tBuCuOHO.S.4). While these energies lie within the range conventionally assigned to CuII,27,28 recent studies have challenged the notion that pre-edge band energies correspond to distinct Cu physical oxidation states.29-33 These effectively isoenergetic bands suggest that each complex bears Cu in a common physical oxidation state, with slight variations plausibly attributed to changes in ligand field strength attending ligand redox changes. The assumption of very small changes in ligand field strength about Cu accord with the very similar spin-Hamiltonian parameters extracted from fitting the EPR spectra of tBuCuOHO.S.2 and tBuCuOHO.S.4. Transitions to higher energy could arise due to excitations to ligand-localized molecular orbitals as well as excitations to Cu 4p. Rising edge inflection points occur at 8991.2 eV (tBuCuOHO.S.2), 8991.4 eV (tBuCuOHO.S.3), and 8992.0 eV (tBuCuOHO.S.4), further indicating minimal participation of Cu in the redox events linking these species.
Figure 4.

Cu K-edge XAS data obtained for tBuCuOHO.S.2–4. Pre-edge features ca. 8979 eV are magnified for clarity.
H-atom Abstraction Reactivity of the CuOH Complexes.
The reactivity of the three members of the electron transfer series toward organic substrates containing weak C─H bonds [dihydroanthracene, xanthene, and 1,4-cyclohexadiene (BDE’s = 75–80 kcal/mol)] was investigated.34 Disappointingly, none of the CuOH species under investigation was able to oxidize these substrates. However, the CuOH complexes reacted with phenols featuring relatively weak O─H bonds.
The 4-substituted 2,6-di-tert-butylphenols (4-X-2,6-DTBP) are typically used as H-atom donors since they generate metastable phenoxyl radicals (see Figure 5B) that can be observed and quantified by UV–vis or EPR (like in the oxidation of 4-methoxy-2,6-di-tert-butylphenol).35 The products derived from coupling of the phenoxyl radicals can be also be analyzed and quantified (e.g., 2,6-di-tert-butylphenol oxidation).36,37 The reactivity of the CuOH cores was studied in DMF at room temperature and the results are summarized in Figure 5A. Overall, tBuCuOHO.S.4 abstracted H atoms from 4-substituted 2,6-di-tert-butylphenols to produce tBuCuOH-(H+)O.S.3 (protonated form of tBuCuOHO.S.3), which also reacted with phenols and was further reduced to tBuCuOH-(H+)2O.S.2. Thus, tBuCuOHO.S.4 can be considered a 2H+/2e− oxidant. However, our experimental evidence suggests that the reduced complex tBuCuOH(H+)2O.S.2 is unstable and undergoes a disproportionation reaction to generate the cuprous complex tBuCuOH(H+)3O.S.1 (that ultimately decomposes to release CuI and tBuLH3; see SI) and tBuCuOH(H+)O.S.3, which can oxidize the phenolic substrates. Hence, the stoichiometry of the reaction dictates that 1 equiv of tBuCuOHO.S.4 can lead to three H-atom transfer events (details given below).
Figure 5.

Summary of the reactivity of CuOH cores with 4-substituted-2,6-di-tert-butylphenols (A) and organic products derived from the H-atom abstraction (B).38
Reactivity with 4-Substituted 2,6-di-tert-Butylphenols: Stoichiometry.
The reaction between tBuCuOHO.S.4 and 50 equiv of 2,6-DTBP was carried out at 298 K and monitored by UV–vis absorption spectroscopy (see Figure 6A). The reaction proceeded in a two-step fashion. In the first step, UV–vis absorption features of tBuCuOHO.S.4 decayed rapidly with concomitant formation of a UV–vis absorption band centered near 625 nm. This resembles the spectrum of tBuCuOHO.S.3, which was assigned as the Cu product derived from the 1H+/1e− reduction/protonation of tBuCuOHO.S.4, tBuCuOH(H+) (green spectrum in Figure 6). tBuCuOH-(H+)O.S.3 could be independently generated by adding 1 equiv of pyridinium triflate (pyr·H+) to a solution of tBuCuOHO.S.3(tBuCuOHO.S.3+ H+ → tBuCuOH(H+)O.S.3; see also Figure 12).38 In the second step of the reaction, tBuCuOH-(H+)O.S.3 reacted with the phenol to produce the diprotonated tBuCuOH(H+)2O.S.2 complex, which is unstable and decays in the presence of phenol to CuI products (see orange spectra in Figure 6A). Addition of 2 equiv of pyr·H+ to tBuCuOHO.S.2 triggered the formation of 0.5 equiv of tBuCuOH(H+)O.S.3 suggesting that tBuCuOHO.S.2 undergoes disproportionation upon protonation (see Figures S78-S80). Both reaction steps exhibited the growth of very absorptive features near 425 nm, which correspond to the formation of 3,3′,5,5′-tetra-tert-butyldiphenoquinone (DPQ, λmax 425 nm, ε = 7.2 × 104 M−1cm−1 in DMF), the C─C coupling product derived from the 2H+/2e− oxidation of 2,6-DTBP.37 This product is formed via H-atom abstraction of 2,6-DTBP to produce a fleeting phenoxyl radical (2,6-DTBP•), which undergoes dimerization to the diphenol H2DPQ (Figure 5B).39 The 2H+/2e− oxidation of the diphenol product leads to the final diphenoquinone DPQ. Under the employed reaction conditions, a UV–vis absorption band appeared corresponding to DPQ, but its high molar absorptivity led to spectral saturation (see Figure 6A).
Figure 6.

Analysis of the stoichiometry of the reaction between tBuCuOHO.S.4 and 2,6-DTBP.
Figure 12.

Bordwell analysis of the reduction/protonation of the tBuCuOH cores (A) and analysis of the protonation tBuCuOHO.S.3 (B) Note: the BDE, pKa and E1/2 values in italics were estimated using the Bordwell equation and were not obtained experimentally (see text below).
In independent experiments under the same conditions (room temperature, [Cu] = 0.25 mM, 50 equiv of 2,6-DTBP), DPQ evolution over time was quantified by UV–vis absorption via dilution of the crude reaction mixture. Meanwhile, H2DPQ was quantified by 1H NMR (Figure 6C and Figure 6D; see SI for details). After the short reaction times, H2DPQ was the main product in solution (0.06 mM) while DPQ was formed in only small quantities (<0.02 mM). Over time, the concentration of H2DPQ decreased slightly (~ 0.04 mM) but DPQ increased substantially (up to 0.11 mM), suggesting that 2,6-DTBP is first oxidized to H2DPQ (via 2,6-DTBP• dimerization), DPQ is formed via subsequent oxidation of H2DPQ. Overall, 1 equiv of tBuCuOHO.S.4 (0.25 mM) produced 0.2 equiv of H2DPQ (0.05 mM) and 0.5 equiv of DPQ (0.11 mM), indicating that tBuCuOHO.S.4 is capable of performing three H-atom transfer reactions (yields ~ 70%). The data suggest that the 2H+/2e− reduction product tBuCuOH(H+)2O.S.2 undergoes a rapid disproportionation to generate a CuI species and tBuCuOH(H+)O.S.3, which can react again with the phenol to produce H2DPQ and DPQ. In support of this hypothesis, the reaction between tBuCuOHO.S.2, pyr·H+ and 2,6-DTBP results in the formation of H2DPQ and DPQ with yields up to 50% (see Figure S25 in SI).
The reaction between tBuCuOHO.S.4 and 50 equiv of 2,6-DTBP was monitored by EPR spectroscopy (Figure 6E). Addition of the substrate caused the disappearance of the tBuCuOHO.S.4 features, suggesting the formation of the EPR silent tBuCuOH(H+)O.S.4 (see also Figure 12). After full decay of tBuCuOH(H+)O.S.3 (t ~ 10000 s), no S = ½ signal corresponding to the fully reduced tBuCuOH(H+)2O.S.2 complex emerged, which is in agreement with the proposed disproportionation of tBuCuOH(H+)2O.S.2 to EPR silent species.
Interestingly, tBuCuOHO.S.3 did not react with 2,6-DTBP. Addition of 1 equiv of pyr·H+ to tBuCuOHO.S.3 generated tBuCuOH(H+)O.S.3, which oxidized 2,6-DTBP (Figure 7). The UV–vis absorption spectra obtained for this reaction (tBuCuOH(H+)O.S.3 + 2,6-DTBP) were very similar to those observed in the second step of the reaction between tBuCuOHO.S.4 and 2,6-DTBP, suggesting that tBuCuOH(H+)O.S.3 is an intermediate in the reaction between tBuCuOHO.S.4 and 2,6-DTBP (Figure 7A). The tBuCuOHO.S.2 complex also reacted with 2,6-DTBP but we found that this transformation entailed the protonation of tBuCuOHO.S.2 without oxidation of the phenol substrate (see Figure S24 and Figure S26 in the SI). We hypothesize that this is due to the high basicity of tBuCuOHO.S.2 (see further discussion below).
Figure 7.

Analysis of the stoichiometry of the reaction between tBuCuOH(H+)O.S.3 and 2,6-DTBP.
The organic products formed in the reaction between tBuCuOH(H+)O.S.3 and 2,6-DTBP were quantified (see SI) and the yields were consistent with tBuCuOH(H+)O.S.3 undergoing two 1H+/1e− events (via reduction of tBuCuOH(H+)O.S.3 to tBuCuOH(H+)2O.S.2 followed by disproportionation and subsequent phenol oxidation).
The reaction between tBuCuOH(H+)O.S.3 and 2,6-DTBP was also monitored using EPR spectroscopy (Figure 7E). Addition of one equiv of pyr·H+ to tBuCuOHO.S.3 produced an EPR silent species, which reacted with the phenol over time to generate EPR silent solutions (Figure 7E). The fact that tBuCuOH(H+)O.S.3 is capable of oxidizing the phenol (as seen by UV–vis absorption) and that it also led to EPR silent mixtures suggests that in both cases, the generation of tBuCuOH(H+)2O.S.2 is followed by fast disproportionation of this putative EPR active complex. Further corroboration was obtained by analyzing EPR samples of the reaction between tBuCuOHO.S.2, pyr·H+ and 2,6-DTBP, which were also silent (see Figure S26 in SI).
The reaction of tBuCuOHO.S.4 with 4-methoxy-2,6-di-tert-butylphenol (4-MeO-2,6-DTBP) was analyzed (Figure 8). Similar UV–vis absorption changes were found to those observed during the reaction between tBuCuOHO.S.4 and 2,6-DTBP, in which the tBuCuOHO.S.4 was sequentially reduced to tBuCuOH(H+)O.S.3 and the products derived from tBuCuOH(H+)2O.S.2 disproportionation (Figure 8A). UV–vis absorption spectra revealed the formation of phenoxyl radical derived from H-atom abstraction (4-MeO-2,6-DTBP·, λmax = 408 nm, ε = 3.0 × 103 M−1cm−1 in DMF). Similarly, the reaction between tBuCuOH(H+)O.S.3 and 4-MeO-2,6-DTBP produced phenoxyl radical, but in lower amounts (the intensity of the UV–vis features for 4-MeO-2,6-DTBP• produced in this reaction were ~50% lower than in the reaction between tBuCuOHO.S.4 and 4-MeO-2,6-DTBP; see Figure 9A). In both cases, we analyzed the organic products derived from the oxidation of with 4-MeO-2,6-DTBP and we observed that 2,6-di-tert-butyl-1,4-benzoquinone (BQ in Figure 5) was produced in excellent yields.
Figure 8.

Analysis of the stoichiometry of reaction between tBuCuOHO.S.4 and 4-MeO-2,6-DTBP.
Figure 9.

Analysis of the stoichiometry of reaction between tBuCuOH(H+)O.S.3 and 4-MeO-2,6-DTBP.
The equivalents of phenoxyl radical formed in the reaction between tBuCuOHO.S.4(or tBuCuOH(H+)O.S.3) and 4-MeO-2,6-DTBP were quantified using EPR (see SI for details). As expected, the oxidation of 4-MeO-2,6-DTBP by tBuCuOHO.S.4 produced higher quantities of the phenoxyl radical than the reaction between tBuCuOH(H+)O.S.3 and 4-MeO-2,6-DTBP (0.25 mM vs 0.11 mM; see Figure 8D and Figure 9D). In both reactions, reduction of tBuCuOH(H+)O.S.3 produced the phenoxyl radical but the S = ½ signal from the putative tBuCuOH(H+)2O.S.2 was not observed (Figure 8E and Figure 9E). As with the reactions with 2,6-DTBP described above, it appears that the putative tBuCuOH(H+)2O.S.2 species undergoes disproportionation to generate EPR-silent complexes. Full accumulation of the phenoxyl radical in these reactions was precluded due to its instability at room temperature (solutions of pure 4-MeO-2,6-DTBP radical in DMF decay at room temperature to produce BQ see the SI). The reaction between tBuCuOHO.S.4 and 4-MeO-2,6-DTBP was carried out at −40 °C and was followed by UV–vis absorption and EPR (see Figures S42-S44 in the SI) to give the tBuCuOH(H+)O.S.3 species, which did not react further with the phenol. This enabled quantification of the phenoxyl radical produced in the 1H+/1e− reduction of tBuCuOHO.S.4 (yields up to 85%).
Reactivity with 4-Substituted 2,6-di-tert-Butylphenols: Kinetics.
The kinetics of the reaction between 4-substituted 2,6-di-tert-butyl phenols (X = MeO, Me, tBu, H, NO2) and tBuCuOHO.S.4 were analyzed (Figure 10). Under pseudo-first order conditions ([tBuCuOHO.S.4] ≪ [phenol]), the reaction rates (kobs) increased linearly upon increasing the concentration of phenol, from which the second-order rate constants were calculated (Figure 10; see SI for details on the exponential fitting of the decay of tBuCuOHO.S.4). tBuCuOHO.S.4 reacted faster with phenols containing weaker O─H bonds (e.g., k(4-MeO-2,6-DTBP) = 4.6 M−1 s−1 vs k(4-NO2-2,6-DTBP) = 0.013 M−1 s−1).
Figure 10.

Kinetic analysis of the reaction between tBuCuOHO.S.4 and 4-X-2,6-DTBP.
A Marcus plot for the reactivity of tBuCuOHO.S.4 against 4-substituted 2,6-di-tert-butyl phenols was obtained (Figure 11B) by plotting (RT/F) ln k vs E0(PhOH/PhO.) where k is the second-order rate constant and E0(PhOH/PhOH.) is the redox potential of 4-X-2,6-DTBP.36,37 These plots are commonly utilized to analyze the reaction pathways by which phenols are oxidized (Figure 11A).36,40-42 Slopes with values ca. −0.5 are indicative of single electron transfer (ET) during the rate-determining step.41 Meanwhile, slopes near −1.0 are found in reactions where proton transfer (PT) is rate limiting. Values close to 0.0 are indicative of hydrogen atom transfer or concerted proton electron transfer (HAT or CPET). The slope observed for tBuCuOHO.S.4 (−0.12) is consistent with a concerted H-atom transfer and is similar to that found in the oxidation of 4-X-2,6-DTBP by a “pure” hydrogen atom abstractor such as cumylperoxyl radical (slope = −0.05).36 Similar slope values have been reported for the H-atom transfer from phenols to formally manganese(IV)-hydroxo,42 copper(II)-superoxo,36 nickel(III),43 and copper(III) complexes.37
Figure 11.

Mechanistic analysis of the reaction between tBuCuOHO.S.4 and 4-X-2,6-DTBP to generate tBuCuOH(H+)O.S.3 and phenoxyl radical.
A Bell-Evans-Polanyi plot was obtained by plotting ΔG‡ vs BDEO─H, where ΔG‡ is the Gibbs energy of activation calculated from the k values, and BDEO─H is strength of the O─H bonds for the phenols.44,45 This data suggests that the reaction between tBuCuOHO.S.4 and 4-substituted 2,6-di-tert-butyl phenols occurs via hydrogen atom transfer (slope = 0.48; note: the theoretical value for ideal HAT is 0.5;34,46 see Figure 11C). The negative slope found in the Hammett plot is also in agreement with the proposed H-atom abstraction mechanism (Figure 11D).47 The primary kinetic isotope effect measured (KIE ca. 2.1 for 2,6-DTBP vs 2,6-DTBP-OD; see Figure 11E) is consistent with O─H cleavage during the rate-determining step of the reaction. The modest KIE value is not unusual and has also been observed in the H-atom abstraction reactivity of phenols with metal-peroxo,48 metal-oxo,40,49 and metal-hydroxo systems.42
Bordwell Analysis – BDE of the Reduction/Protonation of tBuCuOHO.S.4.
The Bordwell equation50 can be used to calculate the driving force associated with the protonation/reduction of oxidants (Figure 12A). Popularized by Mayer and co-workers, this analysis permits the determination of the thermodynamic parameters associated with the reactivity of oxidants; oxidants with higher BDE values are capable of oxidizing stronger C─H and O─H bonds, and usually with faster rates.46,51 The BDE associated with the protonation/reduction of tBuCuOHO.S.4 to tBuCuOH(H+)O.S.3 was calculated using the E1/2 for the reduction of tBuCuOHO.S.4 (see Figure 4 above) and pKa of the protonation of tBuCuOHO.S.3 to tBuCuOH(H+)O.S.3 (pKa = 10.9), obtained by spectrometric titration via deprotonation of tBuCuOH(H+)O.S.3 with triethylamine (Figure 12; see also SI). The BDE value for the reductive protonation of tBuCuOHO.S.4 (83 kcal/mol in DMF) is very similar to the BDE values of the phenols that can be oxidized by tBuCuOHO.S.4 (BDE0─H = 78–85 kcal/mol in DMSO).52,53
Bordwell Analysis – BDE of the Reduction/Protonation of tBuCuOHO.S.3.
As mentioned, tBuCuOHO.S.3 was not able to oxidize 4-susbtituted-2,6-DTBP substrates (BDEO─H = 78–85 kcal/mol in DMSO). tBuCuOHO.S.3 reacted with 1,2-diphenylhydrazine PhNHNHPh (BDEN─H = 71.7 kcal/mol in DMSO34) to generate the corresponding PhN═NPh product (see SI). On the basis of this reactivity and using the Bordwell thermodynamic cycle, we can estimate that the pKa for the protonation of tBuCuOHO.S.2 is between 16 and 20 (see SI for details). In fact, tBuCuOHO.S.2 underwent protonation and subsequent disproportionation with the addition of pyr·H+ (pKa = 3.4 in DMSO54), but also upon addition of 4-NO2-2,6-DTBP (pKa = 7.3 in DMSO55), 2,6-DTBP (pKa = 17.3 in DMSO55), or 4-MeO-2,6-DTBP (pKa = 18.2 in DMSO55), suggesting that the pKa for the protonation of tBuCuOHO.S.2 is at least 19 in DMF (see SI).52 Likewise, the pKa obtained for the protonation of tBuCuOHO.S.3 (pKa ~ 11) was also in agreement with the fact that tBuCuOHO.S.3 was not protonated by 4-MeO-2,6-DTBP or 2,6-DTBP (pKa > 17) but it reacted with 4-NO2-2,6-DTBP (pKa = 7.3) to form tBuCuOH(H+)O.S.3 (see SI). On the basis of the reactivity of tBuCuOHO.S.3 (capable of oxidizing PhNHNHPh but not 2,6-DTBP) and the pKa of its protonation, we estimate the E0 for the reduction of tBuCuOH(H+)O.S.3 to be between −0.5 and −0.8 V (see calculations in the SI). To validate this estimation experimentally, several 1e− reductants were added to tBuCuOH(H+)O.S.3 and the reactions were monitored by UV–vis spectroscopy. tBuCuOH(H+)O.S.3 was reduced by CoCp2 (E1/2 = −1.29 V vs Fc0/+ in DMF) and by Me10Fc (E1/2 = −0.50 V vs Fc0/+ in DMF) but not by Me8Fc (E1/2 = −0.41 V vs Fc0/+ in DMF) or Me2Fc (E1/2 = −0.07 V vs Fc0/+ in DMF).
Bordwell Analysis – BDE of the Reduction/Protonation of tBuCuOH(H+)O.S.3.
In the reaction between tBuCuOHO.S.4 and the phenols, we observed the accumulation of tBuCuOH(H+)O.S.3 which also oxidized phenols but with slower rates.56 These findings suggest that the thermodynamic driving force (BDE) of the reaction between tBuCuOH(H+)O.S.3 and H-atom donors should be similar, but probably slightly lower to the one calculated for tBuCuOHO.S.4 + H• (i.e., lower reaction rates, lower BDE).46 tBuCuOH(H+)O.S.3 could also be reversibly protonated to generate tBuCuOH(H+)2O.S.3 (Figure 12B). Titration of tBuCuOH(H+)2O.S.3 with proton sponge (1,8-bis(dimethylamino)naphthalene) allowed us to determine the pKa for the protonation of tBuCuOH(H+)O.S.3 (pKa = 8.9).57 On the basis of the reactivity of tBuCuOH(H+)O.S.3 (capable of oxidizing 2,6-DTBP and 4-MeO-2,6-DTBP), the BDE for its protonation/reduction could be estimated at 78–83 kcal mol−1. This approximation was used to calculate the E1/2 for the reduction of tBuCuOH(H+)2O.S.3, which we estimate to be between −0.1 and −0.4 V. To validate this calculation experimentally, several 1e− reductants were added to tBuCuOH(H+)2O.S.3 and the reactions were monitored by UV–vis absorption spectroscopy, which revealed that reductants with E0 more negative than −0.4 V (CoCp2, Me10Fc and Me8Fc) could reduce tBuCuOH(H+)2O.S.3 but Me2Fc could not (E1/2 = −0.07 V vs Fc0/+ in DMF).
Bordwell Analysis – Comparison of the BDEs.
The enhanced oxidative power of tBuCuOHO.S.4 compared to tBuCuOHO.S.3 can be rationalized in terms of the higher reduction potential of tBuCuOHO.S.4 (ΔE1/2(tBuCuOHO.S.4/tBuCuOHO.S.3) = 0.77 V; 18 kcal/mol), whereas the change in the pKa for the reduced products does not contribute as significantly to the overall BDE (ΔpKa (tBuCuOHO.S.2/ tBuCuOHO.S.3 = 5–9; 9–12 kcal/mol). Protonation of tBuCuOHO.S.3 to generate tBuCuOH(H+)O.S.3 led to small changes in the basicity of the complex (ΔE1/2(tBuCuOHO.S.3/ tBuCuOH(H+)O.S.3) = 2; 3 kcal/mol) but a substantial increase of the reduction potential of the protonated counterparts (ΔE1/2(tBuCuOH(H+)2O.S.3/tBuCuOH(H+)O.S.3) ~ 0.4 V; 9 kcal/mol), which enables tBuCuOH(H+)O.S.4 to oxidize phenolic substrates like tBuCuOHO.S.4. Overall, the versatility of the tBuCuOH platform, which is able to reversibly reach three physical oxidation states and three different protonation states, is fundamental to achieving the unprecedented 2H+/2e− oxidation reactivity described in this work.
Comparison of tBuCuOHO.S.4 with Other “High-Valent” Copper Complexes.
Previous reports extensively studied the reactivity of mononuclear [CuOH]2+ complexes toward C─H and O─H bonds and it was found that these species act as strong 1H+/1e− oxidants (BDE of 90 kcal/mol), abstracting hydrogen atoms from the substrates via an HAT mechanism (Figure 13A).13,58 Recent studies have also described that analogous “high-valent” [CuX]2+ complexes also behave as a 1H+/1e− oxidants.37,59 Zhang and co-workers have recently reported that a [CuF]2+ species carried out the fluorination of C─H bonds via H-atom abstraction (performed by one equiv of [CuF]2+) followed by radical rebound (performed by a second equiv of [CuF]2+), suggesting that this “high-valent” complex acted as 1e− oxidant.5 This differs from the 2e− reactivity of tBuCuOHO.S.4, capable of performing two consecutive H-atom abstractions.
Figure 13.

2e− oxidations performed by the putative Cu-oxyl intermediate of LPMOs compared with the 1e− reactivity of [CuOH]2+ complexes (A).13,58 2e− reactivity found for the [CuOH]1+ cores reported in this article (B).
We hypothesize that the superior oxidative power of an LCuIIIOH complex58 (depicted in Figure 13A) compared to tBuCuOHO.S.4 (90 kcal/mol in THF vs 83 kcal/mol in DMF) results from the stronger basicity character of the reduced complex LCuIIOH (pKa in THF similar to the strong base DBU) and from the slightly higher redox potential of LCuIIIOH (see Figure 13A). The inclusion of the H-bonding donors on the ligand scaffold, which we believe allow for stabilizing the CuOH cores in different molecular oxidation states, could also be preventing a richer oxidative reactivity toward external substrates. Karlin and co-workers have recently reported that fine-tuning of these H-bonding interactions can stabilize mononuclear Cu/O2 entities but also enhance their oxidative reactivity.35,60
CONCLUSIONS
Recent studies have proposed that the oxidation of C─H bonds in LPMO occurs via formation of a Cu-oxyl complex that can perform the 2e− oxidation of strong C─H bonds via H-atom abstraction followed by OH rebound (Figure 13).5,7 In this report, we describe that the [CuOH]1+ cores bearing a redox-active ligand react with phenolic O─H bonds via two consecutive 1H+/1e− events. This differs from the reactivity of galactose oxidase model systems in which Cu complexes bound by redox-active ligands (L•CuII) typically perform the oxidation of alcohols (RCH2OH) to aldehydes (RCHO) via alkoxide coordination to the Cu center (L•CuIIOCH2R) followed by an intramolecular 1H+/2e− oxidation (L•CuIIOCH2R → LHCu1 + RCHO).25,61 Interestingly, the H• abstraction reactivity of the CuOH complexes reported is not triggered by the oxidation of the CuOH core, which maintains its physical oxidation state, but rather by oxidation and/or protonation of the ligand scaffold, a strategy that might also be utilized by some Cu-dependent monooxygenase enzymes.5,62 Ongoing studies are focused on modifying our copper complexes by tuning the electronics of the redox-active backbone, altering the stereoelectronics of the H-bond donors, and utilizing various anions, which we believe will boost the oxidative power of these reactive M─O(H) cores and allow for monooxygenase-like 2e− functionalization of C─H substrates (H• abstraction followed by •OH rebound).
Supplementary Material
ACKNOWLEDGMENTS
We thank the Robert A. Welch Foundation (grant N-1900-20190330 to I.G.B.) and the National Institutes of Health (NIH award number R15GM128078 to I.G.B.) for financial support. K.M.L. thanks NSF (CHE-1454455) for support. We thank Prof. Pia Vogel (Dept. of Biology at Southern Methodist University) for help with the EPR measurements. XAS data were obtained at SSRL, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy’s Office of Biological and Environmental Research, and by NIH/HIGMS (including P41GM103393).
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c03867.
Physical methods and materials; synthesis and characterization of ligand scaffold tBuLH3; synthesis and characterization of [tBuCuOH]2−, electrochemistry; reactivity with phenols; stoichiometry; reactivity with phenols; kinetics; BDE, pKa, and E1/2 calculations (PDF) (CIF)
Contributor Information
Tong Wu, Department of Chemistry, Southern Methodist University, Dallas, Texas 75275, United States.
Samantha N. MacMillan, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Khashayar Rajabimoghadam, Department of Chemistry, Southern Methodist University, Dallas, Texas 75275, United States.
Maxime A. Siegler, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Kyle M. Lancaster, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Isaac Garcia-Bosch, Department of Chemistry, Southern Methodist University, Dallas, Texas 75275, United States.
REFERENCES
- (1).Elwell CE; Gagnon NL; Neisen BD; Dhar D; Spaeth AD; Yee GM; Tolman WB Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev 2017. 117, 2059–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of dioxygen by copper metalloproteins and insights from model complexes. JBIC, J. Biol. Inorg. Chem 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Frandsen KEH; Simmons TJ; Dupree P; Poulsen J-CN; Hemsworth GR; Ciano L; Johnston EM; Tovborg M; Johansen KS; von Freiesleben P; Marmuse L; Fort S; Cottaz S; Driguez H; Henrissat B; Lenfant N; Tuna F; Baldansuren A; Davies GJ; Lo Leggio L; Walton PH The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat. Chem. Biol 2016, 12, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Span EA; Suess DLM; Deller MC; Britt RD; Marletta MA The Role of the Secondary Coordination Sphere in a Fungal Polysaccharide Monooxygenase. ACS Chem. Biol 2017, 12, 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ciano L; Davies GJ; Tolman WB; Walton PH Bracing copper for the catalytic oxidation of C─H bonds. Nat. Catal 2018, 1, 571–577. [Google Scholar]
- (6).Meier KK; Jones SM; Kaper T; Hansson H; Koetsier MJ; Karkehabadi S; Solomon EI; Sandgren M; Kelemen B Oxygen Activation by Cu LPMOs in Recalcitrant Carbohydrate Polysaccharide Conversion to Monomer Sugars. Chem. Rev 2018, 118, 2593–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bissaro B; Streit B; Isaksen I; Eijsink VGH; Beckham GT; DuBois JL; Røhr ÅK Molecular mechanism of the chitinolytic peroxygenase reaction. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 1504–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Paradisi A; Johnston EM; Tovborg M; Nicoll CR; Ciano L; Dowle A; McMaster J; Hancock Y; Davies GJ; Walton PH Formation of a Copper(II)–Tyrosyl Complex at the Active Site of Lytic Polysaccharide Monooxygenases Following Oxidation by H2O2. J. Am. Chem. Soc 2019, 141, 18585–18599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Singh RK; Blossom BM; Russo DA; Singh R; Weihe H; Andersen NH; Tiwari MK; Jensen PE; Felby C; Bjerrum MJ Detection and Characterization of a Novel Copper-Dependent Intermediate in a Lytic Polysaccharide Monooxygenase. Chem. - Eur. J 2020, 26, 454–463. [DOI] [PubMed] [Google Scholar]
- (10).Whittaker JW Free radical catalysis by galactose oxidase. Chem. Rev 2003, 103, 2347–2363. [DOI] [PubMed] [Google Scholar]
- (11).Rajabimoghadam K; Darwish Y; Bashir U; Pitman D; Eichelberger S; Siegler MA; Swart M; Garcia-Bosch I Catalytic Aerobic Oxidation of Alcohols by Copper Complexes Bearing Redox-Active Ligands with Tunable H-Bonding Groups. J. Am. Chem. Soc 2018, 140, 16625–16634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rajabimoghadam K; Darwish Y; Bashir U; Pitman D; Eichelberger S; Siegler MA; Garcia-Bosch I Tunable intramolecular multicenter H-bonding interactions in first-row metal complexes bearing bidentate redox-active ligands. J. Coord. Chem 2019. 72, 1335–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Donoghue PJ; Tehranchi J; Cramer CJ; Sarangi R; Solomon EI; Tolman WB Rapid C─H Bond Activation by a Monocopper(III)–Hydroxide Complex. J. Am. Chem. Soc 2011, 133, 17602–17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dhar D; Yee GM; Spaeth AD; Boyce DW; Zhang H; Dereli B; Cramer CJ; Tolman WB Perturbing the Copper(III)–Hydroxide Unit through Ligand Structural Variation. J. Am. Chem. Soc 2016, 138, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Dhar D; Yee GM; Markle TF; Mayer JM; Tolman WB Reactivity of the copper(iii)-hydroxide unit with phenols. Chem. Sci 2017, 8, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Munhá RF; Zarkesh RA; Heyduk AF Tuning the Electronic and Steric Parameters of a Redox-Active Tris(amido) Ligand. Inorg. Chem 2013, 52, 11244–11255. [DOI] [PubMed] [Google Scholar]
- (17).Cook SA; Borovik AS Molecular Designs for Controlling the Local Environments around Metal Ions. Acc. Chem. Res 2015, 48, 2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Addison AW; Rao TN; Reedijk J; van Rijn J; Verschoor GC Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2[prime or minute]-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc., Dalton Trans 1984, 1349–1356. [Google Scholar]
- (19).Tubbs KJ; Fuller AL; Bennett B; Arif AM; Berreau LM Mononuclear N3S(thioether)-Ligated Copper(II) Methoxide Complexes: Synthesis, Characterization, and Hydrolytic Reactivity. Inorg. Chem. 2003, 42, 4790–4791. [DOI] [PubMed] [Google Scholar]
- (20).Berreau LM; Mahapatra S; Halfen JA; Young VG; Tolman WB Independent synthesis and structural characterization of a mononuclear copper-hydroxide complex previously assigned as a copper-superoxide species. Inorg. Chem 1996, 35, 6339–6342. [Google Scholar]
- (21).Manabu H; Koichiro J; Hideki M; Hisahiko E A Unique Copper(II)-Hydroxide Complex Derived from Copper(II)-Superoxide. Chem. Lett 1996, 25, 813–814. [Google Scholar]
- (22).Jitsukawa K; Harata M; Arii H; Sakurai H; Masuda H SOD activities of the copper complexes with tripodal polypyridylamine ligands having a hydrogen bonding sites. Inorg. Chim. Acta 2001, 324, 108–116. [Google Scholar]
- (23).Cook SA; Bogart JA; Levi N; Weitz AC; Moore C; Rheingold AL; Ziller JW; Hendrich MP; Borovik AS Mononuclear complexes of a tridentate redox-active ligand with sulfonamido groups: structure, properties, and reactivity. Chem. Sci 2018, 9, 6540–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tabbi G; Giuffrida A; Bonomo RP Determination of formal redox potentials in aqueous solution of copper(II) complexes with ligands having nitrogen and oxygen donor atoms and comparison with their EPR and UV–Vis spectral features. J. Inorg. Biochem 2013, 128, 137–145. [DOI] [PubMed] [Google Scholar]
- (25).Chaudhuri P; Hess H; Weyhermuller T; Wieghardt K Aerobic Oxidation of Primary Alcohols by a New Mononuclear CuII-Radical Catalyst. Angew. Chem., Int. Ed 1999, 38, 1095–1098. [DOI] [PubMed] [Google Scholar]
- (26).(a) Evans DF Magnetic Moment Determination by NMR. J. Chem. Soc 1959, 2003. [Google Scholar]; (b) Evans DF; Jakubovic DA Water-soluble hexadentate Schiff-base ligands as sequestrating agents for iron(III) and gallium(III). J. Chem. Soc., Dalton Trans 1988, 2927. [Google Scholar]; (c) Schubert EM Utilizing the Evans method with a super-conducting NMR spectrometer in the undergraduate laboratory. J. Chem. Educ. 1992, 69, 62. [Google Scholar]
- (27).Kau LS; Spira-Solomon DJ; Penner-Hahn JE; Hodgson KO; Solomon EI X-ray absorption edge determination of the oxidation state and coordination number of copper. Application to the type 3 site in Rhus vernicifera laccase and its reaction with oxygen. J. Am. Chem. Soc 1987, 109, 6433–6442. [Google Scholar]
- (28).DuBois JL; Mukherjee P; Collier AM; Mayer JM; Solomon EI; Hedman B; Stack TDP; Hodgson KO Cu K-Edge XAS Study of the [Cu2(μ-O)2] Core: Direct Experimental Evidence for the Presence of Cu(III). J. Am. Chem. Soc 1997, 119, 8578–8579. [Google Scholar]
- (29).Tomson NC; Williams KD; Dai X; Sproules S; DeBeer S; Warren TH; Wieghardt K Re-evaluating the Cu K pre-edge XAS transition in complexes with covalent metal–ligand interactions. Chem. Sci 2015, 6, 2474–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Walroth RC; Uebler JWH; Lancaster KM Probing CuI in homogeneous catalysis using high-energy-resolution fluorescence-detected X-ray absorption spectroscopy. Chem. Commun 2015, 51, 9864–9867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Walroth RC; Lukens JT; MacMillan SN; Finkelstein KD; Lancaster KM Spectroscopic Evidence for a 3d10 Ground State Electronic Configuration and Ligand Field Inversion in [Cu(CF3)-4]1–. J. Am. Chem. Soc 2016, 138, 1922–1931. [DOI] [PubMed] [Google Scholar]
- (32).Walroth RC; Miles KC; Lukens JT; MacMillan SN; Stahl SS; Lancaster KM Electronic Structural Analysis of Copper(II)–TEMPO/ABNO Complexes Provides Evidence for Copper(I)–Oxoammonium Character. J. Am. Chem. Soc 2017, 139, 13507–13517. [DOI] [PubMed] [Google Scholar]
- (33).DiMucci IM; Lukens JT; Chatterjee S; Carsch KM; Titus CJ; Lee SJ; Nordlund D; Betley TA; MacMillan SN; Lancaster KM The Myth of d8 Copper(III). J. Am. Chem. Soc 2019, 141, 18508–18520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Diaz DE; Quist DA; Herzog AE; Schaefer AW; Kipouros I; Bhadra M; Solomon EI; Karlin KD Impact of Intramolecular Hydrogen Bonding on the Reactivity of Cupric Superoxide Complexes with O─H and C─H Substrates. Angew. Chem., Int. Ed 2019, 58, 17572–17576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lee JY; Peterson RL; Ohkubo K; Garcia-Bosch I; Himes RA; Woertink J; Moore CD; Solomon EI; Fukuzumi S; Karlin KD Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric–Superoxo Complex. J. Am. Chem. Soc 2014, 136, 9925–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Unjaroen D; Gericke R; Lovisari M; Nelis D; Mondal P; Pirovano P; Twamley B; Farquhar ER; McDonald AR High-Valent d7 NiIII versus d8 CuIII Oxidants in PCET. Inorg. Chem 2019, 58, 16838–16848. [DOI] [PubMed] [Google Scholar]
- (38).Note: We propose that the protonation of tBuCuOHO.S.3-produces a Cu-aqua complex but protonation of the tBuL3- ligand could also be occurring. Similarly, we propose that tBuCuOH(−H+)2O.S.2 is an aqua complex with one of the amines of the tridentate ligand protonated but other protonation isomers are possible.
- (39).Türk H; Çimen Y Oxidation of 2,6-di-tert-butylphenol with tert-butylhydroperoxide catalyzed by cobalt(II) phthalocyanine tetrasulfonate in a methanol–water mixture and formation of an unusual product 4,4′-dihydroxy-3,3′,5,5′-tetra-tert-butylbiphenyl. J. Mol. Catal A: Chem 2005, 234, 19–24. [Google Scholar]
- (40).Osako T; Ohkubo K; Taki M; Tachi Y; Fukuzumi S; Itoh S Oxidation mechanism of phenols by dicopper-dioxygen (Cu2O2) complexes. J. Am. Chem. Soc 2003, 125, 11027–11033. [DOI] [PubMed] [Google Scholar]
- (41).Trammell R; Rajabimoghadam K; Garcia-Bosch I Copper-Promoted Functionalization of Organic Molecules: from Biologically Relevant Cu/O2Model Systems to Organometallic Transformations. Chem. Rev 2019, 119, 2954–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zaragoza JPT; Siegler MA; Goldberg DP A Reactive Manganese(IV)–Hydroxide Complex: A Missing Intermediate in Hydrogen Atom Transfer by High-Valent Metal-Oxo Porphyrinoid Compounds. J. Am. Chem. Soc 2018, 140, 4380–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).McManus C; Mondal P; Lovisari M; Twamley B; McDonald AR Carboxamidate Ligand Noninnocence in Proton Coupled Electron Transfer. Inorg. Chem 2019, 58, 4515–4523. [DOI] [PubMed] [Google Scholar]
- (44).Brigati G; Lucarini M; Mugnaini V; Pedulli GF Determination of the Substituent Effect on the O─H Bond Dissociation Enthalpies of Phenolic Antioxidants by the EPR Radical Equilibration Technique. J. Org. Chem 2002, 67, 4828–4832. [DOI] [PubMed] [Google Scholar]
- (45).Note: The BDFEO─H values for these substrates in DMF were not available. We have plotted BDEO─H (an enthalpy) against ΔG‡ (free energies). We believe that the differences between BDE and BDFE values are small enough to make that comparison valid.
- (46).Mayer JM Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res 2011, 44, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bailey WD; Dhar D; Cramblitt AC; Tolman WB Mechanistic Dichotomy in Proton-Coupled Electron-Transfer Reactions of Phenols with a Copper Superoxide Complex. J. Am. Chem. Soc 2019, 141, 5470–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Garcia-Bosch I; Ribas X; Costas M Electrophilic Arene Hydroxylation and Phenol O─H Oxidations Performed by an Unsymmetric μ-η1:η1-O2-Peroxo Dicopper(II) Complex. Chem. - Eur. J 2012, 18, 2113–2122. [DOI] [PubMed] [Google Scholar]
- (49).Garcia-Bosch I; Cowley RE; Diaz DE; Peterson RL; Solomon EI; Karlin KD Substrate and Lewis Acid Coordination Promote O─O Bond Cleavage of an Unreactive L2CuII2(O22-) Species to Form L2CuIII2(O)2 Cores with Enhanced Oxidative Reactivity. J. Am. Chem. Soc 2017, 139, 3186–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bordwell FG; Cheng J; Ji GZ; Satish AV; Zhang X Bond dissociation energies in DMSO related to the gas phase values. J. Am. Chem. Soc 1991, 113, 9790–9795. [Google Scholar]
- (51).Mayer JM Proton-coupled electron transfer: A reaction chemist’s view. Annu. Rev. Phys. Chem 2004, 55, 363–390. [DOI] [PubMed] [Google Scholar]
- (52).Maran F; Celadon D; Severin MG; Vianello E Electrochemical determination of the pKa of weak acids in N,N-dimethylformamide. J. Am. Chem. Soc 1991, 113, 9320–9329. [Google Scholar]
- (53).Note: we estimate that the BDEO─H of the CuOH complexes and the phenols are similar in DMF and DMSO, since the pKa values of weak acids are comparable in both solvents (pKaDMF = 1.56 + 0.96 pKaDMSO) and the redox potentials of our complexes in DMSO are identical to the ones found in DMF.
- (54).Tshepelevitsh S; Kütt A; Lõkov M; Kaljurand I; Saame J; Heering A; Plieger PG; Vianello R; Leito I On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem 2019, 2019, 6735–6748. [Google Scholar]
- (55).Bordwell FG; Zhang X-M Acidities and homolytic bond dissociation enthalpies of 4-substituted-2,6-di-tert-butylphenols. J. Phys. Org. Chem 1995, 8, 529–535. [Google Scholar]
- (56).Note: The kinetics of the reaction between tBuCuOH(H+)O.S.3 and 4-X-2,6-DTBPs are complex (since tBuCuOH(H+)O.S.3 is regenerated during the oxidation of the phenols via disproportionation of tBuCuOH(H+)2O.S.2) and are currently being studied in detail.
- (57).Note: While the pKa’s were measured at −40 °C, the reactivity and electrochemistry of tBuCuOHO.S.4 was analyzed at room temperature. However, we believe that the difference in temperature will have a minor impact on the calculated BDEs.
- (58).Dhar D; Tolman WB Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex. J. Am. Chem. Soc 2015, 137, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bower JK; Cypcar AD; Henriquez B; Stieber SCE; Zhang S C(sp3)─H Fluorination with a Copper(II)/(III) Redox Couple. J. Am. Chem. Soc 2020, 142, 8514. [DOI] [PubMed] [Google Scholar]
- (60).Bhadra M; Lee JYC; Cowley RE; Kim S; Siegler MA; Solomon EI; Karlin KD Intramolecular Hydrogen Bonding Enhances Stability and Reactivity of Mononuclear Cupric Superoxide Complexes. J. Am. Chem. Soc 2018, 140, 9042–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Wang Y; DuBois JL; Hedman B; Hodgson KO; Stack TDP Catalytic Galactose Oxidase Models: Biomimetic Cu(II)-Phenoxyl-Radical Reactivity. Science 1998, 279, 537–540. [DOI] [PubMed] [Google Scholar]
- (62).Crespo A; Marti MA; Roitberg AE; Amzel LM; Estrin DA The Catalytic Mechanism of Peptidylglycine alpha-Hydroxylating Monooxygenase Investigated by Computer Simulation. J. Am. Chem. Soc 2006, 128, 12817–12828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.