

Abstract
Evasion of host immunity is a hallmark of cancer, however mechanisms linking oncogenic mutations and immune escape are incompletely understood. Through loss-of-function screening of 1,001 tumor suppressor genes, we identified DAPK3 as a previously unrecognized driver of anti-tumor immunity through the STING pathway of cytosolic DNA sensing. Loss of DAPK3 expression or kinase activity impaired STING activation and interferon-β (IFN-β)-stimulated gene induction. DAPK3 deficiency in IFN-β-producing tumors drove rapid growth and reduced infiltration of CD103+CD8α+DCs and cytotoxic lymphocytes, attenuating response to cancer chemo-immunotherapy. Mechanistically, DAPK3 coordinated post-translational modifications of STING. In unstimulated cells, DAPK3 inhibited STING K48-linked poly-ubiquitination and proteasome-mediated degradation. After cGAMP stimulation, DAPK3 was required for STING K63-linked poly-ubiquitination and STING-TBK1 interaction. Comprehensive phospho-proteomics uncovered a DAPK3-specific phosphosite on the E3 ligase LMO7, critical for LMO7-STING interaction and STING K63-linked poly-ubiquitination. Thus, DAPK3 is an essential kinase for STING activation that drives tumor-intrinsic innate immunity and tumor immune surveillance.
Introduction
Innate immune sensing of immune-stimulatory DNA through the stimulator of interferon genes (STING) pathway is essential for eliciting anti-tumor immune responses against a variety of cancers1, 2. Upon recognition of cellular DNA from genomic, mitochondrial, micronuclear or retroelement origins3, the innate sensor/enzyme cGAMP synthase (cGAS) produces cGAMP, a unique nucleotide second messenger that binds and activates STING. STING dimerizes and traffics from the endoplasmic reticulum (ER) to the trans-Golgi network to recruit the regulatory kinase Tank Binding Kinase 1 (TBK1). The STING-TBK1 complex recruits and phosphorylates the transcription factor IRF3, which trans-activates the IFNB1 promoter and other early immune response genes4. In the tumor microenvironment, sensing of tumor-derived DNA by antigen-presenting cells drives STING activation and downstream T cell-dependent anti-tumor immune responses5. Accordingly, STING expression in host immune cells is necessary for success of immune checkpoint blockade targeting PD-1/PD-L1 or CTLA-46.
Our understanding of tumor-intrinsic STING-IFN-β signaling and how it impacts tumor immune surveillance is evolving. Recent studies show that cGAS directly recognizes micronuclei induced by genomic instability7, 8, and tumor irradiation or treatment with certain chemical inhibitors9, 10, 11 triggers IFN-β via STING. Thus, productive STING signaling is active in normal cells during cellular transformation and in tumor cells during chemotherapy, and the selective pressure may drive specific genetic mutations to favor immune evasion, a key hallmark of cancer12. However, mechanisms linking specific oncogenic mutations to innate immune escape are poorly understood.
To shed new insights into tumor-intrinsic innate immunity, we performed genetic loss-of-function screening of 1,001 tumor suppressor genes in primary human cells, identifying death-associated protein kinase 3 (DAPK3) as an essential regulatory kinase in the STING-IFN-β pathway. Tumor-specific loss of DAPK3 expression or kinase activity accelerated the natural growth of IFN-β-producing tumors in immunocompetent and STING-deficient hosts, and increased tumor resistance to STING agonists and STING-activating chemo-immunotherapies, attenuating the efficacy of combination therapy with anti-PD-1. At the molecular level, DAPK3 coordinated STING ubiquitination. Loss of DAPK3 expression, but not its kinase activity, enhanced STING K48-linked poly-ubiquitination and diminished steady-state STING protein levels. DAPK3 deficiency also diminished K63-linked STING poly-ubiquitination following cGAMP stimulation, due to hypophosphorylation of the E3 ligase LMO7, which was directly phosphorylated by DAPK3 on S863. Studies establish DAPK3 as an essential activating kinase in the core STING signaling cascade, and a driver of tumor-intrinsic innate immunity and tumor immune surveillance. Thus, DAPK3 loss-of-function observed in several human tumor types13 drives evasion of host immunity and diminished efficacy of cancer immunotherapy.
Results
DAPK3 is a positive regulator of STING signaling
To examine tumor-intrinsic regulation of STING, we performed genetic loss-of-function screening of 1,001 human tumor suppressor genes (Supplementary Table 1, Extended Data Fig. 1a). Inducible nuclear IRF3 translocation was quantified in RNAi-treated human umbilical vein endothelial cells (HUVEC) by in situ immunofluorescence after 3 h of stimulation with poly (dA:dT) DNA (Extended Data Fig. 1b), which requires expression of cGAS, STING and TBK114. From these studies DAPK3 emerged as a positive regulator of DNA-stimulated IRF3 nuclear translocation (Fig. 1a). The DAPK family of serine/threonine kinases includes DAPK1, DAPK2 and DAPK3, which regulate cellular apoptosis and autophagy15. Quantitative reverse transcription (qRT)-PCR indicated broad expression of DAPK1/Dapk1 and DAPK3/Dapk3 in human and mouse cells (Extended Data Fig. 1c, d), while DAPK2/Dapk2 was low or undetectable, consistent with its restricted expression in a sub-population of hematopoietic cells16.
Fig. 1 |. DAPK3 regulates DNA-stimulated STING signaling.
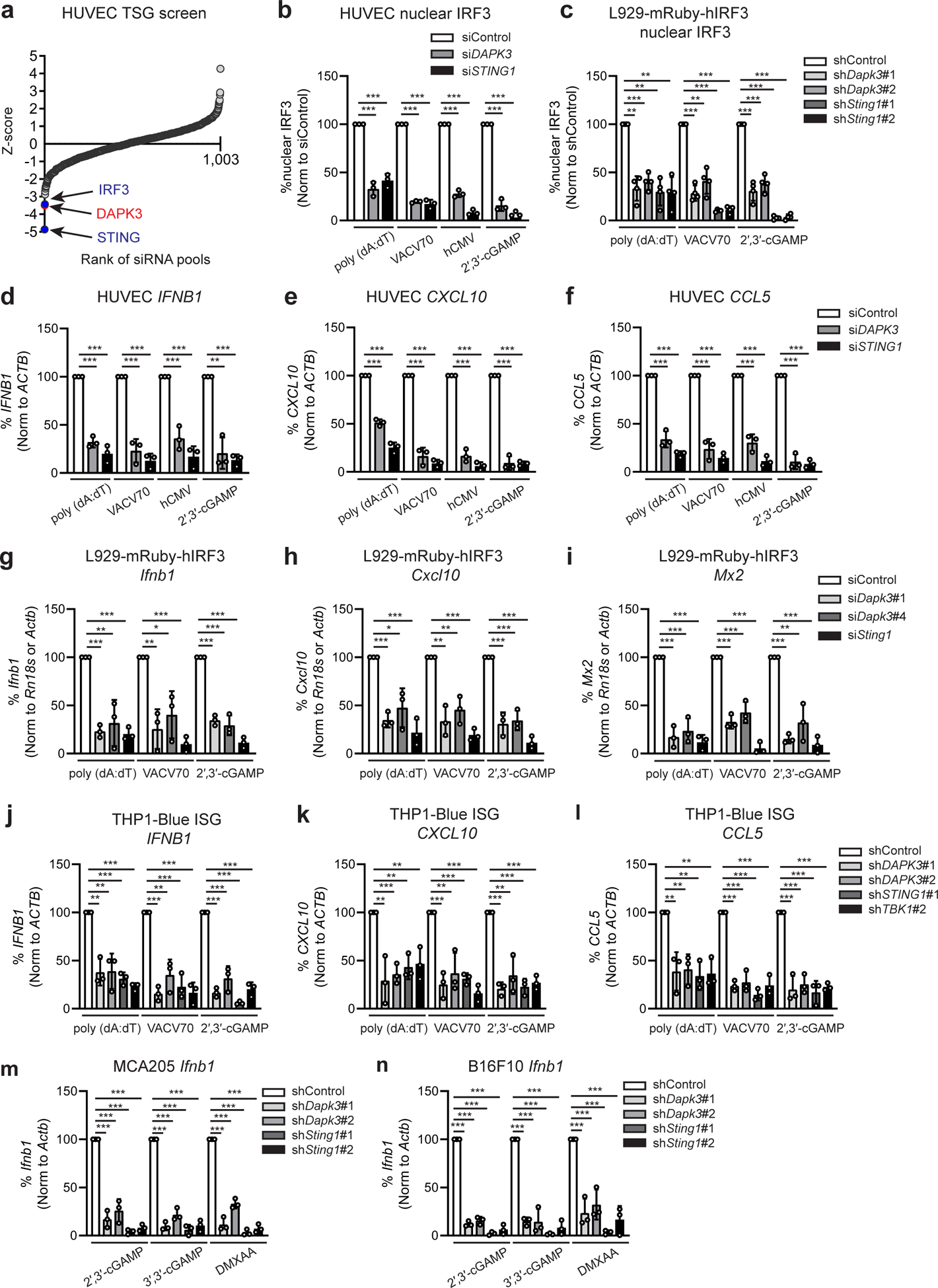
a, RNAi screen of 1,001 tumor suppressor genes in HUVEC, represented as ranked mean Z-score for poly (dA:dT)-induced IRF3 nuclear translocation (0.5 μg/ml) for 3 h. b, c, IRF3 nuclear translocation in (b) HUVEC and (c) L929-mRuby-hIRF3 stimulated with poly (dA:dT) (0.5 μg/ml), VACV70 (2 μg/ml), 2′,3′-cGAMP (10 μg/ml) or infected with hCMV (MOI=5) (b) for 3 h. d-f, qRT-PCR of (d) IFNB1, (e) CXCL10 and (f) CCL5 in HUVEC transfected with indicated siRNA and stimulated with poly (dA:dT) (0.5 μg/ml), VACV70 (2 μg/ml), 2′,3′-cGAMP (10 μg/ml), or infected with hCMV (MOI=5) for 4 h. g-i, qRT-PCR of (g) Ifnb1, (h) Cxcl10, and (i) Mx2 in L929-mRuby-hIRF3 transfected with indicated siRNA and stimulated with poly (dA:dT) (0.5 μg/ml), VACV70 (2 μg/ml), 2′,3′-cGAMP (10 μg/ml) for 4 h. j-l, qRT-PCR of (j) IFNB1, (k) CXCL10 and (l) CCL5 in THP1-Blue ISG transduced with indicated shRNA and stimulated with poly (dA:dT) (0.5 μg/ml), VACV70 (2 μg/ml), 2′,3′-cGAMP (10 μg/ml) for 4 h. m, n, qRT-PCR of Ifnb1 in (m) MCA205 and (n) B16F10 transduced with the indicated shRNA and stimulated with 2′,3′-cGAMP (10 μg/ml), 3′,3′-cGAMP (10 μg/ml) or DMXAA (50 μg/ml) for 4 h. mRNA levels were normalized to values of ACTB (human) and Rn18s or Actb (mouse), and percent expression calculated from stimulated control values. Data in (b-n) represent the mean of three independent experiments. Values represent mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001. Statistical comparisons were conducted using two-tailed t-test (b-n).
To validate screen results, HUVEC were transfected with siRNAs targeting DAPK3 or STING1, and IRF3 activation further assessed. DAPK3 depletion diminished IRF3 activation in response to poly (dA:dT) DNA and VACV70 DNA, the natural STING agonist 2′,3′-cGAMP or infection with human cytomegalovirus (hCMV), a DNA-encoding virus that engages cGAS-STING signaling14 (Fig. 1b). In mouse L929-mRuby-hIRF3 reporter cells (hereafter called L929) (Extended Data Fig. 1e), depletion of DAPK3 using two sequence-independent lentiviral shRNAs impaired IRF3 nuclear translocation induced by DNA and 2′,3′-cGAMP (Fig. 1c). Consistent with these findings, induction of IFNB1/Ifnb1 and ISGs CXCL10/Cxcl10, CCL5/Ccl5, MX2/Mx2 and IL6/Il6 were significantly impaired in HUVEC, L929 and THP1-Blue ISG reporter cells (hereafter called THP1) after DAPK3 depletion (Fig. 1d–l, Extended Data Fig. 1j, k), with effects comparable to STING. Similar results for Ifnb1 were obtained in mouse tumor cell lines MCA205 and B16F10 (Fig. 1m, n). DAPK1 was described as a negative regulator of cytosolic dsRNA sensing through RIG-I17. In HUVEC and mouse bone marrow-derived macrophages (BMDM), which express both DAPK1/Dapk1 and DAPK3/Dapk3, IFNB1/Ifnb1 was increased upon DAPK1 depletion in response to poly (dA:dT) DNA (Extended Data Fig. 1f–i), indicating that DAPK3 and DAPK1 exert distinct effects on IFN-β induction by DNA. In DAPK3-depleted L929 and THP1, IFNB1/Ifnb1 induced by cytosolic dsRNA ligands was unaffected (Extended Data Fig. 1l, m, p, q). However, DAPK3 positively regulated TLR3 and TLR4 signaling in THP1 (Extended Data Fig. 1n, o), suggesting a pleiotropic role in innate immune signaling. Results identify DAPK3 as a new activating kinase in the STING-IRF3-IFN-β pathway in human and mouse cells.
Tumor-expressed DAPK3 shapes immune surveillance
Dapk3 whole body knockout in mice is embryonic lethal18 and we were consistently unable to maintain knockout lines generated using CRISPR/Cas9, suggesting an essential role in cellular growth. However, loss-of-function DAPK3 mutations are frequently observed in human tumors13, and lower DAPK3 expression was associated with significantly shorter survival times in patients with pancreatic carcinoma, esophageal carcinoma, and uterine corpus endometrial carcinoma (Extended Data Fig. 2). Growth phenotypes were analyzed in MCA205 and B16F10 depleted of DAPK3 using lentiviral shRNAs. In both lines, in vitro proliferation was impaired upon loss of DAPK3 but not STING (Extended Data Fig. 3a, b), likely due to specific effects upon cytokinesis19. Despite inhibitory effects in vitro, growth of MCA205 tumors depleted of DAPK3 was significantly accelerated in vivo (Fig. 2a), while B16F10 tumors were unaffected (Fig. 2b). In MCA205 tumors depleted of DAPK3 or STING, significant reduction in numbers of anti-tumor NK cells, CD8+T cells and CD103+CD8α+DCs was accompanied by significant increase in numbers of pro-tumor regulatory T cells and M2 macrophages (Fig. 2c, Extended Data Fig. 4). To examine the contribution of host IFN-β signaling to MCA205 tumor growth, type I IFN receptor (Ifnar1)-knockout mice were examined. Compared to wild type mice, growth advantage of DAPK3-depleted MCA205 tumors was eliminated in the absence of host IFNAR signaling (Fig. 2d), suggesting that tumor-derived IFN-β activates paracrine signaling in host immune cells to drive spontaneous immune rejection of MCA205 tumors. Accordingly, numbers of tumor-infiltrating CD8+ T cells and CD103+CD8α+DCs were significantly reduced in Ifnar1-knockout mice, and differences between control and DAPK3- or STING-depleted MCA205 tumors attenuated (Extended Data Fig. 3c).
Fig. 2 |. Tumor-expressed DAPK3 shapes immune surveillance.
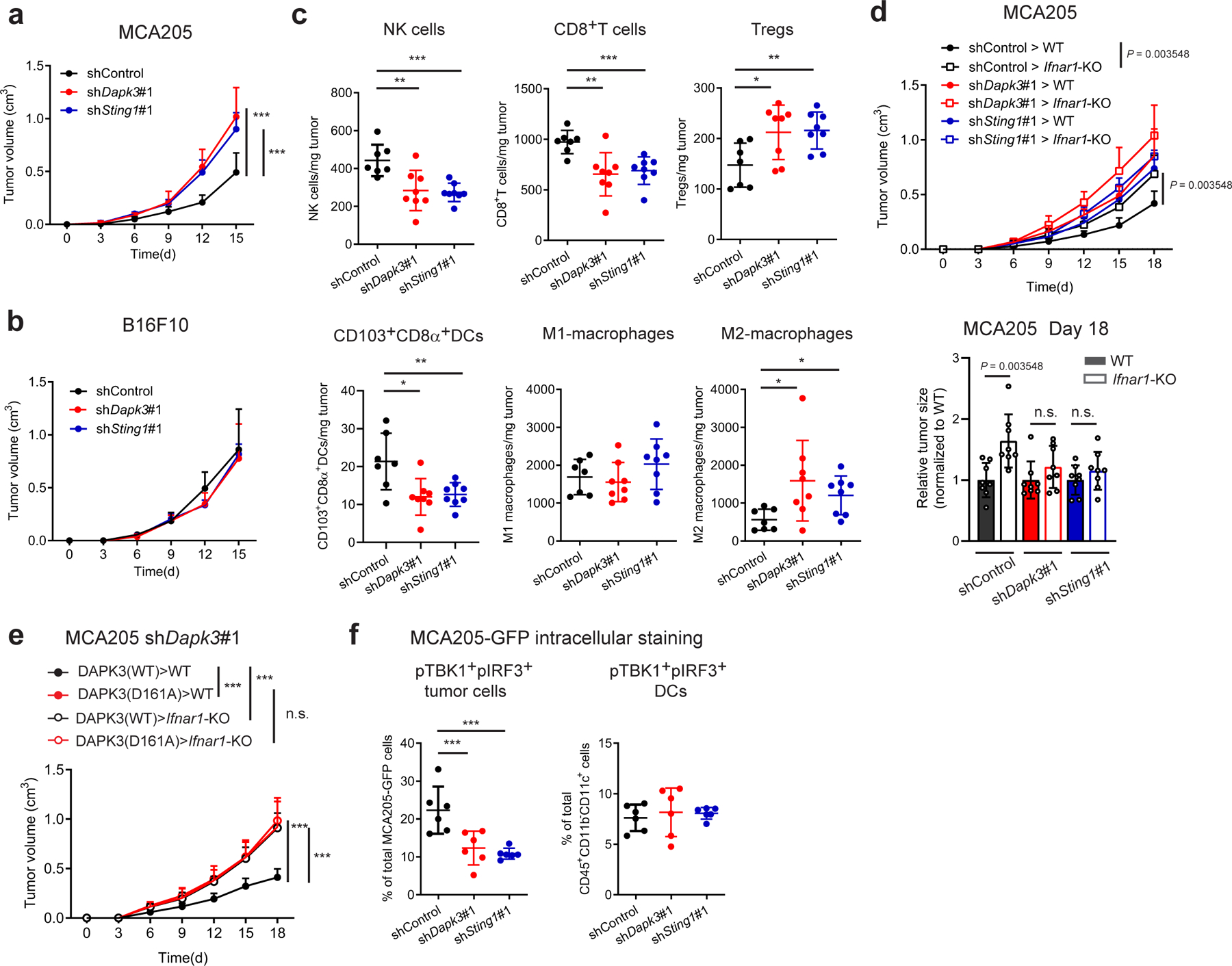
a, b, Tumor volume of shRNA-transduced (a) MCA205 and (b) B16F10 subcutaneously transplanted into C57BL/6J wild type (WT) mice (n=8 per group). c, Flow cytometry of tumor-infiltrating leukocytes in MCA205 tumor suspensions isolated from WT mice on Day 6 (n=7 for shControl, n=8 for shDapk3#1 and shSting1#1). d, Tumor volume of shRNA-transduced MCA205 subcutaneously transplanted into WT and Ifnar1-KO mice (n=8 per group). Tumor size on Day 18 is represented (right panel). e, Tumor volume of shDapk3#1-transduced MCA205 rescued with lentiviral DAPK3 (WT) or DAPK3 (D161A) prior to sub-cutaneous transplantation into WT mice and Ifnar1-KO mice (n=8 per group). f, Flow cytometry of intracellular pTBK1 and pIRF3 in MCA205-GFP tumor cells (CD45−GFP+) and tumor-infiltrating DCs (CD45+CD11b−CD11c+) in MCA205-GFP tumor suspensions isolated from WT mice on Day 6 (n=6 per group). Values represent percentage of each total cell population. Data in (a-f) are representative of three independent experiments. Values represent mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001. Statistical comparisons were conducted using two-tailed t-test (a-f).
MCA205 derive from fibrosarcomas in mice treated with the DNA alkylating agent methylcholanthrene. Due to dependence of natural MCA205 tumor growth upon intrinsic DAPK3-STING-IFN-β signaling, which was not observed in B16F10, we reasoned that genomic instability might drive spontaneous activation of this axis. Confocal microscopy of MCA205-cGAS-Clover reporter cells showed constitutive co-localization of cGAS with γH2AX, a marker of DNA damage-induced micronuclei formation8 in unstimulated cells (Extended Data Fig. 3d). Accordingly, baseline Ifnb1 levels in unstimulated MCA205 were sensitive to DAPK3 or STING depletion, in contrast to B16F10 (Extended Data Fig. 3e). Further, ectopic expression of a kinase-dead DAPK3 D161A point mutant initially identified in human ovarian tumors13 diminished baseline and stimulated Ifnb1 expression (Extended Data Fig. 3f). To assess the functional impact of oncogenic DAPK3 mutations on tumor immunology, DAPK3-depleted MCA205 were transduced with lentiviruses encoding human DAPK3(WT) or kinase dead DAPK3(D161A) prior to engraftment into mice. Expression of DAPK3(D161A) significantly accelerated in vivo tumor growth compared to DAPK3(WT), which depended upon host IFNAR1-signaling (Fig. 2e). Intracellular staining for double positive phospho-TBK1 and phospho-IRF3 MCA205-GFP tumor cells confirmed that tumor-intrinsic IRF3 activation was attenuated by DAPK3 depletion (Fig. 2f), while tumor-infiltrating DCs were comparable to controls. Results demonstrate that functional DAPK3 expression is necessary for STING-driven immune surveillance in IFN-β-producing tumors like MCA205.
STING depletion did not affect natural growth of B16F10 tumors in vivo (Fig. 2b), consistent with relatively low levels of basal Ifnb1 produced in culture (Extended Data Fig. 3e). Treatments that boost anti-tumor immunity by activating tumor-intrinsic cGAS-STING signaling have been reported. Cyclic di-nucleotide (CDN) STING agonists enhance T cell-dependent anti-tumor immunity by targeting innate cells in the tumor microenvironment5, and are currently being explored in clinical cancer trials in combination with immune checkpoint blockade (ID; NCT03010176, NCT03937141). To activate STING, bacterial CDN 3′,3′-cGAMP was injected into B16F10 tumors. While control tumors showed growth delay, anti-tumor effects of CDN were partially blunted in tumors depleted of DAPK3 or STING (Fig. 3a), suggesting that CDN target tumor cells and host immune cells in the tumor microenvironment5, 20. Accordingly, when transplanted in STINGgt/gt mice, DAPK3-depleted tumors lost all CDN responsiveness (Fig. 3a).
Fig. 3 |. DAPK3 regulates response to cancer chemo-immunotherapy.
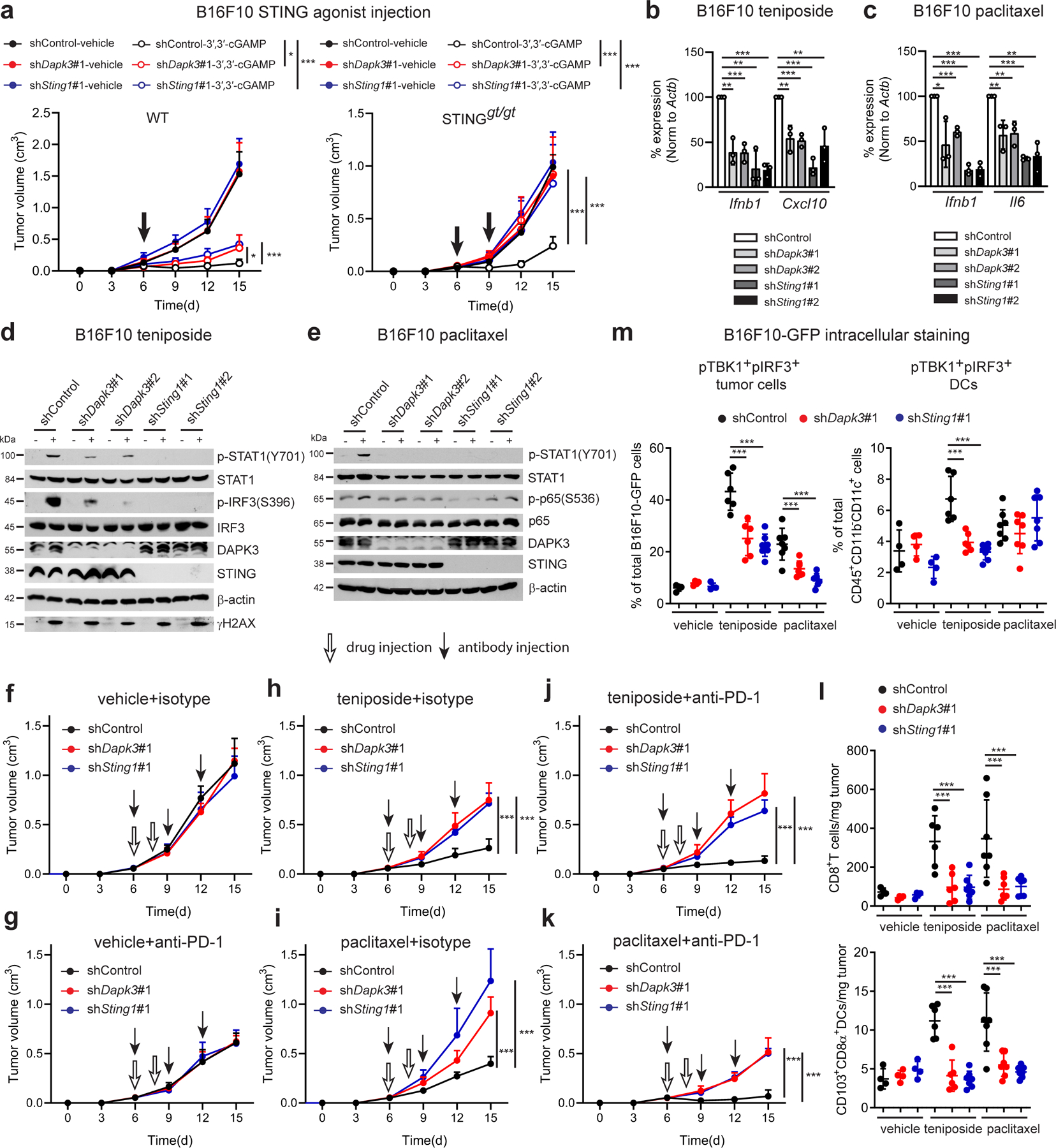
a, (Left) Tumor volume of shRNA-transduced B16F10 subcutaneously transplanted into WT (n=7 per group) or (right) STINGgt/gt mice (n=6 per group) treated using intratumoral injection of 3′,3′-cGAMP. cGAMP injections were performed on Day 6 in WT mice (5 μg per mouse), and Days 6 and 9 in STINGgt/gt mice (10 μg per mouse). b, c, qRT-PCR of Ifnb1, Cxcl10 or Il6 in shRNA-transduced B16F10 treated with (b) teniposide (10 μM) for 24 h or (c) paclitaxel (100 nM) for 48 h. d, e, Immunoblot of shRNA-transduced B16F10 treated with (d) teniposide (10 μM) for 12 h or (e) paclitaxel (1 μM) for 12 h (p-STAT1, STAT1, DAPK3, and STING) or 72 h (p-p65 and p65). Data from two gels are presented (d, e). f-k, Tumor volume of shRNA-transduced B16F10 subcutaneously transplanted into WT mice and treated with indicated chemical and antibody (n=6 per group). l, (Upper) Flow cytometry of tumor-infiltrating CD8+T cells and (lower) CD103+CD8α+DCs in B16F10 tumor suspensions isolated from WT mice on Day 10 (n=4 for vehicle, n=6 for teniposide-shControl and shDapk3#1, n=7 for teniposide-shSting1#1 and paclitaxel). m, Flow cytometry of intracellular pTBK1 and pIRF3 in B16F10-GFP tumor cells (CD45−GFP+) and tumor-infiltrating DCs (CD45+CD11b−CD11c+) in B16F10-GFP tumor suspensions isolated from WT mice on Day 10. Values represent percentage of each total cell population. Data in (a, d-m) are representative of or (b, c) are the mean of three independent experiments. Values represent mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001. Statistical comparisons were conducted using two-tailed t-test (a-c, f-m).
Recent studies indicate that chemo-immunotherapeutic agents activate cGAS-STING signaling in tumors. The Topoisomerase II inhibitor teniposide and the anti-microtubule agent paclitaxel drive mitotic arrest and micronuclei formation and directly activate cGAS11, 21, 22, 23. Confocal analysis of B16F10 cGAS-Clover treated with either teniposide or paclitaxel confirmed co-localization of cGAS and γH2AX (Extended Data Fig. 2g), and depletion of DAPK3 or STING impaired Ifnb1 and Cxcl10 or Il6 induction by teniposide or paclitaxel, respectively (Fig. 3b, c). Teniposide-induced phosphorylation of IRF3 (Fig. 3d), paclitaxel-induced phosphorylation of p65 (Fig. 3e) and STAT1 phosphorylation at Y70111, 23 (Fig. 3d, e) were significantly attenuated by depletion of DAPK3 or STING, while inducible γH2AX levels and cell death were comparable (Extended Data Fig. 3h). In B16F10 tumors, single administration of teniposide or paclitaxel showed significant anti-tumor effects in a host IFNAR-dependent manner (Extended Data Fig. 3i), which was attenuated by tumor-intrinsic loss of DAPK3 or STING (Fig. 3f, h, i). Single administration of anti-PD-1 demonstrated comparable anti-tumor effects in DAPK3- or STING-depleted tumors (Fig. 3g), however the synergistic effects of anti-PD-1 and teniposide or paclitaxel was abolished (Figure. 3j, k). Tumor-infiltrating CD8+T cells and CD103+CD8α+DCs were significantly increased by teniposide or paclitaxel treatment, and blunted by loss of DAPK3 or STING (Fig. 3l). Chemo-immunotherapeutic agents increased percentage of double positive phospho-TBK1 and phospho-IRF3 in B16F10-GFP tumor cells, which was reduced by DAPK3 or STING depletion (Fig. 3m). Collectively, data show that functional DAPK3 expression is critical for tumor-intrinsic cGAS-STING-driven responses during natural and chemo-immunotherapy-induced immune rejection of tumors.
DAPK3 inhibits STING K48-linked poly-ubiquitination
We next examined molecular mechanisms by which DAPK3 regulates STING. Immunoblotting demonstrated that DAPK3 depletion in HUVEC and MCA205, but not in BMDM or B16F10, reduced steady-state STING protein levels (Extended Data Fig. 1g, i, Extended Data Fig. 3a, b). shDapk3-transduced L929 also displayed low levels of STING protein, with no effect upon RIG-I, MAVS or cGAS (Fig. 4a). Sting1 levels were unaffected by DAPK3 depletion (Extended Data Fig. 5a). A strong correlation existed between DAPK3 expression, STING protein levels and IRF3 nuclear translocation in cells treated with DAPK3-specific shRNAs (Extended Data Fig. 5b, c) or siRNAs (Fig. 1c, Extended Data Fig. 5e). DAPK3 depletion also impaired DNA-induced NFκB p65 nuclear translocation (Extended Data Fig. 5d). Consistent with loss of STING protein, DAPK3 depletion in L929 inhibited VACV70-induced TBK1 and IRF3 phosphorylation (Extended Data Fig. 5f). Results indicate that DAPK3 maintains steady-state STING protein levels in some cell types, including HUVEC, MCA205, and L929.
Fig. 4 |. DAPK3 inhibits STING K48-linked poly-ubiquitination.
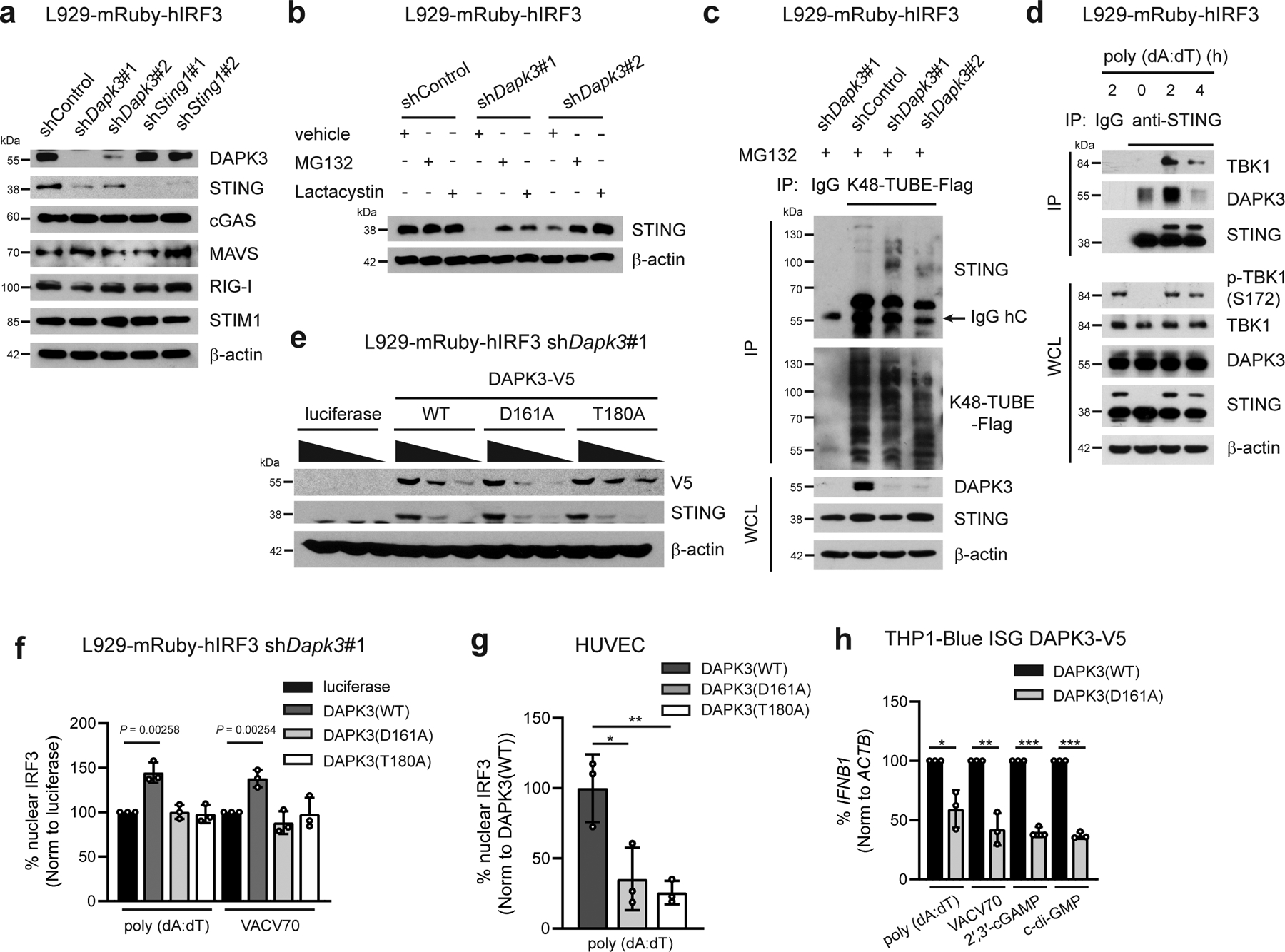
a, Immunoblot of L929-mRuby-hIRF3 transduced with indicated shRNA. Data from two gels are presented. b, Immunoblot of L929-mRuby-hIRF3 transduced with indicated shRNA treated with MG132 (20 μM), Lactacystin (5 μM), or vehicle for 4 h. c, (Upper) Immunoprecipitation and immunoblot of L929-mRuby-hIRF3 transduced with indicated shRNA treated with MG132 (20 μM) for 4 h, and (lower) immunoblot of whole cell lysates (WCL). Endogenous K48-linked poly-ubiquitination chains were immunoprecipitated using K48-TUBE-Flag. d, (Upper) Immunoprecipitation and immunoblot of L929-mRuby-hIRF3 with or without poly (dA:dT) stimulation for 2 h and 4 h and (lower) immunoblot of WCL. e, f, (e) Immunoblot and (f) IRF3 nuclear translocation in shDapk3#1-transduced L929-mRuby-hIRF3 expressing V5-tagged DAPK3(WT), DAPK3(D161A), or DAPK3(T180A). For (e), lentiviral transduction of V5-tagged DAPK3 was performed at MOI=5, 2, or 1. For (f), cells were stimulated with poly (dA:dT) (0.5 μg/ml) and VACV70 (2 μg/ml) for 3 h. g, IRF3 nuclear translocation in HUVEC expressing V5-tagged DAPK3(WT), DAPK3(D161A), and DAPK3(T180A) stimulated with poly (dA:dT) (0.5 μg/ml) for 3 h. Nuclear IRF3-positive cells were counted in >200 V5-positive single-cells in biological replicate wells. h, qRT-PCR of IFNB1 in THP1-Blue ISG ectopically expressing V5-tagged DAPK3(WT) or DAPK3(D161A) stimulated with poly (dA:dT) (0.5 μg/ml), VACV70 (2 μg/ml), 2′,3′-cGAMP (10 μg/ml), or c-di-GMP (10 μg/ml) for 4 h. Data in (a-e, g) are representative of (f, h) or are the mean of three independent experiments. Values represent mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001. Statistical comparisons were conducted using two-tailed t-test (f, h).
Treatment of DAPK3-depleted L929 with proteasome inhibitors MG132 and Lactacystin restored STING protein levels (Fig. 4b), suggesting that proteasome-mediated degradation of STING is enhanced in DAPK3-depleted cells. We examined K48-linked poly-ubiquitination, which controls steady-state STING degradation24. K48-linked poly-ubiquitinated proteins immunoprecipitated from lysates prepared from unstimulated DAPK3-depleted L929 were enriched for K48-ubiquitinated STING (Fig. 4c). Accordingly, endogenous DAPK3 interacted with endogenous STING in unstimulated L929, which was enhanced by DNA stimulation (Fig. 4d). To examine kinase activity, DAPK3-depleted L929 were transduced with lentiviruses encoding human DAPK3(WT), kinase-dead DAPK3(D161A) or kinase-deficient DAPK3(T180A)25. Expression of kinase mutants restored STING protein levels to wild type in L929 (Fig. 4e) and MCA205 (Extended Data Fig. 3f), indicating that DAPK3 kinase activity is dispensable for maintaining steady-state STING protein levels. However, DNA-induced IRF3 nuclear translocation was still impaired in cells expressing kinase mutants (Fig. 4f). Concordant with this, although STING protein levels in D161A-expressing HUVEC and THP1 were comparable to WT (Extended Data Fig. 5g, h), dominant negative effects were observed on IRF3 nuclear translocation (Fig. 4g) and IFNB1 induction after DNA or STING agonist treatment (Fig. 4h), respectively. Further, IFNB1/Ifnb1 induction in L929 and THP1 was inhibited by pharmacological inhibitors of DAPK kinase activity26 (Extended Data Fig. 5i, j). In vitro DAPK3 kinase assays failed to demonstrate phosphorylation of recombinant STING C-terminal peptides (Extended Data Fig. 5k) or full-length kinase-dead TBK1(K38M) (Extended Data Fig. 5l), suggesting that DAPK3 phosphorylates other essential regulatory proteins in the STING cascade. Together, results show that DAPK3 kinase activity is dispensable for maintaining steady-state STING levels, but critical for STING activation.
DAPK3 promotes STING K63-linked poly-ubiquitination
We examined the mechanism of STING activation in THP1, in which steady-state STING protein levels were unaffected by loss of DAPK3 (Fig. 5a). Upon 2′,3′-cGAMP stimulation, inducible phosphorylation of TBK1, STING, and IRF3 were significantly down-regulated by DAPK3 depletion (Fig. 5b). Co-immunoprecipitation showed that interaction between endogenous STING and TBK1 was disrupted by loss of DAPK3 (Fig. 5c), while steady-state localization and cGAMP-induced translocation of STING from ER to Golgi was unaffected (Extended Data Fig. 6a, b). Inducible K63-linked poly-ubiquitination of STING drives interaction with TBK127, 28. Upon 2′,3′-cGAMP stimulation, this was markedly decreased in DAPK3-depleted THP1 stably expressing HA-tagged Lys63-specific ubiquitin (HA-Ub(K63O)) (Fig. 5d). cGAMP-induced K63-linked poly-ubiquitination of endogenous STING was also abolished in D161A-expressing THP1 (Fig. 5e). Results demonstrate that functional DAPK3 positively regulates cGAMP-induced K63-linked poly-ubiquitination of STING.
Fig. 5 |. DAPK3 promotes STING K63-linked poly-ubiquitination.
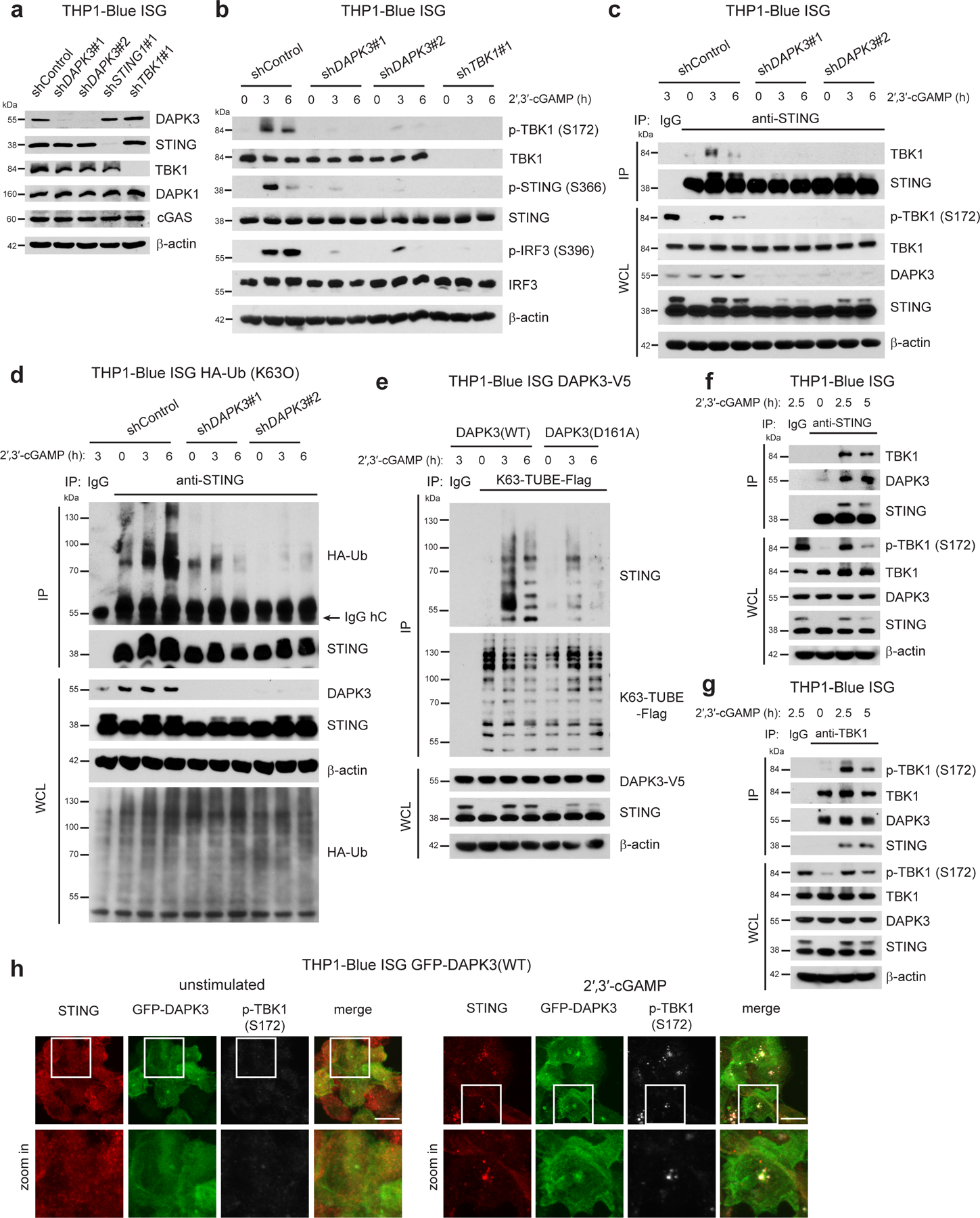
a, Immunoblot of THP1-Blue ISG transduced with indicated shRNA. b, Immunoblot of THP1-Blue ISG transduced with indicated shRNA and stimulated with 2′,3′-cGAMP (10 μg/ml) for 3 h and 6 h. c, (Upper) Immunoprecipitation and immunoblot of THP1-Blue ISG transduced with indicated shRNA stimulated with 2′,3′-cGAMP (10 μg/ml) for 3h and 6 h, and (lower) immunoblot of whole cell lysates (WCL). d, (Upper) Immunoprecipitation and immunoblot of shRNA-transduced THP1-Blue ISG stably expressing HA-Ub(K63O) stimulated with 2′,3′-cGAMP (10 μg/ml) for 3 h and 6 h, and (lower) immunoblot of WCL. e, (Upper) Immunoprecipitation and immunoblot of THP1-Blue ISG stably expressing DAPK3(WT) or DAPK3(D161A) stimulated with 2′,3′-cGAMP (10 μg/ml) for 3 h and 6 h, and (lower) immunoblot of WCL. Endogenous K63-linked poly-ubiquitin chains were immunoprecipitated using K63-TUBE-Flag. f, g, (Upper) Immunoprecipitation and immunoblot of THP1-Blue ISG stimulated with 2′,3′-cGAMP (10 μg/ml) for 2.5 h and 5 h, and (lower) immunoblot of WCL. (f) Anti-STING antibody or (g) anti-TBK1 antibody was used for immunoprecipitation. h, Confocal fluorescence microscopy of THP1-Blue ISG stably expressing GFP-tagged DAPK3(WT) unstimulated or stimulated with 2′,3′-cGAMP (50 μg/ml) for 3 h. Localization of GFP-DAPK3, STING, and phospho-TBK1 examined. Scale bars, 10 μm. Data in (a-h) are representative of three independent experiments.
Co-immunoprecipitation of endogenous proteins in native THP1 lysates revealed interaction between STING, DAPK3, and TBK1 or pTBK1 following cGAMP stimulation (Fig. 5f, g). In unstimulated native THP1 lysates, DAPK3 co-immunoprecipitated with TBK1 (Fig. 5g). Similarly, GFP-DAPK3 co-localized with pTBK1 and STING (Fig. 5h) or TBK1 and STING (Extended Data Fig. 6c) after cGAMP stimulation. To further probe the DAPK3-STING interaction, truncated STING proteins and STING phosphorylation mutants were assessed (Extended Data Fig. 6d). Mutations at the TBK1 phospho-acceptor sites Ser355, Ser358 and Ser36629, 30 did not affect STING interaction with DAPK3 or TBK131. However, truncated STING lacking the C-terminal region (amino acids 341-379), which interacts directly with TBK1 and is required for IRF3 activation31, 32, failed to co-immunoprecipitate DAPK3 in HEK293T (Extended Data Fig. 6e). Collectively, results show that DAPK3 interacts with the C-terminal effector domain of STING and forms a tripartite complex with pTBK1 and STING after cGAMP stimulation.
Phospho-proteomic profiling uncovers DAPK3 targets
Functional DAPK3 kinase activity is required for STING-driven IRF3 activation and IFN-β induction (Fig. 4f–h, Extended Data Fig. 3f, Extended Data Fig. 5i, j) and cGAMP-induced STING K63-linked poly-ubiquitination (Fig. 5e). To identify DAPK3 targets, comprehensive phospho-proteomic profiling was performed using tandem mass tag (TMT)-labeling-based mass spectrometry33. THP1 transduced with shDAPK3, shTBK1 or shControl lentiviruses were stimulated with 2′,3′-cGAMP prior to quantification of phosphorylated peptides. Phospho-proteins were prioritized by identifying hypo-phosphorylation in shRNA-treated cell lysates (Fig. 6a). 330 protein targets were commonly altered (301 at the same phospho-site, 29 at a different phospho-site) in both shDAPK3 cells and shTBK1 cells (Fig. 6b, Supplementary Table 2). To examine DAPK3 targets, we focused on 196 phospho-sites identified in 165 proteins demonstrating hypo-phosphorylation at DAPK3 consensus sequence R/K-X-X-S/T34, 35. The DAPK3-specific cluster overlapped with 16 proteins hypo-phosphorylated at the IKK consensus sequence S-X-X-X-S/T36 (Fig. 6b, Supplementary Table 2). Ingenuity Pathway Analysis (IPA) of genes in the DAPK3-specific cluster demonstrated enrichment of key regulatory kinases (e.g. ERL/MAPK, mTOR and SAPK/JNK) and innate immune response genes specific for cytokine and IRF signaling (Fig. 6c). Rho signaling, actin remodeling, and autophagy pathways were also highlighted, consistent with involvement of DAPK3 in these processes37, 38. Collectively, results uncover a network of innate immune response proteins as substrates for DAPK3 in cGAMP-stimulated THP1.
Fig. 6 |. Phospho-proteomic profiling uncovers DAPK3 targets.
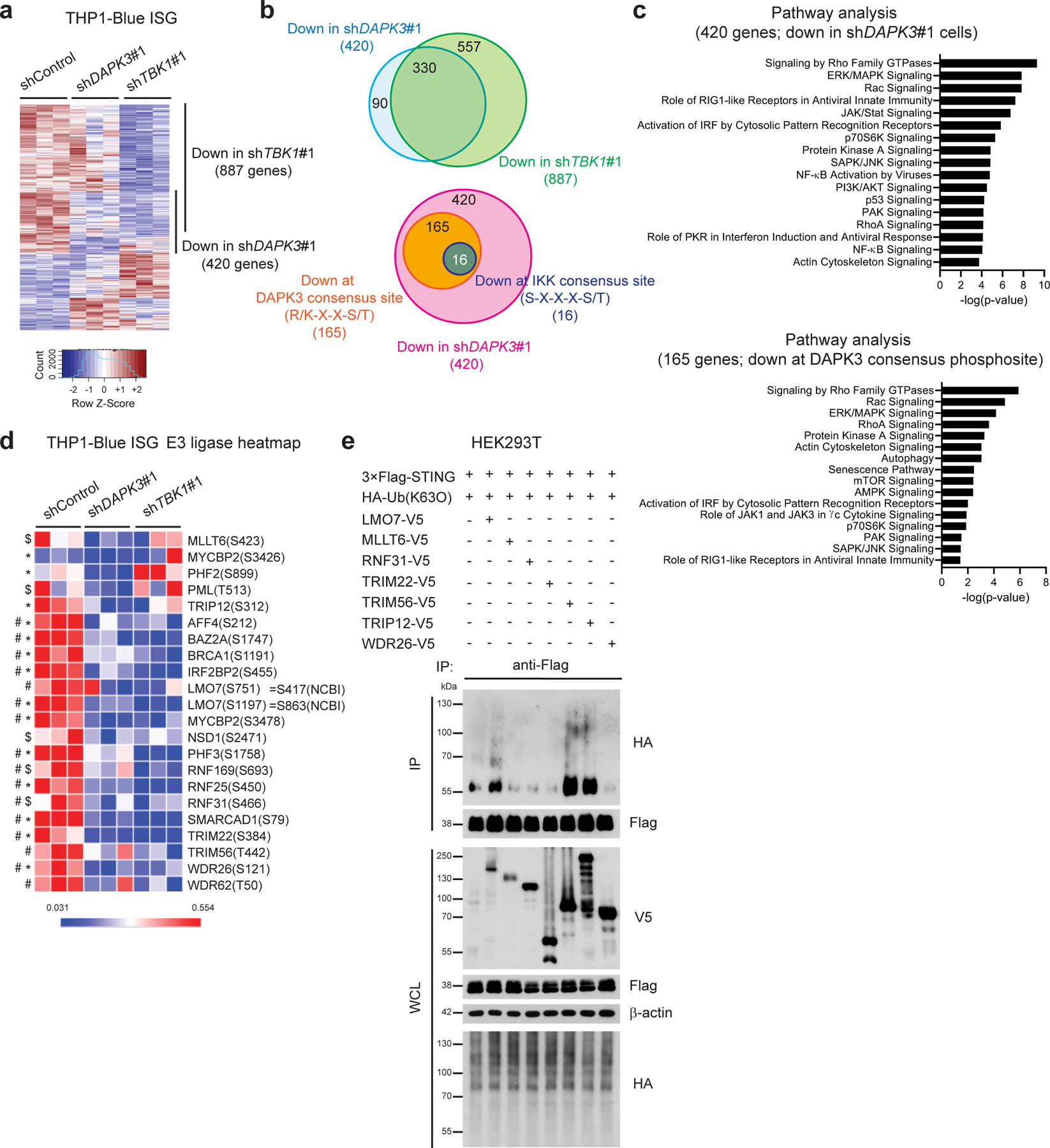
a, Heat map of phosphosites in shControl, shDAPK3#1 or shTBK1#1-transduced THP1-Blue ISG upon 2′,3′-cGAMP stimulation (10 μg/ml) for 3 h. b, (Upper) Overlap of hypo-phosphorylated proteins (330) detected in shDAPK3#1 cells (420) and shTBK1#1 cells (887), and (lower) 165 proteins hypo-phosphorylated at DAPK3 consensus phosphorylation site (R/K-X-X-S/T), of which 16 proteins showed overlap hypo-phosphorylation at IKK consensus site (S-X-X-X-S/T). (c) Ingenuity Pathway Analysis (IPA) of (Upper) 420 genes encoding proteins hypo-phosphorylated in shDAPK3#1 cells, and (lower) 165 genes encoding proteins hypo-phosphorylated at DAPK3 consensus phosphorylation site in shDAPK3#1 cells. (d) Heat map of E3 ligases phosphorylated at DAPK3 consensus site. Amino acids numbering is based upon UniProt ID, and S751 and S1197 of LMO7 correspond to S417 and S863, respectively, of the NCBI ID. *P<0.05, log2FC<−0.4 for shDAPK3#1, $P<0.1, log2FC<−0.4 for shDAPK3#1, #P<0.05, log2FC<−0.4 for shTBK1#1. (e) (Upper) Immunoprecipitation and immunoblot of HEK293T transfected with plasmids encoding 3×Flag-tagged human STING, HA-tagged Ub(K63O) and V5-tagged human E3 ligase, and (lower) immunoblot of whole cell lysates (WCL). Data in (e) are representative of four independent experiments. Statistical comparisons were conducted using two-tailed t-test (d).
To explore STING ubiquitination, we examined 20 candidate E3 ligases identified as potential targets for direct phosphorylation by DAPK3 (Fig. 6d, Supplementary Table 2). Genes encoding these candidates and others E3 ligases implicated in STING K63-linked poly-ubiquitination27, 28, 39, 40 were functionally assessed in THP1 by siRNA experiments analyzing ISG reporter activity after 2′,3′-cGAMP stimulation (Extended Data Fig. 7a). Knockdown of 11 novel E3 ligase-encoding gene candidates significantly impaired ISG reporter activity, 6 of which were prioritized as robust regulators (Extended Data Fig. 7b). Prioritized candidates were selected for STING ubiquitination studies, using human TRIM56 as a positive control for catalyzing K63-linked ubiquitination of human STING28. Immunoprecipitation of lysates from HEK293T transfected with Flag-tagged human STING, HA-tagged Ub (K63O) and individual V5-tagged human E3 ligases revealed that LMO7 and TRIP12 promoted K63-linked STING poly-ubiquitination (Fig. 6e). Notably, somatic mutation and genetic alterations in DAPK3, LMO7 and TRIP12 are frequent in multiple human tumor types (Extended Data Fig. 8a–c, Supplementary Table 3). Results show that DAPK3 regulates phosphorylation of the E3 ligases LMO7 and TRIP12, which catalyze K63-linked STING ubiquitination.
LMO7 phosphorylation on S863 is critical for STING ubiquitination
We next performed in vitro kinase assays of DAPK3 and TBK1 (Extended Data Fig. 7c, d). TRIP12 was phosphorylated by DAPK3 or TBK1, and TRIM56 by TBK1. However, phosphorylation mutants of TRIM56 and TRIP12 did not affect STING-TBK1 interaction or STING K63-linked poly-ubiquitination (Extended Data Fig. 7e–h). DAPK3 phosphorylated LMO7 at S863, the site identified by phospho-proteomic profiling (Fig. 7a). TBK1 also phosphorylated LMO7 at other sites within aa 810-910 and aa 360-460 regions (Extended Data Fig. 9a). Notably, DAPK3 depletion specifically attenuated LMO7-mediated STING K63-linked ubiquitination (Extended Data Fig. 9b) and impaired STING-LMO7 interaction (Fig. 7b), consistent with inability of the phospho-deficient LMO7 S863A mutant to interact with STING (Fig. 7c). Accordingly, S863>A mutation or deletion of LMO7 LIM or PDZ domains, which are responsible for protein-protein interactions, impaired STING K63-linked poly-ubiquitination (Fig. 7d, e). Thus, phosphorylation of LMO7 at S863 by DAPK3 is critical for STING-LMO7 interaction and STING K63-linked poly-ubiquitination. Depletion of LMO7 or TRIP12 in THP1 significantly impaired cGAMP-induced K63 poly-ubiquitination of endogenous STING (Fig. 7f, g) and STING-TBK1 interaction (Extended Data Fig. 9d), resulting in impaired TBK1 activation (Extended Data Fig. 9e, f). STING activation by VACV70 or STING agonists was also abolished in LMO7- or TRIP12-depleted THP1 and HUVEC (Fig. 7h, i, Extended Data Fig. 9g–j). Data show that direct phosphorylation of K63-linked poly-ubiquitination E3 ligases such as LMO7 by DAPK3 is critical for STING interaction and ubiquitination. Collectively, results support a model whereby DAPK3 is a pleiotropic regulator of STING ubiquitination and activity in unstimulated and stimulated cells (Extended Data Fig. 10)
Fig. 7 |. DAPK3 phosphorylation of LMO7 is necessary for STING K63-linked poly-ubiquitination.
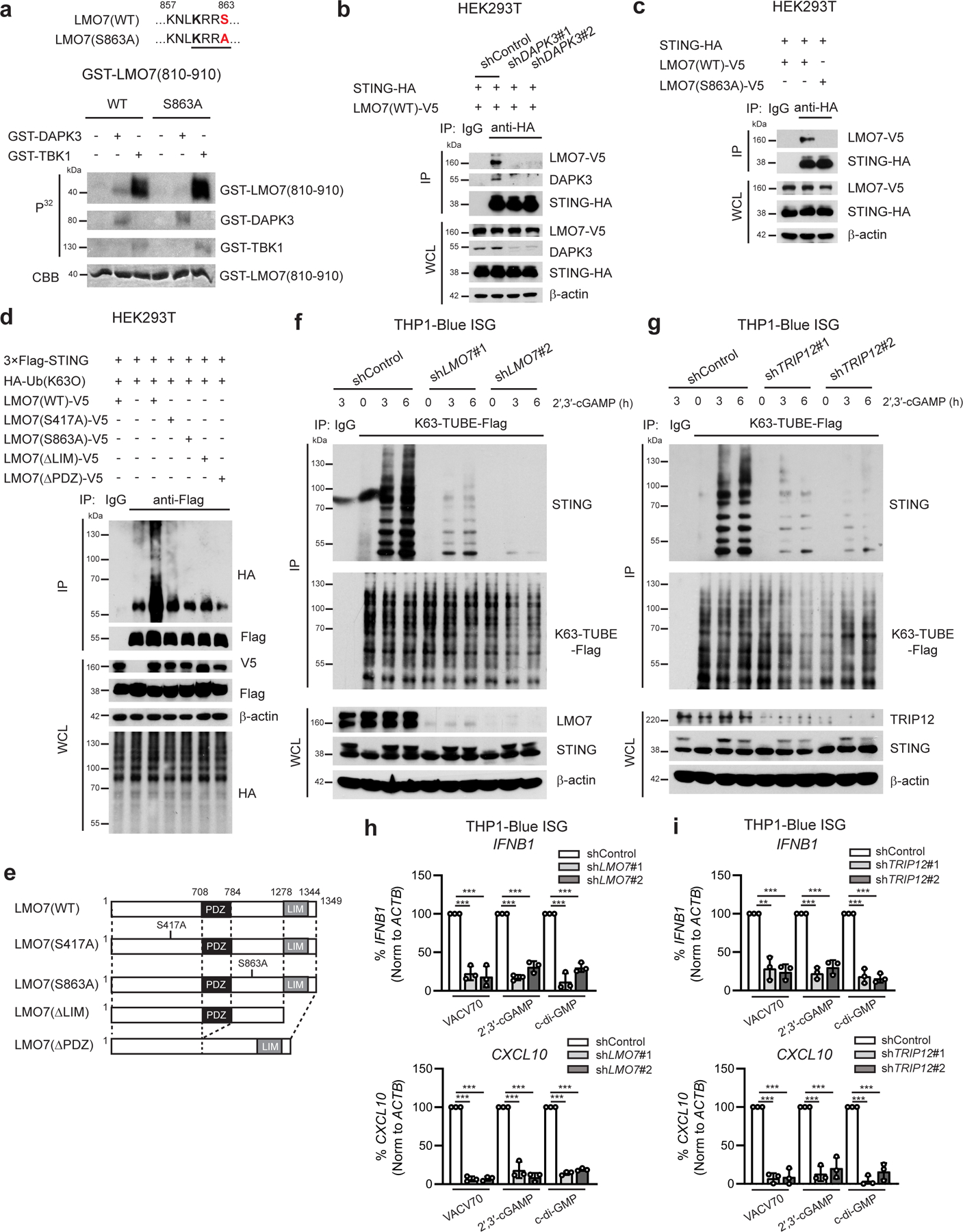
a, In vitro kinase assay of GST-tagged human LMO7 (aa 810-910). Peptides were incubated with GST-tagged DAPK3 or TBK1 in the presence of [γ-32P] ATP. b, (Upper) Immunoprecipitation and immunoblot of HEK293T transduced with indicated shRNA prior to transfection with plasmids encoding HA-tagged human STING and V5-tagged human LMO7(WT) and (lower) immunoblot of whole cell lysates (WCL). c, (Upper) Immunoprecipitation and immunoblot of HEK293T transfected with plasmids encoding HA-tagged human STING and V5-tagged human LMO7(WT) or phosphor-deficient LMO7 (S863A) and (lower) immunoblot of WCL. d, (Upper) Immunoprecipitation and immunoblot of HEK293T transfected with plasmids encoding 3×Flag-tagged human STING, HA-tagged Ub(K63O), and V5-tagged human LMO7 mutants, and (lower) immunoblot of WCL. e, Schematic representation of human LMO7 mutants. f, g, (Upper) Immunoprecipitation and immunoblot of THP1-Blue ISG transduced with two distinct (f) shLMO7 or (g) shTRIP12 and stimulated with 2′,3′-cGAMP (10 μg/ml) for 3 h and 6 h, and (lower) immunoblot of WCL. Endogenous K63-linked poly-ubiquitin chains were immunoprecipitated using K63-TUBE-Flag. h, i, qRT-PCR analysis of IFNB1 and CXCL10 in THP1-Blue ISG transduced with two distinct (h) shLMO7 sequences or (i) shTRIP12 sequences and stimulated with VACV70 (4 μg/ml), 2′,3′-cGAMP (10 μg/ml), or c-di-GMP (10 μg/ml) for 4 h. Data in (a-d, f, g) are representative of or (h, i) the mean of three independent experiments. Values represent mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001. Statistical comparisons were conducted using two-tailed t-test (h, i).
Discussion
IFN-β is a multifunctional cytokine that coordinates anti-tumor immunity by stimulating anti-tumor myeloid and T cell populations, tumor antigen cross-presentation and cytotoxic lymphocyte activity1, 2. STING has emerged as a promising target for boosting IFN-β-driven anti-tumor immunity in multiple pre-clinical models, with STING agonists currently under active pursuit in cancer immunotherapy trials as combinatorial agents with immune checkpoint blockade41. It remains incompletely understood how tumor suppressor mutations impact STING-IFN-β responses and facilitate immune evasion. Here we show that DAPK3 is a critical activating kinase for STING via two distinct processes: 1) maintaining steady-state STING protein levels by attenuating STING K48-linked poly-ubiquitination; 2) promoting agonist-induced STING K63-linked poly-ubiquitination via direct phosphorylation of E3 ubiquitin ligases.
Tumor cells, stromal cells, and immune cells within the tumor microenvironment promote immune surveillance via STING-IFN-β signaling. Accordingly, tumor-infiltrating myeloid cells and tumor endothelial cells20 produce IFN-β upon uptake of tumor-derived DNA or CDN. Recent studies indicate that tumor cells mount robust cell-intrinsic innate immune responses to damaged genomic DNA during neoplastic transformation, irradiation or chemotherapy. Teniposide and paclitaxel are chemo-immunotherapy agents that preferentially activate cGAS-STING signaling in tumor cells, and we show that their anti-tumor effects mainly depend upon host IFNAR signaling rather than apoptosis. Collectively, we show that productive tumor-intrinsic DAPK3-STING-IFN-β signaling is a critical component of immune surveillance.
Reduced expression of DAPK3 and STING in several human cancers42, 43, 44 occurs due to genomic hypermethylation45, and low DAPK3 expression was associated with shorter survival times. We show that functional DAPK3 kinase activity is critical for the full complement of anti-tumor immunity in IFN-β-producing MCA205 tumors, and loss of DAPK3 in B16F10 tumors impaired anti-tumor effects of STING agonists and eliminated anti-tumor effects of chemo-immunotherapies. Our findings suggest that functional expression of DAPK3 in tumors is a key determinant of natural immunogenicity and cancer chemo-immunotherapy response. Mechanistic insights into DAPK3 tumor-suppressor function previously focused upon proliferation and apoptosis13. Here we show a new mechanism whereby DAPK3 functions as a regulatory kinase of innate immune signaling that complexes with TBK1 and STING. These findings represent a paradigm shift in understanding how oncogenic mutations modulate the landscape of the tumor microenvironment to favor tumor growth by immune escape. This is relevant for developing cancer immunotherapies aimed at simultaneously boosting STING-IFN-β activation and curbing T cell exhaustion.
Kinase-active DAPK3 was essential for human STING K63-linked poly-ubiquitination, which enhances STING-TBK1 interaction and subsequent TBK1 auto-phosphorylation. Notably, human STING ubiquitination is catalyzed by several different E3 ligases27, 28, 39, which can exert distinct effects on its activity. For example, K63-linked poly-ubiquitination of human STING by human TRIM56 is dispensable for STING trafficking from the ER to Golgi28, but specifically required for STING-TBK1 interaction. In contrast, human MUL1 positively regulates STING trafficking, which indirectly affects STING-TBK1 interaction39. Global phospho-proteomic profiling in THP1 cells uncovered a diverse network of phospho-proteins co-regulated by DAPK3 and TBK1, consistent with our finding that these key kinases interact. We identified the DAPK3 substrates LMO7 and TRIP12 as positive regulators of cGAMP-stimulated IRF3 activation and IFN-β production. LMO7 and TRIP12 were shown to catalyze K63-linked poly-ubiquitination of STING, with DAPK3 phosphorylation of LMO7 on S863 critical for LMO7-STING interaction, STING K63-linked poly-ubiquitination, and STING-TBK1 interaction. To our knowledge, this is the first demonstration that dynamic phosphorylation controls STING ubiquitination. Missense mutations and gene deletions within LMO7, TRIP12 or DAPK3 loci are frequently observed human cancers. Notably, our mechanistic studies of STING ubiquitination were performed in human cells or using human proteins, underscoring the fact that K63-linked poly-ubiquitination of human STING is well delineated compared to the mouse protein27, 28. Although E3 ligases positively regulate STING in mouse cells28, 40, the relationship between STING ubiquitination and functional activity less clear39, and the role of specific E3 ligases such as TRIM56 appears to differ46, 47. It is possible that STING regulation by murine DAPK3 may involve different regulatory proteins.
Co-immunoprecipitation and confocal microscopy showed that DAPK3 associates with TBK1. Like TBK1, DAPK3 regulates IFN-β induction downstream of TLR3 and TLR4 signaling, and it is likely that DAPK3 regulates post-translational modifications of other innate signaling proteins interacting with the TBK1-DAPK3 complex. DAPK3 also regulates NFκB activation induced by STING, TLR3, and TLR4, which occurs independently of TBK148, 49. Further studies are required to fully delineate the pleiotropic role of DAPK3 in innate immune signaling.
Loss of DAPK3 expression correlated with reduced STING protein levels in non-hematopoietic cells. DAPK3 knockdown increased K48-linked poly-ubiquitination of STING in L929, and DAPK3 may recruit K48 de-ubiquitinase(s) (DUB) to stabilize STING. A recent study showed that iRhom2 recruits the DUB EIF3S5 to STING, leading to inhibition of proteasomal degradation24. More recently, TOLLIP was identified as a key factor stabilizing STING protein in resting cells by inhibiting IRE1α-driven lysosomal degradation50. However, expression of kinase-dead DAPK3 cDNA in DAPK3-depleted L929 and MCA205 restored STING protein expression but not responsiveness to STING agonists, supporting the notion that DAPK3 is a pleotropic regulator of STING and innate immunity.
This study uncovers a previously unknown role for the tumor-suppressor kinase DAPK3 in activating the STING-IFN-β response. We uncovered a new molecular driver of natural cancer immunity and response to cancer immunotherapy, based upon functional DAPK3 expression, and identified new E3 ligase targets that control STING activation. Ultimately, results underscore the importance of tumor cell-intrinsic innate immunity in immune surveillance, establishing a new link between oncogenic mutations and evasion of host innate immunity. Since DAPK3 expression was required for IFN-β production induced by hCMV infection, it is likely that DAPK3 also regulates anti-viral immunity, and future studies examining this are of significance.
Methods
Cells
HUVEC (Lonza) were cultured at 37°C and 5% CO2 in EGM2 Bulletkit media (Lonza). NHDF (from C. Benedict (LJI)) and HEK293T, A549, HeLa and L929 (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and 10% heat-inactivated fetal bovine serum (hi-FBS), 2 mM L-glutamine, 100 U/ml Penicillin-Streptomycin and 10 mM HEPES (pH 8.0). 5 μg/ml Insulin (Sigma-Aldrich) and 1 ng/ml bFGF (Sigma-Aldrich) were added to NHDF cultures. Mouse embryonic fibroblasts (MEFs), B16F10, LLC-RFP, MC38, and MCA205 obtained from C. Benedict (LJI), S. Schoenberger (LJI), C.C. Hedrick (LJI), J. Schlom (NCI), and N.P. Restifo (NCI), respectively, were cultured in DMEM and 10% hi-FBS, 2 mM L-glutamine, 0.1 mM non-essential amino acids (NEAA), 0.1 mM sodium pyruvate, and 50 μg/mL gentamicin sulfate. BMDM were cultured in RPMI1640 and 10% hi-FBS, 2 mM L-glutamine, 0.1 mM NEAA, 55 μM 2-mercaptoethanol, 0.1 mM sodium pyruvate and cultured L929 supernatant. THP1-Blue ISG (Invivogen) were cultured in RPMI1640 and 10% hi-FBS, 55 μM 2-mercaptoethanol, 50 μg/mL gentamicin sulfate and 100 μg/mL Zeocin (Invivogen).
For nucleic acid stimulation, cells were plated at 1×104 cells per well in 96-well plates for immunostaining, 1×105 cells per well for RNA extraction, or 5×105 cells per well in 6-well plates for immunoblotting one day before stimulation. THP1-Blue ISG were plated at 3×105 cells per well for RNA extraction or 2 × 106 cells per well in 6-well plates for immunoblot. Nucleic acids were transfected using Lyovec for HUVEC, Jetprime for L929-mRuby-hIRF3 or Lipofectamine 2000 for THP1-Blue ISG and murine tumor lines, per manufacturer’s instructions. cGAMP stimulation was performed using Lipofectamine 2000, or in cGAMP buffer containing 10 μg/ml saponin for HUVEC, 10 μg/ml digitonin for HEK293T or 15 μg/ml digitonin for L929-mRuby-hIRF3 in 50 mM HEPES (pH 7.4), 100 mM KCl, 3 mM MgCl2, 0.1 mM DTT, 85 mM sucrose, 0.2% BSA,1 mM ATP and 0.1 mM GTP. Cells were incubated in cGAMP buffer for 15 min at 25°C, then fresh growth media replaced for HUVEC or overlayed for HEK293T and L929-mRuby-hIRF3. IRF3 nuclear translocation was assessed 3 h post-stimulation, and RNA extraction performed 4 h post-stimulation.
Mouse experiments
C57BL/6J mice (wild-type, 000664) and STING-goldenticket (STINGgt/gt) mice (017537) were obtained from Jackson Laboratory. Ifnar1 gene-null mice on C57BL/6 background were obtained from S. Shresta (LJI). Mice were bred in pathogen-free conditions at the LJI vivarium. 7–10 week old female mice were used and experiments conducted without blinding using age-matched and sex-matched mice. All experiments were approved by the LJI animal care committee and performed in accordance with LJI experimental guidelines.
3×105 MCA205 or 3–7×105 B16F10 cells were subcutaneously injected into the flank. Tumor size was measured in two dimensions using digital calipers every 2 days until a maximum of 1.0–1.5 cm3. Tumor volume was calculated using modified ellipsoid formula V = (Lxl2)/2, where L = widest and l = smallest diameter. For intra-tumor injection of 3′,3′-cGAMP20, tumors were injected with 100 μl of PBS containing 5 μg or 10 μg of 3′3′-cGAMP (Invivogen) with 3 μl Lipofectamine 2000 (Life Technologies) starting Day 6. PBS containing Lipofectamine 2000 was used as vehicle. 10 mg/kg teniposide (Sigma-Aldrich)11 or 10 mg/kg paclitaxel (Selleck Chemicals)23 was dissolved in DMSO, diluted with 10% Cremophor EL (Sigma Aldrich)/1× PBS for intraperitoneal injection on Day 6 and Day 8. 100 μg anti-PD-1 (BE0033-2, BioXcell) or 100 μg isotype control was intraperitoneally injected on Day 6, 9, and 12.
Antibodies and Reagents
Antibodies are listed in Supplementary Table 4. Poly (dA:dT) (P0883) and MG132 (474787) were obtained from Sigma-Aldrich; VACV70 from IDT; Lyovec, 2′,3′-cGAMP, 3′,3′-cGAMP, DMXAA, c-di-GMP, poly (I:C)(LMW), poly (I:C)(HMW), LPS-EB, and FSL-1 from Invivogen; Jetprime from VWR; Lipofectamine 2000 from Life Technologies; 10× RIPA buffer (9806S) from Cell Signaling Technology; Lactacystin (70890) from Cayman Chemical; CellTiter Blue and CellTiter Glo from Promega; K48-TUBE-Flag (UM607), K63-TUBE-Flag (UM604), PR-619 (SI9619), and 1,10-phenanthroline (SI9619) from Life Sensors; FITC Annexin-V apoptosis detection kit (556547) from BD Biosciences; DAPK inhibitor #1 ((4Z)-4-(3-Pyridylmethylene)-2-styryl-oxazol-5-one) and DAPK inhibitor #2 ((4Z)-2-phenyl-4-(pyridin-3-ylmethylidene)-1,3-oxazol-5(4H)-one) from EMD Millipore (324788) and Interbioscreen (STOCK1N-35684), respectively26. hCMV MOLD obtained from M. Raftery (Charite Medical School) was prepared as described14.
Flow cytometry
Tumors were dissociated in DMEM with 400 U/ml Collagenase type IV (Sigma- Aldrich) and 20 μg/ml DNase I (Worthington Biochemical) for 30 min at 37°C. Tumor cell suspensions were washed 2× with 2% FBS/0.01% sodium azide/1× PBS staining buffer, and treated with red blood cell lysis buffer (Sigma-Aldrich) for 3 min at 25°C. Cells were washed 2× with staining buffer, and filtered through a 70 μm cell strainer. Fcγ receptors were blocked with CD16/32-blocking antibody (553141, BD Biosciences) for 10 min and surface antigens stained for 30 min at 4°C. LIVE/DEAD Fixable Blue Dead Cell Stain (L23015, Invitrogen) was used to assess viability, and forward- and side-scatter (FSC-A vs FSC-W, SSC-A vs SSC-W) to exclude doublets. Flow cytometry antibodies are listed in Supplementary Table 4. For intracellular staining, eBioscience Foxp3 / Transcription Factor Staining Buffer Set (Invitrogen) was used for Foxp3, pTBK1, and pIRF3 staining after surface staining, per manufacturer’s instructions. Fluorochrome-conjugated control IgG was used to calculate percentage of double positive pTBK1 and pIRF3 cells. WT tumor suspensions were used as compensation controls for intracellular staining in GFP-expressing tumors. Alternatively, cells were fixed with 4% PFA at 25°C for 20 min and permeabilized with 0.1% Triton X-100/1× PBS for 10 min prior to intracellular CD206 staining. Cell fluorescence was assessed using LSR-II or LSR Fortessa (BD Biosciences) and data analyzed using FlowJo (TreeStar, Ashland, OR).
siRNA transfection
siRNAs sequences (Dharmacon) are listed in Supplementary Table 5. siRNA pools or individual oligonucleotide were transfected into HUVEC (5×105 cells) or L929-mRuby-hIRF3 (2×105 cells), plated into 6-well plates 1 day before transfection using DharmaFECT 4 (Horizon Discovery), per manufacturer’s instructions, at final concentration 25 nM or 40 nM, respectively. 48 h after siRNA transfection, cells were trypsinized and replated at 1×104 cells per well in 96-well plates for immunostaining or 1×105 cells per well in 12-well plates for RNA extraction. Neon (Thermo Fisher Scientific) was used for BMDM and THP1-Blue ISG siRNA transfection. On Day 5, BMDM were resuspended in R buffer (5×106 cells/ml) and electroporated with 50 nM siRNA at 1,400 V/20 ms/2 pulse. THP1-Blue ISG resuspended in R buffer (2.5×107 cells/ml) were electroporated with 100 nM siRNA at 1,625 V/10 ms/3 pulse.
Total RNA extraction and quantitative RT-PCR
Total cellular RNA was extracted using Quick-RNA Miniprep Plus Kit (Zymoresearch) and cDNA synthesized using qScript cDNA synthesis kit (Quanta), per manufacturer’s instructions. Quantitative reverse transcription-PCR (qRT-PCR) was performed using CFX96 or CFX384 Touch Detection System (Bio-Rad), with Taqman Universal PCR master mix (Applied Biosystems) or FastStart SYBR Green master mix (Roche). mRNA abundance of each gene was normalized to ACTB levels for human and Actb or Rn18s (encoding 18S rRNA) for mouse in Taqman assay, and ACTB/Actb for human and mouse in SYBR assay. Primer sequences are listed in Supplementary Table 6.
Immunoprecipitation and Immunoblot
For whole-cell lysates, cells were lysed in 1× RIPA buffer (Cell Signaling Technology) with 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate and 10 mM sodium fluoride (Sigma-Aldrich). For immunoprecipitation, HEK293T or THP1-Blue ISG were lysed in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 10% glycerol NP-40 lysis buffer with protease and phosphatase inhibitors. After rotation for 30 min at 4°C, lysates were centrifuged at 15,000 rpm for 15 min to collect supernatants. Protein concentration in supernatants was quantified using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Supernatants were immunoprecipitated with antibody-conjugated protein G sepharose (Sigma-Aldrich) overnight at 4°C.
For STING ubiquitination, 3×106 HEK293T were transfected with expression plasmids. 24 h post-transfection, cells were lysed in NP-40 buffer and lysates boiled with 1% SDS for 10 min at 95°C, diluted 1:10 with NP-40 lysis buffer and centrifuged 15,000 rpm for 5 min at 4°C. Supernatants were immunoprecipitated with Flag antibody-conjugated Protein G dynabeads (Life Technologies) for 2 h at 4°C. For STING ubiquitination, 2×107 THP1-Blue ISG HA-Ub(K63O) were lysed in RIPA buffer containing 0.1% SDS, protease and phosphatase inhibitors, and 20 mM N-Ethylmaleimide (Sigma-Aldrich E3876). Lysates were boiled with 1% SDS for 10 min at 95°C, followed by 1:10 dilution in RIPA buffer and centrifugation at 15, 000 rpm for 5 min at 4°C. Lysates were incubated with STING antibody (Cell Signaling Technology, 1:50) overnight at 4°C, followed by Protein G Sepharose (Sigma-Aldrich) for 2 h at 4°C. Magnetic beads or Protein G Sepharose were washed 5× with 50 mM Tris-HCl (pH7.4)/1M NaCl/1 mM EDTA/1% NP-40 buffer, resuspended with Pierce Elution Buffer (pH 2.0) containing 4× SDS sample buffer and 10× reducing buffer (Life Technologies) and boiled for 10 min at 70°C. Endogenous STING ubiquitination was examined in immunoprecipitated lysates using K48-TUBE-Flag or K63-TUBE-Flag, per manufacturer’s instructions. SDS-poly acrylamide gel electrophoresis (SDS-PAGE) and immunoblotting were performed as described14, 51.
Plasmids and recombinant proteins
Human DAPK3 from pDONR223-DAPK3 (Addgene #23436) was Gateway cloned (Thermo Fisher Scientific) into pLX304 (Addgene #25890). DAPK3 (D161A) and DAPK3 (T180A) were constructed by site-directed mutagenesis. Luciferase from pLenti-CMV-Puro-LUC (w168-1) (Addgene #17477) was ligated into pLX304 with or without V5 tag. pEF-BOS-huTBK1-FlagHis (#27241) and pEF-BOS-huTBK1-K38M-Flag-His (#27240) were obtained from Addgene. Human STING and cGAS clones were obtained from Precision LentiORF Collection (GE Healthcare). STING was ligated into pLX304 with N-terminal 3× Flag tag with stop codon inserted before V5 tag, or ligated into pLX304 with C-terminal HA tag with stop codon inserted before V5 tag. cGAS-Clover was ligated into pLV-EF1α. Human ORF clones for LMO7, MLLT6, RNF31, TRIM22, TRIM56, TRIP12, and WDR26 were obtained from GenScript (catalog numbers listed in Supplementary Table 5) and cloned into pCI vector (Promega) with N-terminal V5 tag (for RNF31 and TRIP12), or pCMV6 vector (Origene) with C-terminal V5 tag (for LMO7, TRIM22, TRIM56, WDR26). LMO7 ORF clone corresponds to transcript 1 NM_005358.5, encoding 1349 amino acids, and differs from UniProt ID: Q8WWI1, encoding 1673 amino acids. Thus, Ser751 and Ser1197 in the UniProt sequence correspond to Ser417 and Ser863, respectively, in the NCBI sequence. MLLT6 was Gateway cloned into pDONR221(Invitrogen) and ligated into pLX304. pEF-neo-HA-ubiquitin was a gift from Y.C. Liu (LJI). HA-ubiquitin-K63O was synthesized (IDT) and cloned into pEF-neo. Lentiviral shRNAs from the MISSION shRNA Library (Sigma-Aldrich) are listed in Supplementary Table 5. psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) were used 3:1 as packaging mix.
For recombinant protein expression in Escherichia coli, cDNA encoding human STING (aa 149-379), full-length human TBK1 (K38M), human LMO7 (aa 360-460 and aa 810-910), human TRIM56 (aa 400-500), or human TRIP12 (aa 260-360) was inserted into pGEX-4T-2 (a gift from D. Zajonc, LJI) in-frame with N-terminal GST tag. Primers are listed in Supplementary Table 6. Vectors were transformed into BL21 (Life Technologies), and single colony cultured with 100 μg/ml Ampicillin and 0.5 μM IPTG (Thermo Fisher Scientific) for 16 h at 16°C. Bacterial pellet was resuspended with 50 mM Tris-HCl (pH 7.4), 50 mM NaCl, 5 mM EDTA, 1 mM DTT lysis containing Complete ULTRA Tablets (Roche) and lysed using a microfludizer. Lysates were centrifuged at 20,000 rpm for 1 h at 4°C, and filtered with a 0.45 μm syringe filter. 1 ml Glutathione Sepharose beads (GE healthcare) prewashed with lysis buffer was added to filtrated lysates and rotated overnight at 4°C. Beads were washed 5× with lysis buffer, then 2× with 50 mM Tris-HCl (pH 8.0), 200 mM NaCl Elution Buffer (−). GST-tagged proteins were eluted 3× with 1.5 ml 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 25 mM reduced glutathione Elution Buffer (+). Pooled fractions were concentrated using Pierce Protein Concentrator (Thermo Fisher Scientific), per manufacturer’s instruction and proteins visualized by SDS-PAGE and Coomassie Brilliant Blue staining or immunoblot using anti-GST (Z-5) (sc-459, Santa Cruz Biotechnology).
Lentiviral transduction
HEK293T (6×105 cells per well) were transfected with 375 ng shRNA vector and 375 ng packaging mix. Medium was replaced 24 h post-transfection with DMEM with 30% hi-FBS without antibiotics, and supernatants collected 2× every 24 h, pooled and filtered with 0.45 μm filter. Viral titer was determined by titration in A549 or measuring HIV Type I p24 Antigen ELISA 2.0 (Zeptometrix, #0801008). L929-mRuby-hIRF3, MCA205, and B16F10 (3×105 cells per well), or THP1-Blue ISG (1×106 cells per well) were plated in 6-well plates one day before transduction. Cells were infected with shRNA lentivirus with 10 μg/ml polybrene (TR-1003-G, EMD Millipore) by spin infection at 2,000 rpm for 30 min. Cells were selected with 4 μg/ml puromycin for L929-mRuby-hIRF3, 2.5 μg/ml for B16F10, or 3 μg/ml for THP1-Blue ISG and MCA205. Blasticidin selection was performed at 5 μg/ml for L929-mRuby-hIRF3 or MCA205, and 10 μg/ml for THP1-Blue ISG.
To generate L929-mRuby-hIRF3, L929 were transduced with pLV-EF1α-mRuby-hIRF3 lentiviral supernatants, single clones selected for analysis. MCA205 and B16F10 were transduced with pLV-EF1α-cGAS-Clover or pLV-EF1α-GFP lentiviral supernatants. THP1-Blue ISG were transduced with pLX304-GFP-DAPK3(WT) lentiviral supernatants. All Clover- or GFP-positive cells were sorted by FACSAria Cell Sorter (BD Biosciences). THP1-Blue ISG-HA-Ub (K63O) were generated by transduction of THP1-Blue ISG with pLX304-HA-Ub (K63O) lentiviral supernatants. Cells were selected with 10 μg/ml Blasticidin.
In vitro kinase assay
GST-DAPK3 (Sigma-Aldrich, D7194) and GST-TBK1 (Sigma-Aldrich, SRP5089) were resuspended with 2× kinase buffer containing 40 mM HEPES (pH7.4), 40 mM MgCl2, 2 mM DTT, 0.2 mM sodium orthovanadate, 40 mM β-glycerophosphate, 20 mM sodium fluoride. 2.5 μg of GST-tagged recombinant C-terminal human STING (aa 149-379), full-length human TBK1(K38M), human LMO7(aa 360-460), human LMO7 (aa 810-910), human TRIM56 (aa 400-500) or human TRIP12 (aa 260-360) were incubated with 100 ng of GST-DAPK3 or GST-TBK1 at 30°C for 30 min in a 40 μl final volume of 1× kinase buffer in the presence of 20 μM ATP, 2 μCi [γ-32P] ATP (PerkinElmer), and then boiled at 95°C for 5 min in 5× sample buffer. Phosphorylation was assessed by 10% SDS-PAGE and Coomassie Brilliant Blue staining followed by autoradiography.
siRNA screening
HUVEC were transfected with siRNA pools (2007 Human siGENOME siRNA library, Dharmacon) targeting 1,001 tumor suppressor genes52, STING or TBK1. 1.25 pmol of siRNA was mixed with Dharmafect 4 (Horizon Discovery) in OptiMEM (Thermo Fisher Scientific), and aliquoted to 384-well optical imaging plates (Corning 3985). HUVEC were plated at 800 cells per well and incubated at 37°C 5% CO2 for 24 h. Media was changed after 24 h and cells cultured for 48 h prior to stimulation with poly(dA:dT)-Lyovec complexes (Invivogen) for 3 h and analysis of IRF3 nuclear translocation14, 51. siTBK1, siSTING, and non-targeting siControl were included on each plate. Z score was calculated as described53, 54. THP1-Blue ISG were transfected with 100 nM siRNA using Neon. 72 h post transfection, cells were plated at 6×104 cells per well in 96-well plates and stimulated with 10 μg/ml 2′,3′-cGAMP for 6 h. Culture supernatants were assessed for SEAP activity using QUANTI-Blue (Invivogen), per manufacturer’s instructions. Cell viability was assessed by CellTiter Glo (Promega) to normalize SEAP activity.
Microscopy
Fluorescence microscopy for endogenous IRF3 in HUVEC and L929-mRuby-hIRF3 was performed, as described14. For confocal microscopy, THP1-Blue ISG (1×106 cells/ml) were stimulated with 25 μg/ml 2′,3′-cGAMP in 1 μg/ml digitonin-containing buffer or transfected with 50 μg/ml 2′,3′-cGAMP using Lipofectamine 2000 for 3 h. For GFP-DAPK3 localization, THP1 were pre-treated with 0.5 μM PMA (Sigma-Aldrich) for 3 h, washed 4× with PBS, and 1×106 cells plated on glass coverslips one day before cGAMP stimulation. MCA205-cGAS-Clover and B16F10-cGAS-Clover were plated on glass coverslips at 1.5×105 cells per well and 0.7×105 cells per well, respectively. After stimulation, cells were fixed with 4% paraformaldehyde/1× PBS for 20 min at 25°C, washed 4× with PBS, and permeabilized with 0.2% Triton X-100/1× PBS for 10 min. Cells were blocked with 5% BSA in PBS for 30 min at 25°C and stained with primary antibody in 1% Donkey serum /1% Goat serum/0.2% Triton X-100/1× PBS buffer overnight at 4°C. Cells were washed 3× with PBS and incubated with secondary antibody in 1% Donkey serum/1% Goat serum/0.2% Triton X-100/1× PBS buffer for 1 h at 25°C. Cells were washed 3× with 1× PBS and counterstained with DAPI. Slides were mounted with ProLong Gold (Life Technologies) and images acquired with Olympus FV10i confocal microscope with a 60× objective. Co-localization was analyzed with Just Another Co-localization Plugin (JACoP) on Image J.
Phospho-proteomics
THP1-Blue ISG (1×108 cells per sample, n=3 biological replicates) transduced with shRNAs were stimulated with 10 μg/ml 2′,3′-cGAMP for 3 h. Cells were washed 3× with PBS, and pellets stored at −80°C. Samples were immersed in 1 ml lysis buffer containing protease and phosphatase inhibitors, 1 ml of 8 M Urea and 50 mM HEPES (pH 8.5). Cells were subjected to probe sonication for 3 cycles of 15 sec at 20% amplitude. Disulfide bonds were reduced in 5 mM DTT at 56°C for 30 min. Reduced disulfide bonds were capped using 15 mM iodoacetamide and incubated for 20 min. Reaction quenching was performed using the original volume of DTT and incubation at 25°C for 15 min. Protein was pelleted using 6 ml of HPLC-grade methanol, 1.5 ml of chloroform, and 4 ml of HPLC-grade water was added to each tube. Tubes were subjected to rapid agitation and centrifugation at 4,000 rpm for 3 min. Liquid phases were removed and pellet washed in 6 ml methanol, then centrifuged. Supernatant was removed and tubes kept on ice for subsequent precipitation. Pellets were washed 2× with 3 ml of ice-cold acetone. Pellets were air-dried and rehydrated in 900 μl of 1 M urea with 50 mM HEPES (pH 8.5). Protein digestion proceeded in 2 steps, each repeated once. Pellets were digested overnight at 25°C via addition of LysC, and digested for 7 h in sequencing-grade trypsin at 37°C. Following digest, reaction quenching was performed by addition of 10% TFA and peptide desalting on SepPack C18 columns, per manufacturer’s instructions. Following desalting, peptide abundance was quantified using Pierce Quantitative Colorimetric Assay. 50 μg was separated for labeling, and pooled internal standard consisting of equal peptide abundances from each sample was prepared and separated until TMT labeling step. Remaining peptides were subjected to phosphosite enrichment using TiO2 beads33. Enriched phospho-peptides were dried under vacuum overnight. Samples for proteomics and phospho-proteomics runs were labeled using an identical protocol. TMT labels resuspended in dry acetonitrile were added to resuspended peptide samples and incubated at 25°C for 1 h. Reaction quenching was performed by addition of hydroxylamine and incubating 25°C for 15 min. Samples were acidified via addition of 1% TFA and multiplexed within experiment (proteomics and phospho-proteomics here) before desalting. Samples were dried under vacuum overnight. Multiplexed samples were fractionated using basic reverse-phase liquid chromatography. For proteomics experiments, samples were concatenated as described55, and 12 of 24 fractions analyzed on an Orbitrap Fusion Mass Spectrometer with in-line nano-LC. Multiplexed phospho-proteomics samples were similarly fractionated and concatenated. 24 of 24 fractions were analyzed on an Orbitrap Fusion Mass Spectrometer.
Spectral data were searched against Homo sapiens reference proteome downloaded on 5/22/2019 from Uniprot.com. Spectra were searched using a decoy database generated in SEQUEST56. Phospho-proteomics data were searched using identical parameters as above, except phosphorylation was specified as a variable modification on Serine, Tyrosine, and Threonine residues, and PhosphoRS was used to assign modification site probabilities to phosphor-peptides57. Data were exported and manually filtered such that only “High” confidence peptide spectral matches (PSMs) and no “Rejected” PSMs were used for subsequent analysis. Blank quantitation cells were replaced with a value of “1” to represent noise background. Spectra were filtered such that average quantitation value could not be lower than 10 and such that quantified isolation interference could not exceed 25. Proteomics quantitation values were summed to unique protein level and phospho-proteomics data were summed to unique phosphopeptide level. Data from each experiment were normalized against bridge channel values normalized against median bridge channel value. Quantitation values were normalized against median quantitation values for each channel which were normalized against overall quantitation median value. A final normalization step was applied to phospho-proteomic data, wherein quantitation values for phospho-peptides were normalized against corresponding protein abundance values in proteomics data. This step is applied to prevent quantitation bias associated with increased or decreased protein abundance in absence of increased or decreased phosphorylation.
Heat map and IPA analysis
Phosphopeptide heat map was created with heatmap.2 function in gplots v3.0.1 library under RStudio v3.6.1. log2Fold Change-cutoff of 0.4 and pValue-cutoff of 0.05 in either comparison, shDAPK3#1 vs shControl or shTBK1#1 vs shControl, to select genes. Heat map of E3 ligases was generated with Morpheus (https://software.broadinstitute.org/morpheus/). Raw data is in Supplementary Table 2. QIAGEN Ingenuity Pathway Analysis (IPA) was used for gene dataset analysis.
TCGA data analysis
RNA-seq FPKM data and survival curve data of TCGA pancreatic adenocarcinoma, uterine corpus endometrial carcinoma, and esophageal carcinoma patients were downloaded from The Cancer Genome Atlas (TCGA) (http://tumorsurvival.org/)58. All patients were divided into three subgroups (tertiles) according to DAPK3 expression levels and top (high) and bottom (low) tertiles were compared in survival analysis.
Cancer genomics analysis
DAPK3, LMO7, and TRIP12 alteration data and graph were obtained from cBioPortal for Cancer Genomics (http://cbioportal.org)59, 60. TCGA case study is listed in Supplementary Table 3.
Statistical analysis
Statistical analyses were performed with Excel (Microsoft) and Prism (GraphPad). Two or three independent experiments were replicated, and error bars represent mean ± s.d. Statistical comparisons were evaluated using two-tailed Student’s t-test (2 groups), with P values indicated in figure legends and source data.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data Availability
Screening results are presented in Supplementary Table 1 and phosphoproteomics results in Supplementary Table 2. Mass spectrometry proteome and phosphoproteome data were deposited in MassIVE (identifier PXD023639) and ProteomeXchange (identifier PXD023637). Source data and uncropped immunoblot images are provided in the manuscript. Additional data will be made available from the corresponding author upon reasonable request.
Extended Data
Extended Data Fig. 1. DAPK3 is a positive regulator of STING signaling and some TLR pathways.
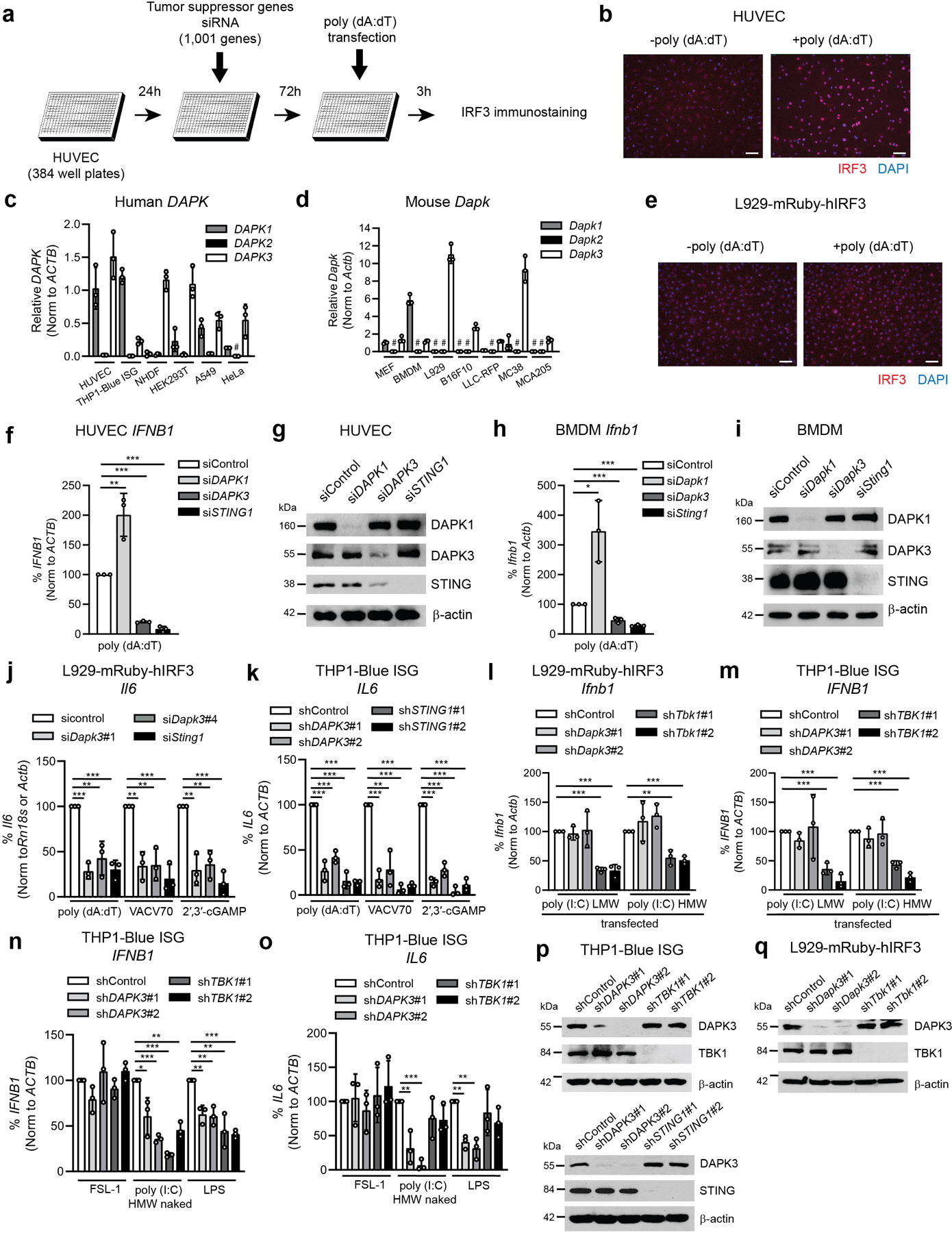
a, Schematic representation of the RNAi screen for IRF3 nuclear translocation. b, Immunostaining of IRF3 in HUVEC stimulated with poly (dA:dT) (1 μg/ml) (right panel) for 3 h. Scale bar, 100 μm. c, d, qRT-PCR of DAPK1/Dapk1, DAPK2/Dapk2, and DAPK3/Dapk3 in (c) human and (d) mouse cell lines. #, Not detected. e, Images of IRF3 localization in L929-mRuby-hIRF3 stimulated with poly (dA;dT) (1 μg/ml) for 3 h. Scale bar, 100 μm. f, g, (f) qRT-PCR of IFNB1 and (g) immunoblot of HUVEC transfected with indicated siRNA. h, i, (h) qRT-PCR of Ifnb1 and (i) immunoblot of BMDM transfected with indicated siRNA. (f, h) Cells were stimulated with poly (dA:dT) (0.2 μg/ml) for 4 h. j, k, (j) qRT-PCR of Il6 in L929-mRuby-hIRF3 and (k) IL6 inTHP1-Blue ISG transduced with indicated shRNA and stimulated as indicated in Fig. 1g–l. l, m, qRT-PCR of (l) Ifnb1 in L929-mRuby-hIRF3 and (m) IFNB1 inTHP1-Blue ISG transduced with indicated shRNA and transfected with poly (I:C)(LMW) and poly (I:C)(HMW)(0.1 μg/ml for L929-mRuby-hIRF3, 0.5 μg/ml for THP1-Blue ISG). n, o, qRT-PCR of (n) IFNB1 and (o) IL6 in THP1-Blue ISG transduced with indicated shRNA and stimulated with FSL-1(100 ng/ml), naked poly (I:C)(LMW)(10 μg/ml), or LPS(100 ng/ml) for 4 h. p, q, Immunoblot of (p) THP1-Blue ISG and (q) L929-mRuby-hIRF3 transduced with indicated shRNA. Data in (b-e, g, i, p, q) are representative or (f, h, j-o) the mean of three independent experiments. Values represent mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001. Statistical comparisons were conducted using two-tailed t-test (f, h, j-o).
Extended Data Fig. 2. Association of DAPK3 with outcomes in human cancer.
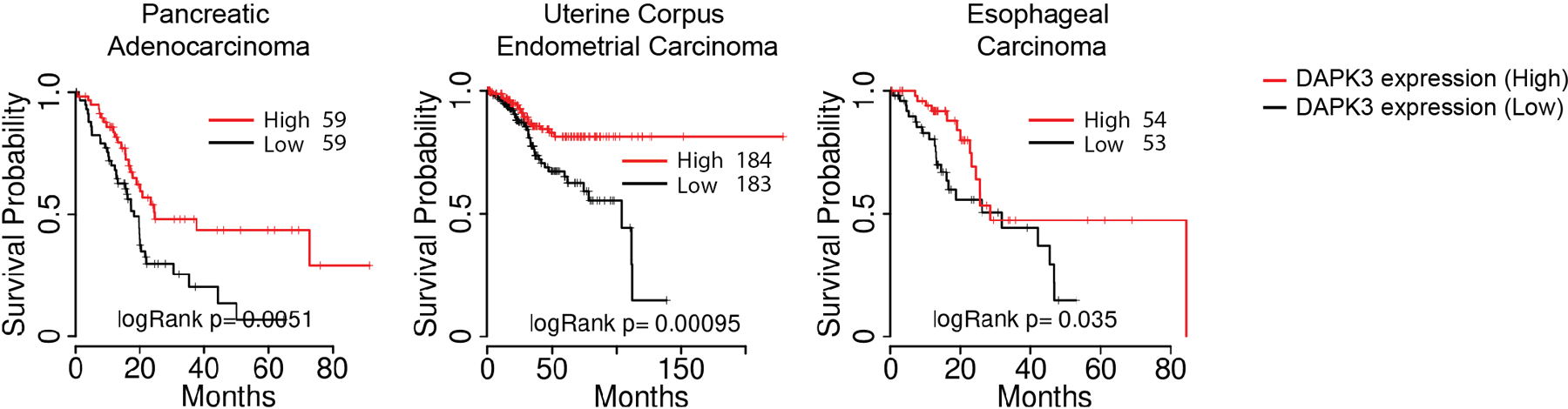
Kaplan-Meier survival analysis of pancreatic adenocarcinoma, uterine corpus endometrial carcinoma, and esophageal carcinoma comparing the top (high) and bottom (low) tertiles of patients with respect to DAPK3 expression levels as reported by TCGA data portal. Statistical comparisons were conducted using two-sided log-rank test58.
Extended Data Fig. 3. Teniposide and paclitaxel induce micronuclei formation and anti-tumor immunity to B16F10 tumors in a type I IFN signaling-dependent manner.
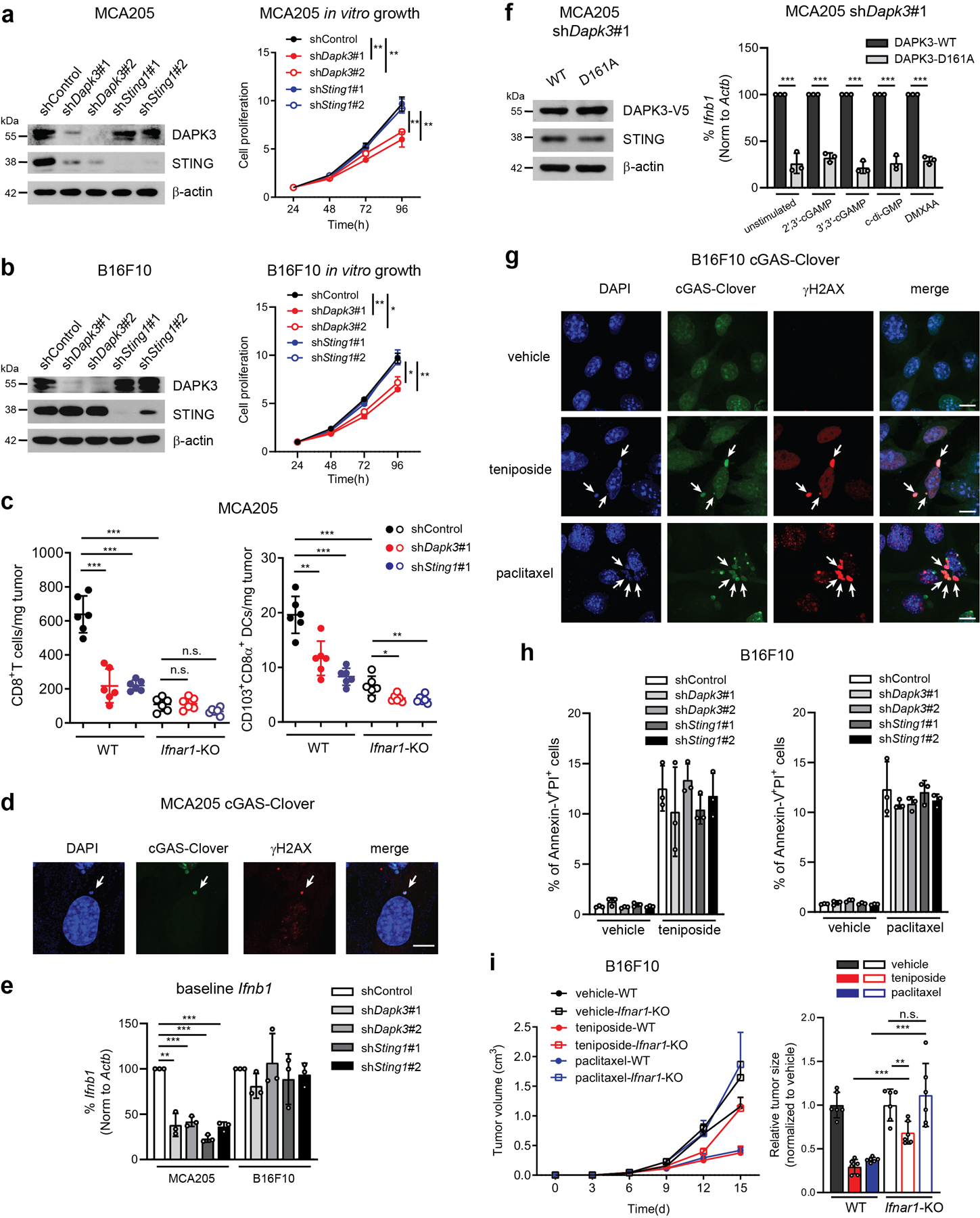
a, b, (Left) Immunoblot and (right) in vitro cell growth of (a) MCA205 and (b) B16F10 transduced with indicated shRNA. c, Flow cytometry of tumor-infiltrating CD8+T cells and CD103+CD8α+DCs in MCA205 tumor suspensions isolated from WT or Ifnar1-KO mice on Day 6 (n=6 per group). d, Confocal fluorescence microscopy of MCA205 stably expressing cGAS-Clover. Scale bar, 10 μm. e, qRT-PCR of Ifnb1 in unstimulated MCA205 and B16F10 transduced with indicated shRNA. f, (Left) Immunoblot and (right) qRT-PCR of Ifnb1 in shDapk3#1-transduced MCA205 ectopically expressing V5-tagged DAPK3(WT) or DAPK3(D161A). Cells were stimulated with 2′,3′-cGAMP, 3′,3′-cGAMP, c-di-GMP (20 μg/ml for all three agonists) or DMXAA (50 μg/ml) for 4 h. g, Confocal fluorescence microscopy of B16F10 stably expressing cGAS-Clover. Cells were treated with teniposide (10 μM) for 24 h or paclitaxel (100 nM) for 72 h. Scale bar, 10 μm. h, (Left) Apoptosis measured in shRNA-transduced B16F10 treated with teniposide (10 μM) for 24 h or (right) paclitaxel (100 nM) for 48 h. i, Tumor volume of B16F10 subcutaneously transplanted into WT and Ifnar1-KO mice and treated with teniposide or paclitaxel (n=6 for vehicle, n=7 for teniposide and paclitaxel). Tumor size on Day 15 is represented (right panel). Data are representative (a-d, g-i) or the mean (a, b, e, f) of three independent experiments. Values represented mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001. Statistical comparisons were conducted using two-tailed t-test (a-c, e, f, h, i).
Extended Data Fig. 4. Flow cytometry gating strategy for tumor-infiltrating leukocytes.
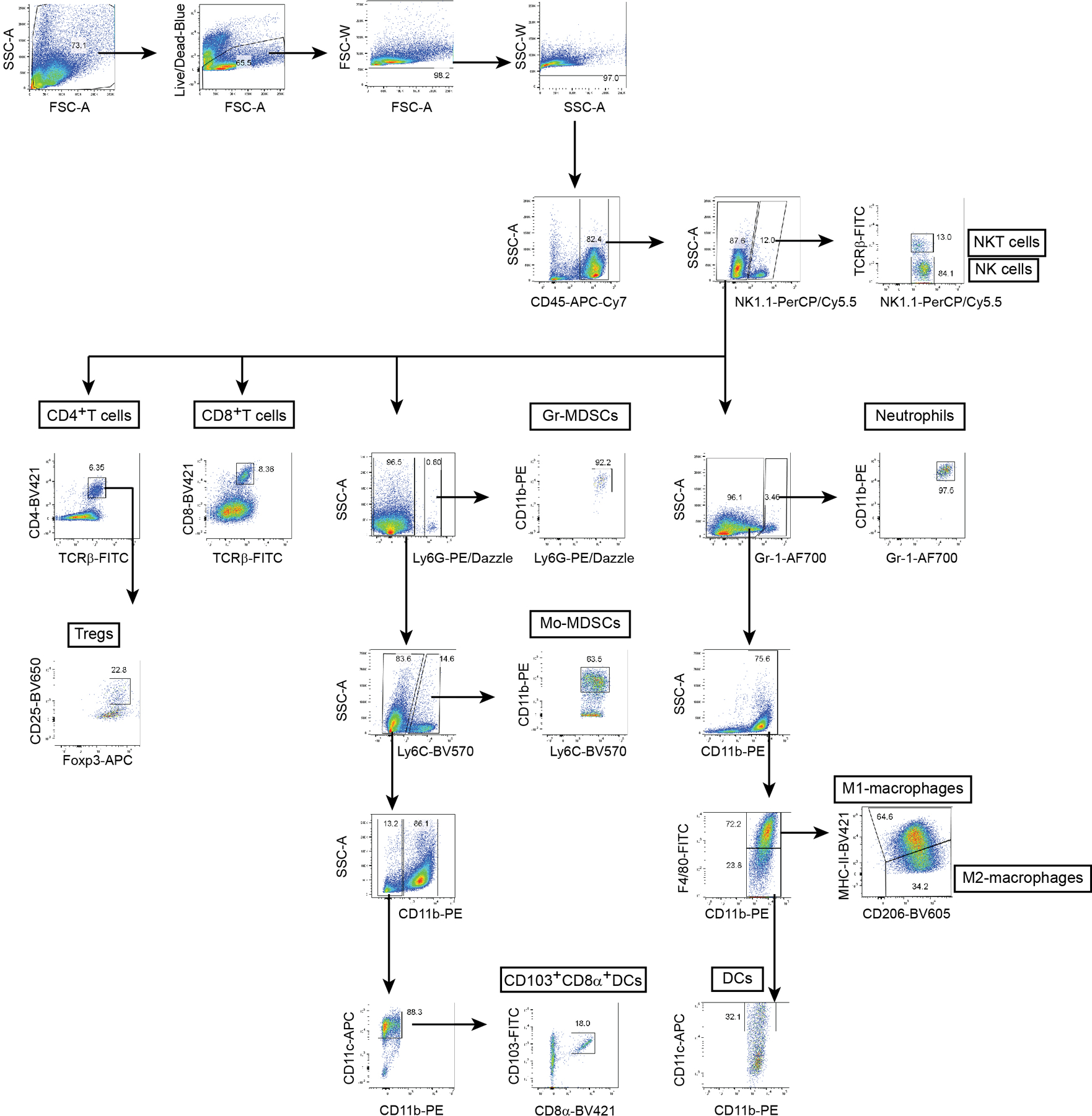
Tumor single cell suspensions were stained with different fluorophore-conjugated antibodies and analyzed by flow cytometry.
Extended Data Fig. 5. DAPK3 does not directly phosphorylate STING or TBK1.
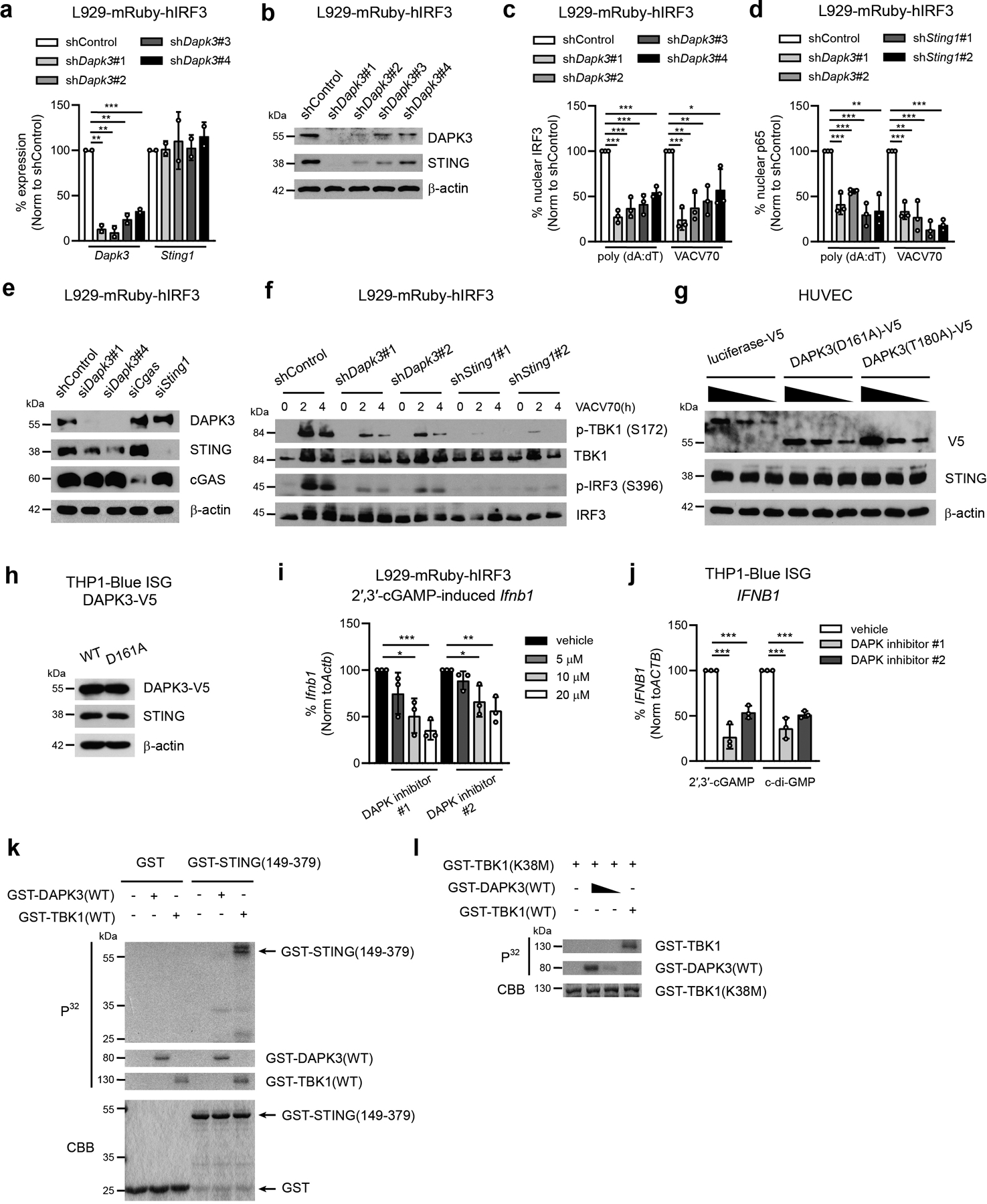
a-d, (a) qRT-PCR of Sting1 and Dapk3, (b) immunoblot, (c) IRF3 nuclear translocation, and (d) p65 nuclear translocation in L929-mRuby-hIRF3 transduced with indicated shRNA stimulated with poly (dA:dT) (0.5 μg/ml) or VACV70 (2 μg/ml) for 3 h. e, Immunoblot of L929-mRuby-hIRF3 transfected with indicated siRNA. f, Immunoblot of L929-mRuby-hIRF3 transduced with indicated shRNA stimulated with VACV70 (2 μg/ml) for 2 h and 4 h. g, Immunoblot of HUVEC stably expressing V5-tagged DAPK3(D161A), DAPK3(T180A), or luciferase. Cells were infected at MOI=5, 2, or 1. h, Immunoblot of THP1-Blue ISG stably expressing V5-tagged DAPK3(WT) or DAPK3(D161A). i, qRT-PCR of Ifnb1 in L929-mRuby-hIRF3 pre-treated with DAPK inhibitors for 3 h prior to 2′,3′-cGAMP stimulation (10 μg/ml) for 4 h. j, qRT-PCR of IFNB1 in THP1-Blue ISG pre-treated with DAPK inhibitors (50 μM) for 6 h prior to 2′,3′-cGAMP or c-di-GMP stimulation (10 μg/ml for both) for 4 h. k, l, In vitro kinase assay of (k) GST-tagged human STING C-terminus (aa 149-379) and (l) GST-tagged human TBK1(K38M). Peptides were incubated with GST-tagged DAPK3 or TBK1 in the presence of [γ-32P] ATP. Data in (b, e-h, k, l) are representative or (a, c, d, i, j) mean of three independent experiments. Values represent mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001. Statistical comparisons were conducted using two-tailed t-test (a, c, d, i, j).
Extended Data Fig. 6. DAPK3 is not involved in STING trafficking from ER to Golgi.
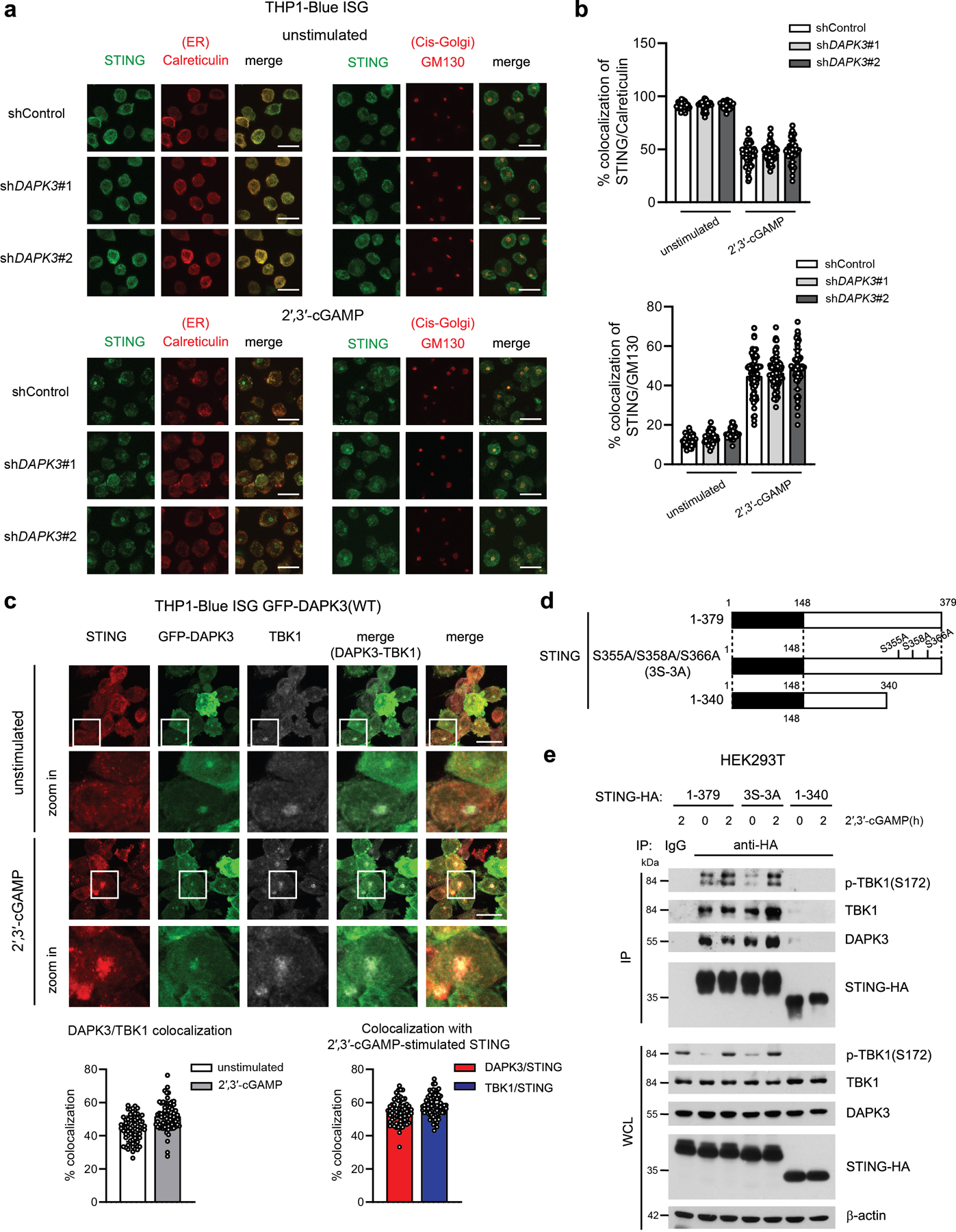
a, Confocal fluorescence microscopy of THP1-Blue ISG transduced with indicated shRNA and unstimulated or stimulated with 2′,3′-cGAMP (25 μg/ml) for 3 h. Scale bar, 15 μm. b, (Upper) Co-localization of STING/Calreticulin and (lower) STING/GM130 analyzed using Image J software. Data are pooled from three independent experiments (n>1,500 cells for unstimulated 32 images and cGAMP-stimulated 73 images). c, (Upper) Confocal fluorescence microscopy of THP1-Blue ISG stably expressing GFP-tagged DAPK3(WT) unstimulated or stimulated with 2′,3′-cGAMP (50 μg/ml) for 3 h. Localization of GFP-DAPK3, STING, and TBK1. (Lower) Co-localization of GFP-DAPK3/TBK1, GFP-DAPK3/STING, and TBK1/STING was analyzed using Image J software. Data are pooled from three independent experiments (n>1,500 cells for unstimulated and cGAMP-stimulated 70 images). Scale bars, 15 μm. d, Schematic representation of human STING mutants. e, (Upper) Immunoprecipitation and immunoblot of HEK293T transfected with plasmid encoding HA-tagged human STING (WT, 1-379), phospho-deficient mutant (3S-3A), or C-terminal deletion mutant (aa 1-340) unstimulated or stimulated with 2′,3′-cGAMP (5 μg/ml) for 2 h, and (lower) immunoblot of whole cell lysates (WCL). Values represented as mean ± s.d. Data in (a, c, e) are representative of three independent experiments.
Extended Data Fig. 7. Phosphorylation of TRIP12 on S312 or TRIM56 on T442 are not involved in STING K63-linked poly ubiquitination.
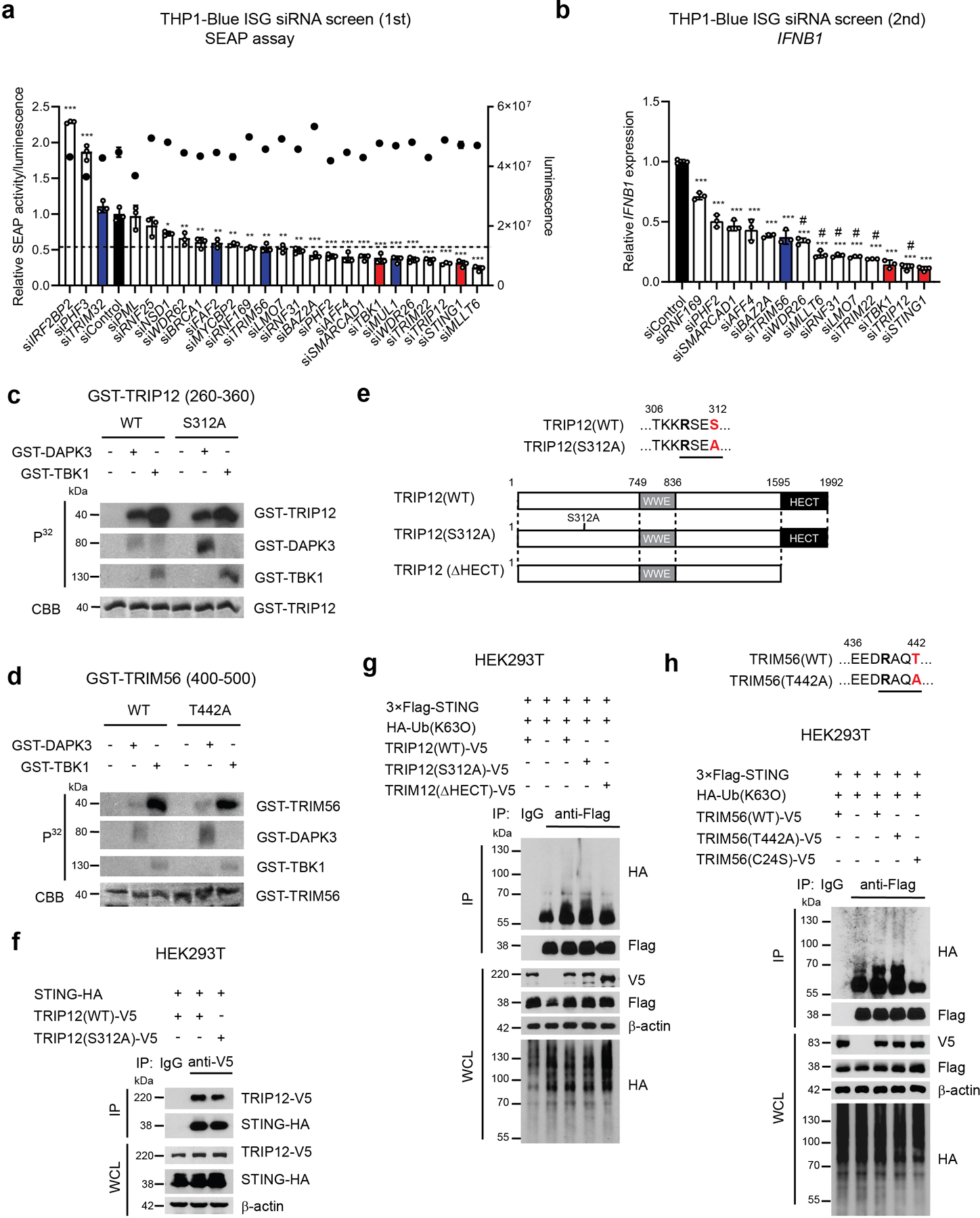
a, Primary RNAi screen of E3 ligases in THP1-Blue ISG transfected with indicated siRNA. SEAP activity was measured after normalization with CellTiter-Glo. Black; siControl, Blue; previously reported E3 ligases for K63-linked poly-ubiquitination of STING, Red; positive control (e.g. siSTING1 and siTBK1). siTRIM56 value was used for determining cut-off. b, Secondary RNAi screen of E3 ligases in THP1-Blue ISG transfected with indicated siRNA. qRT-PCR of IFNB1 was performed. #; candidates for subsequent analysis. c, d, In vitro kinase assay of (c) GST-tagged human TRIP12 peptide (aa 260-360) and (d) GST-tagged human TRIM56 peptide (aa 400-500). Peptides were incubated with GST-tagged DAPK3 or TBK1 in the presence of [γ-32P] ATP. e, Schematic representation of human TRIP12 mutants. f, (Upper) Immunoprecipitation and immunoblot of HEK293T transfected with plasmids encoding HA-tagged human STING and V5-tagged human TRIP12 (WT) or phospho-deficient TRIP12 (S312A), and (lower) immunoblot of whole cell lysates (WCL). g, (Upper) Immunoprecipitation and immunoblot of HEK293T transfected with plasmids encoding 3×Flag-tagged human STING, HA-tagged Ub(K63O), and V5-tagged human TRIP12(WT), phospho-deficient TRIP12(S312A), or HECT domain-deficient TRIP12(ΔHECT), and (lower) immunoblot of WCL. h, (Upper) Immunoprecipitation and immunoblot of HEK293T transfected with plasmids encoding 3×Flag-tagged human STING, HA-tagged Ub(K63O), and V5-tagged human TRIM56(WT), phospho-deficient TRIM56(T442A), or enzyme-inactive TRIM56(C24S), and (lower) immunoblot of WCL. Data in (c, d, f-h) are representative of three independent experiments. Values represent mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001 (compared to siControl) (a, b). Statistical comparisons were conducted using two-tailed t-test (a, b).
Extended Data Fig. 8. DAPK3, LMO7, and TRIP12 are highly mutated in human cancers.
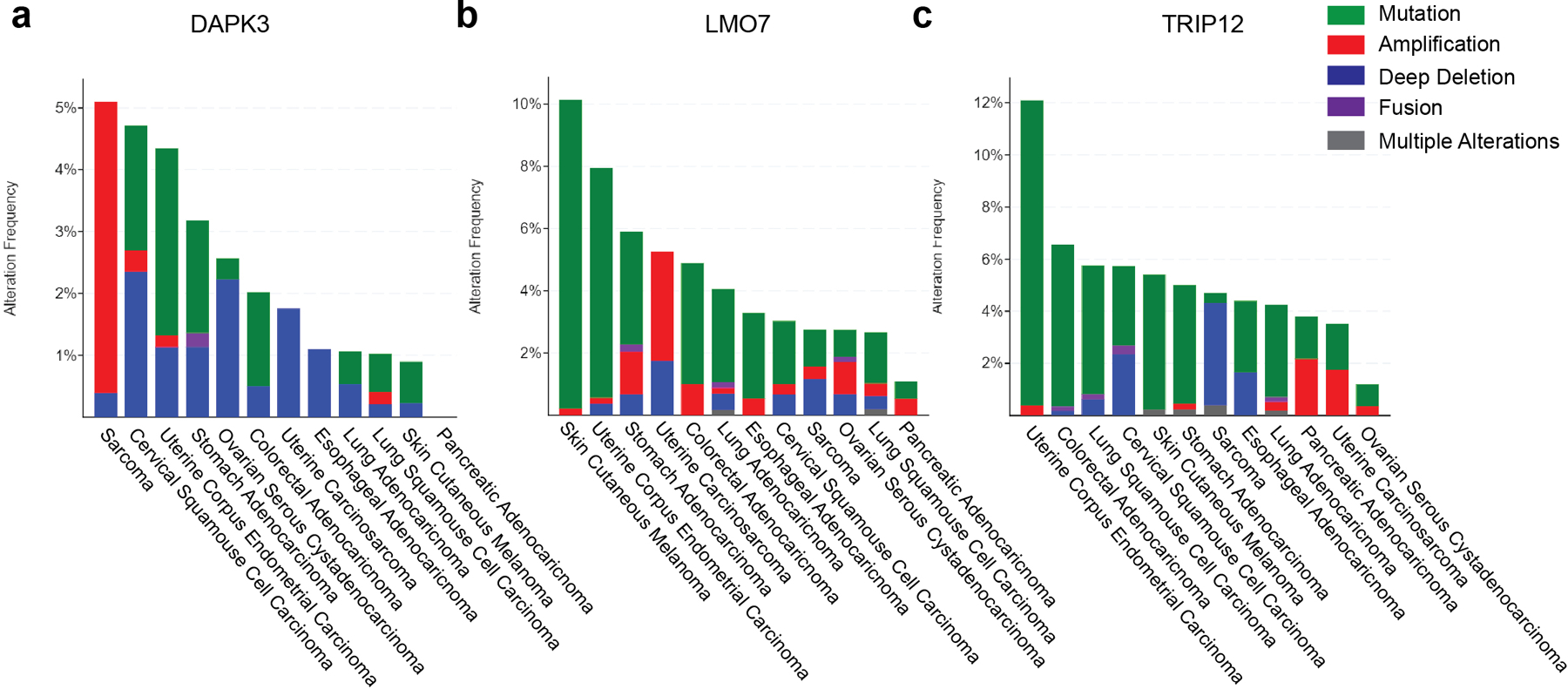
a-c, Genomic alterations of (a) DAPK3, (b) LMO7 and (c) TRIP12 in human cancers from cBioportal.
Extended Data Fig. 9. LMO7 and TRIP12 are positive regulators of STING-IFNβ signaling in THP1 and HUVEC.
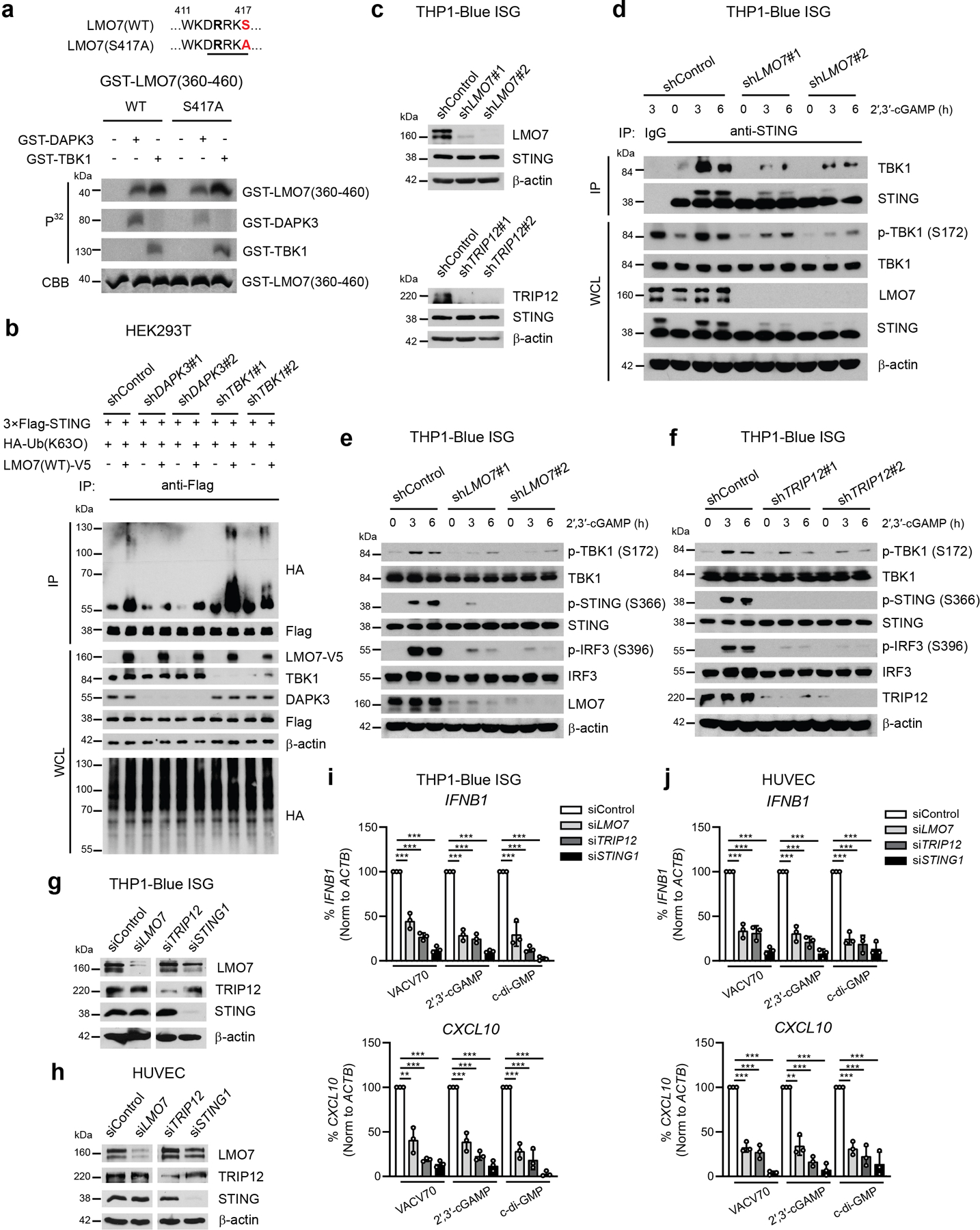
a, In vitro kinase assay of GST-tagged human LMO7 (aa 360-460). Peptides were incubated with GST-tagged DAPK3 or TBK1 in the presence of [γ-32P] ATP. b, (Upper) Immunoprecipitation and immunoblot of HEK293T transduced with indicated shRNA prior to transfection with plasmids encoding 3×Flag-tagged human STING, HA-tagged Ub(K63O), and V5-tagged human LMO7(WT) and (lower) immunoblot of whole cell lysates (WCL). c, (Upper) Immunoblot of THP1-Blue ISG transduced with two distinct shLMO7 or (lower) shTRIP12 sequences. d, (Upper) Immunoprecipitation and immunoblot of THP1-Blue ISG transduced with indicated shRNA and stimulated with 2′,3′-cGAMP (10 μg/ml) for 3 h and 6 h, and (lower) immunoblot of WCL. e, f, Immunoblot of THP1-Blue ISG transduced with two distinct (e) shLMO7 or (f) shTRIP12 sequences and stimulated with 2′,3′-cGAMP (10 μg/ml) for 3 h and 6 h. g, h, Immunoblot of (g) THP1-Blue ISG and (h) HUVEC transfected with indicated siRNA. i, j, qRT-PCR of IFNB1 and CXCL10 in (i) THP1-Blue ISG and (j) HUVEC transfected with indicated siRNA stimulated with VACV70 (2 μg/ml), 2′,3′-cGAMP (10 μg/ml), and c-di-GMP (10 μg/ml). Data in (a-h) are representative or (i, j) mean of three independent experiments. Values represent mean ± s.d. *P<0.05, **P<0.01, and ***P<0.001. Statistical comparisons were conducted using two-tailed t-test (i, j).
Extended Data Fig. 10. Schematic model of the DAPK3-STING axis.
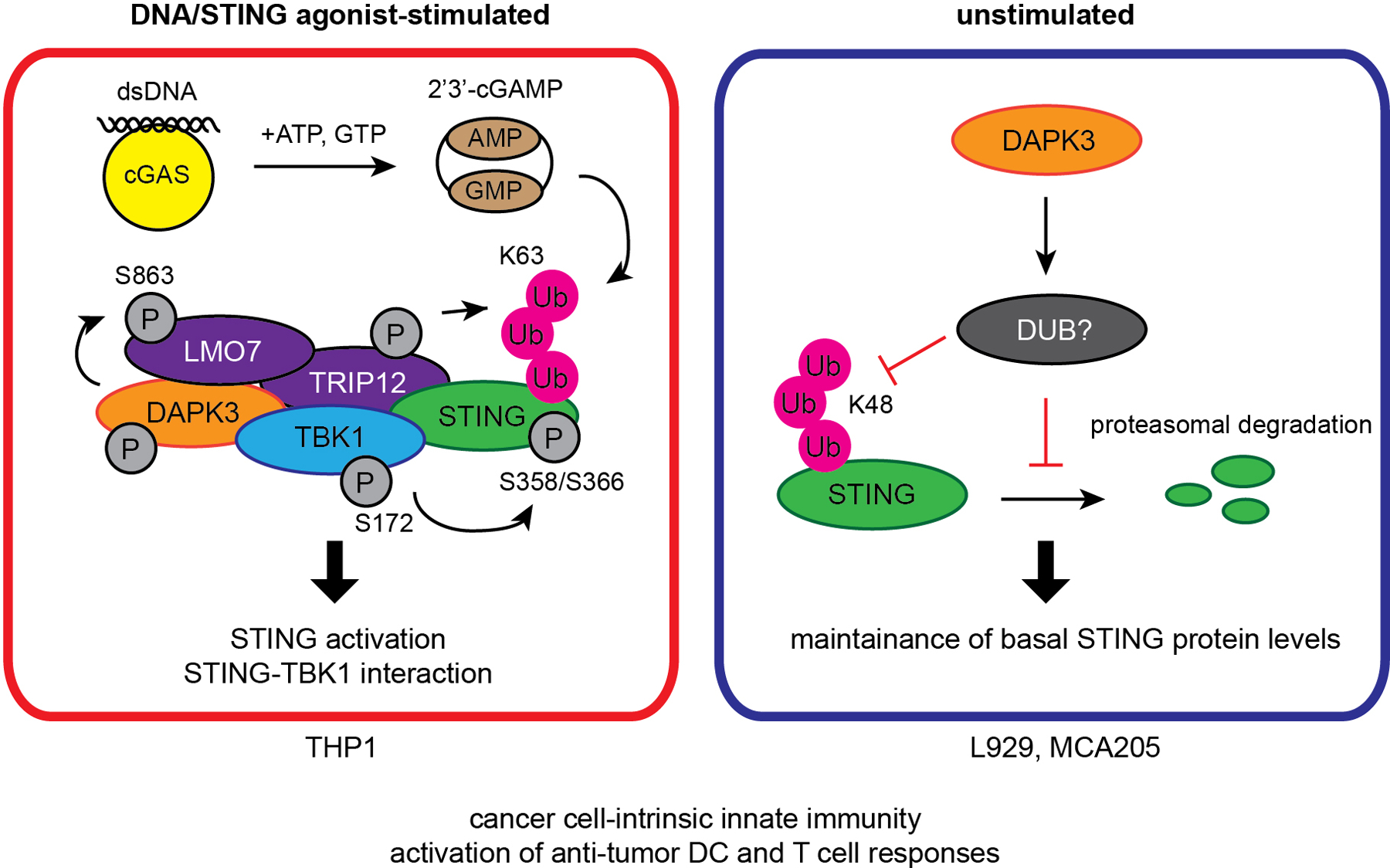
In unstimulated cells (L929 and MCA205), DAPK3 maintains steady-state STING levels by inhibiting STING K48-linked poly-ubiquitination and proteasome-mediated degradation. In DNA-stimulated cells (THP1), DAPK3 promotes STING activation by phosphorylating the E3 ligase LMO7 at S863, enabling LMO7-STING interaction, STING K63-linked poly-ubiquitination, and recruitment of TBK1.
Supplementary Material
Acknowledgements
We thank J. Huang (LJI) for support for the HUVEC siRNA screen; S. Shresta (LJI) for Ifnar1-null mice; C. Benedict (LJI) for MEFs, NHDFs and hCMV; NP. Restifo (NCI) for MCA205; J. Schlom (NCI) for MC38; S. Schoenberger (LJI) for B16F10; CC. Hedrick (LJI) for LLC-RFP; YC. Liu (LJI) for pEF-neo-HA-Ub; D. Zajonc (LJI) for pGEX-4T-2; J. Day and K. Tanguay (LJI) for shRNA and siRNA constructs, and siRNA screen support; D. Freeman, B. McDonald and R. El Morabiti (LJI) for hCMV propagation; M. Diep, K. Foos, and M. Kaur (LJI) for technical assistance; Z. Mikulski (LJI Microscopy Core Facility) for confocal microscopy support; A. Sethi (LJI Bioinformatics Core Facility) for pathway analysis support; LJI Flow Cytometry Core Facility for cell sorting (FACS Aria-II Cell Sorter; supported by the Shared Instrumentation Grant (SIG) program S10 RR027366); D. Araujo (LJI) for proofreading the manuscript. This work was supported by National Institutes of Health (NIH) grants R01CA199376, U01DE028227 and U54CA260591 (S. Sharma); NIH S10OD020025 and R01ES027595 (M. Jain). C.J. Lio was supported by Cancer Research Institute (CRI) Irvington Postdoc Fellowship. A. Campeau was supported by the UCSD Microbial Sciences Initiative Graduate Research Fellowship and by the UCSD Graduate Training Program in Cellular and Molecular Pharmacology, through an institutional training grant from the National Institute of General Medical Sciences, T32 GM007752.
Footnotes
Competing interests
The authors state that they have no competing interests. Correspondence and requests for materials should be addressed to S.S. (soniasharma@lji.org).
References
- 1.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ & Kroemer G Type I interferons in anticancer immunity. Nat Rev Immunol 15, 405–414 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Vanpouille-Box C, Demaria S, Formenti SC & Galluzzi L Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 34, 361–378 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Kwon J & Bakhoum SF The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 10, 26–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Q, Sun L & Chen ZJ Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17, 1142–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Corrales L, McWhirter SM, Dubensky TW Jr. & Gajewski TF The host STING pathway at the interface of cancer and immunity. The Journal of Clinical Investigation 126, 2404–2411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo SR et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harding SM et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackenzie KJ et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahn J et al. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun 5, 5166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H, Wang H, Ren J, Chen Q & Chen ZJ cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 114, E4612–E4620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J Clin Invest 129, 4850–4862 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Brognard J, Zhang YW, Puto LA & Hunter T Cancer-associated loss-of-function mutations implicate DAPK3 as a tumor-suppressing kinase. Cancer Res 71, 3152–3161 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lio CW et al. cGAS-STING Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J Virol 90, 7789–7797 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farag AK & Roh EJ Death-associated protein kinase (DAPK) family modulators: Current and future therapeutic outcomes. Med Res Rev 39, 349–385 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Fang J et al. Attenuation of EPO-dependent erythroblast formation by death-associated protein kinase-2. Blood 112, 886–890 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willemsen J et al. Phosphorylation-Dependent Feedback Inhibition of RIG-I by DAPK1 Identified by Kinome-wide siRNA Screening. Mol Cell 65, 403–415 e408 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Kocher BA, White LS & Piwnica-Worms D DAPK3 suppresses acini morphogenesis and is required for mouse development. Mol Cancer Res 13, 358–367 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hosoba K et al. Phosphorylation of myosin II regulatory light chain by ZIP kinase is responsible for cleavage furrow ingression during cell division in mammalian cultured cells. Biochem Biophys Res Commun 459, 686–691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demaria O et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A 112, 15408–15413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchison TJ, Pineda J, Shi J & Florian S Is inflammatory micronucleation the key to a successful anti-mitotic cancer drug? Open Biol 7, 170182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zierhut C et al. The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 178, 302–315.e323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohard S et al. STING-dependent paracriny shapes apoptotic priming of breast tumors in response to anti-mitotic treatment. Nat Commun 11, 259 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo WW et al. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol 17, 1057–1066 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Graves PR, Winkfield KM & Haystead TA Regulation of zipper-interacting protein kinase activity in vitro and in vivo by multisite phosphorylation. J Biol Chem 280, 9363–9374 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Okamoto M et al. Identification of death-associated protein kinases inhibitors using structure-based virtual screening. J Med Chem 52, 7323–7327 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Hu MM, Wang YY & Shu HB TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J Biol Chem 287, 28646–28655 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuchida T et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Liu S et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Tsuchiya Y, Jounai N, Takeshita F, Ishii KJ & Mizuguchi K Ligand-induced Ordering of the C-terminal Tail Primes STING for Phosphorylation by TBK1. EBioMedicine 9, 87–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao B et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 569, 718–722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka Y & Chen ZJ STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5, ra20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lapek JD Jr., Lewinski MK, Wozniak JM, Guatelli J & Gonzalez DJ Quantitative Temporal Viromics of an Inducible HIV-1 Model Yields Insight to Global Host Targets and Phospho-Dynamics Associated with Protein Vpr. Mol Cell Proteomics 16, 1447–1461 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burch LR, Scott M, Pohler E, Meek D & Hupp T Phage-peptide display identifies the interferon-responsive, death-activated protein kinase family as a novel modifier of MDM2 and p21WAF1. J Mol Biol 337, 115–128 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Arif A, Chatterjee P, Moodt RA & Fox PL Heterotrimeric GAIT complex drives transcript-selective translation inhibition in murine macrophages. Mol Cell Biol 32, 5046–5055 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmid JA & Birbach A IkappaB kinase beta (IKKbeta/IKK2/IKBKB)--a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev 19, 157–165 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Nehru V, Almeida FN & Aspenström P Interaction of RhoD and ZIP kinase modulates actin filament assembly and focal adhesion dynamics. Biochem Biophys Res Commun 433, 163–169 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Tang HW et al. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J 30, 636–651 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ni G, Konno H & Barber GN Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci Immunol 2, eaah7119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang L et al. UBXN3B positively regulates STING-mediated antiviral immune responses. Nat Commun 9, 2329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mullard A Can innate immune system targets turn up the heat on ‘cold’ tumours? Nature Reviews Drug Discovery 17, 3–5 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Mallipeddi R et al. Reduced expression of insulin-like growth factor-binding protein-3 (IGFBP-3) in Squamous cell carcinoma complicating recessive dystrophic epidermolysis bullosa. The Journal of investigative dermatology 122, 1302–1309 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Bi J et al. Downregulation of ZIP kinase is associated with tumor invasion, metastasis and poor prognosis in gastric cancer. Int J Cancer 124, 1587–1593 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Song S et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci Rep 7, 39858 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puto LA & Reed JC Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes & development 22, 998–1010 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seo GJ et al. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat Commun 9, 613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Kawai T & Akira S Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 13, 460–469 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Balka KR et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Rep 31, 107492 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Pokatayev V et al. Homeostatic regulation of STING protein at the resting state by stabilizer TOLLIP. Nature immunology 21, 158–167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dhanwani R et al. Cellular sensing of extracellular purine nucleosides triggers an innate IFN-β response. Sci Adv 6, eaba3688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao M, Sun J & Zhao Z TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res 41, D970–976 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma S & Rao A RNAi screening: tips and techniques. Nat Immunol 10, 799–804 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma S et al. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 499, 238–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elias JE & Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Wozniak JM & Gonzalez DJ PTMphinder: an R package for PTM site localization and motif extraction from proteomic datasets. PeerJ 7, e7046 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu S, Feng Y & Zhao S Proteins with Evolutionarily Hypervariable Domains are Associated with Immune Response and Better Survival of Basal-like Breast Cancer Patients. Comput Struct Biotechnol J 17, 430–440 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Screening results are presented in Supplementary Table 1 and phosphoproteomics results in Supplementary Table 2. Mass spectrometry proteome and phosphoproteome data were deposited in MassIVE (identifier PXD023639) and ProteomeXchange (identifier PXD023637). Source data and uncropped immunoblot images are provided in the manuscript. Additional data will be made available from the corresponding author upon reasonable request.