

Abstract
Simple Summary
Epigenetic modifications can regulate gene expression by altering chromatin structure. Since the beginning of comprehensive research into changes in gene expression due to early DNA methylation and histone acetylation, numerous experimental studies have revealed that the regulation of gene expression by histone methyltransferases plays an important role in cancer development, metastasis, and drug resistance. The enzyme responsible for H3K4 methylation, which is highly correlated with active transcription, has been studied in detail via impaired regulation of gene expression, following rearrangement of the mixed-lineage leukemia 1 (MLL1) gene. Other H3K4 methyltransferases have also been identified and have been shown to play a role in various cancers. In this review, we have examined the overall role of histone H3K4 methyltransferase in the development and progression of various cancers and its specific role in the development of drug-resistant cancers commonly encountered during chemotherapy. Additionally, we have discussed the H3K4-specific methyltransferase inhibitors currently under development for cancer treatment as well as their mechanisms of action.
Abstract
The KMT2 (MLL) family of proteins, including the major histone H3K4 methyltransferase found in mammals, exists as large complexes with common subunit proteins and exhibits enzymatic activity. SMYD, another H3K4 methyltransferase, and SET7/9 proteins catalyze the methylation of several non-histone targets, in addition to histone H3K4 residues. Despite these structural and functional commonalities, H3K4 methyltransferase proteins have specificity for their target genes and play a role in the development of various cancers as well as in drug resistance. In this review, we examine the overall role of histone H3K4 methyltransferase in the development of various cancers and in the progression of drug resistance. Compounds that inhibit protein–protein interactions between KMT2 family proteins and their common subunits or the activity of SMYD and SET7/9 are continuously being developed for the treatment of acute leukemia, triple-negative breast cancer, and castration-resistant prostate cancer. These H3K4 methyltransferase inhibitors, either alone or in combination with other drugs, are expected to play a role in overcoming drug resistance in leukemia and various solid cancers.
Keywords: histone H3K4, methyltransferase, resistant cancer, MLLs, inhibitors
1. Introduction
Epigenetics refers to heritable alterations in gene expression that are not caused by changes in the DNA sequence [1]. The main epigenetic modifications include DNA methylation, histone modification, miRNA-mediated modulation, and modulation of gene expression by alteration of chromatin structure [2,3]. Histone modification is classified into histone methylation, acetylation, ubiquitination, phosphorylation, and sumoylation [4,5]. Early epigenetic studies focused on changes in gene expression resulting from DNA methylation and histone acetylation. Extensive research has also been conducted on histone methylation, revealing that the regulation of bilateral gene expression by histone methylation plays an important role in cancer development, metastasis, and drug resistance [6,7,8,9,10].
Histone methylation, first discovered in the 1960s, occurs primarily at the arginine (R) and lysine (K) residues located in the histone H3, H4, and H2A tails [11,12]. Arginine residues can be mono- or di-methylated symmetrically or asymmetrically at the amine group sites of the H3R17, H3R26, H3R2, H4R3, H2AR3, and H3R8 residues [13,14,15,16,17,18,19]. Lysine residues can be modified by mono-, di-, or tri-methylation of H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20 residues [20,21,22,23,24,25,26]. Histone methylation is controlled by site-specific histone methyltransferases (HMTs) and can be reversed by specific demethylases. Numerous experimental studies have suggested that the regulation of gene expression by HMTs and demethylases is closely associated with cancer development [27,28,29].
Resistance to anticancer drugs remains a significant challenge in tumor treatment and recent studies have recommended targeting HMTs and demethylases to counter drug resistance. In this review, we examine the overall role of histone H3K4-specific methyltransferase in cancer and specifically explore the role of HMT in drug-resistant cancers. H3K4 methyltransferase inhibitors are currently being developed for cancer treatment, and their mechanisms of action are also discussed in this review. The HMTs and inhibitors discussed in this review are cited from primary literature. Data on inhibitors used in clinical studies were retrieved from ClinicalTrials.gov, a database which is maintained by the U.S. National Library of Medicine.
2. Classification of H3K4 Methyltransferases
2.1. KMT2/MLL Family
The major histone H3K4 methyltransferase in mammals is in the KMT2 (MLL) family, which has six members (KMT2A-D, KMT2F, and KMT2G) [30]. The mixed-lineage leukemia (MLL) protein has been studied in detail due to the dysregulation of gene expression caused by the rearrangement of the MLL1 (KMT2A) gene in acute leukemia [31,32]. Binding of DOT1L (KMT4) or the super elongation complex (SEC) occurs via a fusion partner (AF4, AF9, AF10, ELL, or ENL) newly bound to the N-terminus of the MLL1 protein, resulting in the introduction of the H3K79me mark at the specific site in the target gene (e.g., HOXA9 or MEIS1) [33]. In contrast, the wild-type KMT2 protein family exists as large complexes with common interacting subunit proteins (WRADs) such as WD-repeat protein 5 (WDR5), retinal blastoma binding protein 5 (RBBP5), ASH2L, and DPY30 [22,34,35,36]. The SET [Su(var)3-9, enhancer-of-zeste, and trithorax] domains exert major enzyme activities against KMT2 proteins. Additionally, binding to WDR5 via a conserved arginine-containing WDR5 interaction (Win) motif located at the N-terminus of the SET domain of the MLL family is essential for the assembly and enzymatic activity of the MLL core complex [37,38]. Additional biochemical experiments have also shown that the stable heterodimer formed by ASH2L and RbBP5 promotes the interaction between the SET domain and the substrate, thereby indicating increased overall methyltransferase activity of the KMT2 protein complex which is attributable to the interaction [39].
The KMT2 family can be divided into the following three subgroups based on the type of domain it contains: KMT2A and KMT2B (MLL1 and MLL2), KMT2C and KMT2D (MLL3 and MLL4), and KMT2F and KMT2G (SETD1A and SETD1B, Figure 1). KMT2A and KMT2B have taspase 1 cleavage sites. The two protein fragments are produced as a result of taspase 1-induced cleavage and are linked by FY-rich N-terminal (FYRN) and FY-rich C-terminal (FYRC) domains to form a functional heterodimeric complex [40]. MLL1–4 contain the plant homeodomain (PHD), an epigenetic reader of H3K4me3 [41]. Additionally, the CXXC zinc-finger domain is present in MLL1 and MLL2 and the AT-hook region of MLL1–3, and the high mobility group (HMG) box of MLL3 and MLL4 is associated with DNA binding [42]. The SETD1A and SETD1B proteins characteristically contain an RNA recognition motif (RRM) [43].
Figure 1.

Structures of the members of the KMT2/MLL1 protein family. The numbers represent amino acids in each protein. AT-hook, adenosine-thymidine-hook; Bromo, Bromodomain; CXXC, Zinc-finger-CXXC domain; FYRN, Phe/Tyr-rich N-terminal domain; FYRC, Phe/Tyr-rich C-terminal domain; HMG, high mobility group; PHD, plant homeodomain; RRM, RNA recognition motif; SET, Su(var)3-9, Enhancer-of-zeste and trithorax.; BRC, breakpoint common region. Adapted from [44].
Despite structural similarities due to shared key subunits, members of the KMT2 protein family have specific enzymatic reactions. In vitro studies have shown that the MLL1 and MLL2 core complexes have mono-, di-, and low tri-methylation activity on H3K4 [45,46]. The MLL3 and MLL4 core complexes exhibit mainly mono-methyl transfer activity [47,48]. In contrast, the SETD1A and SETD1B core complexes can perform all mono-, di-, and tri-methylation processes on H3K4 [48,49]. However, in cells, including mouse embryonic fibroblasts (MEFs) and mouse embryonic stem (ES) cells, MLL1 and MLL2 have been shown to primarily implement H3K4me3 at gene promoters [50,51]. MLL3 and MLL4 are responsible for the bulk of mono- and di-methylation of H3K4 in the enhancer region [47,52]. It has been suggested that SETD1A and SETD1B contribute mainly to di- and tri-methylation of H3K4 in cells [53,54]. These enzymatic properties in cells can also be confirmed by tracking the location of the KMT2 protein complex distributed throughout the genome. Consistent with the selective methylation properties of KMT2 enzymes at the cellular level, a series of chromatin immunoprecipitation sequencing (ChIP-seq) experiments demonstrated that MLL1 and MLL2 were recruited to both gene promoters and enhancers [55,56], while MLL3 and MLL4 were highly enriched in enhancers [55]. Furthermore, SETD1A and SETD1B were generally found to be bound near transcription start sites (TSSs) [57].
In addition to the shared common core subunits, the proteins in the KMT2 family also have several unique complex-specific subunits. These structural features provide the KMT2 complex with functional diversity for recruitment to the chromatin, in addition to enzymatic activity. For example, MLL1 and menin form an interaction surface for lens epithelial-derived growth factor (LEDGF), which can bind to chromatin via the PWWP domain [58,59,60]. The additional subunits associated with the MLL3 and MLL4 complexes are PTIP (PAX transactivation domain-interacting protein) and PA1 (PTIP-associated 1), which have robust HMT activity on histone H3K4 [61]. An extensive interactome study also revealed that CXXC-finger protein 1 (CFP1) and WD-repeat domain 82 (WDR82) interact only with SETD1A and SETD1B [40].
2.2. SMYD Family
Among the SET (suppressor of variegation, enhancer of zeste, trithorax) and MYND (myeloid-nervy-DEAF1) family proteins (SMYD1-5), SMYD1, SMYD2, and SMYD3 have methyltransferase activity specific to histone H3K4 [62,63,64]. These three members have a common tetratricopeptide (TPR)-like domain, which is an important motif for protein–protein interactions, at their C-termini [62,65,66]. The split-SET (N-SET and C-SET) domains and the MYND domains located between them form their characteristic active enzyme sites; particularly, the intermediate SET spacer (I-SET) located at the C-terminus of MYND is a linker region that plays an important role in regulating substrate selectivity [66].
SMYD1 was initially shown to regulate cardiac differentiation and morphogenesis by binding to skeletal NACA (skNAC), a cardiac and muscle-specific transcription factor [67,68]. The methyltransferase activity of SMYD1, which targets histone H3 lysine4, was first identified in 2006, and the important role of SMYD1 in muscle fiber maturation has since been elucidated [69]. SMYD1 mediates di- and tri-methylation of H3K4 residues [70]. SMYD2 was first identified as a methylation enzyme of histone H3K36 [71]; subsequently, proteomic and genomic studies have demonstrated its ability to catalyze the methylation of H3 lysine 4 and several non-histone targets [63]. HSP90α regulates the selective enzymatic reaction between SMYD2 and two different substrates (H3K36 and H3K4). In the presence of HSP90α, SMYD2 binds to HSP90α, enhancing its methyltransferase activity and specificity towards H3K4 [63]. In the absence of HSP90α, SMYD2 acts as a H3K36 methyltransferase [63,71]. SMYD2 also acts on non-histone proteins. When lysine 370 of p53 is mono-methylated by SMYD2, p53-mediated transcriptional regulation is inhibited [72,73]. The retinoblastoma tumor suppressor gene, RB, is also a substrate for SMYD2, and the methylation of the lysine 860 residue facilitates the binding of RB to the transcriptional repressor L3MBTL histone methyl-lysine binding protein 1 (L3MBTL1) [74]. SMYD3 plays a pivotal role in human carcinogenesis and methylates H3K4, H4K5, and H4K20, which are involved in gene regulation [66,75]. Additionally, it has recently been reported that SMYD3 can methylate vascular endothelial growth factor receptor 1 (VEGFR1) at lysine 831, thus enhancing VEGFR1′s kinase activity in HEK293 cells [76].
2.3. Other H3K4 Methyltransferases
In addition to the two main H3K4 methyltransferase families discussed above, other lysine methyltransferases can also methylate H3K4. SET7/9 (KMT7) is a SET domain-containing lysine methyltransferase that has been shown to monomethylate H3K4 in vitro and in vivo and activates gene expression [77,78,79]. SET7/9 monomethylates various non-histone proteins, including p53 [80], DNA cytosine methyltransferase 1 (DNMT1) [81], E2 promoter-binding factor 1 (E2F1) [82], hypoxia-inducible factor 1α (HIF1α) [83], and Yes-associated protein (YAP) [84], which participate in a series of cellular processes. PRDM9 (KMT8B/MEISETZ), a member of the PRDI-BF1 and RIZ homology domain-containing family (PRDM), has also been shown to methylate H3K4 and H3K36 [85,86,87]. PRDM9 contains an N-terminal KRAB (Krüppel-associated box)-related domain for protein–protein interaction, an SSX repression domain (SSXRD) involved in nuclear localization, a canonical SET domain, and a C-terminal tandem array of C2H2 zinc-finger motifs with sequence-specific DNA binding activity [88].
2.4. Distribution of H3K4 Methylation and H3K4 Readers
Although all types of H3K4 methylations have a strong correlation with active transcription [22], the distribution of H3K4 methylation types on chromosomes is distinctly different [89,90,91,92,93,94]. For example, H3K4me1 is rich in active and priming enhancer regions. H3K4me2 is also an enhancer marker, although predominantly toward the 5′ end of transcribing genes. In contrast, H3K4me3 is considered a hallmark of promoters and is predominantly localized near TSSs where it is responsible for RNA pol II recruitment for the activation of gene expression.
Each state of H3K4 methylation must be recognized by other cellular proteins known as “readers” to recruit distinct downstream effectors and regulate gene expression [95]. Numerous chromatin modifiers have specialized domains that allow them to act as H3K4 methylation readers. Most methylated H3K4 reader proteins contain a plant homeodomain (PHD), and some chromodomains and Tudor domains also appear to bind to H3K4me. For example, BPTF (bromodomain and PHD finger transcription factor), which is a subunit of the ATP-dependent chromatin-remodeling complex nucleosome remodeling factor (NURF), is preferentially associated with H3K4me3 tails [96]. The TAF3 subunit of the TFIID transcription factor also recognizes H3K4me3, directing TFIID recruitment [97]. CxxC-finger protein 1 (CFP1), the histone demethylase plant homeodomain finger 2 (PHF2), spindlin1 (SPIN1), and NUP98 fusion partners plant homeodomain finger 23 (PHF23) are also specifically recognized and interact with H3K4me3 via the PHD domain, thus participating in gene expression [98,99,100,101]. pygopus family PHD finger 2 (PYGO2) has been reported to bind di- and tri-methylated lysine 4 of H3 [102]. CHD1, an ATP-dependent chromatin-remodeling enzyme, recognizes H3K4me2 and H3K4me3 through its two N-terminal chromodomains [103]. The chromo-barrel domain of Tip60 acetyltransferase is recruited to the enhancer region by binding to H3K4me1, thus inducing estrogen-induced transcription [104]. Researchers recognize that the H3K4me mark sometimes invokes the binding of other executor proteins. For example, Sgf29 selectively interacts with H3K4me2/3 marks via the Tudor domain to recruit the SAGA complex and mediate histone H3 acetylation [105]. Additionally, ING4, a subunit of the HBO1 histone acetyltransferase (HAT) complex, recognizes H3K4me3 via the PHD domain, enhancing HBO1 acetylation activity and resulting in increased H3 acetylation in the ING4 target promoter [106].
3. H3K4 Methyltransferases in Drug-Resistant Cancers
Controlling epigenetic factors, including histone methyltransferases and demethylases, has recently emerged as a way of overcoming drug resistance, which is often encountered in chemotherapy [7,107]. In the next section, we investigate the function of H3K4 methyltransferase in cancer, specifically drug-resistant cancer.
3.1. H3K4 Methyltransferase in Breast Cancer
Extensive evidence supports the crucial role of H3K4 methyltransferase in breast cancer development. MLL2 interacts with estrogen receptor α (ERα) and regulates ERα target gene expression to mediate breast cancer growth [108]. Moreover, MLL2 levels are elevated in cell lines and in invasive carcinomas in breast and colon cancer patients [109]. MLL3, one of the most commonly mutated proteins in breast cancer, functions as another major regulatory factor for ERα expression [110]. Recent studies have shown that enhanced MLL3 and SETD1A expression promotes ERα expression to support tamoxifen-resistant breast cancer proliferation, and genome-wide histone methylation studies have revealed that MLL3 is required for acetylation of H3K27 and monomethylation of H3K4 in the ERα enhancer [111]. Similarly, depletion of MLL4 decreased H3K4me3 levels and increased H3K27me3 levels in MMP9, MMP11, and SIX1 genes in MDA-MD-231 cells [112]. These results suggest that H3K4 methyltransferase may link H3K4 methylation and H3K27 acetylation via a specific mechanism in breast cancer cells, similar to ES cells [112,113,114]. This finding was explained by a study of MLL4 expression in breast cancer cells. MLL4 coupled with histone H3 lysine 27 (H3K27) demethylase UTX (KDM6A) showed coordinated regulation of breast cancer proliferation and invasion [112]. Thereafter, studies revealed that the UTX-MLL4 complex remarkably increased binding of H3K27 acetyltransferase p300 to the target chromatin region, further enhancing H3K27 acetylation and resulting in increased enhancer activity for gene activation [115]. The relationship between H3K4 methylation and H3K27 acetylation has been demonstrated in various cell differentiation processes such as adipogenesis, myogenesis, and ESC differentiation [52,116,117].
The frequent duplication or overexpression of MLL1 in breast cancer cells shows its potential as a target molecule for breast cancer treatment [118]. In MCF-7 cells, MLL1 and SETD1A have been reported to be responsible for H3K4 methylation during estrogen-induced transcription of ER target genes [104]. One study also showed that SETD1A regulated breast cancer metastasis by activating MMP expression [113], while another reported that the amplification of SETD1A in mixed ductal and lobular breast cancer (MDLC) regulated the mitotic process by increasing the H3K4me3 marker in the promoter region of mitosis-and-DNA-damage response genes. Additionally, SETD1A regulates the expression of several genes that orchestrate mitosis, the cell cycle, and DNA damage responses through H3K4 methylation of promoters in breast and lung cancer cells. Depletion of SETD1A induces the tumor-suppressing effect of senescence; therefore, SETD1A appears to be essential for maintaining mitosis and proliferation of cancer cells [114]. We recently examined the function of SETD1A in tamoxifen-resistant breast cancer [119]. SETD1A activated a subset of ER-positive target genes by increasing H3K4 methylation and accessibility to the chromatin region of the ERα target gene in ER-positive breast cancer cells, supporting ERα recruitment. Additionally, SETD1A induced the proliferation and migration of ER-positive breast cancer cells by regulating the expression of survival- and migration-related genes in an ER-independent manner. These results suggest that SETD1A may play an important role in acquiring resistance to hormone therapy drugs in breast cancer, independent of ER signaling [119]. Notably, the intracellular protein levels of SETD1A are upregulated in other breast cancer subtypes, including ER-positive, HER2-positive, and TNBC breast cancer compared with normal breast cancer cells, and miR-1915-3p regulates this process. We also found an overlap between genes regulated by SETD1A and tamoxifen resistance-specific genes in ER-positive breast cancer cells, suggesting that SETD1A may be involved in tamoxifen resistance. Indeed, in tamoxifen-resistant breast cancer, SETD1A silencing dramatically reduces the expression of cell growth genes (e.g., EGFR and MYC) required for the development of tamoxifen resistance, and inhibits cell proliferation by inducing cell cycle arrest [119]. Interestingly, SETD1B, which is structurally similar to SETD1A, plays an important role in the survival and pathogenesis of TNBC, regardless of H3K4 methyltransferase activity, e.g., by forming COMPASS complexes in the cytoplasm and regulating ADIPOR1 signaling by interacting with BOD1 [116].
SMYD2 overexpression promotes the progression of triple-negative breast cancer and is clinically associated with poor prognosis [117]. The ability of SMYD3 to increase the expression of the oncogene WNT10B and promote the epithelial–mesenchymal transition (EMT) process is thought to contribute to metastatic breast cancer [120,121]. A previous study on breast cancer reported that excessive expression of SMYD3 increased cell tolerance to cisplatin treatment in MCF-7 cells [122]. Additionally, SMYD3 depletion, in conjunction with cisplatin treatment, inhibited cell viability and mitochondrial membrane potential. These data suggest that SMYD3 is a key factor in determining cisplatin sensitivity and plays an important role in cisplatin resistance in cancer.
In addition to regulating the expression of RUNX2 and some redox enzymes to maintain redox homeostasis [123,124], SET7/9 promotes breast cancer development by stabilizing the ER through methylation of the ER K302 residue, aiding in efficient recruitment to and transactivation of the target genes [125]. However, another study showed a negative feedback loop between SET7/9 and DNMT1. In contrast to DNMT, SET7/9 is significantly less expressed in TNBC and inhibits EMT when overexpressed in these cells. Moreover, loss of SET7/9 increases breast cancer stem cell-like properties and enhances the EMT process, which is relevant to tumor resistance, suggesting that SET7/9 acts as a suppressor in breast cancer [126]. This function of SET7/9 is caused by the negative regulation of stability by methylation of DNMT and E2F1. These results suggest that SET7/9 could be used as a biomarker to predict the potential for metastasis and resistance to anti-estrogen therapy in breast cancer patients.
3.2. H3K4 Methyltransferase in Colorectal Cancer
Previous reports have shown that the restoration of MLL3 expression enhances the H3K4me1 profile and represses colorectal cancer (CRC) growth, suggesting that MLL3 functions as a tumor suppressor [127]. MLL3 and MLL4 are coactivators of p53 and act as tumor suppressors by labeling H3K4me3 to express endogenous p53-target genes induced by DNA-damaging agents, such as doxorubicin [128]. Additionally, SET7/9 has a tumor suppressor function which involves it interacting with HDAC6 and suppressing HDAC6-mediated activation of the ERK signaling pathway in colon cancer [129]. In contrast, MLL4 directly interacts with mutated p53 (p53R273H,P309S) to increase H3K4me1 and histone H3 lysine 27 acetylation (H3K27ac) levels, thus activating the chronic TNFα signaling pathway which accelerates colon cancer invasion [130]. In colorectal cancer, SETD1A has been reported to interact with β-catenin to regulate Wnt signaling and promote cancer cell growth [131]. When the YAP protein, a SETD1A non-histone substrate, is methylated by SETD1A, its migration out of the nucleus is inhibited and, consequently, YAP-TEAD transcriptional activity is increased, leading to colon cancer cell proliferation and tumor formation [132]. Furthermore, SETD1B frameshift mutations play an important role in the tumorigenesis of colorectal and gastric cancers with high microsatellite instability [133]. Increased levels of SMYD2 upregulate MDR1/P-glycoprotein expression via the MEK/ERK/AP-1 pathway in colon cancer, which promotes oxaliplatin resistance in colon cancer [134]. SMYD3 interacts with the RNA helicase HELZ, forming a complex with RNA polymerase II and promoting cancer cell proliferation in colorectal and hepatocellular carcinomas [64].
3.3. H3K4 Methyltransferase in Prostate Cancer
A limited number of H3K4 methyltransferases have been studied in prostate cancer. The MLL1 complex binds directly to the androgen receptor (AR) through a menin subunit, acting as a co-activator of AR signaling. Additionally, menin expression is upregulated in castration-resistant prostate cancer and is correlated with low overall survival in individuals diagnosed with prostate cancer [135]. MLL2 is also considered a potential therapeutic target because it activates the PI3K/EMT process [136] and induces DNA damage in prostate cancer [137]. SMYD3 depletion inhibits prostate cancer progression through a mechanism that blocks the transcription of AR or cyclin D2 [138,139]. SET7/9 interacts directly with AR and enhances AR transcriptional activity by methylating the K632 residue of AR. It not only plays a proliferative role in prostate cancer but is also involved in TNFR and PTEN/PI3K/AKT signaling [140,141]. Whole transcriptome sequencing revealed that SET7/9 has a frameshift mutation that leads to changes in chromatin accessibility during co-transcriptional RNA processing in castration-resistant prostate cancer [142].
A study conducted by our group has recently revealed that SETD1A, which is present at higher levels in metastatic castrate-resistant prostate cancer (mCRPC) than in primary prostate cancer cells, contributes to the tri-methylation of H3K4 by binding to E2F1 in the promoter region of the FOXM1 gene, a cancer cell proliferation-specific transcription factor. SETD1A is also essential for the expression of stem cell factors, e.g., octamer-binding transcription factor 4 (OCT4) and plays an important role in the proliferation of prostate cancer stem cells, which is important in metastatic CRPC tumor formation. Particularly, the expression of SETD1A is significantly correlated with the survival rate of prostate cancer patients, and the high expression of the SETD1A-FOXM1 pair is highly correlated with a poor prognosis, suggesting that it can be used as an important marker for predicting the proliferation and prognosis of mCRPC [143].
3.4. H3K4 Methyltransferase in Leukemia
MLL1 was initially associated with acute myeloid and lymphoblastic leukemia (AML, ALL) through chromosomal rearrangement [144,145]. MLL1 rearrangements on chromosome 11q23 result in a chimeric gene consisting of the N-terminus of MLL1 fused to the C-terminus of fusion partners. Four frequent fusion partners, AF4, AF9, AF10, and ENL, accounted for more than 70% of all observed rearrangements in patients. Importantly, MLL1-rearranged leukemia predicts a poor prognosis in patients [146]. Biological studies have suggested a major mechanism by which MLL1 fusion proteins drive leukemogenic gene expression dependent on its binding with chromatin-associated protein complexes, including menin, SEC, and DOT1L (disruptor of telomeric silencing 1-like). Binding of menin to the MLL1 portion of the fusion protein is critical for MLL1 fusion-mediated transformation [147]. Interaction with SEC is involved in the activation of RNA Pol II for transcriptional elongation [148]. DOT1L, which binds to the fusion partner site of the rearrangement protein, is a histone H3 lysine 79 (H3K79) methyltransferase, wherein abnormal methylation occurs at the H3K79 site at the MLL1 fusion target gene sites, leading to abnormal expression of genes responsible for cell proliferation [149,150].
Apart from MLL1-rearranged leukemias, there are reports on the function of wild-type H3K4 methyltransferase in leukemia. The wild-type MLL1 protein, along with the MLL1-AF9 fusion protein, participates in abnormal H3K4 and H3K79 methylation in the HOX gene region, causing leukemogenesis [151]. Additionally, SETD1A acts as an essential regulator of gene expression required for DNA damage reactions through interaction with cyclin K using the non-catalytic site, the FLOS domain. This SETD1A/cyclin K pathway supports MLL1-rearranged and non-MLL1-rearranged leukemia cell growth [152]. The loss of wt MLL2 significantly reduced MLL-AF9-transformed cell survival, and co-deletion with MLL1 further reduced leukemia cell proliferation by regulating major AML survival pathways [153]. However, MLL2 appears to have a completely different function in chronic myelogenous leukemia (CML) that is resistant to tyrosine kinase inhibitors (TKIs). The expression level of MLL2 decreased in (TKI)-resistant CML. Furthermore, when TKI-sensitive CML cells are treated with TKIs, such as dasatinib or nilotinib, MLL2 expression, which can increase p21 expression, is induced while the expression of CDK2, CDK4, and cyclin B1 is attenuated, resulting in a higher rate of cell death [154]. MLL3 is often deleted from AML, and its function as a tumor suppressor has been proposed; however, the exact mechanism is unknown [155].
SMYD2 overexpression promotes disease progression and is correlated with poorer outcomes in childhood acute lymphoblastic leukemia (ALL) [156]. Specifically, the level of SMYD protein in AML appears to correlate with the level of the SET7/9 protein [157]. When SMYD2 is downregulated, leukemia cells transition to a quiescent state after anti-leukemia chemotherapy treatment, resulting in increased resistance to anticancer drugs, which is thought to be due to increased SET7/9 expression in response to decreasing SMYD2 levels. In this case, a pharmacological SET7/9 inhibitor was effective at low-expressing SMYD2 AML. However, downregulation of SET7/9 through hypermethylation of the SET7/9 promoter was also observed in AML patient cells, and further studies are required to identify the target mechanism underlying the regulation of SMYD2 and SET7/9 [158].
There are no detailed mechanistic studies on the relationship between PRDM9 and cancer. Aberrant PRDM9 expression appears in 32 different cancer types and differential RPDM9 expression is linked to genomic instability and transcriptomic landscape in tumors [159]. Excess rare PRDM9 allelic forms were found in B-cell precursor acute lymphoblastic leukemia (B-ALL) in children, which might influence genomic instability leading to aneuploidies formation related to childhood leukemogenesis [160]. As we have seen so far, alterations in H3K4 methyltransferases either promoted or suppressed various cancers via multiple mechanisms which need to be explored further.
3.5. H3K4 Methyltransferase in Gastric Cancer
In gastric cancer, the functions of H3K4 methyltransferases are contradictory depending on the type of protein. MLL2 is an oncogene that modulates proliferation and apoptosis in gastric cancer [161]. In contrast, restoring MLL3 repressed the proliferation of diffuse-type gastric adenocarcinoma (DGA) by targeting the EMT pathway [162]. Most recently, SETD1A overexpression was also found in HIF1α-related gastric cancer, enhancing glycolysis and promoting gastric cancer progression [163]. Similarly, SMYD2 is overexpressed in gastric cancer and promotes gastric cancer progression, which is associated with worse clinical outcomes [164]. SMYD3 promotes cell proliferation, migration, and invasion in gastric cancer by mediating the ATM-CHK2-p53/Cdc25C pathway [165,166]. In contrast, SET7/9 expression was downregulated in gastric cancer, thus it is considered a tumor suppressor, acting by either activating SREK1IP1 expression or inhibiting three MMP genes (MMP1, MMP7, and MMP9) [167].
3.6. H3K4 Methyltransferase in Other Cancers
Overexpression of P-glycoprotein (Pgp) encoded by human multidrug resistance 1 (MDR1) is closely linked with cancer drug resistance by serving as an ATP-dependent efflux pump which reduces the anticancer agents in resistant cells [168,169]. Recently, it was demonstrated that MLL1-mediated H3K4me3 can activate MDR1 transcription, and MLL1 depletion conferred cells sensitive to multiple chemotherapeutic agents such as paclitaxel, vinblastine, and daunorubicin [170]. In salivary gland squamous and head and neck carcinomas, MLL1 was shown to associate with Wnt/β-catenin signaling to regulate tumor initiation and proliferation [171,172]. In another study, the mRNA and protein levels of MLL1 were higher in pancreatic cancer cells than in normal pancreatic cells, and this phenomenon was also correlated with the transcription level of PD-L1. The higher expression of MLL1 in pancreatic cancer enhances PD-L1(CD274) expression by increasing H3K4me3 status at the CD274 promoter region, thereby helping pancreatic cancer escape anti-PD-L1/PD-1 immunotherapy [173].
Elevated MLL2 expression in esophageal squamous cell cancers (ESCC) and pancreatic ductal adenocarcinoma (PDAC) predicts poor prognosis in both tumors [174,175]. Additionally, a truncation mutation in MLL2 found in small cell lung cancer (SCLC) is known to interfere with the control of transcription enhancers, potentially contributing to the promotion of SCLC [176]. MLL3 is one of the most commonly mutated genes and has diverse functions in different cancers [110,127,162,177,178]. It has been suggested that MLL3 may function as a prognostic predictor of pancreatic cancer because it affects the cell cycle and DNA replication pathway in pancreatic ductal adenocarcinoma (PDAC) [174]. Similar to MLL1 in pancreatic cancer, overexpression of MLL3 in prostate cancer can also activate PD-L1 expression via regulation of the PD-L1 enhancer region, suggesting its potentially vital role in overcoming immune evasion in cancer cells [179]. In contrast, MLL3 is also considered a tumor suppressor that regulates DNA damage in bladder cancer [177].
SETD1A expression was upregulated in hepatocellular carcinoma compared with normal tissues, and its expression levels and sensitivity to sorafenib were negatively correlated. Functionally, it acts as an initiator to augment primary resistance to sorafenib by impairing the phosphorylation of Hippo signaling key factor YAP at serine 127 and enhancing YAP activation in hepatocellular carcinomas [180]. The pRB1-SETD1A complex formed by the action of CUDR in liver cancer cells labels H3K4me3 in the TRF2 promoter region, and ultimately, overexpressed TRF2 binds to the telomere repeat DNA to extend telomere length [181].
Upregulation of SETD1B was found to activate iNOS expression in tumor-induced myeloid-derived suppressor cells, which could provide a foundation for establishing an effective approach for improving the efficacy of cancer immunotherapy by targeting SETD1B [182]. SETD1B is also overexpressed in clear cell renal cell carcinoma (ccRCC), has been associated with cancer metastasis and has been reported as a promising prognostic indicator for ccRCC [183].
Recurrent mutations in SMYD1 have been identified in splenic marginal zone lymphoma using whole-exome sequencing (WES) and copy number variation analysis [184]. SMYD2 overexpression promotes disease progression and correlates with worse outcomes in other types of tumors, including esophageal squamous cell carcinoma [185], primary hepatocellular carcinoma (HCC) [186], and papillary thyroid carcinoma [187]. In non-small cell lung cancer (NSCLC), SMYD2 regulates the P53 signaling pathway to promote cisplatin resistance in NSCLC [188]. Notably, SMYD2 and miR-125b repression improved the sensitivity of multiple anticancer drugs by MDR 1 expression [189]. In contrast, low SMYD2 expression is associated with low survival in patients with renal cell carcinoma (RCC) [190] associated with complex karyotype acquisition and cancer progression [191].
SMYD3 is amplified in various tumor types. SMYD3 regulates the MEK/ERK kinase signaling pathway via methylation of mitogen-activated protein kinase 2 (MEKK2) in Ras-driven pancreatic ductal adenocarcinoma and lung adenocarcinoma cancers [192]. In ovarian cancer, SMYD3 regulates tumor proliferation and apoptosis by downregulating CDKN2A through tri-methylation of H4K20 and upregulating BIRC3 through tri-methylation of H3K4 [193]. SMYD3 was also reported to stimulate homologous recombination (HR)-related genes to modulate DNA repair in many cancer cell lines [194]. Most recently, overexpressed SMYD3 enhanced MMP2 and CDK2 expression in hepatocellular carcinoma and upregulated Bcl-2, Bcl-xl, MMP-2, and MMP-9 in NSCLC to facilitate tumorigenicity and cancer progression [195,196]. Under hypoxic conditions, SET7/9 has been shown to mediate the survival of various cancer cells against hypoxic stress by promoting HIF-1α protein stability and enhancing HIF-1-mediated gene transcription in osteosarcoma, hepatoma, and renal cell carcinoma [197]. In contrast, SET7/9 was shown to interact with, and methylate, β-catenin at lysine 180 to decrease β-catenin stability, which influences Wnt signaling and inhibits cervical cancer cell proliferation [198]. In that study, they showed that the depletion of SET7/9 in HeLa cells promoted the expression of Wnt/β-catenin target genes such as c-myc and cyclin D1 and the growth of cancer cells. The mechanisms underlying the action of H3K4 HMTs on drug-resistant cancers are summarized in Table 1.
Table 1.
Potential role of H3K4 HMTs in drug-resistant cancers.
| Enzyme | Cancer Types | Proposed Mechanism |
|---|---|---|
| MLL1 | Chemotherapy resistant MLL leukemia Castration-resistant prostate cancer Anti-PD-L1/PD-1 resistant pancreatic cancer |
Increases MDR-1 expression [170] Activates androgen receptor signaling [135] Increases PD-L1 expression [173] |
| MLL3 | Tamoxifen-resistant breast cancer Anti-PD-L1/PD-1 resistant prostate cancer |
Increases ERα expression [111] Increases PD-l expression [179] |
| SETD1A | Tamoxifen-resistant breast cancer Tamoxifen-resistant breast cancer Triple-negative breast cancer Castration-resistant prostate cancer Sorafenib resistant hepatocarcinoma |
Increases ERα expression [111] Activates ERα signaling and EGFR expression [119] Activates MMP expression [113] Activates FOXM1 signaling via binding with E2F1 [143] Activates Yes-associate protein [180] |
| SETD1B | Triple-negative breast cancer | Regulates adiponectin receptor 1 signaling [116] |
| SMYD2 | Triple-negative breast cancer Oxaliplatin-resistant colon cancer Cisplatin-resistant non-small cell lung cancer Chemotherapy resistant renal cell carcinoma |
Activates STAT3 and the p65 [117] Increases MDR-1 expression [134] Inhibits p53 signaling [188] Increases MDR-1 expression [189] |
| SMYD3 | Cisplatin-resistant breast cancer | Increases WNT10B expression and promote the EMT [120,121] |
| SET7/9 | Anti-estrogen-resistant breast cancer Castration-resistant prostate cancer |
Controls the stability of E2F1 and DNMT1 [126] Alters chromatin accessibility via frameshift mutation [142] |
4. Inhibitors Targeting H3K4-Specific HMTs for Anticancer Therapy
4.1. WDR5 Inhibitors
Thus far, only a limited number of selective small-molecule compounds that can directly inhibit the active sites of enzymes of specific KMT2 family proteins have been reported [199]. Perhaps apart from the establishment of a high-efficiency screening method using the core SET domain, this is because the function of several subunits constituting the KMT2 complex contributes significantly to the overall H3K4 methylation activity. As mentioned above, H3K4 methyltransferase on its own has very weak methyl transfer catalytic activity, but binding of the RBBP5, WDR5, ASH2L, and DPY30 subunits leads to a significant increase in methyltransferase activity. Therefore, attempts have been made to inhibit the function of the MLL complex subunits. Among these methyltransferase adapter proteins, WDR5 is considered an attractive target because of its great importance within the MLL complex, which is responsible for its interaction with chromatin-remodeling proteins, transcription factors, and long non-coding RNAs. Additionally, by forming protein complexes with other proteins such as MYC, WDR5 induces the expression of key oncogenes, leading to tumor initiation, cell cycle progression, DNA replication, invasion, and cancer metastasis. Therefore, WDR5 inhibitors have great potential as anticancer drugs. Based on the protein interaction information, peptide inhibitors were initially reported, and later, structural analysis led to the development of peptidomimetic inhibitors that inhibit WDR5–MLL1 binding (Table 2). MM-102, MM-401, and MM-589, designed as inhibitors targeting the interaction of WDR5/MLL1, effectively inhibited histone H3K4 methyltransferase activity and reduced oncogenic gene transcription, inhibiting the growth and cell cycle of MLL-rearranged leukemia cells. MM-102 had a Ki < 1 nM in the WDR5 binding assay. MM-102 was also shown to inhibit MLL1 H3K4 methyltransferase activity (IC50 = 0.4 μM) and acute leukemia cell growth in a dose-dependent manner [200].
Table 2.
Potential therapeutic inhibitors targeting H3K4-specific HMTs.
| Inhibitor | Structure | Mode of Action | Kd or Ki | Methylation IC50 |
PPI IC50 | GI50 | Cancer Cell Type | In Vivo Validation |
Ref |
|---|---|---|---|---|---|---|---|---|---|
| MM-102 |
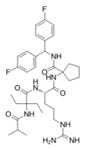
|
WDR5–MLL | <1 nM | 400 nM | 2.4 nM | 25 μM | MLL1-rearranged leukemia | [200] | |
| MM-401 |
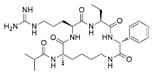
|
WDR5–MLL | <1 nM | 320 nM | 0.9 nM | 5.9~12.6 μM | MLL1-rearranged leukemia | [201] | |
| MM-589 |
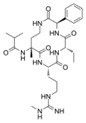
|
WDR5–MLL | <1 nM | 12.7 nM | 0.9 nM | 0.21~0.25 μM | MLL1-rearranged leukemia | [202] | |
| WDR5-0103 |

|
WDR5–MLL | 450 nM | 39 μM | [203] | ||||
| OICR-9429 |
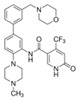
|
WDR5–MLL | 30 nM | ~5 μM | C/EBPα-mutant AML Ovarian cancer (with topotecan) Prostate cancer (with cisplatin) |
Yes Yes |
[204,207] [208] [209] |
||
| Piribedil |
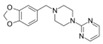
|
WDR5–MLL | 180 nM | 65~92 μM | MLL1-rearranged AML | Yes | [205] | ||
| Win6mer |
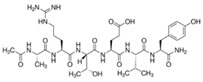
|
WDR5–MLLWDR5–SETD1A | 2.9 nM | 2.2 nM2.5 nM | [206] | ||||
| Compound C6 |
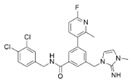
|
WDR5–MLL | 0.1 nM | 20 nM | 2.5~6.4 μM | MLL1-rearranged leukemia | [210] | ||
| Compound 16 |
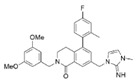
|
WDR5–MLL | <0.02 nM | 2.2 nM | 38~78 nM 0.26~0.49 μM |
MLL1-rearranged leukemia Neuroblastoma and Burkitt’s lymphoma | [211] |
||
| MCP-1 |
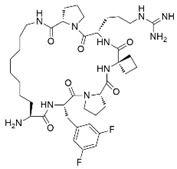
|
Menin–MLL | 4.7 nM | 18.5 nM | MLL1-rearranged leukemia | [212] | |||
| MI-2 |
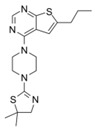
|
Menin–MLL | 158 nM | 446 nM | 7.2~18 μM | MLL1-rearranged leukemia | [213] | ||
| MI-2-2 |
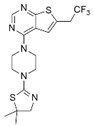
|
Menin–MLL | 22 nM | 46 nM | 3 μM | MLL1-rearranged leukemia (with DOT1L inhibitor) | Yes | [214,215] | |
| MI-463 |
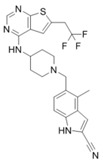
|
Menin–MLL | 9.9 nM | 15.3 nM | 0.23 μM | MLL1-rearranged leukemia | Yes | [216] | |
| MI-503 |
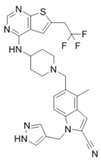
|
Menin–MLL | 9.3 nM | 14.7 nM | 0.22 μM 1.5~11.7 μM |
MLL1-rearranged leukemia Castration-resistant prostate cancer |
Yes Yes |
[216] [135] |
|
| MI-538 |
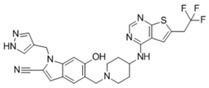
|
Menin–MLL | 6.5 nM | 21 nM | 83 nM | MLL1-rearranged leukemia | Yes | [217] | |
| MI-3454 |
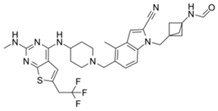
|
Menin–MLL | 0.51 nM | 7.6~27.1 nM | MLL1-rearranged or NPM1-mutated leukemia | Yes | [218] | ||
| BAY-155 |
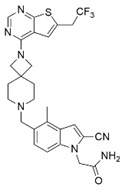
|
Menin–MLL | 75 nM | 8 nM | 90~140 nM | MLL1-rearranged leukemia | Yes | [219] | |
| VTP50469 |

|
Menin–MLL | 104 pM | 13~37 nM | MLL1-rearranged leukemia | Yes | [220] | ||
| M-89 |
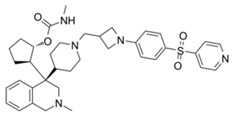
|
Menin–MLL | 1.4 nM | 5 nM | 25~55 nM | MLL1-rearranged leukemia | [221] | ||
| M-525 |
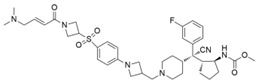
|
Menin–MLL | 3.3 nM | 2.3~10.3 nM | The first irreversible Menin inhibitor | [222] | |||
| M-808 |
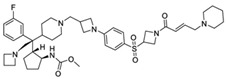
|
Menin–MLL | 2.6 nM | 1~4 nM | MLL1-rearranged leukemia | Yes | [223] | ||
| AZ505 |

|
SMYD2 | 500 nM | 120 nM | MDR-clear cell renal cell carcinoma (ccRCC) Triple-negative breast cancer |
Yes | [224] [117] |
||
| LLY-507 |
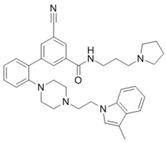
|
SMYD2 | <15 nM | 1.5~6 μM 1.77~2.9 μM |
Esophageal, liver, and breast cancer cells High-grade serous ovarian carcinomas (HGSOCs). |
[225] [226] |
|||
| A-893. |
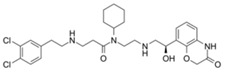
|
SMYD2 | 2.8 nM | Lung cancer | [227] | ||||
| BAY598 |

|
SMYD2 | 1.1~1.2 nM | 27 nM | <10 μM | Esophageal cancer (combi w/doxorubicin) | Yes | [228] | |
| EPZ031686 |
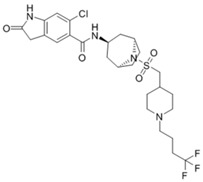
|
SMYD3 | 1.3~4.7 nM | 3 nM | [229] | ||||
| EPZ028862 |
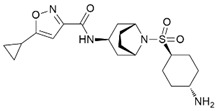
|
SMYD3 | 1.8 nM | >40 μM | Esophageal squamous cell carcinoma | [230] | |||
| GSK2807 |
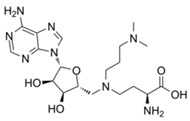
|
SMYD3 | 14 nM | 130 nM | [231] | ||||
| Compound 29 |
|
SMYD3 | 440 nM | 11.7 nM | 17.7 μM (2D) 1.04 μM (3D) |
Hepatocarcinoma | [232] | ||
| (R)-PFI-2 |
|
SETD7/9 | 0.33 nM | 2 nM | Breast cancer cells | [233] |
The high binding affinity of MM-401 to WDR5 (Ki < 1 nM) was detected by the label-free BioLayer Interferometry (BLI/OctetRED) assay and confirmed by competitive fluorescence polarization (FP) experiments. MM-401 interferes with the WDR5–MLL1 interaction (IC50 = 0.9 nM) and inhibits the activity of MLL1 (IC50 = 0.32 µM) in the in vitro HMT assay [201]. MM-589, another peptidomimetic WDR5 inhibitor (Ki <1 nM), has been shown to inhibit H3K4 HMT activity of MLL1 with an IC50 value of 12.7 nM and potently inhibits the growth of human leukemia cell lines containing MLL translocation at sub-micromolar concentrations, which is > 40 times superior to that of MM-401 [202].
At the same time, a nonpeptide small-molecule compound was also discovered that targets the bond between MLL and WDR5. WDR5-0103 was reported to specifically and effectively antagonize the interaction between WDR5 and the Win motif of MLL, which could further compromise MLL catalytic activity [203]. Structural and biophysical analyses showed that this antagonist binds to the WDR5 peptide-binding pocket with a Kd of 450 nM and inhibits the catalytic activity of the MLL core complex in vitro. At concentrations up to 100 μM, WDR5-0103 showed no inhibitory effect on the human H3K4 methyltransferase SETD7 or six other HMTs (G9a, EHMT1, SUV39H2, SETD8, PRMT3, and PRMT5). The specificity of this low-molecular weight compound for the MLL complex shows the potential for further development of WDR5-dependent enzyme inhibitors associated with MLL-rearranged leukemia or other cancers through inhibition of protein–protein interactions.
OICR-9429, which was designed through additional modifications of WDR5-0103 series compounds, showed high-affinity binding with WDR5 and occupied the MLL1-binding pocket with WDR5, resulting in disruption of the WDR5–MLL1 interaction. Grebien et al. have shown that OICR-9429 can decrease the viability of C/EBPα-mutated AML cells, supporting its potential anticancer effect on non-MLL-rearranged leukemia and solid tumors [204]. Zhang et al. screened a library of 592 FDA-approved drugs by measuring changes in the HTRF signal to identify compounds that inhibit the histone methylase activity of MLL1 methylation [205]. Piribedil, previously used to treat Parkinson’s disease, exerted extraordinary activity in inhibiting MLL methyltransferase activity (EC50 = 0.18 μM). An in vitro assay for histone methyltransferase/acetyltransferase activity showed that piribedil had little effect on the activity of MLL4 (IC50 > 100 μM) and less potent effects on EZH2, NSD2, PRMT4, p300, and SMYD3, suggesting that piribedil specifically targets MLL1 as opposed to other SET domain-containing enzymes and effectively inhibits MLL-rearranged AML proliferation by interfering with the MLL1–WDR5 interaction [205].
The Win motif, located on the N-flanking region of the SET domain, is required by all six MLL family members for binding to WDR5. A previous report illustrated that a six-residue Win motif peptidomimetic (Win6mer) that binds to WDR5 and selectively inhibits the SETD1A and MLL1 complex was previously designed [206].
The six-residue peptidomimetic Win6mer, designed selectively in the MLL1 and SETD1A core complex, destroys the Win motif–WDR5 interface required to stabilize contact with the RBBP5/ASH2L heterodimer. Win6mer binds to WDR5 with a Kd of 2.9 nM.
Recently, Aho et al. identified compound C6, which inhibits WDR5–MLL1 binding with picomolar binding affinity through fragment-based screening of 13,800 compounds and structure optimization [210]. Additionally, further optimization of compound C6 led to the generation of compound 16, which contained a stronger dihydrothioquinolinone structure [211]. This compound also has a picomolar binding affinity and inhibits the proliferation of MYC-induced cancer cells (neuroblastoma and Burkitt’s lymphoma) and MLL1-rearranged leukemia by reducing MYC recruitment to the MYC/WDR5 target gene.
4.2. Menin Inhibitors
The protein menin binds to the N-terminal fragment of MLL retained in all MLL fusion proteins, which plays a critical role as an oncogenic cofactor of MLL fusion proteins in leukemia [147]. Thus, inhibiting the interaction between menin and histone methyltransferases could be a novel therapeutic strategy. Initially, macrocyclic peptidomimetic inhibitors (MCP-1) were designed to inhibit the menin−MLL1 interaction. The most potent MCP-1 binds to menin with a Ki value of 4.7 nM and is >600 times more potent than the corresponding acyclic peptide. However, the disadvantage is that cell permeability must be secured to maintain the in vivo activity of MCP-1. Optimization of MCP-1 may ultimately yield a class of potent and cell-permeable small-molecule inhibitors of the menin−MLL1 interaction as potential new therapeutics to treat acute leukemia with MLL1 rearrangements [212].
MI-2-2 is a thienopyridine-based compound optimized from MI-2 [214,215]. Despite its excellent ability to inhibit the binding of menin–MLL1, its in vivo activity is limited due to its poor metabolic stability. However, it has inspired the development of a variety of low-molecular-weight compounds with improved activity. MI-463 and MI-503 directly bind to menin with low nanomolar binding affinities and effectively block the menin–MLL interaction [216]. Pharmacological inhibition of the menin–MLL interaction using small molecules blocks the progression of MLL leukemia in vivo without impairing normal hematopoiesis. These compounds can reach the target protein in mammalian cells and effectively inhibit the menin–MLL–AF9 interaction at sub-micromolar concentrations, as assessed in the co-immunoprecipitation experiment. Treating murine bone marrow cells (BMC) transformed with the MLL-AF9 oncogene with MI-463 or MI-503 resulted in significant growth inhibition, with half-maximal growth inhibitory concentration (GI50) values of 0.23 μM and 0.22 μM, respectively. Both compounds have an appropriate pharmacokinetic profile and high oral bioavailability, and their in vivo administration results in a substantial extension of survival in a mouse model of MLL leukemia through the on-target mechanism of action. Importantly, these compounds do not impair normal hematopoiesis in mice, providing evidence that a sufficient therapeutic window can be achieved [216]. The effectiveness of MI-505 has been proved, not only in leukemia, but also in castration-resistant prostate cancer [135]. MI-503 induces apoptosis by effectively inhibiting the interaction between menin and MLL in VCaP and LNCaP cells, thereby reducing the expression of PSA protein and AR-target genes. Additionally, treatment with MI-503 in the LNCaP-AR xenograft model and the VCaP xenograft mouse castration model showed marked tumor growth inhibition. These findings suggest that inhibiting the menin–MLL1 interaction using MI-503 may be a novel treatment for hormone-refractory prostate cancer.
BAY-155 was recently reported to be an improved menin inhibitor [219]. BAY-155 has an inhibitory IC50 of 8 nM in a time-resolved fluorescence resonance energy transfer (TR-FRET) assay to interfere with menin–MLL binding, which more strongly interferes with menin–MLL binding compared to MI-503. Isothermal titration calorimetry (ITC) experiments confirmed that BAY-155 binds menin protein in a 1:1 ratio, with a binding affinity of 75 nM. Additionally, BAY-155 showed a significantly improved selectivity profile compared to that of MI-503. In an assay panel covering 77 pharmacologically relevant safety targets, including G-protein coupled receptors (GPCRs), ion channels, and transporters, 10 µM BAY-155 inhibited only seven tested proteins, while MI-503 inhibited 28 proteins at the same concentration [219].
In most MLL-rearranged leukemia models, the response to DOT1L inhibitors is limited. One of the reasons for this is the role of the wild-type MLL complex and fusion-MLL protein in leukemogenesis. Accordingly, a combination of a DOT1L inhibitor (EPZ004777) and an MLL–menin interaction inhibitor (MI-2-2) was administered and showed dramatically improved differentiation induction and apoptosis in various MLL disease models, including primary leukemia cells, suggesting its potential as a new strategy for the treatment of leukemia in the future [214].
Unlike most WRD5 inhibitors, which exhibit moderate activity in most cells, hence limiting further application of the compound in animal tumor models, there is potential for the development of potent and selective menin inhibitors. MI-3454 is a newly designed structural analog that utilizes the crystal structures of MI-503 and menin [218]. It blocks the interaction between menin and MLL1 at subnanomolar concentrations, resulting in almost 60 times greater improvement over MI-503. Additionally, MI-3454 shows superior activity across MLL leukemia cells compared with all menin–MLL1 inhibitors reported to date. MI-3454 induced complete remission or regression of leukemia in a mouse xenograft model of MLL1 translocation or NPM1 mutant leukemia derived from AML patients. Most recently, KO-539, a structurally related analog of MI-3454, has been approved for clinical trials (Table 3), further amplifying interest in this field. KO-539 is expected to reduce the transcription of HOXA9 and MEIS1 promoters and lead to terminal differentiation of AML blasts following oral administration once daily in patients with MLL1-rearranged or NPM1-mutant AML cells. Other potent and selective menin–MLL inhibitors, JNJ-75276617 and SNDX-5613 (VTP-50469), have entered Phase 1 and Phase I/II clinical trials, respectively, for relapsed/refractory acute leukemia.
Table 3.
Inhibitors of H3K4-specific HMTs in a clinical trial.
| Drug Name | Status | Mechanism | Cancer Type | Administration | ClinicalTrial.gov ID# |
|---|---|---|---|---|---|
| KO-539 | Phase 1/2a | Menin–MLL1 inhibitor | Relapsed or Refractory Acute Myeloid Leukemia | Oral | NCT04067336 |
| JNJ-75276617 | Phase 1 | Menin–MLL1 inhibitor | Acute Leukemias Acute Myeloid Leukemia Acute Lymphoblastic Leukemia |
Oral | NCT04811560 |
| SNDX-5613 | Phase 1/2 | Menin–MLL1 inhibitor | Acute lymphoblastic leukemia (ALL) Mixed phenotype acute leukemia (MPAL). Acute Myeloid Leukemia (AML). NPM1c AML. |
Oral | NCT04065399 |
4.3. SMYD Inhibitors
SMYD2, which has less limited substrate specificity, is attracting attention as an anticancer target molecule because of the negative correlation between expression levels and patient survival rates in various cancer types. SMYD2-specific inhibitor, AZ505, was identified in high-throughput screening, showing an IC50 of 120 nM in an enzyme assay as well as high selectivity indices (>690-fold) against other methyltransferases [224]. LLY-507, another potent small-molecule inhibitor of SMYD2, showed >100-fold selectivity over a broad range of methyltransferase and non-methyltransferase proteins. The ability of LLY-507 to inhibit the catalytic function of SMYD2 was tested by a scintillation proximity assay (SPA) using peptides derived from a previously described p53, an SMYD2 substrate. LLY-507 potently inhibited the ability of SMYD2 to methylate the p53 peptide with an IC50<15 nM. In the cell-based assay, LLY-507 also inhibited monomethylation of the Lys 370 residue of the p53 protein mediated by SMYD2 at sub-micromolar concentrations. However, cellular global histone methylation levels were not significantly affected by the inhibition of SMYD2 with LLY-507. Subcellular fractionation studies indicated that SMYD2 was primarily localized in the cytoplasm, suggesting that SMYD2 targets a very small subset of histones at specific chromatin loci and/or non-histone substrates [225].
Additionally, A-893 is a benzoxazinone-based inhibitor that exhibited a high level of selectivity for SMYD2 in an inhibition assay using 30 methyltransferase panels [227]. An analysis of the cocrystal structure of A-893 highlights the contribution of the newly installed hydroxyl group to an intricate network of hydrogen bonds around the lysine pocket of SMYD. This compound inhibited SMYD2 with an IC50 of 2.8 nM in the SPA assay measuring the methylation of p53 peptide by SMYD2. Furthermore, in A549 cells, A-893 reduced the monomethylation of p53 K370 by SMYD2. However, the cellular activities of these three SMYD2 inhibitors are limited. Eggert et al. designed a potent and selective amino-pyrazoline-based small-molecule SMYD2 inhibitor [228]. BAY-598 inhibited SMYD2 with an IC50 of 27 ± 7 nM in a biochemical SPA assay and an IC50 of 58 nM in a cell-based assay measuring the methylation activity of p53. In the same study, oral administration (100 mg/kg, q.d.) of BAY-598 significantly reduced SMYD2-dependent methylation signals in an esophageal ex vivo model. However, BAY-598 alone failed to show an anti-proliferative effect in cell-based assays and in vivo xenograft models. BAY-598 (500 mg/kg, q.d.) significantly reduced tumor growth in an in vivo esophageal xenograft model only combined with doxorubicin, suggesting the potential for limitations in the application of SMYD2 inhibitors to specific anticancer drugs in response to certain types of cancer [228].
SMYD3 is overexpressed in different types of tumors, including breast, gastric, pancreatic, colorectal, lung, and hepatocellular carcinoma [121,192,234,235]. EPZ031686, the first orally available SMYD3 inhibitor, was identified by screening the Epizyme proprietary histone methyltransferase-biased library. This compound inhibited SMYD3 with an IC50 of 3 nM in the SPA assay, which measured the methylation of MEKK2 by SMYD3 [229]. EPZ031686 competitively inhibits SAM with Ki = 4.7 nM and non-competitively inhibits MEKK2 with a 1.3 affinity. After oral administration of 50 mg/kg of EPZ031686 to mice, plasma concentration remained above IC50 for more than 12 h, highlighting its possible efficacy in future in vivo models [229]. EPZ030456 could not be tested in vivo because of its low solubility [229]. Structure–activity relationship studies of SMYD3 inhibitors have led to identification of the isoxazole sulfonamide series exemplified by EPZ028862 [230] and this compound inhibited SMYD3 with a biochemical IC50 of 1.80 ± 0.06 nM in the SPA assay. Its mechanism of action is mixed-type inhibition of SAM and non-competitive inhibition of MAP3K2. Moreover, EPZ028862 showed selectivity for SMYD3 in an assay using a panel of 16 methyltransferase enzymes [230]. GSK2807 (Ki = 14 nM), a potent and selective SAM competition inhibitor, inhibits the activity of SMYD3 by forming a dead-end ternary complex of SMYD3, GSK2807, and MEKK2 [231]. BAY-6035 is a selective and substrate-competitive SMYD3 inhibitor which was discovered through scientific collaboration between the Structural Genomics Consortium (SCG) and Bayer AG (https://www.thesgc.org/chemical-probes/BAY-6035 (accessed on 24 June 2021)), although the corresponding publication has not been published thus far. BAY-6035 showed nanomolar IC50 values in vitro and in cell-based assays (88 and 70 nM, respectively) for the methylation of MEKK2. Compound 29, a novel class of tetrahydroacridine compounds, exhibited high potency by irreversibly inhibiting SMYD3 enzymatic activity (IC50 = 0.0117 ± 0.0109 μM in the SPA assay) and showed antiproliferative activity against HepG2 cells in 3D cell culture (GI50 = 1.04 μM [232]).
4.4. Other Inhibitors
SET7/9 has been shown to have a very broad target specificity in vitro, including transcriptional regulators such as TAF10, p53, ER, p65, STAT3, Rb, MYPT, Tat, and FOXO3. Based on its properties, SET7/9 may be involved in various molecular pathways related to metabolism, inflammation, and cancer. (R)-PFI-2 is a first-in-class, potent (Ki app = 0.33 nM), selective, and cell-active inhibitor of SET7/9 with an IC50 value of 2.0 ± 0.2 nM [233]. (R)-PFI-2 inhibits SETD7 activity as a cofactor-dependent substrate competition inhibitory mechanism by occupying the substrate peptide-binding groove of SETD7 and direct contact with the donor methyl group of the cofactor. The binding of (R) -PFI-2 to endogenous SETD7 has also been confirmed in MCF7 cells, and the role of SETD7 in the signaling of the Hippo pathway that controls cell growth and organ size has been elucidated. Although anticancer studies using this compound have not been reported thus far, it provides a useful tool that can be used in future studies to confirm the roles of SETD7 and its potential as a target protein for drug development.
PRDM9 is a PR domain-containing protein that trimethylates histone 3 on lysine 4 and 36. PRDM9 is involved in the expression of meiosis-specific cancer/testis genes [236]. There is increasing evidence that PRDM9 may be involved in oncogenesis and/or cancer evolution [237]. MRK-740 is a potent (IC50 = 80 ± 16 nM) selective PRDM9 inhibitor, and its activity has also been demonstrated in cell-based assays. It binds to the substrate-binding pocket and acts as a SAM-dependent substrate competition inhibitor with an abnormally broad interaction with the cofactor S-adenosylmethionine (SAM). In cells, MRK-740 specifically and directly inhibits H3K4 methylation at endogenous PRDM9 target loci [238]. However, MRK-740 treatment does not affect the proliferation of multiple myeloma lines and breast cancer cell lines, and its usefulness as a target molecule in cancer is still uncertain.
4.5. Protein Degraders
Protein silencing has often been used to investigate protein function. Several types of protein silencing methodologies have been developed for therapeutic purposes, including targeting coding genes, transcripts, and translation processes. Direct degradation of the target protein can overcome some of the limitations of previously described methods. The first cases of PROTAC compounds targeting BET family proteins were reported by several groups in 2015 [239,240,241], and a large number of small-molecule proteolytic agents are currently under development.
Although slow compared with other fields, the development of PROTAC, which regulates the function of H3K4 methyltransferase, is also drawing attention. One of the important subunits of the KMT2 complex, WDR5, has a WD-repeat domain that mediates protein interactions. The primary aim of the researcher is to design PROTAC to induce selective degradation of WDR5 using OICR-9429, a low-molecular substance capable of pharmacologically targeting WDR5, hence confirming whether the proliferation of cancer cells is inhibited by destroying the KMT2 complex in cancer cells. Although specific PROTAC design and intracellular activity have not been reported thus far, interesting results are expected in the future, considering the role of WDR5 in H3K4 methyltransferase activity.
5. Conclusions
Anticancer drug resistance is one of the major challenges that needs to be solved, although the mechanisms involved are very complex. Extensive evidence generated over the past decade shows that H3K4 methyltransferases and demethylases mediate the actions of several genes associated with drug resistance. Therefore, targeting H3K4 methyltransferase is expected to exert a synergistic effect, either on its own or in combination with other drugs, to overcome drug resistance in leukemia and various solid cancers. The majority of research supports that H3K4 methyltransferases promote drug resistance, although there is some evidence suggesting their suppressive effect in developing cancers. H3K4 methyltransferase function generally requires enzyme activity. However, non-enzymatic roles have also been reported. Additionally, several members of the H3K4 methyltransferase family have been well identified. Nevertheless, studies on the detailed mechanisms of action of these proteins are limited, and further exploration is required. Importantly, limited inhibitors specifically targeting H3K4 methyltransferase may be clinically available in the near future. However, efforts to design or optimize clinically potent H3K4 methyltransferase inhibitors targeting enzyme activity, binding activity with other factors, or protein stability, must continue. Finally, we need to generate conclusive evidence of the contribution of HMT to drug resistance. These efforts, combined with current treatments for drug-resistant cancers, could provide more treatment options.
Author Contributions
L.Y., M.J. and K.W.J. conceptualized and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2020R1A6A1A03043708, 2020R1I1A1A01051942, and 2017R1D1A1B03031165).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Dupont C., Armant D.R., Brenner C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009;27:351–357. doi: 10.1055/s-0029-1237423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger S.L., Kouzarides T., Shiekhattar R., Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazzio E.A., Soliman K.F. Basic concepts of epigenetics: Impact of environmental signals on gene expression. Epigenetics. 2012;7:119–130. doi: 10.4161/epi.7.2.18764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannister A.J., Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowman G.D., Poirier M.G. Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 2015;115:2274–2295. doi: 10.1021/cr500350x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCabe M.T., Mohammad H.P., Barbash O., Kruger R.G. Targeting histone methylation in cancer. Cancer J. 2017;23:292–301. doi: 10.1097/PPO.0000000000000283. [DOI] [PubMed] [Google Scholar]
- 7.Yang C., Zhang J., Ma Y., Wu C., Cui W., Wang L. Histone methyltransferase and drug resistance in cancers. J. Exp. Clin. Cancer Res. 2020;39:173. doi: 10.1186/s13046-020-01682-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y., Ren B., Yang J., Wang H., Yang G., Xu R., You L., Zhao Y. The role of histone methylation in the development of digestive cancers: A potential direction for cancer management. Signal Transduct. Target. Ther. 2020;5:143. doi: 10.1038/s41392-020-00252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seligson D.B., Horvath S., McBrian M.A., Mah V., Yu H., Tze S., Wang Q., Chia D., Goodglick L., Kurdistani S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009;174:1619–1628. doi: 10.2353/ajpath.2009.080874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawan C., Herceg Z. Histone modifications and cancer. Adv. Genet. 2010;70:57–85. doi: 10.1016/B978-0-12-380866-0.60003-4. [DOI] [PubMed] [Google Scholar]
- 11.Allfrey V.G., Mirsky A.E. Structural modifications of histones and their possible role in the regulation of RNA synthesis. Science. 1964;144:559. doi: 10.1126/science.144.3618.559. [DOI] [PubMed] [Google Scholar]
- 12.Murray K. The occurrence of iε-N-methyl lysine in histones. Biochemistry. 1964;3:10–15. doi: 10.1021/bi00889a003. [DOI] [PubMed] [Google Scholar]
- 13.Schurter B.T., Koh S.S., Chen D., Bunick G.J., Harp J.M., Hanson B.L., Henschen-Edman A., Mackay D.R., Stallcup M.R., Aswad D.W. Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry. 2001;40:5747–5756. doi: 10.1021/bi002631b. [DOI] [PubMed] [Google Scholar]
- 14.Guccione E., Bassi C., Casadio F., Martinato F., Cesaroni M., Schuchlautz H., Luscher B., Amati B. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449:933–937. doi: 10.1038/nature06166. [DOI] [PubMed] [Google Scholar]
- 15.Hyllus D., Stein C., Schnabel K., Schiltz E., Imhof A., Dou Y., Hsieh J., Bauer U.M. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007;21:3369–3380. doi: 10.1101/gad.447007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iberg A.N., Espejo A., Cheng D., Kim D., Michaud-Levesque J., Richard S., Bedford M.T. Arginine methylation of the histone H3 tail impedes effector binding. J. Biol. Chem. 2008;283:3006–3010. doi: 10.1074/jbc.C700192200. [DOI] [PubMed] [Google Scholar]
- 17.Wang H., Huang Z.Q., Xia L., Feng Q., Erdjument-Bromage H., Strahl B.D., Briggs S.D., Allis C.D., Wong J., Tempst P., et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- 18.Pal S., Vishwanath S.N., Erdjument-Bromage H., Tempst P., Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004;24:9630–9645. doi: 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Lorenzo A., Bedford M.T. Histone arginine methylation. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Black J.C., Van Rechem C., Whetstine J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohan M., Herz H.M., Shilatifard A. SnapShot: Histone lysine methylase complexes. Cell. 2012;149:498–498.e1. doi: 10.1016/j.cell.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shilatifard A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shinkai Y., Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner E.J., Carpenter P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steger D.J., Lefterova M.I., Ying L., Stonestrom A.J., Schupp M., Zhuo D., Vakoc A.L., Kim J.E., Chen J., Lazar M.A., et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jorgensen S., Schotta G., Sorensen C.S. Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013;41:2797–2806. doi: 10.1093/nar/gkt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morera L., Lübbert M., Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenet. 2016;8:57. doi: 10.1186/s13148-016-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai C., So C. Epigenetic therapies by targeting aberrant histone methylome in AML: Molecular mechanisms, current preclinical and clinical development. Oncogene. 2017;36:1753–1759. doi: 10.1038/onc.2016.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song Y., Wu F., Wu J. Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 2016;9:49. doi: 10.1186/s13045-016-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCabe N.R., Burnett R.C., Gill H.J., Thirman M.J., Mbangkollo D., Kipiniak M., van Melle E., Ziemin-van der Poel S., Rowley J.D., Diaz M.O. Cloning of cDNAs of the MLL gene that detect DNA rearrangements and altered RNA transcripts in human leukemic cells with 11q23 translocations. Proc. Natl. Acad. Sci. USA. 1992;89:11794–11798. doi: 10.1073/pnas.89.24.11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tkachuk D.C., Kohler S., Cleary M.L. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71:691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 33.Zeisig B.B., Milne T., García-Cuéllar M.P., Schreiner S., Martin M.E., Fuchs U., Borkhardt A., Chanda S.K., Walker J., Soden R., et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol. Cell. Biol. 2004;24:617–628. doi: 10.1128/MCB.24.2.617-628.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dou Y., Milne T.A., Ruthenburg A.J., Lee S., Lee J.W., Verdine G.L., Allis C.D., Roeder R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi Y.H., Westfield G.H., Oleskie A.N., Trievel R.C., Shilatifard A., Skiniotis G. Structural analysis of the core COMPASS family of histone H3K4 methylases from yeast to human. Proc. Natl. Acad. Sci. USA. 2011;108:20526–20531. doi: 10.1073/pnas.1109360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Southall S.M., Wong P.S., Odho Z., Roe S.M., Wilson J.R. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol. Cell. 2009;33:181–191. doi: 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 37.Patel A., Dharmarajan V., Cosgrove M.S. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J. Biol. Chem. 2008;283:32158–32161. doi: 10.1074/jbc.C800164200. [DOI] [PubMed] [Google Scholar]
- 38.Shinsky S.A., Hu M., Vought V.E., Ng S.B., Bamshad M.J., Shendure J., Cosgrove M.S. A non-active-site SET domain surface crucial for the interaction of MLL1 and the RbBP5/Ash2L heterodimer within MLL family core complexes. J. Mol. Biol. 2014;426:2283–2299. doi: 10.1016/j.jmb.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao F., Chen Y., Cierpicki T., Liu Y., Basrur V., Lei M., Dou Y. An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PLoS ONE. 2010;5:e14102. doi: 10.1371/journal.pone.0014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Nuland R., Smits A.H., Pallaki P., Jansen P.W., Vermeulen M., Timmers H.M. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol. Cell. Biol. 2013;33:2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanchez R., Zhou M.-M. The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 2011;36:364–372. doi: 10.1016/j.tibs.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ayton P.M., Chen E.H., Cleary M.L. Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol. Cell. Biol. 2004;24:10470–10478. doi: 10.1128/MCB.24.23.10470-10478.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schlichter A., Cairns B.R. Histone trimethylation by Set1 is coordinated by the RRM, autoinhibitory, and catalytic domains. EMBO J. 2005;24:1222–1231. doi: 10.1038/sj.emboj.7600607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crump N.T., Milne T.A. Why are so many MLL lysine methyltransferases required for normal mammalian development? Cell. Mol. Life Sci. 2019;76:2885–2898. doi: 10.1007/s00018-019-03143-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patel A., Vought V.E., Dharmarajan V., Cosgrove M.S. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J. Biol. Chem. 2008;283:32162–32175. doi: 10.1074/jbc.M806317200. [DOI] [PubMed] [Google Scholar]
- 46.Wu L., Lee S.Y., Zhou B., Nguyen U.T., Muir T.W., Tan S., Dou Y. ASH2L regulates ubiquitylation signaling to MLL: Trans-regulation of H3 K4 methylation in higher eukaryotes. Mol. Cell. 2013;49:1108–1120. doi: 10.1016/j.molcel.2013.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu D., Gao X., Morgan M.A., Herz H.-M., Smith E.R., Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol. 2013;33:4745–4754. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shinsky S.A., Monteith K.E., Viggiano S., Cosgrove M.S. Biochemical reconstitution and phylogenetic comparison of human SET1 family core complexes involved in histone methylation. J. Biol. Chem. 2015;290:6361–6375. doi: 10.1074/jbc.M114.627646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shilatifard A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 50.Wang P., Lin C., Smith E.R., Guo H., Sanderson B.W., Wu M., Gogol M., Alexander T., Seidel C., Wiedemann L.M., et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu D., Garruss A.S., Gao X., Morgan M.A., Cook M., Smith E.R., Shilatifard A. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1093–1097. doi: 10.1038/nsmb.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee J.E., Wang C., Xu S., Cho Y.W., Wang L., Feng X., Baldridge A., Sartorelli V., Zhuang L., Peng W., et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hallson G., Hollebakken R.E., Li T., Syrzycka M., Kim I., Cotsworth S., Fitzpatrick K.A., Sinclair D.A., Honda B.M. dSet1 is the main H3K4 di- and tri-methyltransferase throughout Drosophila development. Genetics. 2012;190:91–100. doi: 10.1534/genetics.111.135863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu M., Wang P.F., Lee J.S., Martin-Brown S., Florens L., Washburn M., Shilatifard A. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell. Biol. 2008;28:7337–7344. doi: 10.1128/MCB.00976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaikkonen M.U., Spann N.J., Heinz S., Romanoski C.E., Allison K.A., Stender J.D., Chun H.B., Tough D.F., Prinjha R.K., Benner C. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guenther M.G., Jenner R.G., Chevalier B., Nakamura T., Croce C.M., Canaani E., Young R.A. Global and Hox-specific roles for the MLL1 methyltransferase. Proc. Natl. Acad. Sci. USA. 2005;102:8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng C., Li Y., Liang S., Cui K., Salz T., Yang H., Tang Z., Gallagher P.G., Qiu Y., Roeder R. USF1 and hSET1A mediated epigenetic modifications regulate lineage differentiation and HoxB4 transcription. PLoS Genet. 2013;9:e1003524. doi: 10.1371/journal.pgen.1003524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hughes C.M., Rozenblatt-Rosen O., Milne T.A., Copeland T.D., Levine S.S., Lee J.C., Hayes D.N., Shanmugam K.S., Bhattacharjee A., Biondi C.A. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell. 2004;13:587–597. doi: 10.1016/S1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 59.Murai M.J., Pollock J., He S., Miao H., Purohit T., Yokom A., Hess J.L., Muntean A.G., Grembecka J., Cierpicki T. The same site on the integrase-binding domain of lens epithelium–derived growth factor is a therapeutic target for MLL leukemia and HIV. Blood J. Am. Soc. Hematol. 2014;124:3730–3737. doi: 10.1182/blood-2014-01-550079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yokoyama A., Cleary M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cho Y.-W., Hong T., Hong S., Guo H., Yu H., Kim D., Guszczynski T., Dressler G.R., Copeland T.D., Kalkum M. PTIP associates with MLL3-and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007;282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sirinupong N., Brunzelle J., Ye J., Pirzada A., Nico L., Yang Z. Crystal structure of cardiac-specific histone methyltransferase SmyD1 reveals unusual active site architecture. J. Biol. Chem. 2010;285:40635–40644. doi: 10.1074/jbc.M110.168187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abu-Farha M., Lambert J.-P., Al-Madhoun A.S., Elisma F., Skerjanc I.S., Figeys D. The tale of two domains: Proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol. Cell. Proteom. 2008;7:560–572. doi: 10.1074/mcp.M700271-MCP200. [DOI] [PubMed] [Google Scholar]
- 64.Hamamoto R., Furukawa Y., Morita M., Iimura Y., Silva F.P., Li M., Yagyu R., Nakamura Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004;6:731–740. doi: 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- 65.Spellmon N., Holcomb J., Trescott L., Sirinupong N., Yang Z. Structure and function of SET and MYND domain-containing proteins. Int. J. Mol. Sci. 2015;16:1406–1428. doi: 10.3390/ijms16011406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Foreman K.W., Brown M., Park F., Emtage S., Harriss J., Das C., Zhu L., Crew A., Arnold L., Shaaban S. Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS ONE. 2011;6:e22290. doi: 10.1371/journal.pone.0022290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gottlieb P.D., Pierce S.A., Sims R.J., Yamagishi H., Weihe E.K., Harriss J.V., Maika S.D., Kuziel W.A., King H.L., Olson E.N. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat. Genet. 2002;31:25–32. doi: 10.1038/ng866. [DOI] [PubMed] [Google Scholar]
- 68.Sims R.J., 3rd, Weihe E.K., Zhu L., O’Malley S., Harriss J.V., Gottlieb P.D. m-Bop, a repressor protein essential for cardiogenesis, interacts with skNAC, a heart- and muscle-specific transcription factor. J. Biol. Chem. 2002;277:26524–26529. doi: 10.1074/jbc.M204121200. [DOI] [PubMed] [Google Scholar]
- 69.Tan X., Rotllant J., Li H., DeDeyne P., Du S.J. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc. Natl. Acad. Sci. USA. 2006;103:2713–2718. doi: 10.1073/pnas.0509503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berkholz J., Orgeur M., Stricker S., Munz B. skNAC and Smyd1 in transcriptional control. Exp. Cell Res. 2015;336:182–191. doi: 10.1016/j.yexcr.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 71.Brown M.A., Sims R.J., Gottlieb P.D., Tucker P.W. Identification and characterization of Smyd2: A split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer. 2006;5:26. doi: 10.1186/1476-4598-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang J., Perez-Burgos L., Placek B.J., Sengupta R., Richter M., Dorsey J.A., Kubicek S., Opravil S., Jenuwein T., Berger S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 73.Wu J., Cheung T., Grande C., Ferguson A.D., Zhu X., Theriault K., Code E., Birr C., Keen N., Chen H. Biochemical characterization of human SET and MYND domain-containing protein 2 methyltransferase. Biochemistry. 2011;50:6488–6497. doi: 10.1021/bi200725p. [DOI] [PubMed] [Google Scholar]
- 74.Saddic L.A., West L.E., Aslanian A., Yates J.R., Rubin S.M., Gozani O., Sage J. Methylation of the retinoblastoma tumor suppressor by SMYD2. J. Biol. Chem. 2010;285:37733–37740. doi: 10.1074/jbc.M110.137612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van Aller G.S., Reynoird N., Barbash O., Huddleston M., Liu S., Zmoos A.-F., McDevitt P., Sinnamon R., Le B., Mas G. Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics. 2012;7:340–343. doi: 10.4161/epi.19506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kunizaki M., Hamamoto R., Silva F.P., Yamaguchi K., Nagayasu T., Shibuya M., Nakamura Y., Furukawa Y. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 2007;67:10759–10765. doi: 10.1158/0008-5472.CAN-07-1132. [DOI] [PubMed] [Google Scholar]
- 77.Wang H., Cao R., Xia L., Erdjument-Bromage H., Borchers C., Tempst P., Zhang Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell. 2001;8:1207–1217. doi: 10.1016/S1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- 78.Xiao B., Jing C., Wilson J.R., Walker P.A., Vasisht N., Kelly G., Howell S., Taylor I.A., Blackburn G.M., Gamblin S.J. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature. 2003;421:652–656. doi: 10.1038/nature01378. [DOI] [PubMed] [Google Scholar]
- 79.Nishioka K., Chuikov S., Sarma K., Erdjument-Bromage H., Allis C.D., Tempst P., Reinberg D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kurash J.K., Lei H., Shen Q., Marston W.L., Granda B.W., Fan H., Wall D., Li E., Gaudet F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell. 2008;29:392–400. doi: 10.1016/j.molcel.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 81.Estève P.-O., Chin H.G., Benner J., Feehery G.R., Samaranayake M., Horwitz G.A., Jacobsen S.E., Pradhan S. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. USA. 2009;106:5076–5081. doi: 10.1073/pnas.0810362106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kontaki H., Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol. Cell. 2010;39:152–160. doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 83.Liu X., Chen Z., Xu C., Leng X., Cao H., Ouyang G., Xiao W. Repression of hypoxia-inducible factor α signaling by Set7-mediated methylation. Nucleic Acids Res. 2015;43:5081–5098. doi: 10.1093/nar/gkv379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oudhoff M.J., Braam M.J., Freeman S.A., Wong D., Rattray D.G., Wang J., Antignano F., Snyder K., Refaeli I., Hughes M.R. SETD7 controls intestinal regeneration and tumorigenesis by regulating Wnt/β-catenin and Hippo/YAP signaling. Dev. Cell. 2016;37:47–57. doi: 10.1016/j.devcel.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 85.Hayashi K., Yoshida K., Matsui Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature. 2005;438:374–378. doi: 10.1038/nature04112. [DOI] [PubMed] [Google Scholar]
- 86.Eram M.S., Bustos S.P., Lima-Fernandes E., Siarheyeva A., Senisterra G., Hajian T., Chau I., Duan S., Wu H., Dombrovski L., et al. Trimethylation of histone H3 lysine 36 by human methyltransferase PRDM9 protein. J. Biol. Chem. 2014;289:12177–12188. doi: 10.1074/jbc.M113.523183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Powers N.R., Parvanov E.D., Baker C.L., Walker M., Petkov P.M., Paigen K. The Meiotic Recombination Activator PRDM9 Trimethylates Both H3K36 and H3K4 at Recombination Hotspots In Vivo. PLoS Genet. 2016;12:e1006146. doi: 10.1371/journal.pgen.1006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu H., Mathioudakis N., Diagouraga B., Dong A., Dombrovski L., Baudat F., Cusack S., de Massy B., Kadlec J. Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep. 2013;5:13–20. doi: 10.1016/j.celrep.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 89.Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A., et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 91.Kim T., Buratowski S. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5′ transcribed regions. Cell. 2009;137:259–272. doi: 10.1016/j.cell.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 93.Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Santos-Rosa H., Schneider R., Bannister A.J., Sherriff J., Bernstein B.E., Emre N.C., Schreiber S.L., Mellor J., Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 95.Biswas S., Rao C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018;837:8–24. doi: 10.1016/j.ejphar.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 96.Wysocka J., Swigut T., Xiao H., Milne T.A., Kwon S.Y., Landry J., Kauer M., Tackett A.J., Chait B.T., Badenhorst P. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 97.Lauberth S.M., Nakayama T., Wu X., Ferris A.L., Tang Z., Hughes S.H., Roeder R.G. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu C., Fan X., Sha Q.-Q., Wang H.-H., Li B.-T., Dai X.-X., Shen L., Liu J., Wang L., Liu K. CFP1 regulates histone H3K4 trimethylation and developmental potential in mouse oocytes. Cell Rep. 2017;20:1161–1172. doi: 10.1016/j.celrep.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 99.Wen H., Li J., Song T., Lu M., Kan P.-Y., Lee M.G., Sha B., Shi X. Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J. Biol. Chem. 2010;285:9322–9326. doi: 10.1074/jbc.C109.097667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang W., Chen Z., Mao Z., Zhang H., Ding X., Chen S., Zhang X., Xu R., Zhu B. Nucleolar protein Spindlin1 recognizes H3K4 methylation and stimulates the expression of rRNA genes. EMBO Rep. 2011;12:1160–1166. doi: 10.1038/embor.2011.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gough S.M., Lee F., Yang F., Walker R.L., Zhu Y.J., Pineda M., Onozawa M., Chung Y.J., Bilke S., Wagner E.K., et al. NUP98-PHF23 is a chromatin modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer Discov. 2014;4:564–577. doi: 10.1158/2159-8290.CD-13-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cantù C., Valenta T., Hausmann G., Vilain N., Aguet M., Basler K. The Pygo2-H3K4me2/3 interaction is dispensable for mouse development and Wnt signaling-dependent transcription. Development. 2013;140:2377–2386. doi: 10.1242/dev.093591. [DOI] [PubMed] [Google Scholar]
- 103.Sims R.J., III, Chen C.-F., Santos-Rosa H., Kouzarides T., Patel S.S., Reinberg D. Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J. Biol. Chem. 2005;280:41789–41792. doi: 10.1074/jbc.C500395200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jeong K.W., Kim K., Situ A.J., Ulmer T.S., An W., Stallcup M.R. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat. Struct. Mol. Biol. 2011;18:1358–1365. doi: 10.1038/nsmb.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bian C., Xu C., Ruan J., Lee K.K., Burke T.L., Tempel W., Barsyte D., Li J., Wu M., Zhou B.O. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. 2011;30:2829–2842. doi: 10.1038/emboj.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hung T., Binda O., Champagne K.S., Kuo A.J., Johnson K., Chang H.Y., Simon M.D., Kutateladze T.G., Gozani O. ING4 mediates crosstalk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol. Cell. 2009;33:248–256. doi: 10.1016/j.molcel.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wilting R.H., Dannenberg J.-H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updates. 2012;15:21–38. doi: 10.1016/j.drup.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 108.Mo R., Rao S.M., Zhu Y.-J. Identification of the MLL2 complex as a coactivator for estrogen receptor α. J. Biol. Chem. 2006;281:15714–15720. doi: 10.1074/jbc.M513245200. [DOI] [PubMed] [Google Scholar]
- 109.Natarajan T.G., Kallakury B.V., Sheehan C.E., Bartlett M.B., Ganesan N., Preet A., Ross J.S., FitzGerald K.T. Epigenetic regulator MLL2 shows altered expression in cancer cell lines and tumors from human breast and colon. Cancer Cell Int. 2010;10:13. doi: 10.1186/1475-2867-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gala K., Li Q., Sinha A., Razavi P., Dorso M., Sanchez-Vega F., Chung Y.R., Hendrickson R., Hsieh J.J., Berger M. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERα enhancer function. Oncogene. 2018;37:4692–4710. doi: 10.1038/s41388-018-0273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim S.-S., Lee M.-H., Lee M.-O. Histone methyltransferases regulate the transcriptional expression of ERα and the proliferation of tamoxifen-resistant breast cancer cells. Breast Cancer Res. Treat. 2020;180:45–54. doi: 10.1007/s10549-019-05517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim J.-H., Sharma A., Dhar S.S., Lee S.-H., Gu B., Chan C.-H., Lin H.-K., Lee M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014;74:1705–1717. doi: 10.1158/0008-5472.CAN-13-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Salz T., Deng C., Pampo C., Siemann D., Qiu Y., Brown K., Huang S. Histone methyltransferase hSETD1A is a novel regulator of metastasis in breast cancer. Mol. Cancer Res. 2015;13:461–469. doi: 10.1158/1541-7786.MCR-14-0389. [DOI] [PubMed] [Google Scholar]
- 114.Tajima K., Matsuda S., Yae T., Drapkin B.J., Morris R., Boukhali M., Niederhoffer K., Comaills V., Dubash T., Nieman L. SETD1A protects from senescence through regulation of the mitotic gene expression program. Nat. Commun. 2019;10:2854. doi: 10.1038/s41467-019-10786-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang S.P., Tang Z., Chen C.W., Shimada M., Koche R.P., Wang L.H., Nakadai T., Chramiec A., Krivtsov A.V., Armstrong S.A., et al. A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Mol. Cell. 2017;67:308–321.e6. doi: 10.1016/j.molcel.2017.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang L., Collings C.K., Zhao Z., Cozzolino K.A., Ma Q., Liang K., Marshall S.A., Sze C.C., Hashizume R., Savas J.N., et al. A cytoplasmic COMPASS is necessary for cell survival and triple-negative breast cancer pathogenesis by regulating metabolism. Genes Dev. 2017;31:2056–2066. doi: 10.1101/gad.306092.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li L.X., Zhou J.X., Calvet J.P., Godwin A.K., Jensen R.A., Li X. Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis. 2018;9:326. doi: 10.1038/s41419-018-0347-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jin M.L., Kim Y.W., Jin H.L., Kang H., Lee E.K., Stallcup M.R., Jeong K.W. Aberrant expression of SETD1A promotes survival and migration of estrogen receptor α-positive breast cancer cells. Int. J. Cancer. 2018;143:2871–2883. doi: 10.1002/ijc.31853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fenizia C., Bottino C., Corbetta S., Fittipaldi R., Floris P., Gaudenzi G., Carra S., Cotelli F., Vitale G., Caretti G. SMYD3 promotes the epithelial–mesenchymal transition in breast cancer. Nucleic Acids Res. 2019;47:1278–1293. doi: 10.1093/nar/gky1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hamamoto R., Silva F.P., Tsuge M., Nishidate T., Katagiri T., Nakamura Y., Furukawa Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006;97:113–118. doi: 10.1111/j.1349-7006.2006.00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang L., Xu M.L., Wang C., Dong Q.Q., Miao Z., Chen X.Y., Wang N., He H.P., Zhang T.C., Luo X.G. SET and MYND domain-containing protein 3 inhibits tumor cell sensitivity to cisplatin. Oncol. Lett. 2020;19:3469–3476. doi: 10.3892/ol.2020.11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Si W., Zhou J., Zhao Y., Zheng J., Cui L. SET7/9 promotes multiple malignant processes in breast cancer development via RUNX2 activation and is negatively regulated by TRIM21. Cell Death Dis. 2020;11:151. doi: 10.1038/s41419-020-2350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang R., Li X., Yu Y., Ma L., Liu S., Zong X., Zheng Q. SETD7 is a prognosis predicting factor of breast cancer and regulates redox homeostasis. Oncotarget. 2017;8:94080. doi: 10.18632/oncotarget.21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Subramanian K., Jia D., Kapoor-Vazirani P., Powell D.R., Collins R.E., Sharma D., Peng J., Cheng X., Vertino P.M. Regulation of estrogen receptor α by the SET7 lysine methyltransferase. Mol. Cell. 2008;30:336–347. doi: 10.1016/j.molcel.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Montenegro M., Sánchez-Del-Campo L., González-Guerrero R., Martínez-Barba E., Piñero-Madrona A., Cabezas-Herrera J., Rodríguez-López J. Tumor suppressor SET9 guides the epigenetic plasticity of breast cancer cells and serves as an early-stage biomarker for predicting metastasis. Oncogene. 2016;35:6143–6152. doi: 10.1038/onc.2016.154. [DOI] [PubMed] [Google Scholar]
- 127.Larsson C., Cordeddu L., Siggens L., Pandzic T., Kundu S., He L., Ali M.A., Pristovšek N., Hartman K., Ekwall K. Restoration of KMT2C/MLL3 in human colorectal cancer cells reinforces genome-wide H3K4me1 profiles and influences cell growth and gene expression. Clin. Epigenet. 2020;12:1–12. doi: 10.1186/s13148-020-00863-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee J., Kim D.H., Lee S., Yang Q.H., Lee D.K., Lee S.K., Roeder R.G., Lee J.W. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. USA. 2009;106:8513–8518. doi: 10.1073/pnas.0902873106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang S.-L., Du X., Tan L.-N., Deng F.-H., Zhou B.-Y., Zhou H.-J., Zhu H.-Y., Chu Y., Liu D.-L., Tan Y.-Y. SET7 interacts with HDAC6 and suppresses the development of colon cancer through inactivation of HDAC6. Am. J. Transl. Res. 2020;12:602. [PMC free article] [PubMed] [Google Scholar]
- 130.Rahnamoun H., Hong J., Sun Z., Lee J., Lu H., Lauberth S.M. Mutant p53 regulates enhancer-associated H3K4 monomethylation through interactions with the methyltransferase MLL4. J. Biol. Chem. 2018;293:13234–13246. doi: 10.1074/jbc.RA118.003387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Salz T., Li G., Kaye F., Zhou L., Qiu Y., Huang S. hSETD1A regulates Wnt target genes and controls tumor growth of colorectal cancer cells. Cancer Res. 2014;74:775–786. doi: 10.1158/0008-5472.CAN-13-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fang L., Teng H., Wang Y., Liao G., Weng L., Li Y., Wang X., Jin J., Jiao C., Chen L., et al. SET1A-Mediated Mono-Methylation at K342 Regulates YAP Activation by Blocking Its Nuclear Export and Promotes Tumorigenesis. Cancer Cell. 2018;34:103–118.e9. doi: 10.1016/j.ccell.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 133.Choi Y.J., Oh H.R., Choi M.R., Gwak M., An C.H., Chung Y.J., Yoo N.J., Lee S.H. Frameshift mutation of a histone methylation-related gene SETD1B and its regional heterogeneity in gastric and colorectal cancers with high microsatellite instability. Hum. Pathol. 2014;45:1674–1681. doi: 10.1016/j.humpath.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 134.Ren H., Wang Z., Chen Y., Liu Y., Zhang S., Zhang T., Li Y. SMYD2-OE promotes oxaliplatin resistance in colon cancer through MDR1/P-glycoprotein via MEK/ERK/AP1 pathway. OncoTargets Ther. 2019;12:2585. doi: 10.2147/OTT.S186806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Malik R., Khan A.P., Asangani I.A., Cieslik M., Prensner J.R., Wang X., Iyer M.K., Jiang X., Borkin D., Escara-Wilke J., et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat. Med. 2015;21:344–352. doi: 10.1038/nm.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lv S., Ji L., Chen B., Liu S., Lei C., Liu X., Qi X., Wang Y., Leung E.L.-H., Wang H. Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene. 2018;37:1354–1368. doi: 10.1038/s41388-017-0026-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lv S., Wen H., Shan X., Li J., Wu Y., Yu X., Huang W., Wei Q. Loss of KMT2D induces prostate cancer ROS-mediated DNA damage by suppressing the enhancer activity and DNA binding of antioxidant transcription factor FOXO3. Epigenetics. 2019;14:1194–1208. doi: 10.1080/15592294.2019.1634985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vieira F.Q., Costa-Pinheiro P., Almeida-Rios D., Graça I., Monteiro-Reis S., Simões-Sousa S., Carneiro I., Sousa E.J., Godinho M.I., Baltazar F. SMYD3 contributes to a more aggressive phenotype of prostate cancer and targets Cyclin D2 through H4K20me3. Oncotarget. 2015;6:13644. doi: 10.18632/oncotarget.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu C., Wang C., Wang K., Liu L., Shen Q., Yan K., Sun X., Chen J., Liu J., Ren H. SMYD3 as an oncogenic driver in prostate cancer by stimulation of androgen receptor transcription. J. Natl. Cancer Inst. 2013;105:1719–1728. doi: 10.1093/jnci/djt304. [DOI] [PubMed] [Google Scholar]
- 140.Gaughan L., Stockley J., Wang N., McCracken S.R., Treumann A., Armstrong K., Shaheen F., Watt K., McEwan I.J., Wang C. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011;39:1266–1279. doi: 10.1093/nar/gkq861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang C., Sargsyan D., Zhang C., Wu R., Yang Y., Kong A.-N. Transcriptomic Analysis of Histone Methyltransferase Setd7 Knockdown and Phenethyl Isothiocyanate in Human Prostate Cancer Cells. Anticancer Res. 2018;38:6069–6083. doi: 10.21873/anticanres.12957. [DOI] [PubMed] [Google Scholar]
- 142.Sowalsky A.G., Xia Z., Wang L., Zhao H., Chen S., Bubley G.J., Balk S.P., Li W. Whole transcriptome sequencing reveals extensive unspliced mRNA in metastatic castration-resistant prostate cancer. Mol. Cancer Res. 2015;13:98–106. doi: 10.1158/1541-7786.MCR-14-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang L., Jin M., Park S.J., Seo S.-Y., Jeong K.W. SETD1A promotes proliferation of castration-resistant Prostate cancer cells via FOXM1 transcription. Cancers. 2020;12:1736. doi: 10.3390/cancers12071736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Djabali M., Selleri L., Parry P., Bower M., Young B.D., Evans G.A. A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat. Genet. 1992;2:113–118. doi: 10.1038/ng1092-113. [DOI] [PubMed] [Google Scholar]
- 145.Ziemin-van der Poel S., McCabe N.R., Gill H.J., Espinosa R., Patel Y., Harden A., Rubinelli P., Smith S.D., LeBeau M.M., Rowley J.D. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA. 1991;88:10735–10739. doi: 10.1073/pnas.88.23.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mohan M., Lin C., Guest E., Shilatifard A. Licensed to elongate: A molecular mechanism for MLL-based leukaemogenesis. Nat. Rev. Cancer. 2010;10:721–728. doi: 10.1038/nrc2915. [DOI] [PubMed] [Google Scholar]
- 147.Yokoyama A., Somervaille T.C., Smith K.S., Rozenblatt-Rosen O., Meyerson M., Cleary M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 148.Smith E., Lin C., Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661–672. doi: 10.1101/gad.2015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Krivtsov A.V., Feng Z., Lemieux M.E., Faber J., Vempati S., Sinha A.U., Xia X., Jesneck J., Bracken A.P., Silverman L.B., et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Okada Y., Feng Q., Lin Y., Jiang Q., Li Y., Coffield V.M., Su L., Xu G., Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 151.Thiel A.T., Blessington P., Zou T., Feather D., Wu X., Yan J., Zhang H., Liu Z., Ernst P., Koretzky G.A., et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell. 2010;17:148–159. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hoshii T., Cifani P., Feng Z., Huang C.-H., Koche R., Chen C.-W., Delaney C.D., Lowe S.W., Kentsis A., Armstrong S.A. A non-catalytic function of SETD1A regulates cyclin K and the DNA damage response. Cell. 2018;172:1007–1021.e17. doi: 10.1016/j.cell.2018.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chen Y., Anastassiadis K., Kranz A., Stewart A.F., Arndt K., Waskow C., Yokoyama A., Jones K., Neff T., Lee Y. MLL2, not MLL1, plays a major role in sustaining MLL-rearranged acute myeloid leukemia. Cancer Cell. 2017;31:755–770.e6. doi: 10.1016/j.ccell.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rabello D.D.A., Ferreira V., Berzoti-Coelho M.G., Burin S.M., Magro C.L., Cacemiro M.D.C., Simões B.P., Saldanha-Araujo F., de Castro F.A., Pittella-Silva F. MLL2/KMT2D and MLL3/KMT2C expression correlates with disease progression and response to imatinib mesylate in chronic myeloid leukemia. Cancer Cell Int. 2018;18:26. doi: 10.1186/s12935-018-0523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chen C., Liu Y., Rappaport A.R., Kitzing T., Schultz N., Zhao Z., Shroff A.S., Dickins R.A., Vakoc C.R., Bradner J.E., et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014;25:652–665. doi: 10.1016/j.ccr.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sakamoto L.H.T., de Andrade R.V., Felipe M.S.S., Motoyama A.B., Silva F.P. SMYD2 is highly expressed in pediatric acute lymphoblastic leukemia and constitutes a bad prognostic factor. Leuk. Res. 2014;38:496–502. doi: 10.1016/j.leukres.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 157.Zipin-Roitman A., Aqaqe N., Yassin M., Biechonski S., Amar M., van Delft M.F., Gan O.I., McDermott S.P., Buzina A., Ketela T., et al. SMYD2 lysine methyltransferase regulates leukemia cell growth and regeneration after genotoxic stress. Oncotarget. 2017;8:16712–16727. doi: 10.18632/oncotarget.15147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gu Y., Wang Y., Wang X., Gao L., Yu W., Dong W.F. Opposite Effects of SET7/9 on Apoptosis of Human Acute Myeloid Leukemia Cells and Lung Cancer Cells. J. Cancer. 2017;8:2069–2078. doi: 10.7150/jca.19143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Houle A.A., Gibling H., Lamaze F.C., Edgington H.A., Soave D., Fave M.-J., Agbessi M., Bruat V., Stein L.D., Awadalla P. Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res. 2018;28:1611–1620. doi: 10.1101/gr.231696.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hussin J., Sinnett D., Casals F., Idaghdour Y., Bruat V., Saillour V., Healy J., Grenier J.-C., De Malliard T., Busche S. Rare allelic forms of PRDM9 associated with childhood leukemogenesis. Genome Res. 2013;23:419–430. doi: 10.1101/gr.144188.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Xiong W., Deng Z., Tang Y., Deng Z., Li M. Downregulation of KMT2D suppresses proliferation and induces apoptosis of gastric cancer. Biochem. Biophys. Res. Commun. 2018;504:129–136. doi: 10.1016/j.bbrc.2018.08.143. [DOI] [PubMed] [Google Scholar]
- 162.Cho S.-J., Yoon C., Lee J.H., Chang K.K., Lin J.-X., Kim Y.-H., Kook M.-C., Aksoy B.A., Park D.J., Ashktorab H. KMT2C mutations in diffuse-type gastric adenocarcinoma promote epithelial-to-mesenchymal transition. Clin. Cancer Res. 2018;24:6556–6569. doi: 10.1158/1078-0432.CCR-17-1679. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 163.Wu J., Chai H., Xu X., Yu J., Gu Y. Histone methyltransferase SETD1A interacts with HIF1α to enhance glycolysis and promote cancer progression in gastric cancer. Mol. Oncol. 2020;14:1397–1409. doi: 10.1002/1878-0261.12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Komatsu S., Ichikawa D., Hirajima S., Nagata H., Nishimura Y., Kawaguchi T., Miyamae M., Okajima W., Ohashi T., Konishi H. Overexpression of SMYD2 contributes to malignant outcome in gastric cancer. Br. J. Cancer. 2015;112:357–364. doi: 10.1038/bjc.2014.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang L., Wang Q.-T., Liu Y.-P., Dong Q.-Q., Hu H.-J., Miao Z., Li S., Liu Y., Zhou H., Zhang T.-C. ATM signaling pathway is implicated in the SMYD3-mediated proliferation and migration of gastric cancer cells. J. Gastric Cancer. 2017;17:295–305. doi: 10.5230/jgc.2017.17.e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wu X., Xu Q., Chen P., Yu C., Ye L., Huang C., Li T. Effect of SMYD3 on biological behavior and H3K4 methylation in bladder cancer. Cancer Manag. Res. 2019;11:8125. doi: 10.2147/CMAR.S213885. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 167.Akiyama Y., Koda Y., Byeon S.J., Shimada S., Nishikawaji T., Sakamoto A., Chen Y., Kojima K., Kawano T., Eishi Y., et al. Reduced expression of SET7/9, a histone mono-methyltransferase, is associated with gastric cancer progression. Oncotarget. 2016;7:3966–3983. doi: 10.18632/oncotarget.6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Stavrovskaya A., Stromskaya T. Transport proteins of the ABC family and multidrug resistance of tumor cells. Biochemistry (Moscow) 2008;73:592–604. doi: 10.1134/S0006297908050118. [DOI] [PubMed] [Google Scholar]
- 169.Eckford P.D., Sharom F.J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009;109:2989–3011. doi: 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- 170.Huo H., Magro P.G., Pietsch E.C., Patel B.B., Scotto K.W. Histone methyltransferase MLL1 regulates MDR1 transcription and chemoresistance. Cancer Res. 2010;70:8726–8735. doi: 10.1158/0008-5472.CAN-10-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhu Q., Fang L., Heuberger J., Kranz A., Schipper J., Scheckenbach K., Vidal R.O., Sunaga-Franze D.Y., Müller M., Wulf-Goldenberg A. The Wnt-driven Mll1 epigenome regulates salivary gland and head and neck cancer. Cell Rep. 2019;26:415–428.e5. doi: 10.1016/j.celrep.2018.12.059. [DOI] [PubMed] [Google Scholar]
- 172.Wend P., Fang L., Zhu Q., Schipper J.H., Loddenkemper C., Kosel F., Brinkmann V., Eckert K., Hindersin S., Holland J.D., et al. Wnt/beta-catenin signalling induces MLL to create epigenetic changes in salivary gland tumours. EMBO J. 2013;32:1977–1989. doi: 10.1038/emboj.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lu C., Paschall A.V., Shi H., Savage N., Waller J.L., Sabbatini M.E., Oberlies N.H., Pearce C., Liu K. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J. Natl. Cancer Inst. 2017;109:djw283. doi: 10.1093/jnci/djw283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dawkins J.B., Wang J., Maniati E., Heward J.A., Koniali L., Kocher H.M., Martin S.A., Chelala C., Balkwill F.R., Fitzgibbon J. Reduced expression of histone methyltransferases KMT2C and KMT2D correlates with improved outcome in pancreatic ductal adenocarcinoma. Cancer Res. 2016;76:4861–4871. doi: 10.1158/0008-5472.CAN-16-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Abudureheman A., Ainiwaer J., Hou Z., Niyaz M., Turghun A., Hasim A., Zhang H., Lu X., Sheyhidin I. High MLL2 expression predicts poor prognosis and promotes tumor progression by inducing EMT in esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2018;144:1025–1035. doi: 10.1007/s00432-018-2625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Augert A., Zhang Q., Bates B., Cui M., Wang X., Wildey G., Dowlati A., MacPherson D. Small cell lung cancer exhibits frequent inactivating mutations in the histone methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance) J. Thorac. Oncol. 2017;12:704–713. doi: 10.1016/j.jtho.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rampias T., Karagiannis D., Avgeris M., Polyzos A., Kokkalis A., Kanaki Z., Kousidou E., Tzetis M., Kanavakis E., Stravodimos K. The lysine-specific methyltransferase KMT 2C/MLL 3 regulates DNA repair components in cancer. EMBO Rep. 2019;20:e46821. doi: 10.15252/embr.201846821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chen X., Zhang G., Chen B., Wang Y., Guo L., Cao L., Ren C., Wen L., Liao N. Association between histone lysine methyltransferase KMT2C mutation and clinicopathological factors in breast cancer. Biomed. Pharmacother. 2019;116:108997. doi: 10.1016/j.biopha.2019.108997. [DOI] [PubMed] [Google Scholar]
- 179.Xiong W., Deng H., Huang C., Zen C., Jian C., Ye K., Zhong Z., Zhao X., Zhu L. MLL3 enhances the transcription of PD-L1 and regulates anti-tumor immunity. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:454–463. doi: 10.1016/j.bbadis.2018.10.027. [DOI] [PubMed] [Google Scholar]
- 180.Wu J., Chai H., Li F., Ren Q., Gu Y. SETD1A augments sorafenib primary resistance via activating YAP in hepatocellular carcinoma. Life Sci. 2020;260:118406. doi: 10.1016/j.lfs.2020.118406. [DOI] [PubMed] [Google Scholar]
- 181.Li T., Zheng Q., An J., Wu M., Li H., Gui X., Pu H., Lu D. SET1A Cooperates With CUDR to Promote Liver Cancer Growth and Hepatocyte-like Stem Cell Malignant Transformation Epigenetically. Mol. Ther. 2016;24:261–275. doi: 10.1038/mt.2015.208. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 182.Redd P.S., Ibrahim M.L., Klement J.D., Sharman S.K., Paschall A.V., Yang D., Nayak-Kapoor A., Liu K. SETD1B Activates iNOS Expression in Myeloid-Derived Suppressor Cells. Cancer Res. 2017;77:2834–2843. doi: 10.1158/0008-5472.CAN-16-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kumar A., Kumari N., Rai A., Singh S.K., Kakkar N., Prasad R. Expression and clinical significance of COMPASS family of histone methyltransferases in clear cell renal cell carcinoma. Gene. 2018;674:31–36. doi: 10.1016/j.gene.2018.06.063. [DOI] [PubMed] [Google Scholar]
- 184.Peveling-Oberhag J., Wolters F., Döring C., Walter D., Sellmann L., Scholtysik R., Lucioni M., Schubach M., Paulli M., Biskup S. Whole exome sequencing of microdissected splenic marginal zone lymphoma: A study to discover novel tumor-specific mutations. BMC Cancer. 2015;15:773. doi: 10.1186/s12885-015-1766-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Komatsu S., Imoto I., Tsuda H., Kozaki K.-i., Muramatsu T., Shimada Y., Aiko S., Yoshizumi Y., Ichikawa D., Otsuji E. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis. 2009;30:1139–1146. doi: 10.1093/carcin/bgp116. [DOI] [PubMed] [Google Scholar]
- 186.Zuo S.-R., Zuo X.-C., He Y., Fang W.-J., Wang C.-J., Zou H., Chen P., Huang L.-F., Huang L.-H., Xiang H. Positive expression of SMYD2 is associated with poor prognosis in patients with primary hepatocellular carcinoma. J. Cancer. 2018;9:321. doi: 10.7150/jca.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Xu W., Chen F., Fei X., Yang X., Lu X. Overexpression of SET and MYND domain-containing protein 2 (SMYD2) is associated with tumor progression and poor prognosis in patients with papillary thyroid carcinoma. Med. Sci. Monit. 2018;24:7357. doi: 10.12659/MSM.910168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shang L., Wei M. Inhibition of SMYD2 sensitized cisplatin to resistant cells in NSCLC through activating p53 pathway. Front. Oncol. 2019;9:306. doi: 10.3389/fonc.2019.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yan L., Ding B., Liu H., Zhang Y., Zeng J., Hu J., Yao W., Yu G., An R., Chen Z. Inhibition of SMYD2 suppresses tumor progression by down-regulating microRNA-125b and attenuates multi-drug resistance in renal cell carcinoma. Theranostics. 2019;9:8377. doi: 10.7150/thno.37628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Pires-Luís A.S., Vieira-Coimbra M., Vieira F.Q., Costa-Pinheiro P., Silva-Santos R., Dias P.C., Antunes L., Lobo F., Oliveira J., Gonçalves C.S. Expression of histone methyltransferases as novel biomarkers for renal cell tumor diagnosis and prognostication. Epigenetics. 2015;10:1033–1043. doi: 10.1080/15592294.2015.1103578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Oliveira-Santos W., Rabello D.A., Lucena-Araujo A.R., de Oliveira F.M., Rego E.M., Silva F.P., Saldanha-Araujo F. Residual expression of SMYD2 and SMYD3 is associated with the acquisition of complex karyotype in chronic lymphocytic leukemia. Tumor Biol. 2016;37:9473–9481. doi: 10.1007/s13277-016-4846-z. [DOI] [PubMed] [Google Scholar]
- 192.Mazur P.K., Reynoird N., Khatri P., Jansen P.W., Wilkinson A.W., Liu S., Barbash O., Van Aller G.S., Huddleston M., Dhanak D., et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014;510:283–287. doi: 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jiang Y., Lyu T., Che X., Jia N., Li Q., Feng W. Overexpression of SMYD3 in ovarian cancer is associated with ovarian cancer proliferation and apoptosis via methylating H3K4 and H4K20. J. Cancer. 2019;10:4072. doi: 10.7150/jca.29861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chen Y.-J., Tsai C.-H., Wang P.-Y., Teng S.-C. SMYD3 promotes homologous recombination via regulation of H3K4-mediated gene expression. Sci. Rep. 2017;7:3842. doi: 10.1038/s41598-017-03385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wang Y., Xie B.-h., Lin W.-h., Huang Y.-h., Ni J.-y., Hu J., Cui W., Zhou J., Shen L., Xu L.-f. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene. 2019;38:4948–4961. doi: 10.1038/s41388-019-0766-x. [DOI] [PubMed] [Google Scholar]
- 196.Li J., Zhao L., Pan Y., Ma X., Liu L., Wang W., You W. SMYD3 overexpression indicates poor prognosis and promotes cell proliferation, migration and invasion in non-small cell lung cancer. Int. J. Oncol. 2020;57:756–766. doi: 10.3892/ijo.2020.5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Liu Q., Geng H., Xue C., Beer T.M., Qian D.Z. Functional regulation of hypoxia inducible factor-1α by SET9 lysine methyltransferase. Biochim. Biophys. Acta Mol. Cell Res. 2015;1853:881–891. doi: 10.1016/j.bbamcr.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shen C., Wang D., Liu X., Gu B., Du Y., Wei F.Z., Cao L.L., Song B., Lu X., Yang Q., et al. SET7/9 regulates cancer cell proliferation by influencing β-catenin stability. FASEB J. 2015;29:4313–4323. doi: 10.1096/fj.15-273540. [DOI] [PubMed] [Google Scholar]
- 199.Chern T.R., Liu L., Petrunak E., Stuckey J.A., Wang M., Bernard D., Zhou H., Lee S., Dou Y., Wang S. Discovery of Potent Small-Molecule Inhibitors of MLL Methyltransferase. ACS Med. Chem. Lett. 2020;11:1348–1352. doi: 10.1021/acsmedchemlett.0c00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Karatas H., Townsend E.C., Cao F., Chen Y., Bernard D., Liu L., Lei M., Dou Y., Wang S. High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein–protein interaction. J. Am. Chem. Soc. 2013;135:669–682. doi: 10.1021/ja306028q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Cao F., Townsend E.C., Karatas H., Xu J., Li L., Lee S., Liu L., Chen Y., Ouillette P., Zhu J. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell. 2014;53:247–261. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Karatas H., Li Y., Liu L., Ji J., Lee S., Chen Y., Yang J., Huang L., Bernard D., Xu J. Discovery of a highly potent, cell-permeable macrocyclic peptidomimetic (MM-589) targeting the WD repeat domain 5 protein (WDR5)—Mixed lineage leukemia (MLL) protein–protein interaction. J. Med. Chem. 2017;60:4818–4839. doi: 10.1021/acs.jmedchem.6b01796. [DOI] [PubMed] [Google Scholar]
- 203.Senisterra G., Wu H., Allali-Hassani A., Wasney G.A., Barsyte-Lovejoy D., Dombrovski L., Dong A., Nguyen K.T., Smil D., Bolshan Y. Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J. 2013;449:151–159. doi: 10.1042/BJ20121280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Grebien F., Vedadi M., Getlik M., Giambruno R., Grover A., Avellino R., Skucha A., Vittori S., Kuznetsova E., Smil D. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol. 2015;11:571–578. doi: 10.1038/nchembio.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhang X., Zheng X., Yang H., Yan J., Fu X., Wei R., Xu X., Zhang Z., Yu A., Zhou K. Piribedil disrupts the MLL1-WDR5 interaction and sensitizes MLL-rearranged acute myeloid leukemia (AML) to doxorubicin-induced apoptosis. Cancer Lett. 2018;431:150–160. doi: 10.1016/j.canlet.2018.05.034. [DOI] [PubMed] [Google Scholar]
- 206.Alicea-Velázquez N.L., Shinsky S.A., Loh D.M., Lee J.-H., Skalnik D.G., Cosgrove M.S. Targeted disruption of the interaction between WD-40 repeat protein 5 (WDR5) and mixed lineage leukemia (MLL)/SET1 family proteins specifically inhibits MLL1 and SETd1A methyltransferase complexes. J. Biol. Chem. 2016;291:22357–22372. doi: 10.1074/jbc.M116.752626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Getlik M., Smil D., Zepeda-Velazquez C., Bolshan Y., Poda G., Wu H., Dong A., Kuznetsova E., Marcellus R., Senisterra G., et al. Structure-Based Optimization of a Small Molecule Antagonist of the Interaction Between WD Repeat-Containing Protein 5 (WDR5) and Mixed-Lineage Leukemia 1 (MLL1) J. Med. Chem. 2016;59:2478–2496. doi: 10.1021/acs.jmedchem.5b01630. [DOI] [PubMed] [Google Scholar]
- 208.Cao L., Wu G., Zhu J., Tan Z., Shi D., Wu X., Tang M., Li Z., Hu Y., Zhang S., et al. Genotoxic stress-triggered β-catenin/JDP2/PRMT5 complex facilitates reestablishing glutathione homeostasis. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-11696-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zhou Q., Chen X., He H., Peng S., Zhang Y., Zhang J., Cheng L., Liu S., Huang M., Xie R., et al. WD repeat domain 5 promotes chemoresistance and Programmed Death-Ligand 1 expression in prostate cancer. Theranostics. 2021;11:4809–4824. doi: 10.7150/thno.55814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Aho E.R., Wang J., Gogliotti R.D., Howard G.C., Phan J., Acharya P., Macdonald J.D., Cheng K., Lorey S.L., Lu B., et al. Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 2019;26:2916–2928.e13. doi: 10.1016/j.celrep.2019.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tian J., Teuscher K.B., Aho E.R., Alvarado J.R., Mills J.J., Meyers K.M., Gogliotti R.D., Han C., Macdonald J.D., Sai J., et al. Discovery and Structure-Based Optimization of Potent and Selective WD Repeat Domain 5 (WDR5) Inhibitors Containing a Dihydroisoquinolinone Bicyclic Core. J. Med. Chem. 2020;63:656–675. doi: 10.1021/acs.jmedchem.9b01608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhou H., Liu L., Huang J., Bernard D., Karatas H., Navarro A., Lei M., Wang S. Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL1) protein–protein interaction. J. Med. Chem. 2013;56:1113–1123. doi: 10.1021/jm3015298. [DOI] [PubMed] [Google Scholar]
- 213.Grembecka J., He S., Shi A., Purohit T., Muntean A.G., Sorenson R.J., Showalter H.D., Murai M.J., Belcher A.M., Hartley T., et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Dafflon C., Craig V.J., Mereau H., Grasel J., Schacher Engstler B., Hoffman G., Nigsch F., Gaulis S., Barys L., Ito M., et al. Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia. 2017;31:1269–1277. doi: 10.1038/leu.2016.327. [DOI] [PubMed] [Google Scholar]
- 215.Shi A., Murai M.J., He S., Lund G., Hartley T., Purohit T., Reddy G., Chruszcz M., Grembecka J., Cierpicki T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. 2012;120:4461–4469. doi: 10.1182/blood-2012-05-429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Borkin D., He S., Miao H., Kempinska K., Pollock J., Chase J., Purohit T., Malik B., Zhao T., Wang J. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell. 2015;27:589–602. doi: 10.1016/j.ccell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Borkin D., Pollock J., Kempinska K., Purohit T., Li X., Wen B., Zhao T., Miao H., Shukla S., He M., et al. Property Focused Structure-Based Optimization of Small Molecule Inhibitors of the Protein-Protein Interaction between Menin and Mixed Lineage Leukemia (MLL) J. Med. Chem. 2016;59:892–913. doi: 10.1021/acs.jmedchem.5b01305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Klossowski S., Miao H., Kempinska K., Wu T., Purohit T., Kim E., Linhares B.M., Chen D., Jih G., Perkey E., et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020;130:981–997. doi: 10.1172/JCI129126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Brzezinka K., Nevedomskaya E., Lesche R., Haegebarth A., Ter Laak A., Fernández-Montalván A.E., Eberspaecher U., Werbeck N.D., Moenning U., Siegel S. Characterization of the Menin-MLL interaction as therapeutic cancer target. Cancers. 2020;12:201. doi: 10.3390/cancers12010201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Krivtsov A.V., Evans K., Gadrey J.Y., Eschle B.K., Hatton C., Uckelmann H.J., Ross K.N., Perner F., Olsen S.N., Pritchard T., et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell. 2019;36:660–673.e11. doi: 10.1016/j.ccell.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Aguilar A., Zheng K., Xu T., Xu S., Huang L., Fernandez-Salas E., Liu L., Bernard D., Harvey K.P., Foster C., et al. Structure-Based Discovery of M-89 as a Highly Potent Inhibitor of the Menin-Mixed Lineage Leukemia (Menin-MLL) Protein-Protein Interaction. J. Med. Chem. 2019;62:6015–6034. doi: 10.1021/acs.jmedchem.9b00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Xu S., Aguilar A., Xu T., Zheng K., Huang L., Stuckey J., Chinnaswamy K., Bernard D., Fernandez-Salas E., Liu L., et al. Design of the First-in-Class, Highly Potent Irreversible Inhibitor Targeting the Menin-MLL Protein-Protein Interaction. Angew. Chem. 2018;57:1601–1605. doi: 10.1002/anie.201711828. [DOI] [PubMed] [Google Scholar]
- 223.Xu S., Aguilar A., Huang L., Xu T., Zheng K., McEachern D., Przybranowski S., Foster C., Zawacki K., Liu Z., et al. Discovery of M-808 as a Highly Potent, Covalent, Small-Molecule Inhibitor of the Menin-MLL Interaction with Strong In Vivo Antitumor Activity. J. Med. Chem. 2020;63:4997–5010. doi: 10.1021/acs.jmedchem.0c00547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ferguson A.D., Larsen N.A., Howard T., Pollard H., Green I., Grande C., Cheung T., Garcia-Arenas R., Cowen S., Wu J. Structural basis of substrate methylation and inhibition of SMYD2. Structure. 2011;19:1262–1273. doi: 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 225.Nguyen H., Allali-Hassani A., Antonysamy S., Chang S., Chen L.H., Curtis C., Emtage S., Fan L., Gheyi T., Li F. LLY-507, a cell-active, potent, and selective inhibitor of protein-lysine methyltransferase SMYD2. J. Biol. Chem. 2015;290:13641–13653. doi: 10.1074/jbc.M114.626861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kukita A., Sone K., Oda K., Hamamoto R., Kaneko S., Komatsu M., Wada M., Honjoh H., Kawata Y., Kojima M., et al. Histone methyltransferase SMYD2 selective inhibitor LLY-507 in combination with poly ADP ribose polymerase inhibitor has therapeutic potential against high-grade serous ovarian carcinomas. Biochem. Biophys. Res. Commun. 2019;513:340–346. doi: 10.1016/j.bbrc.2019.03.155. [DOI] [PubMed] [Google Scholar]
- 227.Sweis R.F., Wang Z., Algire M., Arrowsmith C.H., Brown P.J., Chiang G.G., Guo J., Jakob C.G., Kennedy S., Li F. Discovery of A-893, a new cell-active benzoxazinone inhibitor of lysine methyltransferase SMYD2. ACS Med. Chem. Lett. 2015;6:695–700. doi: 10.1021/acsmedchemlett.5b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Eggert E., Hillig R.C., Koehr S., Stockigt D., Weiske J., Barak N., Mowat J., Brumby T., Christ C.D., Ter Laak A., et al. Discovery and Characterization of a Highly Potent and Selective Aminopyrazoline-Based in Vivo Probe (BAY-598) for the Protein Lysine Methyltransferase SMYD2. J. Med. Chem. 2016;59:4578–4600. doi: 10.1021/acs.jmedchem.5b01890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Mitchell L.H., Boriack-Sjodin P.A., Smith S., Thomenius M., Rioux N., Munchhof M., Mills J.E., Klaus C., Totman J., Riera T.V., et al. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2016;7:134–138. doi: 10.1021/acsmedchemlett.5b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Thomenius M.J., Totman J., Harvey D., Mitchell L.H., Riera T.V., Cosmopoulos K., Grassian A.R., Klaus C., Foley M., Admirand E.A., et al. Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS ONE. 2018;13:e0197372. doi: 10.1371/journal.pone.0197372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Van Aller G.S., Graves A.P., Elkins P.A., Bonnette W.G., McDevitt P.J., Zappacosta F., Annan R.S., Dean T.W., Su D.S., Carpenter C.L., et al. Structure-Based Design of a Novel SMYD3 Inhibitor that Bridges the SAM-and MEKK2-Binding Pockets. Structure. 2016;24:774–781. doi: 10.1016/j.str.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 232.Huang C., Liew S.S., Lin G.R., Poulsen A., Ang M.J.Y., Chia B.C.S., Chew S.Y., Kwek Z.P., Wee J.L.K., Ong E.H., et al. Discovery of Irreversible Inhibitors Targeting Histone Methyltransferase, SMYD3. ACS Med. Chem. Lett. 2019;10:978–984. doi: 10.1021/acsmedchemlett.9b00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Barsyte-Lovejoy D., Li F., Oudhoff M.J., Tatlock J.H., Dong A., Zeng H., Wu H., Freeman S.A., Schapira M., Senisterra G.A. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. USA. 2014;111:12853–12858. doi: 10.1073/pnas.1407358111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Tsuge M., Hamamoto R., Silva F.P., Ohnishi Y., Chayama K., Kamatani N., Furukawa Y., Nakamura Y. A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5′ flanking region of SMYD3 is a risk factor for human cancers. Nat. Genet. 2005;37:1104–1107. doi: 10.1038/ng1638. [DOI] [PubMed] [Google Scholar]
- 235.Liu Y., Luo X., Deng J., Pan Y., Zhang L., Liang H. SMYD3 overexpression was a risk factor in the biological behavior and prognosis of gastric carcinoma. Tumour Biol. 2015;36:2685–2694. doi: 10.1007/s13277-014-2891-z. [DOI] [PubMed] [Google Scholar]
- 236.Feichtinger J., Aldeailej I., Anderson R., Almutairi M., Almatrafi A., Alsiwiehri N., Griffiths K., Stuart N., Wakeman J.A., Larcombe L., et al. Meta-analysis of clinical data using human meiotic genes identifies a novel cohort of highly restricted cancer-specific marker genes. Oncotarget. 2012;3:843–853. doi: 10.18632/oncotarget.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Stransky N., Egloff A.M., Tward A.D., Kostic A.D., Cibulskis K., Sivachenko A., Kryukov G.V., Lawrence M.S., Sougnez C., McKenna A., et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Allali-Hassani A., Szewczyk M.M., Ivanochko D., Organ S.L., Bok J., Ho J.S.Y., Gay F.P., Li F., Blazer L., Eram M.S. Discovery of a chemical probe for PRDM9. Nat. Commun. 2019;10:5759. doi: 10.1038/s41467-019-13652-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K., Hines J., Winkler J.D., Crew A.P., Coleman K., et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Winter G.E., Buckley D.L., Paulk J., Roberts J.M., Souza A., Dhe-Paganon S., Bradner J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zengerle M., Chan K.H., Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.