

Abstract
Alterations in glutamatergic function are well established in schizophrenia (Sz), but new treatment development is hampered by the lack of translational pathophysiological and target engagement biomarkers as well as by the lack of animal models that recapitulate the pathophysiological features of Sz. Here, we evaluated the rodent auditory steady state response (ASSR) and long-latency auditory event-related potential (aERP) as potential translational markers. These biomarkers were assessed for their sensitivity to both the N-methyl-d-aspartate receptor (NMDAR) antagonist phencyclidine (PCP) and to knock-out (KO) of Serine Racemase (SR), which is known to lead to Sz-like alterations in function of parvalbumin (PV)-type cortical interneurons. PCP led to significant increases of ASSR that were further increased in SRKO−/−, consistent with PV interneuron effects. Similar effects were observed in mice with selective NMDAR KO on PV interneurons. By contrast, PCP but not SRKO reduced the amplitude of the rodent analog of the human N1 potential. Overall, these findings support use of rodent ASSR and long-latency aERP, along with previously described measures such as mismatch negativity (MMN), as translational biomarkers, and support SRKO mice as a potential rodent model for PV interneuron dysfunction in Sz.
Keywords: Schizophrenia, Rodent, Mouse, Auditory steady-state response, ASSR, N1, N-methyl-d-aspartate, NMDA receptor, NMDAR, Phencyclidine
Introduction
N-methyl-d-aspartate-type glutamate receptor (NMDAR) antagonists, such as phencyclidine (PCP) and ketamine, induce a wide range of symptoms and cortical neurocognitive deficits similar to those observed in schizophrenia (Sz), leading to theories of impaired NMDAR function [1–4]. NMDAR are modulated by the endogenous amino acids glycine and d-serine, which bind to an allosteric modulatory site that regulates channel opening [5,6]. Moreover, disturbances in both glycine and d-serine metabolism are reported in Sz (rev. in [7]). To date, however, no FDA-approved glutamate-based treatments are available, in part due to lack of translational biomarkers that permit integration across rodent and human models.
In Sz, deficits are observed in a range of neurophysiological biomarkers including auditory mismatch negativity (MMN), steady-state response (ASSR) and N1 refractoriness (rev. in [8,9]). We have recently shown that rodent MMN is highly sensitive to both NMDAR antagonists and agonists and thus may potentially be used as both a translational and target engagement biomarker [10]. Auditory N1 refractoriness and ASSR, however, have been studied to a lesser degree. Here, we evaluated the sensitivity of these measures both to NMDAR antagonist administration and to genetic manipulations involving d-serine synthesis and parvalbumin interneuron function that potentially recapitulate pathophysiological features of Sz, with the goal of evaluating their utility for translational drug development.
In the ASSR paradigm, a series of repetitive clicks are played at rates of typically between 20 and 100 Hz and elicit an entrained response at the stimulation frequency. Human ASSR shows a natural resonance at 40 Hz, consistent with an interaction between glutamatergic principal neurons and local circuit parvalbumin (PV)-type GABAergic interneurons (rev. in [9,11]). Deficits in ASSR were first reported ~20 yrs ago [12], and have been replicated consistently since that time when relatively short (~1 s) interstimulus intervals are used (rev. in [13,14]). ASSR deficits have been postulated to interrelate with oxidative-stress driven impairments in PV interneuron function [15,16], although increases have been reported in paradigms that use longer (e.g. ~3 s) intervals [17,18].
Deficits are also observed in other aspects of the response, such as phase delay [12,19]. As opposed to other auditory ERP, such as MMN, ASSR appears to be intact in individuals at clinical high risk for Sz [20,21], although it is impaired in chronic stages (rev. in [14]) and in first-degree relatives [22,23]. Thus, it may index downregulation of cortical circuits over time during early stages of the disorder as well as genetic risk and could thus be a target for early intervention.
In healthy human volunteers, NMDAR antagonist administration is reported to increase gamma band (40–85 Hz) neural response to auditory [24,25] or visual [26] stimuli, but to decrease response in delta and theta frequency ranges [24]. In these studies, increases in gamma were associated with clinical symptoms, supporting their relevance to Sz [24]. More recently, however, a reduction in ASSR following ketamine administration was reported [27]. In rodents, both increases [28–30] and decreases [31,32] in ASSR are also reported, potentially explainable based on varying degrees of NMDAR occupancy across studies [33].
In the auditory refractoriness paradigm, individual stimuli (e.g. tones, clicks) are presented repeatedly at intervals of several hundred milliseconds to seconds, with increasing amplitude as a function of interval length. Under these stimulation conditions, the tones elicit a series of long-latency auditory ERP (aERP) components, including the P1, N1 and P2 potentials. Like ASSR, deficits in auditory N1/P2 response have been extensively documented in Sz [9,34–36]. Also like ASSR, Sz-like deficits in auditory N1 generation are induced by NMDAR antagonists in both monkey [37] and rodent [10,31,38,39] models. However, as opposed to ASSR, power associated with the auditory N1/P2 component maps predominantly to the theta frequency range, implicating somatostatin (SOM) interneuron related circuits [40–42]. These measures thus may provide complementary insights into integrity of specific interneuron mechanisms within cortex.
In the present study, we evaluated the integrity of these measures in a putative rodent model of Sz associated with knockout of the serine racemase (SR) gene. SR is the primary enzyme mediating d-serine synthesis in brain. The presence of d-serine in rodent brain was first demonstrated by Hashimoto et al. [43] and was subsequently shown to reflect interconversion of l- to d-serine [44] via SR [45,46]. Moreover, expression-reducing deficits in the non-coding region of the serine racemase (SRR) gene are reported in Sz [47], suggesting potential etiological involvement.
Over recent years, SRKO mice have been extensively phenotyped and have been shown to have the predicted reduction in brain d-serine levels, as well as specific homologies to features of Sz (rev. in [45]). These include most prominently a significant reduction in PV interneuron number [16] and loss of cortical grey matter in both frontal [48] and sensory [49] cortex.
The present study therefore evaluated the degree to which SRKO mice showed abnormalities in auditory aERP, potentially linked to disturbances in PV interneuron function. aERP were obtained both prior to and following administration of the NMDAR antagonist PCP. We hypothesized that SRKO mice would show abnormalities similar to those induced by acute NMDAR antagonist administration. Finally, in order to obtain a non-pharmacological comparison group, we constructed mice in which the NR1 subunit of the NMDAR (GRIN1) was knocked-out specifically in PV interneurons using a Cre/Lox approach. As with SRKO mice, we hypothesized that this would produce a phenotype similar to that observed for acute NMDAR antagonists.
Materials and methods
This study was carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health and approved by Nathan Kline Institute Animal Care and Use Committee. Heterozygous mice mutants (+/−) with a deletion of the gene encoding SR which converts l-Ser to d-Ser [50] were initially obtained from Coyle laboratory of McLean Hospital, Harvard Medical School. At least F10 hybrids were bred in house resulting in wildtype (WT) (+/+), heterozygous (HET) (+/−) and homozygous (HOM) (−/−) littermates with consequent reduction of D-serine level by 50 % and 90 % respectively. The genetic testing was performed by Transnetyx Inc. (Cordova, TN). The animals were maintained under a 12 h/12 h dark/light cycle and were allowed food and water ad libitum.
Approximately 3–4 month old mice underwent stereotaxic implantation of tripolar electrode assemblies (Plastics One Inc., Roanoke, VA) under isoflurane anesthesia. Stereotactic coordinates were AP −2.50 from bregma, ML + 3.50 mm, and depth 1–2.25 mm (15° angle). Three stainless-steel electrodes, mounted in a single pedestal, were aligned along the sagittal axis of the skull (at 1 mm intervals (positive,ground and negative). Positive electrodes (2.25 mm) were placed above the left primary auditory cortex region.
Negative electrodes (1 mm) were placed adjacent to the ipsilateral neocortex. Ground electrodes (1 mm) were located between recording and reference electrodes. The electrode pedestal is secured to the skull with cyanoacrylic cement (PlasticsOne, Roanoke, VA). An analgesic (Buprenex0.3 mg/kg, s.c.) was administered after surgery and every 24 hr thereafter as needed. Following surgery, animals were individually housed and allowed to recover for at least 7 days prior to the recording of evoked potentials. At the completion of all recordings, mice were anesthetized with ketamine hydrochloride and acepromazine maleate 1:1 mixture (1 μl/g i.p.). The brain was fixed with 10 % formaldehyde solution and was stored in 30 % glucose solution for determination of placement of the electrodes. A total of 100 animals were used for this study (31 WT (+/+), 33 HET (+/−) and 36 HOM (−/−)).
Neurophysiology studies
After a recovery period, aERP measures were recorded from freely moving mice in a clear polycarbonate bowl with food ad libitum in a sound proof chamber. Mice were allowed to habituate for an hour to the new surroundings. An initial testing session was performed for each animal to permit assessment of genetic status. Each animal then participated in two additional drug-treatment sessions in random order with at least a 2-week intervening period. Each session consisted of a 2 -h baseline recording, followed by acute administration of either PCP (10 mg/kg) or saline i.p. followed by an additional 2 -h recording.
Stimuli were administered free-field using a speaker located above the recording chamber generated by a Presentation program, calibrated to 80 dB. To assess the N1 refractoriness function mice were exposed to separate blocks of 200 stimuli (6000 KHz 60 ms) at 1, 3 and 6 s interstimulus interval (ISI). To assess ASSR, 500-ms trains of 0.1 ms clicks were delivered at stimulation rates of 20, 25, 40, 50, and 80 Hz in separate blocks (300 stimuli each) with 500-ms interval between click trains (overall stimulus onset asynchrony = 1 s).
Neuroelectric signals were impedance matched with unity gain preamplifiers located near the electrode, and further differentially amplified with an appropriate bandpass (typically 0.3 Hz to 3 kHz). For additional noise reduction we used 50/60 Hz noise eliminator (HumBug, Quest Scientific, North Vancouver, CA). Data were acquired continuously along with digital stimulus identification tags at a digitization rate exceeding 10 kHz using a Neuroscan SCAN system. Epoching, sorting, artifact rejection and averaging were then conducted off-line. For ASSR a FFT spectral analysis was performed, highest amplitudes were recorded in the relevant frequency bands.
PV-specific GRIN1 KO mice
Grin1−/PV mice were generated by crossing female mice with floxed NMDAR (Grin1tm2Stl/J) and male mice with Cre recombinase expressed specifically within PV-interneurons (129P2-Pvalbtm1(cre)Arbr/J mice). Both mouse lines were obtained from Jackson laboratory (Bar Harbor, ME) and were tested relative to mice with the same C57BL/6 J background. Genotype was confirmed by tail-clip (Transnetyx, Inc. Cordova, TN).
Statistical analysis
Data were analyzed across animals using linear mixed-model Analysis of Variance (ANOVA). Separate analyses were performed for 1) the initial (non-drug) session from each animal, and 2) pre/post treatment effects from saline/PCP drug-challenge days. Least significant difference (LSD) analyses were used for post-hoc testing All statistics are two-tailed with pre-specified α level of significant set at p < .05.
Results
For SRKO mice, ASSR and aERP responses were obtained both at pretreatment and following challenge with PCP 10 mg/kg. For PV-selective GRIN1 KO animals, only ASSR was obtained.
Auditory steady state response
Analyses were performed on both the baseline (500-ms pre-stimulus) and stimulus (0–500 ms) period (Fig. 1A,B). During active stimulations, 500-ms click trains were presented at stimulation frequencies of 20, 25, 40, 50, and 80 Hz and peak power was calculated at each frequency. We performed a 2-way mixed-model ANOVA on the initial (non-drug) testing day for each animal to examine the main effects of stimulation frequency and genotype on ASSR peak power as well as any interactions; followed by a 4-way ANOVA on combined data from the two drug challenge days to examine the main effects of stimulation frequency, genotype, drug treatment (saline vs PCP), and pre-or-post-treatment vs ASSR peak power as well as any interactions.
Fig. 1.
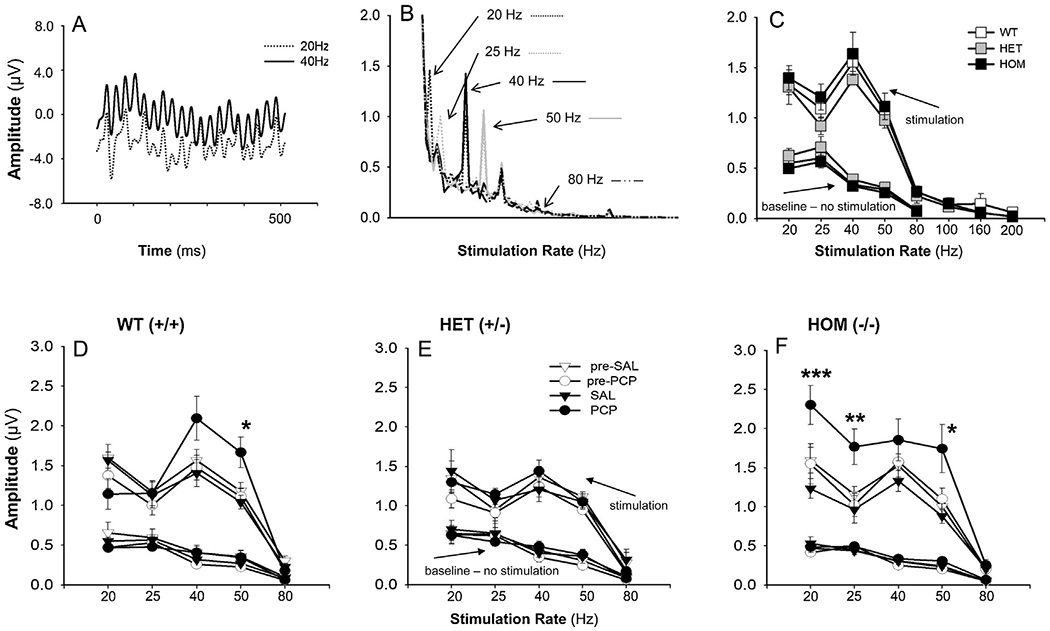
Effect of acute PCP (10 mg/kg) and SAL on auditory steady-state response (ASSR) measured in primary auditory cortex (A1) of SRK Omice. A: Responses (amplitude in μV) to 20 and 40 Hz click train stimuli. B: Average waveform amplitudes to 20, 25, 40, 50 and 80 Hz responses. C: ASSR amplitude by stimulation rate and genotype. Data are the mean ± SEM (n = 14 WT (+/+), n = 12 HET (+/−) and n = 14 HOM (−/−) from the initial testing day for each animal. D-F: Effect of PCP treatment on ASSR amplitude by frequency for WT (D), HET (E), and HOM (F). Data are mean of 7-11 animals per group, across the saline and PCP treatment days. ***p = 0.001, **p ≤ 0.02 and *p ≤ 0.05 for saline vs. PCP groups.
Pre-stimulation baseline:
In the baseline period, there was a significant main effect of frequency (F4,11 = 105.8, p < .0001), reflecting a significant linear 1/f reduction in amplitude from 20 to 80 Hz (F1,22 = 312.6, p < .0001) (Fig. 1C). There were no significant effects of either PCP or genotype on prestimulus (baseline) activity as a function of genotype (Fig. 1C) or PCP treatment (Fig. 1D–F).
Stimulation period, initial testing day:
In initial testing, the effects of stimulation frequency on power were strongly significant (F4,132.5 = 84.7, p < .0001) with the greatest amplitude occurring at 40 Hz (Fig. 1C). The main effect of genotype (F2,35.9 = 1.32, p = .28) on power and the genotype X stimulation-frequency interaction (F8,132.5 = .4, p = .9) were both non-significant.
Stimulation period drug challenge days (Fig. 1D–F):
When analyses were conducted across saline and PCP-challenge days, the main effect of stimulation frequency on power was again highly significant (F4,386.5 = 157.9, p < .0001). The treatment day (sal/PCP) X time (pre/post treatment) interaction was also highly significant (F1,383.2 = 25.2, p < .0001) reflecting an increase in ASSR amplitude across all stimulation frequencies and genotypes by PCP.
The main effect of genotype on power was not significant (F2,19.8 = 23.8, p = .083). However, genotype interacted significantly with both stimulation frequency (F8,386.4 = 2.05, p = .04) and drug treatment (F2,407.6 = 7.27, p = .001). The 3-way genotype X time X drug treatment interaction was also significant (F2,383 = 3.70, p = .026), reflecting differential sensitivity to PCP vs. saline across the WT(+/+), HET(+/−) and HOM(−/−) animals.
In order to further parse the significant time X treatment and genotype X time X treatment effect, we calculated difference values for each session reflecting post-vs-pre-treatment values (i.e. within-session drug effects). As expected, the main effect of treatment was again highly significant (F1,199.7 = 42.9, p < .0001) reflecting a larger change during PCP vs. saline treatment. In addition, a significant drug treatment X stimulation frequency effect emerged (F4,182.7 = 3.18, p = .015). The genotype X drug treatment (F2,199.1 = 6.38, p = .002) and genotype X drug treatment X stimulation frequency (F8,182.7 = 3.06, p = .003) effects were both significant.
These final interactions were parsed by conducting separate one-way analyses at each stimulation frequency by genotype and treatment. In WT (+/+) animals, significant PCP effects were observed at 40/50 Hz, but not at other frequencies (Fig. 1D). In HET(+/−) mice, no significant PCP effects were observed at any stimulation frequency (Fig. 1E). By contrast, in HOM(−/−) mice, significant effects were observed both at lower (20/25 Hz) and higher (40 Hz) stimulation frequencies (Fig. 1F), reflecting a specific genotype effect primarily on the 20 Hz response (F2,23 = 4.12 = .031) with significant post-hoc difference between HOM(−/−) and WT (+/+) animals (LSD p = .009).
N1 refractoriness function
aERPs analogous to the human P1/N1 response were obtained in WT (−/−), HET(+/−) and HOM(−/) SRKO mice in response to tones presented with 1, 3 and 6 s ISIs. Based upon the ERP we evaluated 3 peaks of interest: P20 was defined as the greatest positivity between 15–35 ms post-stimulus; N40 was defined as the greatest negativity between 30–75 ms post-stimulus; and P80 was defined as the greatest positivity between 70–105 ms (Fig. 2). Analogous to ASSR, for each component a 2-way ANOVA with factors of genotype and ISI was conducted on data obtained from the initial testing day for each animal, whereas a 4-way ANOVA with factors of stimulation frequency, genotype, drug treatment (saline vs PCP), and pre-or-post-treatment was conducted across treatment days.
Fig. 2.

Auditory event-related potentials (aERP) from A1 in response to stimuli presented at 1, 3 and 6 sin WT(+/+), and HET(+/−) and HOM (−/−) SRKO mice, showing P20, N40 and P80 peaks of interest. A-C: aERPs shown as averages of n = 12-15 mice. D-F: Values are the mean ± SEM (n = 11-14).
P20:
At initial testing, P20 amplitudes were not significantly affected by ISI (F2,70 = 1.31, p = .3) or genotype (F2,35 = 1.64, p = .2). The genotype X ISI interaction was also non-significant (F4,70 = .8, p = .5) (Fig. 2A and 2D).
When analyses were analyzed across treatment day (sal/PCP) and time (pre/post), the ISI effect was again non-significant (F2,234.3 = 2.24, p = .1). The time (F1,234.3 = .62, p = .4), treatment (F1,244.5 = .1, p = .7) and treatment X time interaction (F2,234.3 = .9, p = .4) were all non-significant. The main effect of genotype (F2,24.1 = 2.05, p = .15) and the genotype X time interaction (F2,234.3 = .1, p = .9) and genotype X treatment interaction (F2,244.0 = 2.41, p = .09) were also all non-significant as were higher order interactions (Table 1).
Table 1.
Mean amplitude of indicated aERP components by saline and PCP treatment.
| EEG component | genotype | ISI s | PRE-SALINE | PRE-PCP | SALINE | PCP |
|---|---|---|---|---|---|---|
| P20 | Wildtype | 1 | 5.674 ± 4.762 (11) | 6.513 ± 3.650 (8) | 6.410 ± 5.179 (11) | 1.762 ± 4.588 (8) * |
| 3 | 4.014 ± 3.282 (11) | 6.525 ± 4.693 (8) | 4.209 ± 6.232 (11) | 5.547 ± 6.405 (8) | ||
| 6 | 5.181 ± 4.030 (11) | 3.904 ± 6.234 (8) | 5.506 ± 5.649 (11) | 6.555 ± 7.383 (8) | ||
| Heterozygous | 1 | 4.697 ± 5.350 (6) | 3.888 ± 8.013 (6) | 3.224 ± 7.567 (6) | 5.009 ± 5.129 (6) | |
| 3 | 3.636 ± 7.966 (6) | 8.609 ± 8.884 (6) | 4.396 ± 4.558 (6) | 5.520 ± 10.969 (6) | ||
| 6 | 7.277 ± 2.885 (6) | 5.777 ± 4.948 (6) | 7.517 ± 5.282 (6) | 6.726 ± 5.442 (6) | ||
| Homozygous | 1 | 9.505 ± 9.007 (8) | 9.346 ± 6.772 (8) | 7.027 ± 6.373 (8) | 6.521 ± 3.499 (8) | |
| 3 | 7.938 ± 11.608 (8) | 7.404 ± 8.512 (8) | 11.093 ± 9.321 (8) | 7.074 ± 5.469 (8) | ||
| 6 | 10.884 ± 9.941 (8) | 9.067 ± 9.778 (8) | 13.455 ± 7.073 (8) | 7.404 ± 8.472 (8) | ||
| N40 | Wildtype | 1 | −5.624 ± 3.630 (11) | −3.046 ± 3.509 (8) | −3.416 ± 9.687 (11) | −8.138 ± 6.856 (8) |
| 3 | −16.420 ± 13.588 (11) | −12.907 ± 11.871 (8) | −13.180 ± 15.316 (11) | −11.674 ± 10.884 (8) | ||
| 6 | −20.443 ± 12.990 (11) | −19.107 ± 13.324 (8) | −21.692 ± 20.048 (11) | −20.177 ± 17.459 (8) | ||
| Heterozygous | 1 | −5.165 ± 5.126 (6) | −3.398 ± 6.554 (6) | −6.414 ± 9.925 (6) | −3.697 ± 9.278 (6) | |
| 3 | −18.356 ± 16.327 (6) | −15.685 ± 15.049 (6) | −15.456 ± 16.006 (6) | −12.983 ± 15.008 (6) | ||
| 6 | −23.093 ± 36.470 (6) | −21.403 ± 21.153 (6) | −19.537 ± 22.896 (6) | −21.150 ± 18.254 (6) | ||
| Homozygous | 1 | −4.482 ± 6.171 (8) | −1.730 ± 5.105 (8) | −1.782 ± 5.617 (8) | −7.453 ± 4.959 (8) * | |
| 3 | −14.040 ± 10.489 (8) | −9.278 ± 6.294 (8) | −9.440 ± 13.298 (8) | −14.010 ± 6.468 (8) | ||
| 6 | −15.966 ± 12.172 (8) | −12.029 ± 4.897 (8) | −14.437 ± 14.775 (8) | −14.251 ± 9.406 (8) | ||
| P80 | Wildtype | 1 | 7.629 ± 6.909 (11) | 6.946 ± 4.994 (8) | 12.659 ± 7.447 (8) | 5.446 ± 9.335 (8) |
| 3 | 19.946 ± 12.595 (11) | 9.137 ± 4.326 (8) | 19.835 ± 11.140 (8) | 13.961 ± 7.771 (8) | ||
| 6 | 22.896 ± 17.402 (11) | 22.424 ± 10.493 (8) | 28.063 ± 19.045 (8) | 7.522 ± 7.901 (8) *** | ||
| Heterozygous | 1 | 8.044 ± 4.569 (6) | 8.507 ± 8.184 (6) | 11.811 ± 4.552 (6) | 6.718 ± 4.823 (6) | |
| 3 | 18.263 ± 17.964 (6) | 16.769 ± 16.578 (6) | 20.767 ± 18.104 (6) | 11.440 ± 10.270 (6) | ||
| 6 | 33.805 ± 31.497 (6) | 25.379 ± 24.228 (6) | 29.607 ± 18.441 (6) | 17.895 ± 15.134 (6) | ||
| Homozygous | 1 | 10.928 ± 6.622 (8) | 10.411 ± 6.121 (8) | 16.654 ± 21.769 (8) | 7.997 ± 7.892 (8) | |
| 3 | 20.169 ± 20.173 (8) | 17.415 ± 21.514 (8) | 24.593 ± 27.613 (8) | 11.073 ± 10.456 (8) | ||
| 6 | 22.343 ± 19.330 (8) | 19.382 ± 20.434 (8) | 31.111 ± 27.824 (8) | 14.463 ± 9.881(8) |
Results are Means ± SD. Number of animals per observation is shown in parentheses
p < 0.05
p < 0.01 compared to baseline levels and to saline treatment.
N40:
At initial testing, N40 amplitudes showed a significant ISI effect (F2,60.8 = 31.9, p < .0001) reflecting large N1 potentials with increasing ISI. The effects of genotype (F2,31.3 = .84,p = .44) and the genotype X ISI interactions (F4,60.7 = .1.16, p = .3) were both non-significant (Fig. 2B and E).
When analyses were analyzed across treatment day (sal/PCP) and time (pre/post), the ISI effect was again strongly significant (F2,233.5 = 43.3, p < .0001). The treatment X time interaction was also significant (F1,233.5 = 4.07, p = .045), reflecting a near-significant decrease in N40 amplitude in the PCP vs. saline groups post- (F1,109.4 = 3.89, p = .051), but not pre- (F1,110.9 = .25), treatment.
The main effect of genotype was non-significant (F2,23.4 = .33, p = .7), as was the genotype interaction with time (F2,233.5 = .03, p = .98) and treatment (F2,240.5 = .7, p = .5), along with higher order interactions (Table 1).
P80:
At initial testing, P80 amplitude also showed a highly significant main effect of ISI (F2,60.9 = 45.6, p < .0001), reflecting increased amplitude with increasing ISI (Fig. 2C and F). The main effect of genotype (F2,31.3 = 1.22, p = .31) and genotype X ISI interaction (F4,60.8 = 1.13, p = .35) were both non-significant.
When analyses were conducted across treatment days, there was a highly significant treatment X time (F1,234.2 = 10.7, p = .001) interaction, reflecting a highly significant reduction in P80 post- vs. pre-PCP (F1,110.6 = 25.7, p < .0001) but not saline (F1,110.0 = .64, p = .4). The main effect of genotype was again non-significant (F2,24.2 = .32, p = .7) as was the genotype interaction with treatment (F2,234.2 = .24, p = .8) and time (F2,240.0 = .86, p = .4) along with higher order interactions (Table 1).
Effect of PV specific GRIN1 KO
In order to further evaluate the mechanism underlying increased (rather than decreased) ASSR response following NMDAR antagonist treatment, we evaluated ASSR in an additional group of animals with PV interneuron-specific KO of the GRIN1 subunit. In order to confirm our previously observed NMDAR antagonist effect, we evaluated saline vs. PCP effect in a group of C57/BL6 animals matching the Cre background.
As with studies of the SRKO mice (Figs. 1D–F) there was a significant main effect of drug treatment (F1,47.9 = 7.13, p = .01), with PCP increasing ASSR peak power. There was also a trend toward a drug treatment X time interaction (F1,46.1 = 3.77, p = .058). When data were split by pre vs. post treatment no significant difference was observed between groups in the pretreatment period (i.e., pre-saline vs pre-PCP administration) (F1,25 = .023, p = .88), whereas a significant difference was observed in the post-treatment period (i.e., post-saline vs post-PCP administration) (F1,24.1 = 16.1, p = .001), reflecting greater ASSR amplitude in the PCP vs. saline treated group (Fig. 3A).
Fig. 3.
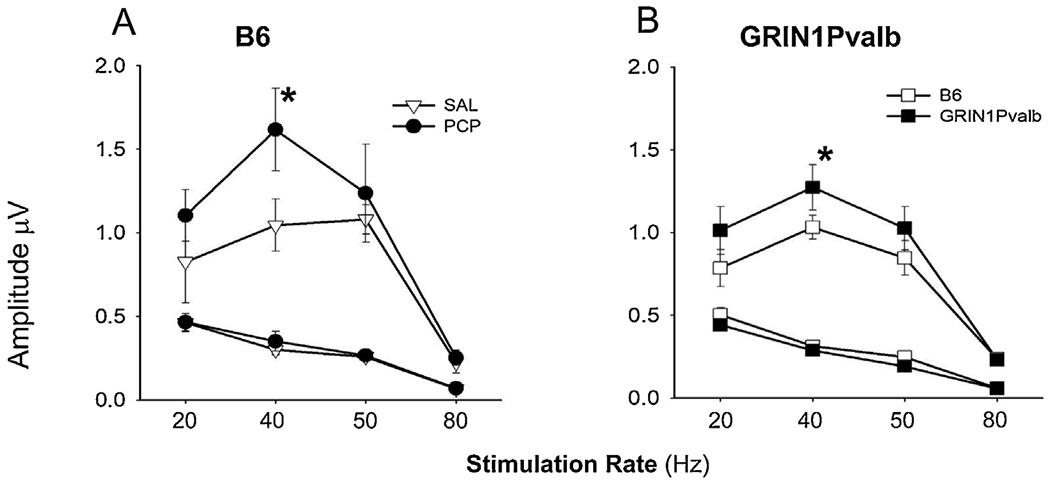
A: Comparison of NMDA antagonism by acute PCP (10 mg/kg) versus SAL on ASSR responses (Amplitude in μV) in B6 mice and B: in GRIN1Pvalb (NR1 subunit of the NMDAR was knocked-out specifically in PV interneurons) versus B6 mice. Values are the mean ± SEM (n = 3-6). *p ≤ 0.05 SAL vs. PCP or B6 vs.GRIN1Pvalb mice.
Similar analyses were conducted comparing GRIN1 KO vs. WT animals. Consistent with our acute PCP effects, GRIN1 KO mice showed enhanced ASSR across frequencies (F1,9 = 5.40, p = .046) as well as at 40 (F1,9 = 8.26, p = .018) and 50 (F1,9 = 8.09, p = .019) Hz individually (Fig. 3B).
Discussion
Treatment development research in Sz is hampered at present both by the lack of validated biomarkers that can be used to translate across species, and the lack of animal models that recapitulate key functional features of the disorder. ERP-based neurophysiological biomarkers have become increasingly appreciated given their physiological homology across rodent, monkey and human species. We [10] and others [51,52] have previously investigated the sensitivity of MMN to NMDAR antagonists across species and have documented highly significant homologies in both frequency content and sensitivity to NMDAR antagonist administration.
Here, we evaluated two additional measures – ASSR and auditory N1 refractoriness – that have been shown to be impaired in Sz and may reflect differential involvement of PV-and SOM-related ensembles within rodent auditory cortex. In parallel, we evaluated the degree to which Sz-like phenotypes were reproduced in SRKO mice, a putative model for Sz that is associated specifically with Sz-like PV interneuron downregulation.
Principal findings were that both acute NMDAR antagonist administration (Figs. 1D, 3A) and selective NMDAR KO on PV interneurons increased ASSR response (Fig. 3B), consistent with the postulated involvement of PV interneurons in ASSR generation. Unperturbed SRKO mice showed ASSR levels that were statistically indistinguishable from WT. However, SRKO significantly modulated the effect of treatment with the NMDAR antagonist PCP, such that HET(+/−) mice showed reduced sensitivity while HOM(−/−) showed significantly enhanced sensitivity (Fig. 1D–F).
By contrast to its effects on ASSR, SRKO did not significantly affect the rodent N40/P80, which may serve as a rodent analog of the human N1. These findings thus especially support the role of ASSR and N1 as rodent translational biomarkers, and of SRKO mice as an animal model that recapitulates PV-specific aspects of NMDAR dysfunction in Sz.
ASSR has been extensively studied before both in Sz and in rodent models. In Sz, the majority of studies have shown a reduction of ASSR power (e.g., [12,22,53–56], although increases are also reported [17,18]. Deficits are observed as well across the psychosis spectrum [57]. Although the reductions have often been attributed to reduced NMDAR dysfunction on PV interneurons themselves, alternative formulations suggest that the high firing rate of PV interneurons is critically dependent upon rapid Ca2+ influx through Ca2+-permeable AMPA receptors, and that the slower kinetics of Ca2+ entry through NMDAR may impede rather than support 40 Hz response [58,59].
The inhibitory effects of high-dose NMDAR antagonists on ASSR may thus reflect the additional effects of NMDAR antagonists on current flow within pyramidal excitatory neurons, rather than PV interneurons [60], consistent with our observation that selective NMDAR KO on PV interneurons increases, rather than decreases, ASSR amplitude. These findings are in contrast to findings in mice with KO of NMDAR on more general GABAergic interneurons, where reduction of ASSR is observed [61].
Our findings of increased ASSR are consistent as well with other studies of acute NMDAR administration in rodents, which then resolves following chronic treatment [28,29]. Regardless of the precise circuitry of ASSR, however, we demonstrate in this study that SRKO mice show increased sensitivity to the augmenting effects of PCP on the ASSR, supporting that it may serve as an effective model of PV-interneuron dysfunction for translational treatment development.
As opposed to the findings with ASSR, SRKO mice showed unchanged sensitivity to disruptive effects of PCP on the rodent N40/P80 potentials, which may be viewed as a rodent analog of the human N1/P2 potential. Like N1, the rodent N40/P80 shows a prolonged refractoriness function, such that full amplitude is not reached until ISI is prolonged to >6 s [62]. In humans, N1 responses map primarily to the theta frequency range, and thus may interrelate with circuit motifs involving non-PV interneurons, especially SOMs [8,40,42]. Our finding of significant aERP response in mice following NMDAR blockade is consistent with prior studies in monkey [37] and rodent [10,38,39,63]. The lack of effect of SRKO on the rodent N1 response may thus suggest that the model recapitulates the PV-related aspects of NMDAR dysfunction in Sz more fully than it captures dysfunction of other neuronal populations.
These findings are also consistent with multi-hit models in which multiple genes contribute in each individual, with each gene only conferring a relatively restricted (1–2 %) proportion of the risk [47]. Here, even complete KO of the SRR gene did not fully recapitulate the NMDAR antagonist-induced phenotype, suggesting that additional genes may also need to be perturbed. At present, relatively little is known about the effects of d-serine on individual interneuron subtypes. Overall, however, these findings are supportive both of use of neurophysiological biomarkers for cross-species comparison, and for KO of genes regulating NMDAR as potential platforms for investigating the effects of different pharmacological agents.
Limitations
In the present study, we used 3–4 month old mice, which equate to a young adult age range. We consider this age mouse to be most relevant to the pathophysiology of Sz, which typically presents in late adolescence and early adulthood. Nevertheless, pathological consequences of SRKO develop more fully between 3 and 8 months [45], suggesting that a more robust phenotype may have been observed in older mice.
Also, in our control experiment we knocked out the GRIN1 subunit of the NMDAR in order to ensure a functional deficit. However, Sz may be specifically associated with disruption of GRIN2A-containing NMDAR [64,65]. Future studies with cell-type specific disruption of GRIN2A and GRIN2B subunits are therefore also required, as well as those targeting other neuronal (e.g. SOM) populations.
Finally, we used only a single acute dose of PCP in the present study, and thus did not confirm the previously reported dose-dependent effects of NMDAR on ASSR [33]. Nevertheless, our dose was sufficient to reduce amplitude of the rodent N1 response, suggesting effective inhibition of NMDAR-mediated neurotransmission.
Conclusions
Development of NMDAR-based biomarkers and animal models for translational research in Sz remains an important priority area. The present results add to the prior literature that disruption of brain d-serine metabolism affects PV interneuron-related function within cortex as reflected in altered ASSR sensitivity to NMDAR antagonists. In addition, we provide additional evidence that PV interneuron-selective NMDAR inhibition increases, rather than decreases, ASSR amplitude in rodents, suggesting that ASSR reductions observed in Sz may reflect dysfunction of NMDAR within other neuronal compartments (e.g. pyramidal neurons, SOM interneurons). We did not observe effects on rodent long-latency aERP components, suggesting that modulation of d-serine metabolism only partially recapitulates the phenotype of Sz. The present results suggest that a combination of aERP measures, including ASSR, auditory N1 and MMN may provide complementary information over the inclusion of any measure in isolation.
Acknowledgements
We would like to acknowledge the contribution of Dr. Joseph Coyle in providing the SRKO mice for these studies.
Funding sources
Funded in part by NIH grants R01MH49334 and R01MH109289 to DCJ.
Footnotes
Declaration of competing interest
Within the last 3 years, Dr. Javitt has received consulting payments from Concert, Lundbeck, Phytec, Pfizer, Cadence, Biogen, SK Life Science, Autifony and Boeringer-Ingelheim and research support from Cerevance he holds equity in Glytech, AASI, and NeuroRx. He serves on the Scientific Advisory Board of NeuroRx and Promentis. He holds intellectual property for the use of d-serine combinations in the treatment of neuropsychiatric disorders, and for NMDAR antagonists in the treatment of depression. Other authors declare no conflicts of interest.
References
- [1].Coyle JT, The glutamatergic dysfunction hypothesis for schizophrenia, Harv. Rev. Psychiatry 3 (5) (1996) 241–253. [DOI] [PubMed] [Google Scholar]
- [2].Javitt DC, Zukin SR, The role of excitatory amino acids in neuropsychiatric illness, J. Neuropsychiatry Clin. Neurosci 2 (1) (1990) 44–52. [DOI] [PubMed] [Google Scholar]
- [3].Javitt DC, Zukin SR, Recent advances in the phencyclidine model of schizophrenia, Am. J. Psychiatry 148 (10) (1991) 1301–1308. [DOI] [PubMed] [Google Scholar]
- [4].Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB Jr., Charney DS, Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses, Arch. Gen. Psychiatry 51 (3) (1994) 199–214. [DOI] [PubMed] [Google Scholar]
- [5].Javitt DC, Balla A, Sershen H, Lajtha A, Reversal of phencyclidine-induced effects by glycine and glycine transport inhibitors A.E. Bennett Research Award, Biol. Psychiatry 45 (6) (1999) 668–679. [DOI] [PubMed] [Google Scholar]
- [6].Javitt DC, Zukin SR, Interaction of [3H]MK-801 with multiple states of the N-methyl-D-aspartate receptor complex of rat brain, Proc. Natl. Acad. Sci. USA 86 (2) (1989) 740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kantrowitz J, Javitt DC, Glutamatergic transmission in schizophrenia: from basic research to clinical practice, Curr. Opin. Psychiatry 25 (2) (2012) 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Javitt DC, Siegel SJ, Spencer KM, Mathalon DH, Hong LE, Martinez A, Ehlers CL, Abbas AI, Teichert T, Lakatos P, Womelsdorf T, A roadmap for development of neuro-oscillations as translational biomarkers for treatment development in neuropsychopharmacology, Neuropsychopharmacology (2020) , doi: 10.1038/s41386-020-0697-9 In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Javitt DC, Spencer KM, Thaker GK, Winterer G, Hajos M, Neurophysiological biomarkers for drug development in schizophrenia, Nature reviews 7 (1) (2008) 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lee M, Balla A, Sershen H, Sehatpour P, Lakatos P, Javitt DC, Rodent mismatch Negativity/theta neuro-oscillatory response as a translational neurophysiological biomarker for N-Methyl-D-Aspartate receptor-based new treatment development in schizophrenia, Neuropsychopharmacology 43 (3) (2018) 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dienel SJ, Lewis DA, Alterations in cortical interneurons and cognitive function in schizophrenia, Neurobiol. Dis 131 (2019) 104208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kwon JS, O’Donnell BF, Wallenstein GV, Greene RW, Hirayasu Y, Nestor PG, Hasselmo ME, Potts GF, Shenton ME, McCarley RW, Gamma frequency-range abnormalities to auditory stimulation in schizophrenia, Arch. Gen. Psychiatry 56 (11) (1999) 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].O’Donnell BF, Vohs JL, Krishnan GP, Rass O, Hetrick WP, Morzorati SL, The auditory steady-state response (ASSR): a translational biomarker for schizophrenia, Suppl. Clin. Neurophysiol 62 (2013) 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tada M, Kirihara K, Koshiyama D, Fujioka M, Usui K, Uka T, Komatsu M, Kunii N, Araki T, Kasai K, Gamma-band auditory steady-state response as a neurophysiological marker for excitation and inhibition balance: a review for understanding schizophrenia and other neuropsychiatric disorders, Clin. EEG Neurosci (2019) 1550059419868872. [DOI] [PubMed] [Google Scholar]
- [15].Gonzalez-Burgos G, Hashimoto T, Lewis DA, Alterations of cortical GABA neurons and network oscillations in schizophrenia, Curr. Psychiatry Rep 12 (4) (2010) 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Steullet P, Cabungcal JH, Coyle J, Didriksen M, Gill K, Grace AA, Hensch TK, LaMantia AS, Lindemann L, Maynard TM, Meyer U, Morishita H, O’Donnell P, Puhl M, Cuenod M, Do KQ, Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia, Mol. Psychiatry 22 (7) (2017) 936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hamm JP, Gilmore CS, Clementz BA, Augmented gamma band auditory steady-state responses: support for NMDA hypofunction in schizophrenia, Schizophr. Res 138 (1) (2012) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim S, Jang SK, Kim DW, Shim M, Kim YW, Im CH, Lee SH, Cortical volume and 40-Hz auditory-steady-state responses in patients with schizophrenia and healthy controls, Neuroimage Clin. 22 (2019) 101732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Roach BJ, Ford JM, Mathalon DH, Gamma band phase delay in schizophrenia, Biol. Psychiatry Cogn. Neurosci. Neuroimaging 4 (2) (2019) 131–139. [DOI] [PubMed] [Google Scholar]
- [20].Lepock JR, Ahmed S, Mizrahi R, Gerritsen CJ, Maheandiran M, Drvaric L, Bagby RM, Korostil M, Light GA, Kiang M, Relationships between cognitive event-related brain potential measures in patients at clinical high risk for psychosis, Schizophr. Res (2019) , doi: 10.1016/j.schres.2019.01.014. [DOI] [PubMed] [Google Scholar]
- [21].Tada M, Nagai T, Kirihara K, Koike S, Suga M, Araki T, Kobayashi T, Kasai K, Differential alterations of auditory gamma oscillatory responses between pre-onset high-risk individuals and first-episode schizophrenia, Cereb. Cortex 26 (3) (2016) 1027–1035. [DOI] [PubMed] [Google Scholar]
- [22].Hong LE, Summerfelt A, McMahon R, Adami H, Francis G, Elliott A, Buchanan RW, Thaker GK, Evoked gamma band synchronization and the liability for schizophrenia, Schizophr. Res 70 (2–3) (2004) 293–302. [DOI] [PubMed] [Google Scholar]
- [23].Rass O, Forsyth JK, Krishnan GP, Hetrick WP, Klaunig MJ, Breier A, O’Donnell BF, Brenner CA, Auditory steady state response in the schizophrenia, first-degree relatives, and schizotypal personality disorder, Schizophr. Res 136 (1–3) (2012) 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hong LE, Summerfelt A, Buchanan RW, O’Donnell P, Thaker GK, Weiler MA, Lahti AC, Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine, Neuropsychopharmacology 35 (3) (2010) 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Plourde G, Baribeau J, Bonhomme V, Ketamine increases the amplitude of the 40-Hz auditory steady-state response in humans, Br. J. Anaesth 78 (5) (1997) 524–529. [DOI] [PubMed] [Google Scholar]
- [26].Grent-’t-Jong T, Rivolta D, Gross J, Gajwani R, Lawrie SM, Schwannauer M, Heidegger T, Wibral M, Singer W, Sauer A, Scheller B, Uhlhaas PJ, Acute ketamine dysregulates task-related gamma-band oscillations in thalamo-cortical circuits in schizophrenia, Brain 141 (8) (2018) 2511–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Curic S, Leicht G, Thiebes S, Andreou C, Polomac N, Eichler IC, Eichler L, Zollner C, Gallinat J, Steinmann S, Mulert C, Reduced auditory evoked gamma-band response and schizophrenia-like clinical symptoms under subanesthetic ketamine, Neuropsychopharmacology 44 (7) (2019) 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Leishman E, O’Donnell BF, Millward JB, Vohs JL, Rass O, Krishnan GP, Bolbecker AR, Morzorati SL, Phencyclidine disrupts the auditory steady state response in rats, PLoS One 10 (8) (2015) e0134979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sullivan EM, Timi P, Hong LE, O’Donnell P, Effects of NMDA and GABA-A receptor antagonism on auditory steady-state synchronization in awake behaving rats, Int. J. Neuropsychopharmacol 18 (7) (2015) pyu118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vohs JL, Chambers RA, O’Donnell BF, Krishnan GP, Morzorati SL, Auditory steady state responses in a schizophrenia rat model probed by excitatory/inhibitory receptor manipulation, Int. J. Psychophysiol 86 (2012) 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schuelert N, Dorner-Ciossek C, Brendel M, Rosenbrock H, A comprehensive analysis of auditory event-related potentials and network oscillations in an NMDA receptor antagonist mouse model using a novel wireless recording technology, Physiol. Rep 6 (16) (2018) e13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sivarao DV, Frenkel M, Chen P, Healy FL, Lodge NJ, Zaczek R, MK-801 disrupts and nicotine augments 40 Hz auditory steady state responses in the auditory cortex of the urethane-anesthetized rat, Neuropharmacology 73 (2013) 1–9. [DOI] [PubMed] [Google Scholar]
- [33].Sivarao DV, Chen P, Senapati A, Yang Y, Fernandes A, Benitex Y, Whiterock V, Li YW, Ahlijanian MK, 40 hz auditory steady-state response is a pharmacodynamic biomarker for cortical NMDA receptors, Neuropsychopharmacology. 41 (2016) 2232–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ford JM, Mathalon DH, Kalba S, Marsh L, Pfefferbaum A, N1 and P300 abnormalities in patients with schizophrenia, epilepsy, and epilepsy with schizophrenialike features, Biol. Psychiatry 49 (10) (2001) 848–860. [DOI] [PubMed] [Google Scholar]
- [35].Shelley AM, Silipo G, Javitt DC, Diminished responsiveness of ERPs in schizophrenic subjects to changes in auditory stimulation parameters: implications for theories of cortical dysfunction, Schizophr. Res 37 (1) (1999) 65–79. [DOI] [PubMed] [Google Scholar]
- [36].Turetsky BI, Bilker WB, Siegel SJ, Kohler CG, Gur RE, Profile of auditory information-processing deficits in schizophrenia, Psychiatry Res. 165 (1–2) (2009) 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Javitt DC, Jayachandra M, Lindsley RW, Specht CM, Schroeder CE, Schizophrenia-like deficits in auditory P1 and N1 refractoriness induced by the psychomimetic agent phencyclidine (PCP), Clin. Neurophysiol 111 (5) (2000) 833–836. [DOI] [PubMed] [Google Scholar]
- [38].Connolly PM, Maxwell C, Liang Y, Kahn JB, Kanes SJ, Abel T, Gur RE, Turetsky BI, Siegel SJ, The effects of ketamine vary among inbred mouse strains and mimic schizophrenia for the P80, but not P20 or N40 auditory ERP components, Neurochem. Res 29 (6) (2004) 1179–1188. [DOI] [PubMed] [Google Scholar]
- [39].Maxwell CR, Ehrlichman RS, Liang Y, Trief D, Kanes SJ, Karp J, Siegel SJ, Ketamine produces lasting disruptions in encoding of sensory stimuli, J. Pharmacol. Exp. Ther 316 (1) (2006) 315–324. [DOI] [PubMed] [Google Scholar]
- [40].Javitt DC, Lee M, Kantrowitz JT, Martinez A, Mismatch negativity as a biomarker of theta band oscillatory dysfunction in schizophrenia, Schizophr. Res 191 (2018) 51–60. [DOI] [PubMed] [Google Scholar]
- [41].Javitt DC, Sweet RA, Auditory dysfunction in schizophrenia: integrating clinical and basic features, Nat. Rev. Neurosci 16 (9) (2015) 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Womelsdorf T, Valiante TA, Sahin NT, Miller KJ, Tiesinga P, Dynamic circuit motifs underlying rhythmic gain control, gating and integration, Nat. Neurosci 17 (8) (2014) 1031–1039. [DOI] [PubMed] [Google Scholar]
- [43].Hashimoto A, Nishikawa T, Hayashi T, Fujii N, Harada K, Oka T, Takahashi K, The presence of free D-serine in rat brain, FEBS Lett. 296 (1) (1992) 33–36. [DOI] [PubMed] [Google Scholar]
- [44].Dunlop DS, Neidle A, The origin and turnover of D-serine in brain, Biochem. Biophys. Res. Commun 235 (1) (1997) 26–30. [DOI] [PubMed] [Google Scholar]
- [45].Coyle JT, Balu DT, The role of serine racemase in the pathophysiology of brain disorders, Adv. Pharmacol 82 (2018) 35–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wolosker H, Blackshaw S, Snyder SH, Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission, Proc. Natl. Acad. Sci. U.S.A 96 (23) (1999) 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schizophrenia Working Group of. the Psychiatric Genomics, C, Biological insights from 108 schizophrenia-associated genetic loci, Nature 511 (7510) (2014) 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].DeVito LM, Balu DT, Kanter BR, Lykken C, Basu AC, Coyle JT, Eichenbaum H, Serine racemase deletion disrupts memory for order and alters cortical dendritic morphology, Genes Brain Behav. 10 (2) (2011) 210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Balu DT, Basu AC, Corradi JP, Cacace AM, Coyle JT, The NMDA receptor co-agonists, D-serine and glycine, regulate neuronal dendritic architecture in the somatosensory cortex, Neurobiol. Dis 45 (2) (2012) 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, Han L, Jiang ZI, Benneyworth MA, Froimowitz MP, Lange N, Snyder SH, Bergeron R, Coyle JT, Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior, Mol. Psychiatry 14 (7) (2009) 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ehrlichman RS, Maxwell CR, Majumdar S, Siegel SJ, Deviance-elicited changes in event-related potentials are attenuated by ketamine in mice, J. Cogn. Neurosci 20 (8) (2008) 1403–1414. [DOI] [PubMed] [Google Scholar]
- [52].Featherstone RE, Melnychenko O, Siegel SJ, Mismatch negativity in preclinical models of schizophrenia, Schizophr. Res 191 (2018) 35–42. [DOI] [PubMed] [Google Scholar]
- [53].Light GA, Hsu JL, Hsieh MH, Meyer-Gomes K, Sprock J, Swerdlow NR, Braff DL, Gamma band oscillations reveal neural network cortical coherence dysfunction in schizophrenia patients, Biol. Psychiatry 60 (11) (2006) 1231–1240. [DOI] [PubMed] [Google Scholar]
- [54].Reilly TJ, Nottage JF, Studerus E, Rutigliano G, Micheli AI, Fusar-Poli P, McGuire P, Gamma band oscillations in the early phase of psychosis: a systematic review, Neurosci. Biobehav. Rev 90 (2018) 381–399. [DOI] [PubMed] [Google Scholar]
- [55].Spencer KM, Niznikiewicz MA, Nestor PG, Shenton ME, McCarley RW, Left auditory cortex gamma synchronization and auditory hallucination symptoms in schizophrenia, BMC Neurosci. 10 (2009) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Thune H, Recasens M, Uhlhaas PJ, The 40-Hz auditory steady-state response in patients with schizophrenia: a meta-analysis, JAMA Psychiatry 73(11) (2016) 1145–1153. [DOI] [PubMed] [Google Scholar]
- [57].Parker DA, Hamm JP, McDowell JE, Keedy SK, Gershon ES, Ivleva EI, Pearlson GD, Keshavan MS, Tamminga CA, Sweeney JA, Clementz BA, Auditory steady-state EEG response across the schizo-bipolar spectrum, Schizophr. Res 209 (2019) 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Goldberg JH, Yuste R, Tamas G, Ca2+ imaging of mouse neocortical interneurone dendrites: contribution of Ca2+−permeable AMPA and NMDA receptors to subthreshold Ca2+dynamics, J. Physiol. (Paris) 551 (Pt 1) (2003) 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gonzalez-Burgos G, Lewis DA, NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia, Schizophr. Bull 38 (5) (2012) 950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA, Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia, Trends Neurosci. 31 (5) (2008) 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nakao K, Nakazawa K, Brain state-dependent abnormal LFP activity in the auditory cortex of a schizophrenia mouse model, Front. Neurosci 8 (2014) 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Javitt DC, Neurophysiological models for new treatment development in schizophrenia: early sensory approaches, Ann. N. Y. Acad. Sci 1344 (2015) 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bickel S, Lipp HP, Umbricht D, Early auditory sensory processing deficits in mouse mutants with reduced NMDA receptor function, Neuropsychopharmacology 33 (7) (2008) 1680–1689. [DOI] [PubMed] [Google Scholar]
- [64].Bitanihirwe BK, Lim MP, Kelley JF, Kaneko T, Woo TU, Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia, BMC Psychiatry 9 (2009) 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM, A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons, J. Neurosci. 26 (5) (2006) 1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]