

Abstract
Purpose:
To determine the pharmacodynamic relationship between target occupancy of Bruton’s tyrosine kinase (BTK) and inhibition of downstream signaling.
Experimental Design:
Patients with Chronic Lymphocytic Leukemia (CLL) enrolled on a phase 2 clinical trial (NCT02337829) with the covalent, selective BTK inhibitor acalabrutinib donated blood samples for pharmacodynamic analyses. Study design included randomization to acalabrutinib 100mg twice daily or 200mg once daily and dose interruptions on day 4 and 5 of the first week. BTK occupancy and readouts of intracellular signaling were assessed sequentially between 4 and 48 hours from last dose.
Results:
Four hours from last dose, BTK occupancy exceeded 96% and at trough, was higher with twice daily, median 95.3%, than with once daily dosing, median 87.6% (p<0.0001). By 48 hours from last dose median free BTK increased to 25.6%. Due to covalent binding of acalabrutinib, free BTK is generated by de novo synthesis. The estimated rate of BTK synthesis varied widely between patients ranging from 3.6% to 31.4% per day. Acalabrutinib reduced phosphorylation of BTK and inhibited downstream BCR and NF-κB signaling. During dosing interruptions up to 48 hours, expression of BCR target genes rebounded, while phosphorylation of signaling molecules remained repressed. In vitro crosslinking of IgM on CLL cells obtained 36 to 48 hours from last dose upregulated CD69, with high correlation between cellular free BTK and response (R=0.7, p≤0.0001).
Conclusions:
Higher BTK occupancy was achieved with twice daily over once daily dosing, resulting in deeper and more sustained inhibition of BCR signaling.
Introduction
Chronic lymphocytic leukemia (CLL) is a mature B-cell malignancy, characterized by defective apoptosis and accumulation of malignant cells in the blood, bone marrow, and lymph nodes [1]. CLL cells depend on survival and proliferation signals from interactions with neighboring cells or soluble factors in their microenvironment [2]. Among several pathways that are implicated in CLL proliferation and survival B-cell receptor (BCR) signaling and anti-apoptotic pathways, in particular B-cell lymphoma 2 protein (BCL2) have emerged as critical[1, 3].
BCR signaling and CLL cell proliferation occur primarily within lymphoid tissues[4]. Gene expression profiling (GEP) of CLL cells purified from lymph node biopsies demonstrated ongoing activation of BCR signaling in lymph node resident cells compared to circulating tumor cells[2]. Ligation of the BCR leads to activation of a signaling network comprising of tyrosine-protein kinase (LYN), spleen tyrosine kinase (SYK), Bruton’s tyrosine kinase (BTK), and Phosphoinositide 3–kinase δ (PI3Kδ). These kinases have been validated in clinical trials as therapeutic targets in CLL[5]. In particular, BTK inhibitors are effective and well-tolerated and have replaced chemoimmunotherapy in all lines of therapy for CLL [6, 7]. BTK, a member of the TEC family of kinases, plays a critical role in the propagation of downstream proliferation and survival signals[2, 8]. BTK signals through phospholipase Cγ2 (PLCγ2) to nuclear factor κB (NF-κB)[3]. BTK also plays a role in chemokine-mediated homing and adhesion of CLL cells to the microenvironment, a critical interaction in CLL pathogenesis[9, 10].
The first in class BTK inhibitor ibrutinib is FDA approved for all lines of therapy in CLL and WM. In addition, ibrutinib is approved for treatment of several types of B-cell Non-Hodgkin lymphomas. Ibrutinib is dosed once daily until disease progression or limiting toxicity. On long-term therapy, toxicity is a common reason for treatment discontinuation[11]. Toxicity, in part, may result from inhibition of kinases other than BTK (including, but not limited to ITK, EGFR, and TEC). Acalabrutinib is a highly selective, potent BTK inhibitor that may have a more favorable safety profile than ibrutinib[12]. Clinical trials have shown acalabrutinib to be efficacious and well-tolerated in CLL and relapsed mantle cell lymphoma[13, 14]. For the latter indication, acalabrutinib was approved by the FDA in 2017.
Both ibrutinib and acalabrutinib, irreversibly inactivate BTK through covalent binding to Cysteine 481 in the ATP binding pocket. Consequently, reactivation of BTK activity requires de novo protein synthesis. The high selectivity for BTK and short half-life of acalabrutinib make twice daily dosing possible and twice daily versus once daily drug administration has been shown to result in higher target occupancy in peripheral blood CLL cells [14]. Whether differences in occupancy translate to more potent inhibition of downstream signaling and at what occupancy threshold signaling can be restored is not known. Herein, we analyzed the in vivo effects of acalabrutinib and investigated the relationship between BTK occupancy and inhibition of BCR signaling.
Patients, materials, and methods
Patients and study design
Patients with relapsed/refractory and high-risk treatment-naïve CLL were enrolled on a phase II, single-center study using acalabrutinib. (www.clinicaltrials.gov; NCT02337829)[15]. Written informed consent was obtained in accordance with the Declaration of Helsinki, applicable federal regulations, and requirements from the local Institutional Review Board. Patients (n=48) were randomized to acalabrutinib 200mg every 24 hours (qd) (n=24) or 100mg every 12 hours (bid) (n=24). Characteristics of 45 patients included in these correlative studies are summarized in Table S1. After dosing for three consecutive days, drug was held for two days followed by continuous daily dosing until disease progression or intolerance. Peripheral blood samples were collected pre-treatment (Pre), at peak (4 hours post dose; day 3), at trough, (12 hours post dose for bid; 24 hours for qd; day 4), at trough+24 hours (36 and 48 hours, respectively, day 5) and at the end of cycle 1 (day 28). Mononuclear cells were isolated using lymphocyte separation medium (ICN Biomedicals, Irvine, CA) and viably frozen in 90% fetal bovine serum (FBS) and 10% dimethyl sulfoxide (DMSO) (Sigma, St. Louis, MO) in liquid nitrogen.
Gene expression
Tumor cells were purified using CD19+ selection (MACS Cell Separation Columns, Miltenyi Biotec, Cambridge, MA). Total RNA was extracted from CD19+ cell pellets using RNeasy (Zymo Research, Irvine, CA), and cDNA was prepared using the High Capacity cDNA RT Kit (Applied Biosystems, Carlsbad, CA). Expression of 11 previously validated BCR and NF-κB target genes (Table S2) was quantified by real-time polymerase chain reaction (RT-PCR) on TaqMan Custom Arrays (384-well plate) on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) and pathway-specific signature scores were determined as previously described[2, 16]. Briefly, the difference in threshold cycle (ΔCt) for each gene of interest was calculated from the Ct of the housekeeping gene (VCP) minus the Ct of the gene of interest. The ΔCt for the pathway-specific genes were averaged into a signature score (five unique genes for BCR, five unique genes for NF-κB, and one gene [CCL4] shared by both pathways).
BTK Target Occupancy ELISA
The percentage of drug-bound BTK in human samples was measured using an ELISA-based method as previously described[17]. OptiPlates (96-well; PerkinElmer) were coated overnight at 4C with anti-BTK antibody (BD Biosciences; Franklin Lakes, NJ) and blocked with BSA the following day for 2 to 3 hours at room temperature. Lysis buffer containing protease inhibitor cocktail (Sigma-Aldrich) was used to lyse PBMC pellets. For each timepoint, the cell lysate was incubated for 1 hour in the presence or absence of acalabrutinib (1μM). Incubation of the cell lysate with an excess of acalabrutinib was used to correct for background signal not related to free BTK. After exogenous treatment with or without acalabrutinib, the cell lysates were incubated with the BTK target occupancy probe ACP-4016 for 1 hour before being plated onto an anti-BTK antibody-coated plate. This probe binds covalently to Cys481 in the ATP pocket in BTK when the ATP pocket is not occupied by a covalent BTK inhibitor. Two-hour incubation was followed by a 1-hour incubation with streptavidin horseradish peroxidase (strep-HRP; ThermoFisher, Waltham, MA) to allow binding between strep-HRP and the probe ACP-4016. Substrate that reacts with the bound strep-HRP was added to allow for measurement of luminescence. The percentage of BTK protein bound by acalabrutinib was calculated using the difference in the luminescence signal of the samples incubated with or without exogenous acalabrutinib (1μM) and is expressed as a percentage of the luminescence signal in the pre-dose sample. The signal of the day 1 (pre-dose) sample without exogenous acalabrutinib represents 100% free BTK, while the signal of the day 1 (pre-dose) sample with 1μM exogenous acalabrutinib represents 0% free BTK (or 100% occupied BTK).
Flow Cytometry
Analysis of phosphoprotein expression was evaluated as previously described[18]. Briefly, anti-CD19 APC was used to define the CLL population. Cells were then fixed with 4% paraformaldehyde (Alfa Aesar, Haverhill, MA), permeabilized with 80% ethanol (Sigma) at −20C and stained with one of the following Alexa Fluor 488-conjugated antibodies (BD Biosciences): IgG1-isotype control, IgG2a-isotype control, pBTKY223, pBTKY551, pPLCg2Y759, pP38Y182, pERKT202/Y204 and pNF-κB pP65S529. Gating strategy and representative histograms are shown in Supplementary Fig. S1. To concurrently analyze two samples in one flow tube, cells were barcoded with pacific blue (Invitrogen), as previously described[18]. Cells were acquired on a BD FACSCanto II and analyzed using FlowJo software (TreeStar, Ashland, OR). MFI is measured after isotype subtraction. Cell surface activation markers were evaluated as previously described[16]. Briefly, PBMCs were stained with: anti-CD19 APC, anti-CD3 FITC, anti-CD5 PE-Cy7 and one of the following PE-conjugated antibodies: IgG1-isotype control, CD69, or CD86 (Fig. S2.)
For direct activation of BCR signaling, PBMCs were stimulated with goat anti-human IgM F(ab’)2 [10μg/mL] + H2O2 [3.3 nM] for 10 minutes at 37°C, as described previously[14]. Samples were fixed with 1.6% paraformaldehyde for 10 minutes at 37°C, and permeabilization with ice cold 90% methanol overnight at −80°C. Samples were washed and stained with rabbit anti-pBTKY223 antibody (Abcam; Cambridge, United Kingdom) plus fluorescently labeled mouse antibodies against human CD20 PerCP-Cy5.5, CD19 PE-Cy7, CD3 BV421, CD5 BV510, and cleaved PARP Alexa Fluor 700 (BD Biosciences). Samples were washed with PBS + 0.5% BSA and incubated with secondary goat anti-rabbit IgG Alexa Fluor 647 (ThermoFisher) for 30 minutes at 4°C. Cells were acquired on a BD FACSVerse flow cytometer and analyzed using FCS Express (De Novo Software, Glendale, CA). The pBTKY223 median fluorescence value (MFI) was collected from the CLL population, defined as cleaved PARP−, CD19+, CD20+, CD5+, CD3− phenotype (Fig. S3.) To measure BCR-induced CD69 expression, PBMCs were stimulated with [10μg/mL] goat anti-human IgM and incubated for 18 hours at 37°C, 5% CO2 in 96-well flat-bottom plates. Cells were stained with anti-CD69 FITC, anti-CD5 Alexa Fluor 700, anti-CD19 BV421, anti-CD3 BV510 antibodies (BD Biosciences) and acquired on FACSVerse, excluding dead cells with 7-AAD viability dye (ThermoFisher). The degree of inhibition was compared to positive controls where a saturating concentration of acalabrutinib (1μM) is incubated with each pre-dose patient sample, prior to ex vivo stimulation (Fig. S4.)
NF-κB activity assay
NF-κB activity was measured using the TransAM NF-κB p50 transcription factor assay kit (Active Motif, Carlsbad, CA) as previously described[19]. Nuclear lysates of purified CLL cells pretreatment, day 3 and 28 were applied to 96-well plates coated with oligonucleotides containing the NF-κB consensus sequence (59-GGGACTTTCC-39). Concentration of p50 was determined by comparing samples with a standard curve of purified p50 protein (Active Motif).
Next generation sequencing (NGS) and IGHV gene analysis
NGS was performed on DNA from isolated B cells using an Illumina TruSeq Custom Amplicon panel of 10 genes associated with CLL on a MiSeq Sequencing instrument. Sequence alignment to hg19 and variant calling were performed using Illumina MiSeq Reporter Software. Average coverage of 500x (bi-directional) and a variant allele frequency >5% was required for reporting. Clinical relevance for each variant was determined according to 2015 ACMG guidelines. IGHV rearrangements were identified by NGS and aligned to reference sequences in the international ImMunoGeneTics (IMGT) database for percent homology (Aegis, Nashville, TN).
Serum levels of CCL4
Serum was stored at −80°C until analysis. Concentrations of CCL4 were measured using the Bio-Plex Pro Human assay (BIO-RAID, Hercules, CA) with the Luminex 200 instrument (Austin, TX) according to the manufacturer’s instructions.
Statistical analysis
To compare measurements in individual patients across time, the Wilcoxon matched-pairs signed-rank test was used. To compare results between the two dosing cohorts, the Wilcoxon rank-sum was used. Correlations were evaluated using the Pearson test. Data were computed using Prism 7 (GraphPad Software, San Diego, CA).
Results
Acalabrutinib demonstrates strong on-target effects with deepening response over time.
We first sought to determine the on-target effects of treatment in circulating CLL cells from patients receiving acalabrutinib, dosed either 200mg once daily or 100mg twice daily. We measured the phosphorylation state of proteins involved in BCR signal transduction by flow cytometry at peak occupancy after three days and after one cycle of treatment (day 28). Compared to baseline, we found significantly reduced levels of phosphorylated BTKY551 on day 3, four hours after acalabrutinib administration (Fig. 1A, median change −16 %; IQR −24 to −6; P=0.001) and on day 28 (Fig. 1A, median change, −25 %; IQR, −33 to −17, P<0.0001). We next evaluated the BTK autophosphorylation site of BTKY223 and PLCγ2Y759, an immediate downstream target of BTK. Both BTKY223 and PLCγ2Y759 phosphorylation were diminished in most patients on day 3 (Fig. 1B–C, respectively) but were only statistically significantly decreased on day 28 (Fig. 1B–C, BTKY223 median change −28%, IQR, −50 to −4; P=0.0004; PLCγ2Y759 median change −23%, IQR, −38 to −7; P=0.0003). Next, we evaluated the activity of MAP kinase signaling. Phosphorylation of P38Y182 and ERKY204 were significantly reduced on both day 3 (Fig. 1D–E, P38Y182 median change −15%, IQR, −33 to 2; P=0.001; ERK T202/Y204 median change −17%, IQR, −21 to −5; P=0.004) and day 28 (Fig. 1D–E, P38Y182 median change, −34%, IQR, −52 to −10; P<0.0001; and ERKY204 median change −27%, IQR, −39 to −15; P<0.0001). Acalabrutinib also significantly reduced phosphorylation of NF-κB P65S529 on day 3 (median change −12%, IQR, −21 to −1; P =0.001) and day 28 (median change −23%, IQR, −35 to −5; P<0.0001; Fig. 1F). Altogether, these results indicate rapid and sustained inhibition of BCR and NF-κB signaling in circulating CLL cells across all patients and both dosing schedules.
Figure 1: Immediate inhibition of BCR signaling with deepening response over time.
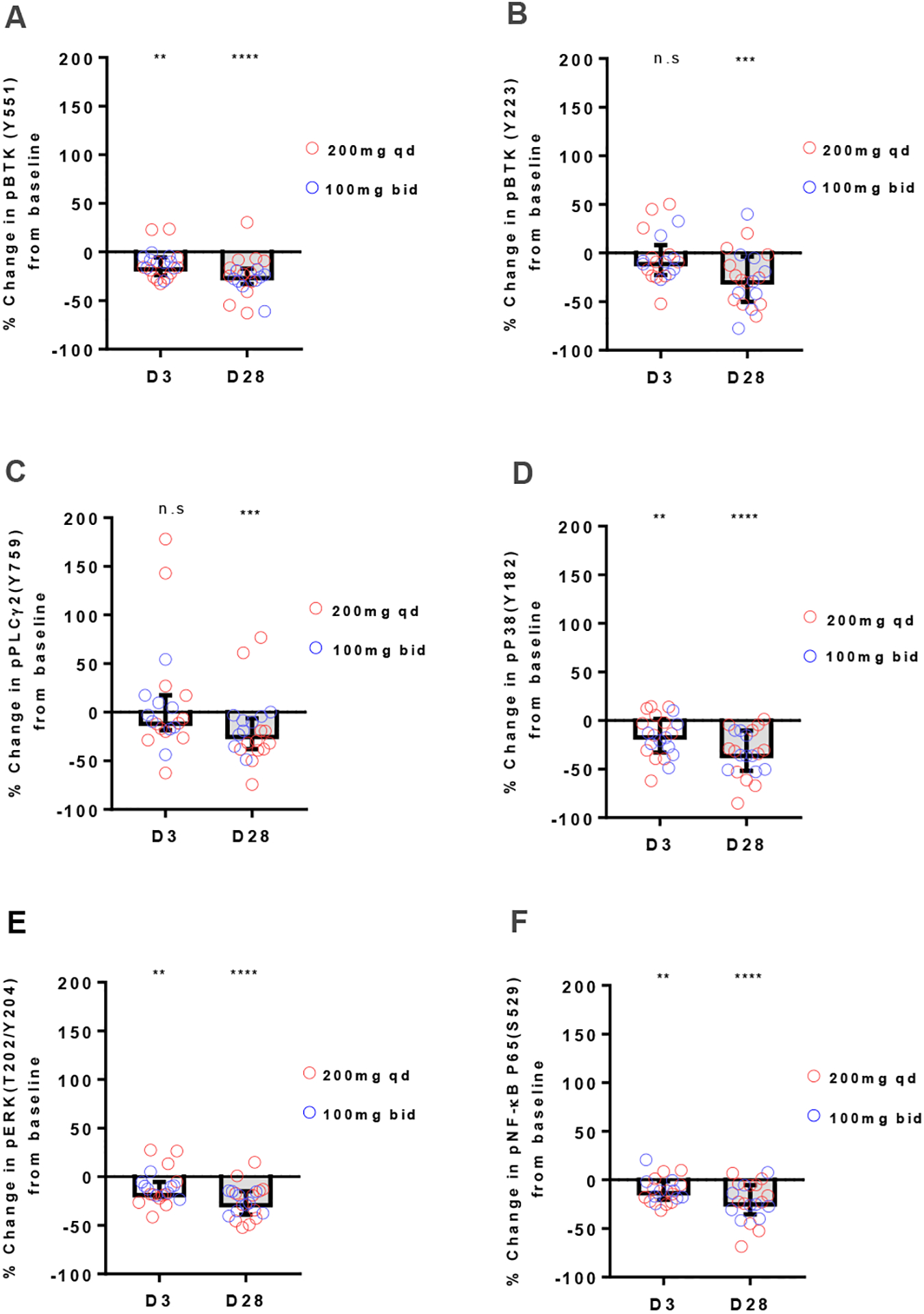
A-F, Levels of phosphorylated signaling molecules measured by flow cytometry on day3 (D3) and day28 (D28) of acalabrutinib therapy are displayed as % change from baseline. Red circles represent 200mg qd dosing; blue circles represent 100mg bid dosing. Box and Whisker plots display median (±IQR) for all patients analyzed (n=21). A, pBTKY551; B, pBTKY223; C, pPLCG2Y759; D, pP38Y182; E, pERKT202/Y204; F, pNF-κB p65S529. Statistical significance by Wilcoxon matched-pairs signed-rank test between baseline and treatment timepoints is indicated: **P<0.01, ***P<0.001, and ****P<0.0001.
Acalabrutinib induces rapid and sustained inhibition of downstream effects of BCR activation.
To assess the cellular response to acalabrutinib, we measured the expression of 11 genes regulated by BCR and NF-κB signaling (Table S2) in purified CLL cells from a representative group of patients using quantitative RT-PCR. As a measure of pathway activation, mRNA levels of the respective target genes were averaged into gene signature scores as previously described[16]. The BCR signature score was significantly reduced on day 3 (median change −2.6, IQR, −3.7 to −2.3; P=0.001) and day 28 (median change −1.8, IQR, −2.8 to −1.1; P=0.002) compared to pre-treatment (Fig. 2A). Consistently, the NF-κB signature score was also significantly reduced on day 3 (median change −1.6, IQR, −2 to −1.5; P=0.001) and day 28 (median change −1.7, IQR, −2.1 to −0.7; P=0.004; Fig. 2B). The reductions in gene signature scores were highly correlated (r=0.8, P<0.0001; Fig. 2C). A significant reduction in nuclear NF-κB p50 was evident on day 28 but not day 3 (Fig. 2D, median change −40%, IQR, −61 to −7, P=0.002). Next, we evaluated serum levels of CCL4, a chemokine secreted by CLL cells in response to BCR and NF-κB activation[20]. Serum levels of CCL4 were significantly decreased by acalabrutinib on day 3 and day 28 (Fig. 2E, P<0.0001) compared to baseline. Lastly, we assessed the effect of acalabrutinib on cellular activation as evidenced by expression of CD69 and CD86 on circulating CLL cells. Cell surface expression of both markers was substantially reduced in all patients on day 3 (CD69 median change −41%, IQR, −65 to −20; P<0.0001; CD86 median change −26%, IQR, −53 to −17; P<0.0001) and day 28 (CD69 median change −62%, IQR, −85 to −48; P<0.0001; CD86 median change −46%, IQR, −60 to −31; P<0.0001; Fig. 1F–G). Taken together, acalabrutinib-mediated inhibition of BTK signaling results in significant changes in gene expression, cell activation, and chemokine secretion. For all these measures of BCR signaling, there was no statistically significant difference between dosing regimens.
Figure 2: Inhibition of downstream effects of BCR activation by acalabrutinib.
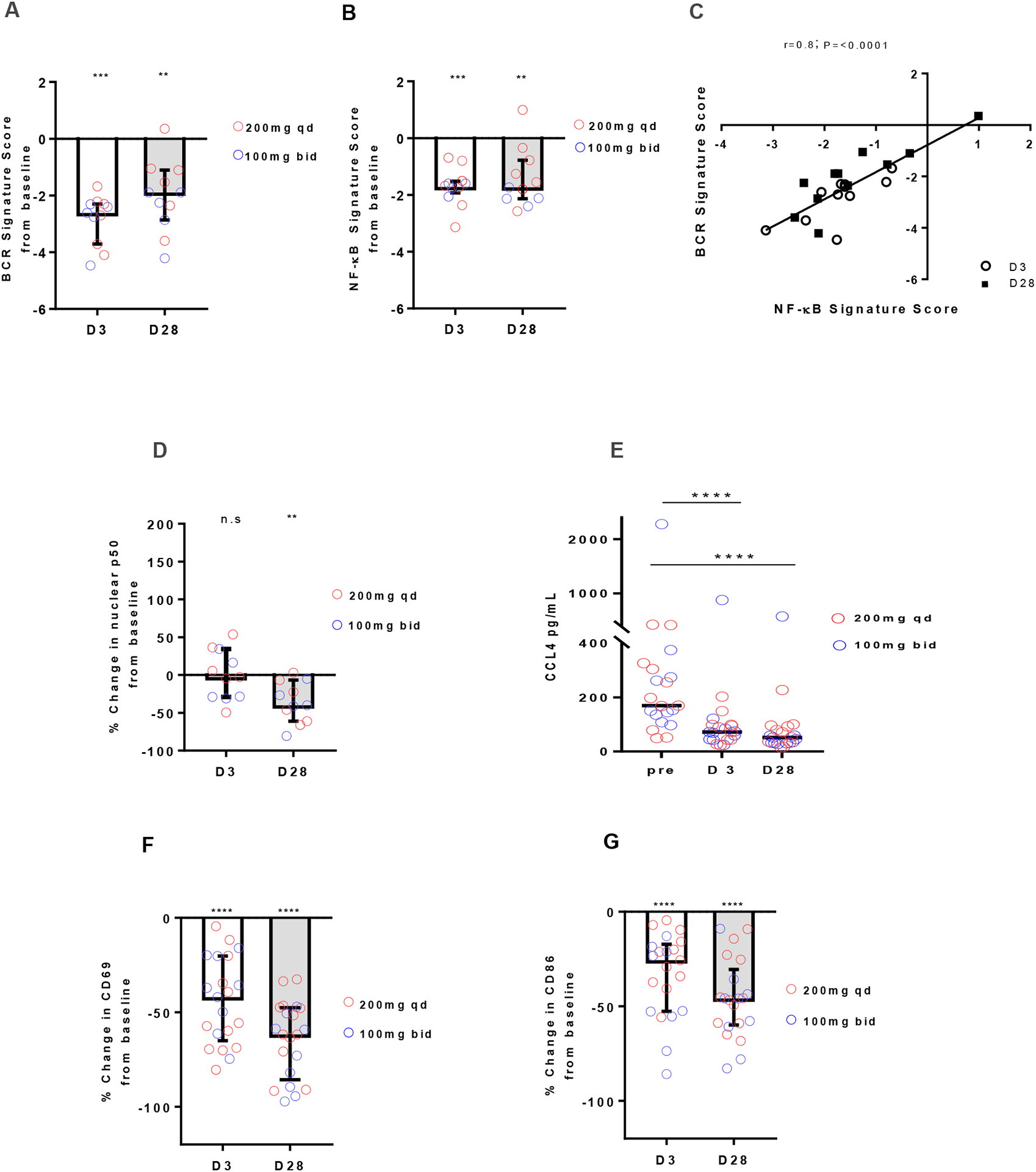
A-B, Change in gene signature score in CD19+ selected CLL cells collected on day 3 (D3) and day 28 (D28) normalized to pre-treatment (n=11). Red circles represent 200mg qd dosing (n=6); blue circles represent 100mg bid dosing (n=5). Bars represent median (±IQR). (A) BCR signature score. (B) NF-κB signature score. C, Pearson correlation between change in BCR and NF-κB signature scores. Each dot represents one patient, circles represents day 3, squares represent day28. R and P values of Pearson correlation are displayed. D, Nuclear expression of NF-κB p50 in CD19+ selected CLL cells by ELISA (n=11). E, CCL4 serum levels measured by Luminex Assays at baseline (Pre), day 3, and day 28 (n=22). F-G Median (±IQR) percent change in MFI measured by flow cytometry (n=21) at day 3 and day 28 compared to baseline for (F) CD69; (G) CD86. Statistical significance by Wilcoxon matched-pairs signed-rank test between baseline and treatment timepoints is indicated: **P<0.01, ***P<0.001 and ****P<0.0001.
BTK synthesis rates are highly variable between patients
Acalabrutinib, like ibrutinib, inhibits BTK by covalently binding to Cys481 in the ATP-binding pocket. Thus, reactivation of signaling requires de novo synthesis of BTK. To estimate the rate of BTK synthesis in vivo we measured free BTK (% unbound by acalabrutinib) in serial blood samples taken on day 3 (peak level), day 4 (trough level); and on day 5 (trough plus 24 hours from last dose of drug). After 3 days of dosing at peak effect (4 hours from dose), virtually all BTK was bound by acalabrutinib and median free BTK levels were ~1.0% in both dosing groups (Fig. 3A). At drug trough time points, the median free BTK was significantly higher in the qd (24 hours post dose) than bid (12 hours post dose) group: (qd median 13.4%, IQR, 10.2% to 16.17% vs bid median: 4.7%, IQR, 3.8 % to 6.3 %; P<0.0001) During the pre-planned treatment interruption there was a further increase in free BTK: at 36 hours (bid cohort) and 48 hours (qd cohort) post dose, median free BTK increased to 19% and 25.6%, respectively. Based on the sequential measurements over time the calculated rate of BTK synthesis was highly variable between patients, ranging from 3.6% to 31.4% per day (Fig. 3B). As expected, there was no difference in BTK synthesis rates between dosing groups (Fig. S5). In addition, we found no correlation between clinical or biologic disease characteristics and the rate of BTK synthesis (Fig. 3B).
Figure 3: Interpatient variability in BTK synthesis rates.
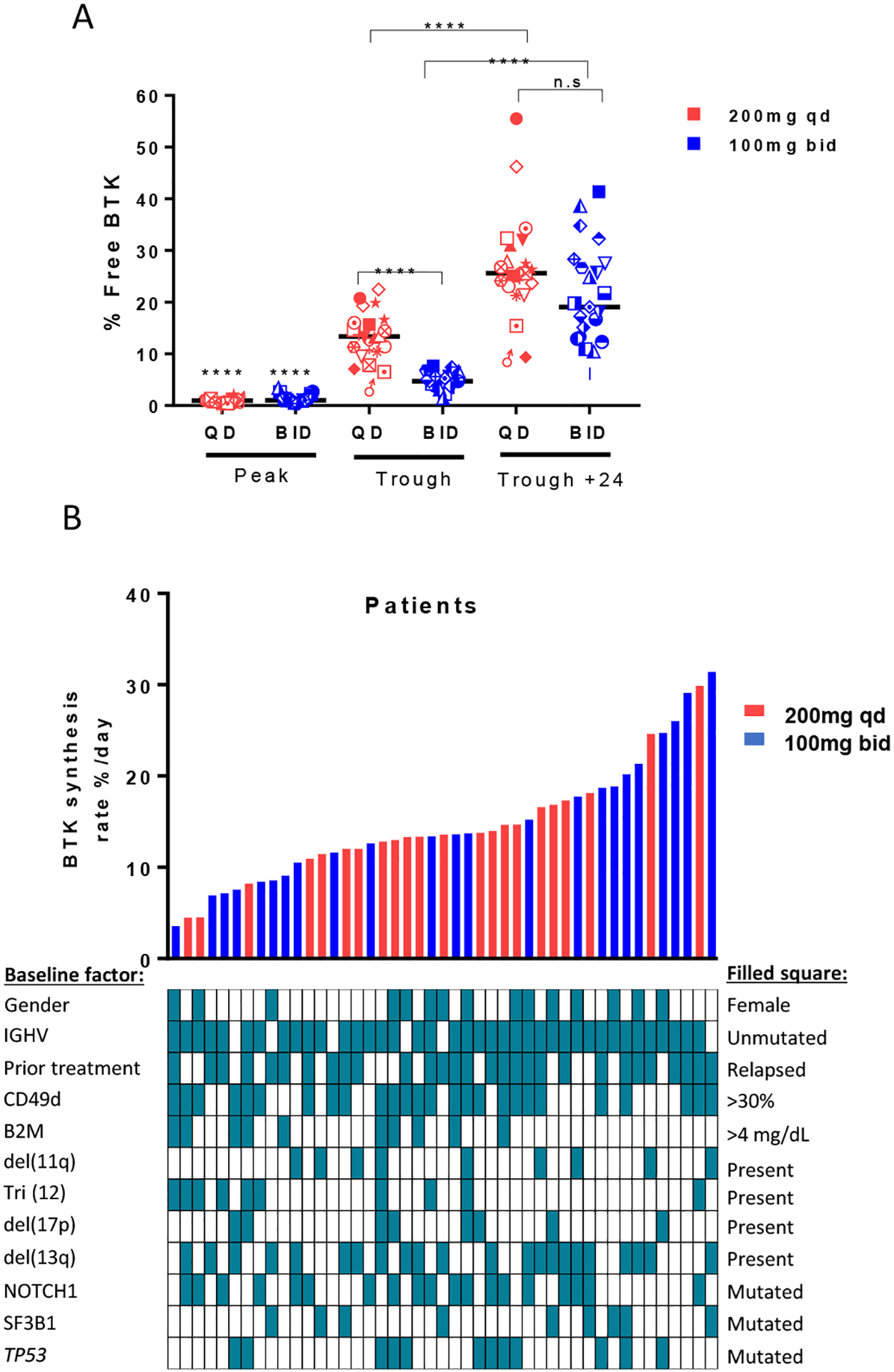
A, percent free BTK on day 3 (Peak), day 4 (Trough) and day 5 (Trough + 24 hours). Black line represents the median. Each symbol represents one patient. Red symbols represent qd dosing; blue symbols represent bid dosing. B, Rate of change in percent free BTK per day for all evaluated patients aligned with different baseline factors; red bars represent 200mg qd patients; blue bars represent 100mg bid patients. Range 3.5 – 31.4%. Linear regression was used to calculate slopes for the return of free BTK and to determine the BTK synthesis rate per day. Statistical significance by Wilcoxon matched-pairs signed-rank test between baseline and treatment timepoints, and Wilcoxon rank-sum test between dose groups is indicated: ****P<0.0001.
Minimal rebound in intracellular signaling in circulating cells up to 48 hours from the last dose
Having observed recovery of free BTK from peak to 48 hours post dose, we sought to investigate the correlation between free BTK and intracellular signaling during the drug withholding period. Samples from subjects showing reduced phosphorylation of signaling molecules at peak were tested at trough plus 24 hours (Table S3). Compared to peak, only pBTK551 increased significantly (Fig. 4A, P=0.03). pBTKY223, pERKY204 and NF-κB pp65S529 remained suppressed up to 48 hours from last dose (Fig. 4B–D). By quantitative RT-PCR, both BCR and NF-κB target gene expression increased from peak to trough levels only in the qd (P=0.03) but not in the bid dosing cohort. Cell surface CD69 expression and CCL4 plasma levels did not rebound from peak inhibition (Fig. 4F–G, respectively). Thus, we found little evidence for reactivation of intracellular signaling in circulating CLL cells up to 48 hours post dose.
Figure 4: On-target effects up to 48 hours from last dose.
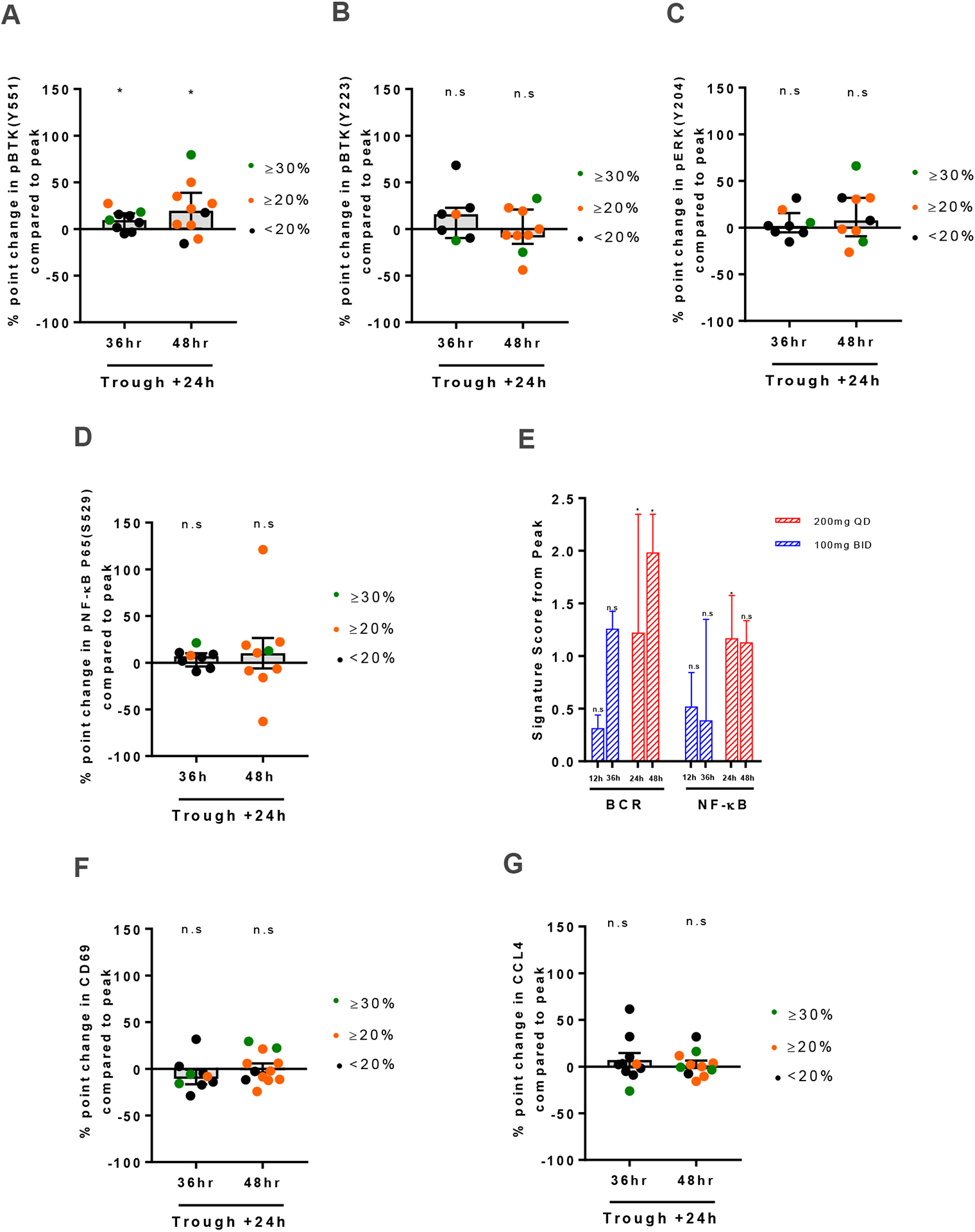
A-D, Levels of phosphorylated signaling molecules measured by flow cytometry at 36 and 48 hours from the last dose of acalabrutinib therapy are displayed as % point change from peak. (A) pBTKY551, (B) pBTKY223 (C) pERKT202/Y204 and (D) pNF-κB p65S529. Only subjects with a significant reduction at peak were included here; Box and Whisker plots display median (±IQR) for all patients analyzed. E, Change in gene signature average in CD19+ selected CLL cells collected 12, 24, 36, and 48 hours from the last dose of acalabrutinib therapy normalized to peak (n=11). Red bars represent 200mg qd dosing (n=6); blue bars represent 100mg bid dosing (n=5). Bars represent median (±IQR). F, Median (±IQR) percent point change in CD69 MFI measured by flow cytometry at 36 and 48 hours from the last dose of acalabrutinib therapy compared to peak (n=21). G, CCL4 serum levels measured by Luminex Assays at 36 and 48 hours from the last dose of acalabrutinib therapy are displayed as % point change from peak (n=22). Green circles represent patients with measured free BTK ≥30%; orange circles represent patients with measured free BTK ≥20% but <30 %; black circles represent patients with measured free BTK <20. Statistical significance by Wilcoxon matched-pairs signed-rank test is indicated: *P<0.05.
Rebound of BCR signaling occurs in the presence of activation signals and correlates with occupancy.
Considering that restoration of BCR signaling may depend not only on free BTK levels, but also the return of circulating cells into the tissue microenvironment where most of the signaling occurs, we tested the response to direct activation of BCR signaling in CLL cells. CLL PBMCs, sampled at different timepoints post-dose, were stimulated with anti-IgM in vitro, and cellular responses were measured by flow cytometry. Compared to samples obtained at peak, samples obtained at trough plus 24 hours (48 hours post- dose), showed partial restoration of BTK signaling as evidenced by increased CD69 expression (Fig.5A, P=0.0005) and pBTKY223 (Fig. 5B, P=0.0006). Samples obtained at trough (24 hours post dose), also showed a statistically significant, albeit more modest rebound from peak inhibition. In contrast, with the bid dosing schedule, only CD69 expression but not pBTKY223 increased significantly in response to IgM engagement, and a response was only detected in samples obtained at trough plus 24 hours but not in samples at trough (Fig. 5C–D). The degree of anti IgM-mediated increase in CD69 correlated with free BTK levels (Fig. 5E, r=0.70; P≤0.000).
Figure 5: Rebound of BCR signaling occurs in the presence of activation signals and correlates with level of free BTK.
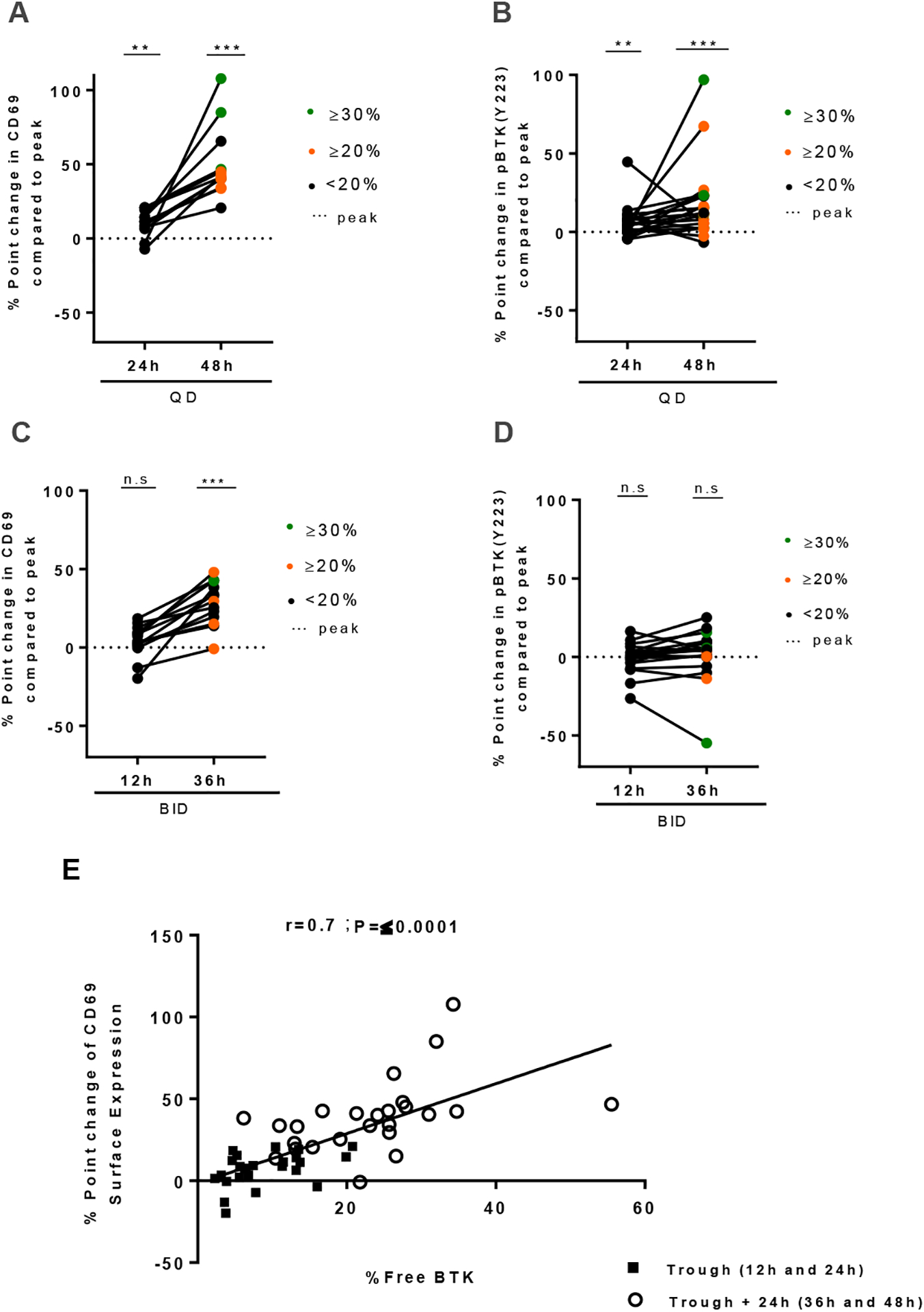
A-B, levels of (A) CD69 expression and (B) pBTKY223 at 24 and 48 hours (qd) from the last dose of acalabrutinib are displayed as % point change from peak. C-D, Levels of (C) CD69 expression and (D) pBTKY223 measured by flow cytometry at 12 and 36 hours (bid) from the last dose of acalabrutinib are displayed as % point change from peak. Statistical significance by Wilcoxon matched-pairs signed-rank test between treatment timepoints is indicated: **P<0.01, ***P<0.001. PBMCs collected at the indicated time points were stimulated with anti-IgM in vitro. Only subjects with day 1 predose ≥ 1.5 fold change over exogenous treatment control (+ 1μM acalabrutinib) were included in the CD69 analysis. In all panels, green circles represent patients with free BTK ≥30%; orange circles represent patients with measured free BTK ≥20% but <30%; black circles represent patients with measured free BTK <20%. E, Pearson correlation of free BTK and % point change in CD69 expression in matched patient samples. Each dot represents one patient, squares represent trough (12h-24h), circles represents trough + 24h (36h-48h).
Discussion
Covalent BTK inhibitors are highly effective in CLL and other B-cell malignancies. Ibrutinib, the first in class, is approved for several indications. More recently, results of clinical trials with two additional covalent BTK inhibitors have been reported: acalabrutinib is approved for the treatment of mantle cell lymphoma [13], and has shown efficacy and excellent tolerability in CLL[12, 14]. Phase I results for zanubrutinib have been reported, and both drugs are being investigated across the spectrum of B-cell malignancies [14, 21]. All three molecules inhibit BTK by covalently binding to Cys481 and achieve sustained kinase inhibition despite short systemic half-life of the drugs. High BTK occupancy has been demonstrated in blood and tissues.[15, 16, 22] Recovery of BTK dependent signaling requires de novo synthesis of BTK. Here we investigated, the relationship between BTK occupancy and inhibition of downstream signaling at different times from last dose. At peak, 4 hours after dose, virtually all BTK molecules were acalabrutinib bound and BTK-dependent signaling was effectively reduced. Levels of free BTK increased with time from last dose, albeit with large interpatient variability. A rebound of cellular signaling as early as 24 hours from last dose was evidenced by increasing expression in genes regulated by BCR or NF-κB signaling. Similarly, in vitro activation of BCR signaling increased pBTKY223 and CD69 expression in cells obtained at least 24 hours from last dose. However, in cells obtained 12 hours from last dose, BTK-dependent signaling remained inhibited. Together, these data demonstrate more potent inhibition of BTK-dependent signaling with twice daily dosing of acalabrutinib and suggest that even minor differences in BTK occupancy could impact efficacy of BTK inhibitors.
On this phase 2 trial, patients were randomly assigned to receive acalabrutinib either 100mg bid or 200mg qd and on day 4 and 5 of the first week were asked to interrupt dosing for 48 hours. This unique design allowed collection of serial samples at peak (4 hours), trough (bid: 12 hours; qd: 24 hours) and trough plus 24 hours from last dose. To measure BTK occupancy, a biotin-tagged analogue probe was used, as previously described[14]. As expected, both dosing regimens completely saturated BTK 4 hours post-dose. However, during the drug withholding period, the two regimens diverged; at trough all patients on bid dosing-maintained occupancy >90%, while only 5 (24%) patients dosed once daily maintained occupancy >90%. Over the next 24 hours, occupancy decreased further. As acalabrutinib binds covalently to C481 in the kinase domain, de novo protein synthesis is required to restore functional BTK. The BTK synthesis rate calculated from sequential occupancy data showed wide interpatient variability with no statistically significant association with biologic or genetic characteristics of the disease. However, all but one of the patients with IGHV mutated CLL had de novo synthesis rates at or below the median. Our study included too few IGHV mutated patients to formally test whether BTK synthesis is higher in IGHV unmutated patients. However, protein synthesis rates have been reported to be higher in BCR signaling competent cells,[23] suggesting that target occupancy might drop off faster in IGHV unmutated CLL.
The relationship between BTK occupancy and the degree of inhibition of downstream signaling is not well understood. At peak and drug trough levels, inhibition of BCR and NF-κB signaling and cell proliferation has been shown for both ibrutinib and acalabrutinib.[14, 16, 24] Here, we conducted a pulse chase type analysis in vivo. As in previous studies,[9, 16, 25] activity of the signaling pathways was estimated from changes in the phosphorylation status of intracellular signaling molecules, expression of activation markers, chemokine secretion, and target gene expression. At peak and trough levels, the expected on-target effects were readily demonstrated in both qd and bid dosing cohorts. However, at trough plus 24 hours, protein-based readouts showed wide variability and only pBTKY551 significantly increased from peak. In contrast, gene signature scores increased significantly at 24 hours but not 12 hours from last dose resulting in statistically significant differences in these measures between the two dosing regimens. The differing impression conferred by gene expression analysis and phospho-flow cytometry is noteworthy. In this context, gene expression may serve as a more sensitive downstream measure of intracellular signaling, due to several advantages: gene signature scores are based on the expression levels of multiple target genes, gene expression is more easily quantified than protein phosphorylation, and transcriptional responses may be more sustained than changes in protein phosphorylation.
A reason for the absence of a more robust rebound in protein phosphorylation in peripheral blood cells during the dose interruption, may be that reconstitution of signaling would require renewed engagement of the BCR within the microenvironment of lymphoid tissues. To test the response of cells obtained at different timepoints from the last dose, we stimulated CLL cells with anti-IgM in vitro. With qd dosing, pBTKY223 levels and CD69 expression showed a modest but significant increase in cells obtained at trough that was even more pronounced 24 hours later (48 hours from the last dose). In contrast, only CD69 expression but not pBTKY223 increased significantly in patients on bid dosing, and this increase was only detected at trough plus 24 hours (36 hours from the last dose). Overall, the extent of reactivation in BCR signaling positively correlated with the cellular free BTK level.
In conclusion, higher BTK target occupancy was achieved with acalabrutinib 100 mg bid than with 200 mg qd. At the respective drug trough levels, inhibition of target gene expression was superior in the bid cohort suggesting that higher BTK occupancy translates into more potent inhibition of pathogenic signals in CLL cells. The response to exogenous BCR activation was correlated with the amount of free BTK in cells and once free BTK reached ~20% of total, response to BCR engagement was at least partially restored. Overall, these data support dosing strategies that maximize BTK occupancy.
Supplementary Material
Translational Relevance.
Covalent inhibitors of Bruton’s tyrosine kinase (BTK) are effective across multiple B-cell malignancies. A measure of on target effects for this class of agents is BTK occupancy. Our pharmacodynamic analyses in patients with chronic lymphocytic leukemia treated with acalabrutinib correlate the level of free BTK with endogenous and exogenously activated BCR and NF-кB signaling. Reactivation of signaling is demonstrable as early as 24 hours from last dose of drug, and responses to exogenous BCR activation are proportional to levels of free BTK generated by de novo synthesis. The rate of BTK re-synthesis shows considerable interpatient variability. Superior inhibition of oncogenic signaling is achieved with twice daily compared to once daily dosing.
Acknowledgements
We thank our patients for participating and donating samples to make this research possible. We thank the NHLBI flow core for help with the Luminex Assays. This work was funded by King Saud bin Abdulaziz University for Health Sciences- Cultural Mission of the Royal Embassy of Saudi Arabia (No:2085374 to A. Alsadhan), the Intramural Research Program of the NHLBI (HL002346-15 to A. Wiestner), and Acerta.
Footnotes
Conflicts of Interest
J.C., M.G., A.H., R.I., E.B., P.P., and T.C. were or currently are full-time employees and shareholders of Acerta Pharma, BV. AW received research funding from Acerta Pharma, a member of the Astra-Zeneca group; Pharmacyclics, an Abbvie company; Merck; and Nurix. All other authors declare no competing interests.
References
- 1.Kipps TJ, et al. , Chronic lymphocytic leukaemia. Nat Rev Dis Primers, 2017. 3: p. 16096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herishanu Y, et al. , The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood, 2011. 117(2): p. 563–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burger JA and Wiestner A, Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer, 2018. 18(3): p. 148–167. [DOI] [PubMed] [Google Scholar]
- 4.Herndon TM, et al. , Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia, 2017. 31(6): p. 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiestner A, The role of B-cell receptor inhibitors in the treatment of patients with chronic lymphocytic leukemia. Haematologica, 2015. 100(12): p. 1495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shanafelt TD, et al. , Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N Engl J Med, 2019. 381(5): p. 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woyach JA, et al. , Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N Engl J Med, 2018. 379(26): p. 2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quiroga MP, et al. , B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel spleen tyrosine kinase inhibitor, R406. Blood, 2009. 114(5): p. 1029–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ponader S, et al. , The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood, 2012. 119(5): p. 1182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Rooij MF, et al. , The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood, 2012. 119(11): p. 2590–4. [DOI] [PubMed] [Google Scholar]
- 11.Maddocks KJ, et al. , Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol, 2015. 1(1): p. 80–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Awan FT, et al. , Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv, 2019. 3(9): p. 1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang ML, et al. , Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med, 2013. 369(6): p. 507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrd JC, et al. , Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med, 2016. 374(4): p. 323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun CC, et al. , Acalabrutinib in Patients with Relapsed/Refractory (R/R) and High-Risk, Treatment-Naive (TN) Chronic Lymphocytic Leukemia (CLL). Blood, 2018. 132(Suppl 1): p. 4424–4424. [Google Scholar]
- 16.Herman SE, et al. , Ibrutinib inhibits BCR and NF-kappaB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood, 2014. 123(21): p. 3286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barf T, et al. , Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J Pharmacol Exp Ther, 2017. 363(2): p. 240–252. [DOI] [PubMed] [Google Scholar]
- 18.Krutzik PO and Nolan GP, Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A, 2003. 55(2): p. 61–70. [DOI] [PubMed] [Google Scholar]
- 19.Duhren-von Minden M, et al. , Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature, 2012. 489(7415): p. 309–12. [DOI] [PubMed] [Google Scholar]
- 20.Herishanu Y, et al. , Biology of chronic lymphocytic leukemia in different microenvironments: clinical and therapeutic implications. Hematol Oncol Clin North Am, 2013. 27(2): p. 173–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tam CS, Zanubrutinib: a novel BTK inhibitor in chronic lymphocytic leukemia and non-Hodgkin lymphoma. Clin Adv Hematol Oncol, 2019. 17(1): p. 32–34. [PubMed] [Google Scholar]
- 22.Tam CSL, et al. , Phase 1 study of selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeomans A, et al. , Engagement of the B-cell receptor of chronic lymphocytic leukemia cells drives global and MYC-specific mRNA translation. Blood, 2016. 127(4): p. 449–57. [DOI] [PubMed] [Google Scholar]
- 24.Cheng S, et al. , BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia, 2014. 28(3): p. 649–57. [DOI] [PubMed] [Google Scholar]
- 25.Herman SE, et al. , Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood, 2011. 117(23): p. 6287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.