

Abstract
Mechanical ventilation (MV) is a life‐saving instrument used to provide ventilatory support for critically ill patients and patients undergoing surgery. Unfortunately, an unintended consequence of prolonged MV is the development of inspiratory weakness due to both diaphragmatic atrophy and contractile dysfunction; this syndrome is labeled ventilator‐induced diaphragm dysfunction (VIDD). VIDD is clinically important because diaphragmatic weakness is an important contributor to problems in weaning patients from MV. Investigations into the pathogenesis of VIDD reveal that oxidative stress is essential for the rapid development of VIDD as redox disturbances in diaphragm fibers promote accelerated proteolysis. Currently, no standard treatment exists to prevent VIDD and, therefore, developing a strategy to avert VIDD is vital. Guided by evidence indicating that activation of the classical axis of the renin‐angiotensin system (RAS) in diaphragm fibers promotes oxidative stress and VIDD, we hypothesized that activation of the nonclassical RAS signaling pathway via angiotensin 1‐7 (Ang1‐7) will protect against VIDD. Using an established animal model of prolonged MV, our results disclose that infusion of Ang1‐7 protects the diaphragm against MV‐induced contractile dysfunction and fiber atrophy in both fast and slow muscle fibers. Further, Ang1‐7 shielded diaphragm fibers against MV‐induced mitochondrial damage, oxidative stress, and protease activation. Collectively, these results reveal that treatment with Ang1‐7 protects against VIDD, in part, due to diminishing oxidative stress and protease activation. These important findings provide robust evidence that Ang1‐7 has the therapeutic potential to protect against VIDD by preventing MV‐induced contractile dysfunction and atrophy of both slow and fast muscle fibers.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Prolonged mechanical ventilation results in ventilator‐induced diaphragm dysfunction (VIDD). This is significant because VIDD is a major risk factor for problems in weaning patients from the ventilator. Currently, no standard treatment exists to prevent VIDD. However, emerging evidence reveals that pharmacological inhibition of the classical axis of the renin‐angiotensin system (RAS) protects against VIDD. Although angiotensin 1‐7 (Ang1‐7) activates the nonclassical arm of the RAS and antagonizes classical RAS signaling, the therapeutic potential of Ang1‐7 to protect against VIDD remains unknown.
WHAT QUESTION DID THIS STUDY ADDRESS?
Is Ang1‐7 a viable therapy to prevent VIDD?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Treatment of animals with Ang1‐7 protected the diaphragm against both MV‐induced diaphragmatic contractile dysfunction and fiber atrophy. Importantly, Ang1‐7 protected against MV‐induced atrophy of both fast and slow‐type fibers and contractile dysfunction.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
These new findings provide a foundation for future testing of Ang1‐7, a potential therapy to protect against VIDD.
INTRODUCTION
Mechanical ventilation (MV) is an important clinical tool used to provide adequate alveolar ventilation to patients during critical illness and surgery. At present, it is estimated that ~40% of patients in the intensive care unit receive MV. 1 Although MV is a life‐saving intervention for patients in respiratory failure, prolonged MV results in the rapid development of inspiratory muscle weakness due to both diaphragmatic atrophy and contractile dysfunction, collectively labeled as ventilator‐induced diaphragm dysfunction (VIDD). 2 The incidence of VIDD is high (~63%) among patients exposed to prolonged MV; this is significant because respiratory muscle weakness is predicted to be a major risk factor for difficulty in weaning patients from ventilator support to independent breathing. 3 The inability to wean patients from MV is an important conundrum that results in extended hospitalization that is associated with increased morbidity and mortality. 3 Unfortunately, a standard treatment to prevent VIDD does not currently exist, and, therefore, developing a therapy to prevent VIDD is vital.
Our understanding of the cell signaling pathways that promote VIDD has increased significantly in recent years. For example, it is now established that MV‐induced increases in the production of reactive oxygen species (ROS) in diaphragm muscle fibers is a required trigger to initiate the MV‐induced increase in diaphragmatic proteolysis, atrophy, and contractile dysfunction. 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 Although MV‐induced ROS production likely occurs at several sites within diaphragm fibers, evidence reveals that both mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase are important sites of ROS emission during prolonged MV. 12 , 13 , 14 , 15 Further, cross‐talk between NADPH oxidase and mitochondria exists whereby accelerated NADPH oxidase‐derived ROS production results in increased mitochondrial ROS emission. 16
A potential nexus between MV and ROS production in diaphragm fibers is the activation of the classical axis of the renin‐angiotensin system (RAS). The RAS is comprised of two signaling pathways known as the classical and the nonclassical axis. Signaling through the classical RAS axis occurs by activation of angiotensin II type I receptors (AT1Rs) located on the cell membrane. 17 Activation of AT1Rs, via circulating angiotensin (Ang)II or mechanical activation of the receptor, triggers a signaling cascade resulting in activation of NADPH oxidase 2 (NOX2) and increased mitochondrial ROS production resulting in protease activation and muscle fiber atrophy. 18 , 19 , 20 , 21 In contrast, the nonclassical arm of the RAS opposes the actions of the classical pathway; this antagonism occurs via Ang1‐7 binding to the G‐protein‐coupled Mas receptor (MasR). Importantly, treatment of animals with Ang1‐7 has been shown to protect locomotor muscles against atrophy due to prolonged inactivity or high circulating levels of AngII. 22 , 23
Recently, our group has confirmed that AT1Rs exist on the sarcolemma of skeletal muscle fibers 24 and we also discovered that activation of the AT1R is required to promote VIDD. Specifically, pharmacological blockade of the AT1R attenuated MV‐induced oxidative stress, contractile dysfunction, and muscle fiber atrophy in the diaphragm. 25 , 26 These results prompted Sigurta et al. to hypothesize that opposing AT1R signaling via infusion of Ang1‐7 can protect against VIDD. 27 The postulate was recently tested by studies concluding that treatment with a low dose of Ang1‐7 does not prevent MV‐induced diaphragm contractile dysfunction but defends against diaphragmatic fiber atrophy. 28 Unfortunately, this investigation suffers from several shortcomings and additional rigorous research is required to confirm the efficacy of Ang1‐7 as a therapy to avert VIDD; this forms the basis for the current experiments. Guided by our preliminary data, we tested the hypothesis that treatment with Ang1‐7 will protect against MV‐induced increases in diaphragmatic oxidative stress, mitochondrial dysfunction, and VIDD.
METHODS
Experimental animals
Our experiments used adult (4–6‐month‐old) female Sprague‐Dawley rats. The experimental animals were housed at the University of Florida Animal Care Center and the experimental protocol was reviewed and approved by the Animal Care and Use Committee at the University of Florida. During the experiments, animals were provided food and water ad libitum and maintained on a 12:12‐h light‐dark cycle.
Experimental design
To address the postulate that Ang1‐7 infusion will lessen MV‐induced upsurges in mitochondrial ROS emission in the diaphragm and defend against VIDD, animals were randomly allocated into one of four experimental groups: (1) control animals (CON; 12 h of spontaneous breathing with saline infusion; n = 11); (2) mechanically ventilated animals, treated with saline infusion and exposed to 12 h of MV (Saline MV, n = 11); (3) spontaneous breathing animals exposed to 12 h of spontaneous breathing with Ang1‐7 infusion (Ang1‐7 SB, n = 10); and (4) mechanically ventilated animals treated with 12 h of MV with Ang1‐7 infusion (Ang1‐7 MV, n = 10).
Mechanical ventilation and spontaneous breathing
Animal surgical procedures followed rigorous aseptic techniques. All animals were anesthetized using an i.p. injection of pentobarbital sodium (60 mg/kg) and after achieving a surgical plane of anesthesia, all animals were tracheostomized. For monitoring blood pressure and sampling of arterial blood, an arterial catheter was placed in the carotid artery. Arterial blood samples were removed every 2–4 h and used to monitor blood pH and partial pressures of arterial oxygen and carbon dioxide. The jugular vein was also cannulated with a catheter attached to a “y” connector to facilitate a continuous infusion of anesthesia (20 mg/kg/h of sodium pentobarbital) and the respective treatment of Ang1‐7 or saline. Animals in SB groups (CON and Ang1‐7 SB) breathed spontaneously for 12 h. Animals in MV groups (Saline MV and Ang1‐7 MV) were connected to a neonatal ventilator (Siemens 300A, Munich, Germany) and full‐support MV was provided for 12 h, as described previously. 29 This duration of MV was chosen because this time interval results in a highly reproducible level of VIDD. 2 To minimize airway secretions, all animals received intramuscular injections of glycopyrrolate (0.04 mg/kg) every 2 h. After 12 h of MV, the full costal diaphragm was harvested and sectioned for assessment of functional, histological, and biochemical properties.
Drug administration
Animals in the Saline MV group received an intravenous infusion of sterile saline throughout the 12 h experimental period. Animals treated with Ang1‐7 received intravenous infusion of Ang1‐7 (300 µg/kg/h) throughout the 12‐h experimental period. This dose of Ang1‐7 was selected for study based upon results from preliminary dose‐response experiments. Briefly, we first determined the highest infusion rate of Ang1‐7 that can be provided to animals during MV while maintaining a mean arterial blood pressure of greater than or equal to 70 mmHg (Figure S1). After determining that 300 µg/kg/h is the highest acceptable rate of Ang1‐7 infusion, we then performed dose‐response experiments infusing Ang1‐7 (i.e., 50, 150, 200, and 300 µg/k/h) to animals (n = 2–6/group) during 12 h of MV. These pilot experiments revealed that the infusion rate of 300 µg/kg/h provided the greatest protection against VIDD (Figure S1).
Measurement of in vitro diaphragmatic contractile properties
Contractile properties of the costal diaphragm were measured in vitro, as previously described. 27 Briefly, to evaluate diaphragmatic contractile properties, a strip of diaphragm muscle was hung upright using two lightweight Plexiglas clamps; the uppermost end of the muscle was attached to a force transducer (model FT‐03; Grass Instruments). The muscle was housed in a temperature‐controlled chamber (maintained at 25°C) containing Krebs‐Hensleit solution equilibrated with 95% O2‐5% CO2 gas. The optimal force producing length of the muscle (L0) was determined in step‐wise increments of length change. After determining L0, the muscle strip was electrically stimulated to contract at stimulation frequencies of 15, 30, 60, and 100 Hz. The muscle force production measurements were captured using a computerized system (National Instruments Corporation). 29 Muscle force production was recorded as specific force (i.e., force production normalized to muscle cross‐sectional area).
Isolation of diaphragm mitochondria
As described previously, ~300 mg of costal diaphragm muscle was homogenized to isolate mitochondria. 12 Briefly, a section of diaphragm muscle was placed in isolation buffer composed of: 100 mM KCl, 40 mM Tris HCl, 10 mM Tris base, 1 mM MgSO4, 0.1 mM EDTA, 0.2 mM ATP, and 2% (wt/vol) free fatty acid bovine serum albumin (pH 7.4). The diaphragm was then minced with scissors, homogenized with a polytron homogenizer, and incubated with trypsin (5 mg/g wet muscle). Trypsin digestion was terminated after 7 min of incubation by addition of an equal volume of isolation buffer. The sample was then centrifuged at 500 × g for 10 min at 4°C and the supernatant was decanted through a layer of cheesecloth followed by another centrifugation step at 3500 × g for 10 min. The mitochondrial pellet was then washed twice with isolation buffer (once with bovine serum albumin [BSA] and once without BSA) followed by centrifugation at 3500 × g for 10 min. The final mitochondrial pellet was suspended in 200 μl of a solution containing 220 mM mannitol, 70 mM sucrose, 10 mM Tris HCl, and 1 mM EGTA (pH 7.40).
Assessment of mitochondrial respiration
Mitochondrial respiration was evaluated using a temperature controlled (37°C) respiration chamber (Hansatech Instruments). Following careful calibration of the oxygen electrode, a 10 μl sample of isolated mitochondria was incubated with 1 ml of respiration buffer Z. Complex I flux was determined using 5 mM pyruvate and 2 mM malate. State 3 respiration (i.e., ADP‐stimulated respiration) was measured by addition of 0.25 mM adenosine diphosphate (ADP) to the chamber. State 4 respiration (basal) was evaluated at the conclusion of state 3 respiration measurements. By convention, the respiratory control ratio (RCR) was computed as the ratio of oxygen consumption during state 3 respiration divided by the oxygen consumption during state 4 respiration.
Measurement of diaphragm mitochondrial ROS production
The rate of mitochondrial H2O2 emission was measured using the oxidant sensitive probe, Amplex UltraRed (Molecular Probes). This method of evaluating H2O2 emission from mitochondria uses horseradish peroxidase to catalyze the H2O2‐dependent oxidation of nonfluorescent AmplexM UltraRed to fluorescent resorufin; this product reflects the accumulation of H2O2 and provides an estimation of superoxide production within mitochondria. Excitation and emission were set at 565 and 600 nm, respectively. Measurements were performed at 30‐s intervals during a 30‐min period; H2O2 production was subsequently established using a standard curve. Mitochondrial protein was determined using the Bradford method and rate of ROS production was expressed as H2O2 emission in nmol/min/mg of mitochondrial protein.
Diaphragm myofiber cross‐sectional area
To determine diaphragm fiber cross‐sectional area (CSA), frozen strips of diaphragm muscle were sectioned into 10 μm segments (HM 550 Cryostat; Thermo Scientific, Waltham, MA). Muscle sections were then stained for dystrophin (Thermo Scientific #RB‐9024‐R7), myosin heavy chain I (Hybridoma Bank A4.840 s IgM 1:15), and myosin heavy chain IIa (Hybridoma Bank SC‐71 IgG 1:50) for CSA analysis. After identifying type I and IIa fibers, the remaining muscle fibers were classified as type IIx/IIb fibers. For each muscle sample, Image J software package was used to determine fiber CSA by measurement of the CSA of 300–600 fibers from two or more different areas of the mid‐region of diaphragm muscle samples (National Institutes of Health).
Western blotting to determine the relative abundance of diaphragmatic proteins
Western blot analysis was performed by homogenizing diaphragm muscle samples in 10 volumes of homogenate buffer (5 mM Tris and 5 mM EDTA with 1% Triton X‐100 at a pH of 7.4) containing a cocktail of protease inhibitors (buffer, Sigma‐Aldrich; protease cocktail, PhosSTOP [Roche]). The homogenized samples were then centrifuged at 1500 × g for 10 min at 4°C. Following 3 washes (with 10 volumes of homogenate buffer), the insoluble pellets were then suspended in 10 volumes of lysis buffer (5 mM Tris; pH = 7.4, 250 mM NaCl, 1% [w/v] sodium dodecyl sulfate [SDS]) and centrifuged again at 17,000 × g for 5 min at 4°C. A sample of homogenate (80 μl) was then added to 20 μl of 5× lysis buffer (5% [w/v] SDS, 1250 mM NaCl) and centrifuged at 17,000 × g for 5 min at 4°C. The fractionated supernatants were removed and the protein concentration was measured using the Bradford technique (Sigma‐Aldrich, B6916). Following adjustment of protein concentrations, the appropriate volume of sample was then added to 2× Laemmli sample buffer (1610747, Bio Rad) containing 5% dithiothreitol.
Western blotting was performed by loading protein samples on 4–20% gradient Criterion TGX gels (Bio‐Rad) and run at 150 V for 65 min. Upon completion, the proteins were transferred onto a low fluorescence polyvinylidene difluoride (LF‐PVDF) membrane that was blocked with 5% non‐fat dry milk (Millipore). Following transfer, all membranes were incubated at 4°C for ~12 h with primary antibodies of interest; αII‐spectrin (1:250; Santa Cruz, sc48382), 4‐hydroxynonenal (4‐HNE; 1:500; Abcam, ab46545), mono‐ and polyubiquitinated conjugates (1:1000; Enzo Therapeutics, Inc., FK2), and Mas receptor (1:1000; Novus Biologicals, NBP1‐78444). After several washes, the membranes were exposed to either IRDye 680RD or 800CW goat anti‐rabbit or mouse IgG secondary antibody (1:10,000; Li‐Cor Biosciences) for 1 h at 25°C. Membranes were then scanned and analyzed using the Li‐Cor Odyssey Infrared Imager (Li‐Cor Biosciences) using Odyssey 2.1 software. All Western blots except for phosphorylated proteins were normalized using the total protein contained within in each lane labeled with Revert (Li‐Cor Biosciences). All analyses were performed using the Image Studio software package (Li‐Cor Biosciences, version 5.2.5).
Total RNA extraction from diaphragm muscle tissue
Using the manufacturer’s recommended protocol, total RNA was extracted from diaphragm muscle fibers (~30 mg) by homogenizing in TRIZOL reagent (Life Technologies). Briefly, diaphragm samples were homogenized in 1 ml of TRIZOL Reagent (Invitrogen) and added to 200 μl of 1‐bromo‐3‐chloropropane. The mixture was then shaken, incubated for 3 min at room temperature, and then centrifuged at 13,000 × g for 15 min (4°C). To achieve RNA precipitation, the supernatant was then mixed with 0.5 ml isopropanol, incubated for 10 min at 22°C, and then centrifuged for 10 min at 13,000 × g. The resulting supernatant was eliminated and 1 ml of 75% ethanol was added to the pellet; this was followed by 10 min of centrifugation at 13,000 × g. The resulting pellet was then washed with 75% ethanol, and centrifuged again for 10 min at 13,000 × g. The resulting pellet was then dried at room temperature and later dissolved in 30 μl of RNase‐free water. The purity and concentration of total RNA was then determined (A260/280 and A260/230, NanoDrop ND‐2000, Thermo Fischer Scientific). Reverse transcription was achieved using SuperScript VILO MasterMix (Invitrogen). Polymerase chain reactions (PCRs) were performed using TaqMan Universal Master Mix II (Thermo Fischer Scientific).
Quantitative PCR
Using the StepOne Plus Real‐Time PCR System, the reaction conditions for PCR conditions were: 95°C for 10 min followed by 40 cycles at 95°C for 15 s, 40 cycles at 60°C for 1 min, followed by a final cycle at 37°C for 30 s (Thermo Fischer Scientific). The mRNA levels for both atrogin‐1/MAFbx (Rn00591730_m1) and MuRF1 (Rn00590197_m1) were determined using a TaqMan gene expression assay (Thermo Fischer Scientific); mRNA values were normalized to GUSB (Rn00667869_m1) mRNA levels. All measurements were performed in duplicate and the 2−ΔΔCt method was applied for data analysis (cycle threshold [C t] = C t [gene of interest] − C t [reference gene]). 30 Finally, the changes (ΔΔC t) in target gene expression were calculated by subtracting the ΔC t of the diaphragm muscle.
Statistical analysis
Statistical comparisons between groups were performed for each dependent variable using a one‐way or two‐way analysis of variance (ANOVA); when indicated, a Sidak’s post hoc test used to determine group differences when indicated. A value of p less than 0.05 was selected a priori as the benchmark for statistical significance. All analyses were performed using a commercial statistical analysis package (Prism, version 6.0; GraphPad Software). Results are presented as mean ± SD.
RESULTS
Body weight and systemic responses to prolonged MV
The body weight and systemic and physiological responses to MV are provided in Table 1 and Figure 1. Notably, no significant differences existed in body weights between the experimental groups at the conclusion of the experiment. However, differences existed in heart rate, arterial blood gases, and pH between the Ang1‐7 SB and both MV groups. Importantly, arterial blood pressure was well maintained during 12 h of MV and did not differ between the two MV groups (Figure 1). Finally, body temperature was well‐maintained within normal ranges (36.5–37.5°C) in all experimental groups during MV (data not shown).
Table 1.
Initial body weight, and blood gas at completion of 12 h of MV
| CON (n = 11) | Saline MV (n = 11) | Ang1‐7 SB (n = 10) | Ang1‐7 MV (n = 10) | |
|---|---|---|---|---|
| Body weight, g | 336.7 (18.1) | 335.3 (18.0) | 335.0 (25.1) | 338.0 (22.1) |
| HR, beat/min | N/A | 327.6 (11.5) | 366.0 (19.0) θ | 336.0 (15.4) |
| pH | N/A | 7.44 (0.02) | 7.39 (0.05) θ | 7.46 (0.02) |
| PCO2, mmHg | N/A | 35.3 (2.5) | 46.3 (7.3) θ | 33.4 (2.9) |
| PO2, mmHg | N/A | 79.2 (4.1) | 51.6 (3.2) θ | 80.4 (3.8) |
| Hct (%) | N/A | 33.4 (2.3) | 34.5 (1.7) | 34.8 (2.7) |
Values are means (SD).
Abbreviations: CON, control; Ang1‐7 MV, angiotensin1‐7 12 h of CMV with Ang1‐7 infusion; Ang1‐7 SB, 12 h of spontaneous breathing (SB) with Ang1‐7 infusion; Hct, hematocrit; HR, heart rate; MV, mechanical ventilation; N/A, not applicable; Saline MV, 12 h CMV with saline.
θ = Ang1‐7 SB different (p < 0.05) from Saline MV and Ang1‐7 MV.
Figure 1.

Changes in the blood pressure during 12 h of mechanical ventilation (MV). Values are presented as means ± SD. CON, control; Saline MV, 12 h MV with saline; angiotensin (Ang)1‐7 SB, 12 h of spontaneous breathing (SB) with Ang1‐7 infusion; Ang1‐7 MV, 12 h of MV with Ang1‐7 infusion. Θ = Ang1‐7 SB different (p < 0.05) from both Saline MV and Ang1‐7 MV
Ang1‐7 provides partial protection against MV‐induced diaphragmatic contractile dysfunction
To determine if Ang1‐7 protects against MV‐induced diaphragmatic contractile dysfunction, we measured diaphragmatic specific force production in vitro at stimulation frequencies ranging from 15 to 160 Hz. Note that peak diaphragmatic force production was achieved at 100 Hz and therefore Figure 2 presents diaphragmatic force development ranging from 15 to 100 Hz. Compared with control, prolonged MV significantly reduced diaphragmatic specific force production by 10–20% at 15, 60, and 100 Hz. Importantly, treatment with Ang1‐7 protected the diaphragm against MV‐induced declines in diaphragmatic specific force production at both submaximal and maximal stimulation frequencies. Specifically, diaphragmatic‐specific force production did not differ between control and the ANG1‐7 MV group at any stimulation frequency (Figure 2).
Figure 2.

Diaphragm specific force. (a) 15, (b) 30, (c) 60, and (d) 100 Hz. Values are presented as means ± SD. * = different (p < 0.05) from CON. # = different (p < 0.05) from Ang1‐7 SB. Ang, angiotensin; CON, control; MV, mechanical ventilation; SB, spontaneous breathing
Ang1‐7 protects against MV‐induced diaphragmatic atrophy
To establish whether Ang1‐7 infusion protects against MV‐induced diaphragmatic atrophy, the size of diaphragmatic myofibers were compared between experimental groups (Figure 3). As expected, prolonged MV resulted in atrophy of all diaphragm fiber types. Importantly, treatment with Ang1‐7 protected diaphragm fibers against MV‐induced atrophy.
Figure 3.

Myofiber cross‐sectional area in the costal diaphragm. (a) Type IIx/b, (b) type IIa, and (c) type I. Values are presented as means ± SD. * = different (p < 0.05) from CON; # = different (p < 0.05) from Ang1‐7 SB; § = different (p < 0.05) from Ang1‐7 MV. Ang, angiotensin; CON, control; MV, mechanical ventilation; SB, spontaneous breathing
Impact of Ang1‐7 infusion on MV‐induced diaphragmatic oxidative stress and mitochondrial function
After determining that Ang1‐7 is protective against VIDD, we then examined the potential mechanisms contributing to this protection. Because mitochondrial uncoupling increased mitochondrial ROS emission, and oxidative stress in the diaphragm are all hallmarks of VIDD, we determined if Ang1‐7 protected the diaphragm against these damaging events. Our results reveal that treatment with Ang1‐7 defended against MV‐induced reductions in the mitochondrial RCR, increases in mitochondrial ROS emission, and prevented MV‐induced increases in the abundance of 4 HNE‐conjugated myofibrillar proteins in the diaphragm (i.e., biomarker of lipid peroxidation and oxidative stress; Figure 4).
Figure 4.
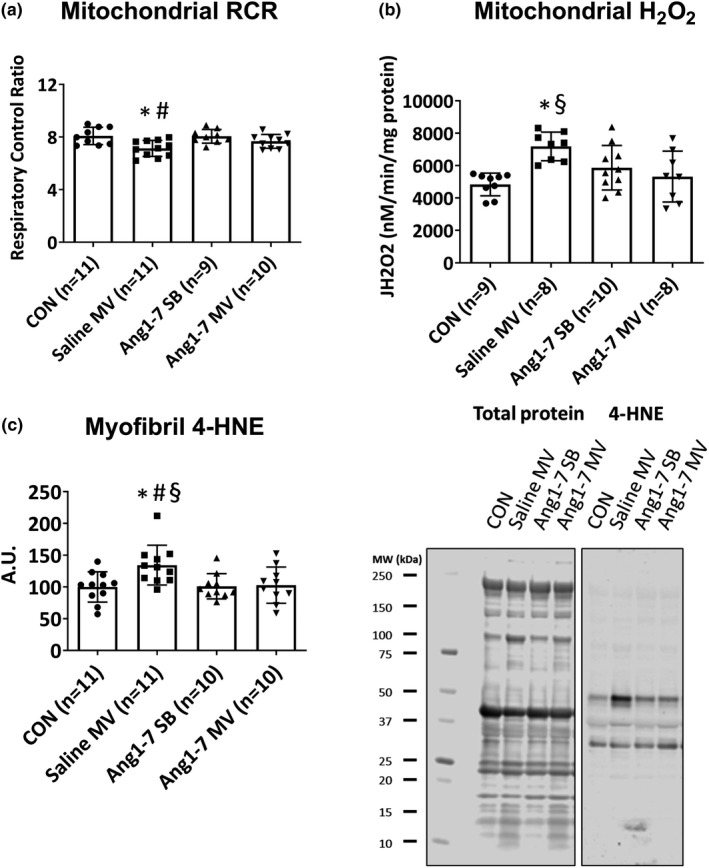
Impact of Ang1‐7 infusion on mitochondrial respiration (a), hydrogen peroxide emission (b), and expression of four hydroxynonenal (c), in the diaphragm. Values are presented as means ± SD. * = different (p < 0.05) from CON; # = different (p < 0.05) from Ang1‐7 SB; § = different (p < 0.05) from Ang1‐7 MV. Ang, angiotensin; CON, control; MV, mechanical ventilation; RCR, respiratory control ratio; SB, spontaneous breathing
Ang1‐7 prevents MV‐induced activation of proteolytic pathways in the diaphragm
Because accelerated proteolysis is a major contributor to VIDD, we determined the impact of Ang1‐7 treatment on MV‐induced activation of major proteases in the diaphragm. First, because prolonged MV is associated with an increase in mRNA of muscle specific E3 ligases and ubiquitinated proteins in the diaphragm, we examined these elements of the ubiquitin‐proteasome system by measuring the mRNA levels of both atrogin‐1/MAFbx and MuRF1 mRNA along with abundance of ubiquitinated myofibrillar proteins within the diaphragm. Our results reveal that treatment of animals with Ang1‐7 did not prevent the MV‐induced increase in both atrogin‐1/MAFbx and MuRF1 mRNA in the diaphragm (Figure 5). Nonetheless, Ang1‐7 diminished the MV‐induced increase in diaphragmatic levels of ubiquitinated myofibrillar proteins within the diaphragm (Figure 5).
Figure 5.

E3 ubiquitin ligase Atrogin‐1 (a) and MuRF1 (b) mRNA and ubiquitinated protein (c) expression in the diaphragm. Values are presented as means ± SD. Log2 fold changes are shown for Atrogin‐1 and MuRF1 mRNA levels. * = different (p < 0.05) from CON; # = different (p < 0.05) from Ang1‐7 SB; § = different (p < 0.05) from Ang1‐7 MV. Ang, angiotensin; CON, control; MV, mechanical ventilation; RCR, respiratory control ratio; SB, spontaneous breathing
To determine if Ang1‐7 protects the diaphragm against MV‐induced activation of calpain and caspase‐3, we measured the abundance of calpain and caspase‐3–specific degradation products of α‐II spectrin. Our findings reveal that Ang1‐7 treatment protected the diaphragm against the MV‐induced activation of caspase‐3, as evidenced by the lower abundance of the caspase‐3 specific 120 kDa α‐II spectrin degradation product in the diaphragm (Figure 6). Although abundance of the calpain‐specific 145 kDa α‐II spectrin degradation product tended to increase in the diaphragm during prolonged MV, this increase was not significantly different from control (i.e., p = 0.064). Nonetheless, together, our findings support the concept that treatment with Ang1‐7 protects the diaphragm against MV‐induced increases in proteolysis via the ubiquitin‐proteasome system and caspase‐3.
Figure 6.

Levels of the 145 kDa (a) and 120 kDa (b) α‐II spectrin breakdown product in the diaphragm. Values are presented as means ± SD. * = different (p < 0.05) from CON; # = different (p < 0.05) from Ang1‐7 SB; § = different (p < 0.05) from Ang1‐7 MV. Ang, angiotensin; CON, control; MV, mechanical ventilation; RCR, respiratory control ratio; SB, spontaneous breathing
Diaphragm Mas receptor levels
Because the cellular effects of Ang1‐7 are mediated via the Mas receptor, we determined the abundance of Mas receptor protein in the costal diaphragm in the experimental animals. Notably, no differences existed between experimental groups in diaphragmatic Mas receptor protein expression (Figure S2).
DISCUSSION
Overview of principal findings
This study tested the hypothesis that infusion of Ang1‐7 protects against MV‐induced diaphragmatic mitochondrial dysfunction, oxidative stress, and VIDD. Our data support this postulate as infusion of Ang1‐7 shielded the diaphragm against both MV‐induced diaphragmatic contractile dysfunction and fiber atrophy. Moreover, Ang1‐7 prevented MV‐induced mitochondrial dysfunction and oxidative stress in diaphragm muscle fibers. Importantly, Ang1‐7 also prevented MV‐induced activation of the ubiquitin‐proteasome system and caspase‐3 in diaphragm muscle. A brief discussion of each of these important findings follows.
Ang1‐7 protects against MV‐induced diaphragm contractile dysfunction
Our data reveal that treatment with Ang1‐7 protects against MV‐induced diaphragmatic contractile dysfunction at a wide range of stimulation frequencies. Our finding that Ang1‐7 protects the diaphragm against MV‐induced contractile dysfunction directly opposes the results of a previous report concluding that Ang1‐7 does not rescue MV‐induced diaphragmatic contractile dysfunction. 28 Several possible explanations exist for this disparity. First, compared with Zambelli et al., the current study treated animals with a fivefold higher dose of Ang1‐7. 28 Second, animals in the current study were not provided with neuromuscular blocking agents. In contrast, Zambelli et al. treated animals with the neuromuscular blocking agent rocuronium bromide that has been shown to decrease diaphragmatic force production at all stimulation frequencies. 31 Last, the diaphragmatic‐specific forces reported by Zambelli et al. are 50–60% lower than other reports 32 , 33 , 34 , 35 ; therefore, these low levels of diaphragmatic force production raise technical concerns about the validity of the diaphragm force measurements in these experiments.
The discovery that Ang1‐7 protects against MV‐induced reductions in diaphragmatic‐specific force production at stimulation frequencies at 15 and 100 Hz is important. Indeed, the motor unit discharge rate for the rat diaphragm ranges from ~27 to 37 Hz during eupnea; this discharge rate increases to ~30–45 Hz during exposure to hypoxia/hypercapnia and peaks at ~60–70 Hz during deep breathing. 36 Therefore, Ang1‐7 protected the rat diaphragm against MV‐induced contractile dysfunction at motor neuron firing frequencies within the physiological range for normal ventilatory behaviors.
Ang1‐7 protects against MV‐induced diaphragmatic atrophy
Zambelli et al. recently reported that Ang1‐7 protects against MV‐induced diaphragmatic atrophy in rodents; however, this study did not examine which diaphragmatic fiber types were protected against atrophy. 28 Therefore, the current study provides the first evidence that Ang1‐7 protects all diaphragm muscle fiber types (i.e., type I, IIa, and IIx/b fibers) against MV‐induced atrophy. Given that a reduction in muscle fiber size translates into a decrease in force‐generating capacity, the rescue of diaphragm fibers from MV‐induced atrophy is physiologically significant.
Mechanisms responsible for Ang1‐7‐mediated protection against VIDD
As discussed earlier, our understanding of the pathogenesis of VIDD has advanced in recent years. Specifically, it is established that MV‐induced increases in mitochondrial ROS production within diaphragm muscle fibers is required to initiate signaling events that trigger protease activation leading to the diaphragmatic fiber atrophy and contractile dysfunction associated with prolonged MV. Hence, to protect against VIDD, a therapeutic agent must disrupt either the MV‐induced increased ROS production and/or protease activation. A short overview of potential mechanisms linking Ang1‐7 to the protection against VIDD follows.
As previously mentioned, stimulation of AT1Rs initiates a signaling cascade that activates NOX2 to increase ROS production. In particular, a crosstalk exists between NOX2 and mitochondria whereby the elevated ROS production by NOX2 results in increased mitochondrial ROS production. 16 Interestingly, this amplified mitochondrial ROS emission acts to further activate NOX2 to produce more ROS. 16 Together, this oxidant production in muscle fibers results in redox disturbances leading to decreased protein synthesis, accelerated proteolysis, and muscle atrophy. 18 , 19 , 20 , 21 In reference to VIDD, evidence confirms that AT1R signaling is required for both MV‐induced diaphragmatic contractile dysfunction and fiber atrophy. 26 Moreover, this AT1R signaling is linked to a mechanical activation of AT1Rs located on the sarcolemma of diaphragm fibers. 24 Hence, a therapeutic agent that prevents VIDD must successfully block AT1R signaling. In this regard, Ang1‐7 binding to the G protein‐coupled MasR opposes the actions of the classical RAS pathway and has been shown to protect locomotor skeletal muscles against atrophy due to prolonged disuse or high circulating levels of AngII. 22 , 23 In the current experiments, our results confirm that infusion of Ang1‐7 is also protective against VIDD; details of the potential mechanisms providing for this protection are debated in the next segments.
A comprehensive understanding of the mechanisms responsible for the MV‐induced diaphragmatic contractile dysfunction remains unknown. However, it is probable that this reduction in diaphragmatic force production results from both impaired cross‐bridge kinetics and a loss of myofibrillar proteins. 2 , 37 The molecular processes liable for the MV‐induced impairment in cross‐bridge kinetics is unclear but active force generation in skeletal muscle fibers is determined by the number of cross‐bridges strongly bound to actin. 38 A potential mechanism to explain the MV‐induced reduction in diaphragm fiber force production is a decrease in myofibrillar calcium sensitivity 39 ; this can occur due to redox‐mediated post‐translational modifications of myofibrillar proteins or in response to the loss of key regulatory proteins, including myosin light chains, troponin‐I, troponin‐T, and tropomyosin. 37 , 39 Given that our findings reveal that Ang1‐7 prevented both MV‐induced oxidative stress and protease activation in diaphragm fibers, it is feasible that Ang1‐7 protects against the MV‐induced reduction in diaphragmatic‐specific force by preventing both oxidative damage and proteolytic degradation of myofibrillar proteins. Nonetheless, future experiments are required to definitively uncover the molecular mechanism(s) responsible for Ang1‐7’s protection against MV‐induced diaphragm contractile dysfunction.
The rapid atrophy of diaphragm muscle fibers during the first 12–24 h of prolonged MV is principally due to accelerated proteolysis. 2 This occurs due to activation of all four of the major proteolytic systems in muscle (i.e., ubiquitin‐proteasome, calpain, caspase‐3, and autophagy). 2 As discussed earlier, hallmarks of MV‐induced VIDD are mitochondrial dysfunction, increased mitochondrial oxidation production, and oxidative modification of muscle proteins. Importantly, our results provide the first evidence that treatment of animals with Ang1‐7 protects diaphragm muscle fibers against MV‐induced mitochondrial uncoupling (i.e., RCR), increases in mitochondrial ROS emission, and oxidative modification of myofibrillar proteins (Figure 4). Moreover, Ang1‐7 prevented the MV‐induced increase in ubiquitinated myofibrillar protein, and elevated abundance of Atrogin‐1/MAFbx and MuRF1 mRNA in diaphragm fibers (Figure 5). Similarly, treatment with Ang1‐7 prevented the ventilator‐induced activation of caspase‐3 in diaphragm fibers; this is significant because activation of caspase‐3 contributes to MV‐induced diaphragmatic atrophy. 40 , 41 Together, these results suggest that Ang1‐7‐mediated protection against diaphragmatic atrophy is due, in part, to the protection against the MV‐induced increase in oxidative stress and proteolysis.
Critique of experimental approach
Because of the ethical constrains of studying VIDD in humans, an animal model is required to perform experiments investigating pharmacological agents aimed at protecting the diaphragm against ventilator‐induced weakness. The current experiments used the rat as a preclinical model because the anatomic and biochemical properties of the rat diaphragm are similar to humans. 42 , 43 Importantly, the time course of ventilator‐induced weakness in the human and rat follow a similar pattern. 2 Hence, the rat is predicted to be an excellent preclinical experimental model to investigate protective strategies to prevent VIDD.
Investigating the impact of Ang1‐7 to protect against VIDD at only one end point is a potential limitation to our experiments because it remains unknown if treatment with Ang1‐7 will protect the diaphragm against VIDD for longer durations of MV. Nonetheless, investigating one duration of prolonged MV provides proof of concept that Ang1‐7 has therapeutic potential in protecting the diaphragm against ventilator‐induced weakness.
CONCLUSIONS
These are the first experiments to demonstrate that infusion of Ang1‐7 protects against MV‐induced diaphragmatic contractile dysfunction. Notably, our study also provides original evidence that treatment with Ang1‐7 prevents MV‐induced atrophy in all diaphragm fiber types. Further, our findings suggest that Ang1‐7 infusion confers protection against VIDD, at least in part, due to a decrease in MV‐induced diaphragmatic oxidative stress and mitochondrial dysfunction. Last, infusion of Ang1‐7 also prevented activation of major proteolytic systems in diaphragm fibers. Collectively, these results support the concept that activation of MasR signaling is a viable therapeutic approach to protect against VIDD. This is significant and may have application in human medicine because the safety and efficacy of Ang1‐7 as a therapeutic treatment for both hypertension and metastatic sarcoma has been investigated in clinical trials (e.g., NCT03604289 and NCT01553539).
DISCLOSURES
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
T.Y., H.H., and S.K.P. wrote the manuscript. T.Y., R.D., H.H., and S.K.P. designed the research. T.Y., R.D., H.H., M.O., and B.N. performed the research. T.Y., R.D., and H.H. analyzed the data.
Supporting information
Figure S1
Figure S2
Funding information
This work was supported by a National Institutes of Health grant (NIAMS R21AR073956) awarded to Scott K. Powers.
REFERENCES
- 1. Wunsch H, Wagner J, Herlim M, Chong DH, Kramer AA, Halpern SD. ICU occupancy and mechanical ventilator use in the United States. Crit Care Med. 2013;41:2712‐2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Powers SK, Wiggs MP, Sollanek KJ, Smuder AJ. Ventilator‐induced diaphragm dysfunction: cause and effect. Am J Physiol Regul Integr Comp Physiol. 2013;305:R464‐R477. [DOI] [PubMed] [Google Scholar]
- 3. Dres M, Dubé B‐P, Mayaux J, et al. Coexistence and impact of limb muscle and diaphragm weakness at time of liberation from mechanical ventilation in medical intensive care unit patients. Am J Respir Crit Care Med. 2017;195:57‐66. [DOI] [PubMed] [Google Scholar]
- 4. Agten A, Maes K, Smuder A, Powers SK, Decramer M, Gayan‐Ramirez G. N‐Acetylcysteine protects the rat diaphragm from the decreased contractility associated with controlled mechanical ventilation. Crit Care Med. 2011;39:777‐782. [DOI] [PubMed] [Google Scholar]
- 5. Betters JL, Criswell DS, Shanely RA, et al. Trolox attenuates mechanical ventilation‐induced diaphragmatic dysfunction and proteolysis. Am J Respir Crit Care Med. 2004;170:1179‐1184. [DOI] [PubMed] [Google Scholar]
- 6. McClung JM, Kavazis AN, Whidden MA, et al. Antioxidant administration attenuates mechanical ventilation‐induced rat diaphragm muscle atrophy independent of protein kinase B (PKB Akt) signalling. J Physiol. 2007;585:203‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McClung JM, Whidden MA, Kavazis AN, Falk DJ, DeRuisseau KC, Powers SK. Redox regulation of diaphragm proteolysis during mechanical ventilation. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1608‐R1617. [DOI] [PubMed] [Google Scholar]
- 8. Powers SK, Hudson MB, Nelson WB, et al. Mitochondria‐targeted antioxidants protect against mechanical ventilation‐induced diaphragm weakness. Crit Care Med. 2011;39:1749‐1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smuder AJ, Sollanek KJ, Nelson WB, et al. Crosstalk between autophagy and oxidative stress regulates proteolysis in the diaphragm during mechanical ventilation. Free Radic Biol Med. 2018;115:179‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Whidden MA, McClung JM, Falk DJ, et al. Xanthine oxidase contributes to mechanical ventilation‐induced diaphragmatic oxidative stress and contractile dysfunction. J Appl Physiol. 2009;106:385‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Whidden MA, Smuder AJ, Wu M, Hudson MB, Nelson WB, Powers SK. Oxidative stress is required for mechanical ventilation‐induced protease activation in the diaphragm. J Appl Physiol. 2010;108:1376‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kavazis AN, Talbert EE, Smuder AJ, Hudson MB, Nelson WB, Powers SK. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic Biol Med. 2009;46:842‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McClung JM, Van Gammeren D, Whidden MA, et al. Apocynin attenuates diaphragm oxidative stress and protease activation during prolonged mechanical ventilation. Crit Care Med. 2009;37:1373‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Picard M, Azuelos I, Jung B, et al. Mechanical ventilation triggers abnormal mitochondrial dynamics and morphology in the diaphragm. J Appl Physiol. 2015;118:1161‐1171. [DOI] [PubMed] [Google Scholar]
- 15. Picard M, Jung B, Liang F, et al. Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am J Respir Crit Care Med. 2012;186:1140‐1149. [DOI] [PubMed] [Google Scholar]
- 16. Ferreira LF, Laitano O. Regulation of NADPH oxidases in skeletal muscle. Free Radic Biol Med. 2016;98:18‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Powers SK, Morton AB, Hyatt H, Hinkley MJ. The renin‐angiotensin system and skeletal muscle. Exerc Sport Sci Rev. 2018;46:205‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cabello‐Verrugio C, Cordova G, Salas JD. Angiotensin II: role in skeletal muscle atrophy. Curr Protein Pept Sci. 2012;13:560‐569. [DOI] [PubMed] [Google Scholar]
- 19. Cabello‐Verrugio C, Morales MG, Rivera JC, Cabrera D, Simon F. Renin‐angiotensin system: an old player with novel functions in skeletal muscle. Med Res Rev. 2015;35:437‐463. [DOI] [PubMed] [Google Scholar]
- 20. Kadoguchi T, Kinugawa S, Takada S, et al. Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle. Exp Physiol. 2015;100:312‐322. [DOI] [PubMed] [Google Scholar]
- 21. Yoshida T, Tabony AM, Galvez S, et al. Molecular mechanisms and signaling pathways of angiotensin II‐induced muscle wasting: potential therapeutic targets for cardiac cachexia. Int J Biochem Cell Biol. 2013;45:2322‐2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cisternas F, Morales MG, Meneses C, et al. Angiotensin‐(1‐7) decreases skeletal muscle atrophy induced by angiotensin II through a Mas receptor‐dependent mechanism. Clin Sci (Lond). 2015;128:307‐319. [DOI] [PubMed] [Google Scholar]
- 23. Morales MG, Abrigo J, Acuña MJ, et al. Angiotensin‐(1‐7) attenuates disuse skeletal muscle atrophy in mice via its receptor, Mas. Dis Model Mech. 2016;9:441‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deminice R, Hyatt H, Yoshihara T, et al. Human and rodent skeletal muscles express angiotensin ii type 1 receptors. Cells. 2020;9(7):1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hall SE, Ahn B, Smuder AJ, et al. Comparative efficacy of angiotensin II type 1 receptor blockers against ventilator‐induced diaphragm dysfunction in rats [published online ahead of print November 22, 2020]. Clin Transl Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwon OS, Smuder AJ, Wiggs MP, et al. AT1 receptor blocker losartan protects against mechanical ventilation‐induced diaphragmatic dysfunction. J Appl Physiol. 2015;119:1033‐1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sigurta A, Zambelli V, Bellani G. Renin‐angiotensin system in ventilator‐induced diaphragmatic dysfunction: potential protective role of angiotensin (1‐7). Med Hypotheses. 2016;94:132‐137. [DOI] [PubMed] [Google Scholar]
- 28. Zambelli V, Sigurtà Anna, Rizzi Laura, et al. Angiotensin‐(1‐7) exerts a protective action in a rat model of ventilator‐induced diaphragmatic dysfunction. Intensive Care Med Exp. 2019;7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smuder AJ, Min K, Hudson MB, et al. Endurance exercise attenuates ventilator‐induced diaphragm dysfunction. J Appl Physiol. 2012;112:501‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 31. Testelmans D, Maes K, Wouters P, Powers SK, Decramer M, Gayan‐Ramirez G. Infusions of rocuronium and cisatracurium exert different effects on rat diaphragm function. Intensive Care Med. 2007;33:872‐879. [DOI] [PubMed] [Google Scholar]
- 32. Maes K, Stamiris A, Thomas D, et al. Effects of controlled mechanical ventilation on sepsis‐induced diaphragm dysfunction in rats. Crit Care Med. 2014;42:e772‐782. [DOI] [PubMed] [Google Scholar]
- 33. Maes K, Testelmans D, Cadot P, et al. Effects of acute administration of corticosteroids during mechanical ventilation on rat diaphragm. Am J Respir Crit Care Med. 2008;178:1219‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Powers SK, Shanely RA, Coombes JS, et al. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol. 2002;92:1851‐1858. [DOI] [PubMed] [Google Scholar]
- 35. Smith IJ, Godinez GL, Singh BK, et al. Inhibition of Janus kinase signaling during controlled mechanical ventilation prevents ventilation‐induced diaphragm dysfunction. FASEB J. 2014;28:2790‐2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seven YB, Mantilla CB, Sieck GC. Recruitment of rat diaphragm motor units across motor behaviors with different levels of diaphragm activation. J Appl Physiol. 2014;117:1308‐1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hussain SN, Cornachione AS, Guichon C, et al. Prolonged controlled mechanical ventilation in humans triggers myofibrillar contractile dysfunction and myofilament protein loss in the diaphragm. Thorax. 2016;71:436‐445. [DOI] [PubMed] [Google Scholar]
- 38. Brenner B. Effect of Ca2+ on cross‐bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci USA. 1988;85:3265‐3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moopanar TR, Allen DG. Reactive oxygen species reduce myofibrillar Ca2+ sensitivity in fatiguing mouse skeletal muscle at 37 degrees C. J Physiol. 2005;564:189‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maes K, Testelmans D, Powers S, Decramer M, Gayan‐Ramirez G. Leupeptin inhibits ventilator‐induced diaphragm dysfunction in rats. Am J Respir Crit Care Med. 2007;175:1134‐1138. [DOI] [PubMed] [Google Scholar]
- 41. Nelson WB, Smuder AJ, Hudson MB, Talbert EE, Powers SK. Cross‐talk between the calpain and caspase‐3 proteolytic systems in the diaphragm during prolonged mechanical ventilation. Crit Care Med. 2012;40:1857‐1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Levine S, Nguyen T, Taylor N, et al. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med. 2008;358:1327‐1335. [DOI] [PubMed] [Google Scholar]
- 43. Powers SK, Demirel HA, Coombes J, et al. Myosin phenotype and bioenergetic characteristics of rat respiratory muscles. Med Sci Sports Exerc. 1997;29:1573‐1579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2