

Abstract
The missense variant, breast cancer resistance protein (BCRP) p.Q141K, which encodes a reduced function BCRP, has been linked to poor response to allopurinol. Using a multifaceted approach, we aimed to characterize the relationship(s) between BCRP p.Q141K, the pharmacokinetics (PK) and pharmacodynamics (PD) of oxypurinol (the active metabolite of allopurinol), and serum uric acid (SUA) levels. A prospective clinical study (NCT02956278) was conducted in which healthy volunteers were given a single oral dose of 300 mg allopurinol followed by intensive blood sampling. Data were analyzed using noncompartmental analysis and population PK/PD modeling. Additionally, electronic health records were analyzed to investigate whether clinical inhibitors of BCRP phenocopied the effects of the p.Q141K variant with respect to SUA. Subjects homozygous for p.Q141K had a longer half‐life (34.2 ± 12.2 h vs. 19.1 ± 1.42 h) of oxypurinol. The PK/PD model showed that women had a 24.8% lower volume of distribution. Baseline SUA was affected by p.Q141K genotype and renal function; that is, it changed by 48.8% for every 1 mg/dl difference in serum creatinine. Real‐world data analyses showed that patients prescribed clinical inhibitors of BCRP have higher SUA levels than those that have not been prescribed inhibitors of BCRP, consistent with the idea that BCRP inhibitors phenocopy the effects of p.Q141K on uric acid levels. This study identified important covariates of oxypurinol PK/PD that could affect its efficacy for the treatment of gout as well as a potential side effect of BCRP inhibitors on increasing uric acid levels, which has not been described previously.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Recently, genomewide association studies have implicated genetic variants within ABCG2, which encodes the efflux pump breast cancer resistant protein (BCRP), as determinants of allopurinol response. Specifically, the missense variant, BCRP p.Q141K (rs2231142), which encodes a reduced function BCRP, has been linked to poor response to allopurinol.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study characterized the relationship(s) between BCRP p.Q141K, oxypurinol pharmacokinetics/pharmacodynamics (PK/PD), and serum uric acid levels.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study was able to identify covariates important in the PK and PD of oxypurinol and to demonstrate the use of real‐world data to validate the effects of BCRP p.Q141K on clinical measurements. That is, clinical inhibitors of BCRP phenocopied laboratory measurements associated with BCRP p.Q141K. Additionally, this study suggests that the mechanism by which BCRP p.Q141K modulates response to allopurinol is complex and may be related to intestinal levels of its active metabolite oxypurinol, ultimately affecting the PD of allopurinol.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The current study provides the first investigation into the relationship between BCRP p.Q141K and allopurinol/oxypurinol PK/PD in a prospective clinical trial and BCRP inhibition and uric acid levels using real‐world data.
INTRODUCTION
Gout, an inflammatory arthritis associated with high serum uric acid (SUA) levels, is a painful disease that is associated with comorbidities. Although there are several drugs on the market for the treatment of gout, such as febuxostat, allopurinol is recommended as first line treatment due to its tolerability, safety, and lower cost. 1 As a xanthine oxidase inhibitor, allopurinol inhibits the formation of uric acid; however, response to the drug is highly variable, with studies showing as few as 21% of patients reaching an acceptable treatment end point. 2 Thus, recognizing features and covariates that affect allopurinol response is vital in determining the right dose, schedule, and patient population.
Many studies have been performed to investigate the relationship between allopurinol and antihyperuricemic response. A spectrum of covariates, such as baseline SUA, diuretic use, creatinine clearance, and body weight, have been examined for use in predicting pharmacodynamic (PD) response, but results have been conflicting. 3 , 4 , 5 Recently, breast cancer resistant protein (BCRP) p.Q141K (rs2231142) has been shown to associate with poor response to allopurinol (i.e., greater than the rheumatologist recommended SUA levels of <6 mg/dl). 6 , 7 , 8
Both allopurinol and oxypurinol, the active metabolite of allopurinol, are substrates of BCRP, which acts as an efflux pump on the apical membrane of the renal proximal tubule and intestinal epithelia and on the canalicular membrane of hepatocytes. 7 Allopurinol is rapidly and extensively metabolized to oxypurinol, as reflected by its short half‐life (1–2 h). Although the major route of elimination for allopurinol is metabolism to oxypurinol, oxypurinol is eliminated in the urine almost entirely as unchanged drug. 9 Thus, BCRP can affect the pharmacokinetics (PK) of allopurinol and/or oxypurinol by reducing absorption of allopurinol following an oral dose, limiting distribution of allopurinol and oxypurinol, and aiding in the elimination of both. BCRP p.Q141K has been shown to alter the PK and PD of many substrates, such as rosuvastatin, and is typically associated with increased systemic levels and improved pharmacologic response. Thus, it is difficult to understand the mechanisms by which the variant associates with poor response to allopurinol. 10
Using a multifaceted approach, we aimed to characterize the relationship(s) between BCRP p.Q141K, oxypurinol PK/PD, and SUA levels. Specifically, we: (1) characterized the PK/PD relationship of oxypurinol and SUA; (2) quantified the effects of covariates affecting the PK/PD of oxypurinol, including the effects of BCRP p.Q141K; and (3) determined whether clinical inhibitors of BCRP phenocopy the effects of the p.Q141K variant on SUA levels.
METHODS
Study participants
Healthy human volunteers between 18 and 50 years of age were recruited for this study and 19 subjects were enrolled. All subjects were evaluated to be healthy on the basis of medical history provided by a study questionnaire. Volunteers were excluded if they were taking any medications that are known to interact with uric acid levels, BCRP function, or allopurinol PK. Individuals with elevated liver enzymes (>1.5× normal range), elevated creatinine concentrations (>1.5× normal range), and abnormal platelet and/or white blood cell count levels were also excluded. Individuals carrying the HLA‐B*58:01 allele were excluded from this study because of the increased risk for allopurinol‐induced Stevens‐Johnson syndrome. All participants gave written, informed consent, and the study was registered on clinicaltrials.gov (NCT02956278).
Genotyping
BCRP p.Q141K (rs2231142) was genotyped by TaqMan assay (Applied Biosystems, assay ID C__15854163_70) in DNA extracted from a cheek swab. The reaction mixture consisted of 5–10 ng DNA, 12.5 μl of TaqMan Genotyping Master Mix (Applied Biosystems), 1.25 μl of TaqMan genotyping assay mix, and 11.25 μl of distilled water. The cycling conditions were as follows: 95°C for 10 min and 40 cycles of 95°C for 15 s then 60°C for 1 min. The reaction was run on a BioRad MyCycler (Bio‐Rad) and allele discrimination determined by ABI 7900 Fast HT Sequence Detection Systems (Applied Biosystems).
Clinical study design
This study protocol was reviewed and approved by Western Institutional Review Board. Healthy individuals of European or Asian heritage were recruited into this study and informed consent was obtained from each subject. Prior to their enrollment in the study, subjects were screened. The screening included two stages: (1) a questionnaire on health, medications, and self‐reported ethnicity, as well as a cheek swab to determine BCRP p.Q141K genotype, and (2) a blood sample to measure complete blood count, hepatic function, renal function, uric acid, and HLA‐B*58:01 genotype.
Subjects were enrolled under two protocols. The first protocol incorporated a multidose design, in which subjects were asked to arrive at the site following an overnight fast. An initial blood sample was taken to establish baseline SUA and serum creatinine (SCr) before drug was administered. An oral dose of 300 mg of allopurinol was administered to all subjects, followed by blood sampling at: 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, and 24 h postdose. The subjects were then asked to take allopurinol 300 mg once daily and return to the site on day 6 for their final dose of allopurinol. Blood samples were drawn at 0, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 24, 48, and 72 h after the final dose.
For the second protocol, subjects were asked to arrive at the site following an overnight fast. An initial blood draw was taken to establish baseline SUA and SCr before drug was administered. A single dose of 300 mg of allopurinol was administered to all subjects, followed by blood sampling at: 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 24, 48, and 72 h postdose.
Samples collected following a single dose of allopurinol (from both protocols) were used for subsequent analyses.
Serum was isolated from blood samples using clotting and centrifugation and stored at −80°C for analysis of uric acid, creatinine, allopurinol, and oxypurinol concentrations.
Bioanalytical methods
Allopurinol and oxypurinol were analyzed by Quintara Discovery using a validated liquid chromatography tandem mass spectrometry (LC‐MS/MS) method. Samples were diluted as needed. An aliquot of 20 μl of serum samples was treated with 200 μl of 25% Methanol 75% acetonitrile containing internal standard (Fulvestrant). The mixture was vortexed on a shaker for 15 min and subsequently centrifuged at 1600 x g for 15 min. The supernatant was transferred to a microtiter plate for the injection to the LC‐MS/MS. Calibration standards and quality control samples were prepared by spiking the test compound into corresponding blank matrix and processed with the unknown samples. The quantification limit was 2 ng/ml for serum.
Uric acid and creatinine levels were analyzed by Open Medicine Institute according to standard spectrophotometry protocol.
Noncompartmental PK analysis
The concentration‐time profiles of allopurinol and oxypurinol were plotted using GraphPad Prism 8.4 (GraphPad Software). PK parameters of allopurinol and oxypurinol were determined by noncompartmental analysis using the PKNCA package 11 in R (version 3.4.0). Values below the limit of quantification were excluded from the analysis.
Data are expressed as mean ± SE unless otherwise noted. Differences in demographics, clinical characteristics, and PK parameters relative to genotype were analyzed in R using a one‐way ANOVA followed by Tukey’s post hoc test for multiple test correction (TukeyHSD). The p values less than 0.05 were considered statistically significant.
Collection of mouse luminal contents
All animal experiments were approved by the University of California, San Francisco (UCSF) Institutional Animal Care and Use Committee. Experiments were performed using 12‐week‐old male C57BL/6 mice (Charles River). The method of collection was described previously 12 with modifications. In brief, mouse intestine was evenly divided into four segments and opened longitudinally. The contents were gently removed using forceps without scraping the surface and resuspended in 1 ml of solution (50 mM Tris‐HCl [pH 7.0], 50 mM Mannitol, 2 mM EGTA, 8 μg/ml Aprotinin, 10 μg/ml Leupeptin, and 2 mM DTT). Protein concentration was quantified by the BCA assay (Thermo Scientific).
Xanthine oxidase assay
Xanthine oxidase activities in the mouse luminal contents were determined according to the manufacturer’s protocol (Abcam, ab102522). In brief, in a black wall 96‐well plate, 10 μl of sample solution was added in 50 μl of Reaction Mix containing assay buffer, hypoxanthine, and OxiRed probe, with or without oxypurinol (1 mM), an inhibitor of xanthine oxidase. Then, ddH2O was added to adjust the total volume to 100 μl. Then, 10 mU of xanthine oxidase enzyme was used as the positive control. The plate was incubated at 37°C for 1 h, and protected from light. The fluorescent signal was measured at Ex/Em = 535/590 nm.
Population PK/PD model
Longitudinal oxypurinol PK and SUA data were linked using the population approach using nonlinear mixed effects modeling software NONMEM version 7.3.0 (ICON Development Solutions). The analysis was performed sequentially. First, the PK analysis was performed. In this analysis, Fm was used to represent the fraction of the allopurinol dose systemically available as oxypurinol and Kfm denotes the formation rate constant, similar to a previous model for oxypurinol. 13 Values below the limit of quantification were excluded from the analysis. Nine of 250 samples were either missing/not collected or below the limit of quantification.
Second, a maximum effect model was used to link the predicted individual oxypurinol PK concentration data with the SUA data (PD). Genotype (discrete), gender (discrete), race (discrete), serum creatine (continuous), and weight (continuous) were all assessed as potential covariates for every PK/PD parameter in both the PK and PK/PD model. Covariate selection was guided by using the stepwise covariate modeling approach with the PsN software (version 4.8.1). This method consists of stepwise testing different covariate‐parameter relationships with forward inclusion and backward exclusion approaches with significance levels of 0.05 and 0.01, respectively. The inclusion of covariates in the final model was done by considering scientific plausibility, significance, and clinical relevance following stepwise covariate selection.
Model parameters were estimated using first order conditional estimation method with the option for interaction. Between subject variability (BSV) was modeled exponentially and residual variability was modeled using a combination of additive and proportional error. Model selection was based on goodness‐of‐fit (GOF) plots and the difference in the objective function value between nested models. Final models were internally validated through simulation‐based diagnostics using visual predictive check (VPC) plots (1000 simulations). The precision of the final parameter estimates was evaluated using 1000 simulated bootstrap datasets using PsN software. 14
Model development, diagnostics, and graphing were done using R (version 3.4.0) including packages such as ggplot2 and Xpose. 15 Percent coefficient of variation was calculated as follows: sqrt(OMEGA) * 100. The final PK/PD model was used to simulate (n = 500) SUA profiles after 30 days of once daily dosing of 300 mg of allopurinol as a post hoc analysis of covariate effects.
Electronic health record analyses to evaluate uric acid levels in patients prescribed clinical inhibitors of BCRP
To investigate whether drugs that are clinical inhibitors of BCRP can phenocopy the effect of p.Q141K on baseline SUA, we mined the electronic health records (EHRs) at UCSF and identified subjects both on and off BCRP inhibitors with SUA levels in the database. The UCSF Research Data Browser was utilized to search for patients who had a numeric serum/plasma uric acid laboratory test value reported, giving a total of 26,328 patients and 120,570 laboratory values. Values reported as an inequality were changed to a numerical value (i.e. ,<0.5 mg/dl = 0.5 mg/dl). Laboratory values with missing values (i.e., DE‐IDENTIFIED) and laboratory values without a laboratory collection date were excluded.
Patients were divided into two groups depending on their medication prescriptions. Specifically, patients prescribed cyclosporine and/or eltrombopag, both of which are identified as clinical inhibitors of BCRP by the US Food and Drug Administration (https://www.fda.gov/drugs/drug‐interactions‐labeling/drug‐development‐and‐drug‐interactions‐table‐substrates‐inhibitors‐and‐inducers), were grouped into the “on” drug group. Search terms included: “cyclosporine,” “sandimmune,” “neoral,” “gengraf,” “eltrombopag,” and “promacta.” Only medication orders with an oral route of administration and with a medication order start date were included in the analysis, which resulted in 1785 patients. The remaining patients (i.e., individuals who were never prescribed cyclosporine or eltrombopag) were grouped into the “off” drug group. Only patients with one uric acid level reported in their EHR were included in the “off” drug group, which reduced the sample size to 15,042 individuals. Additionally, for both groups, patients with a prescription to any uric acid lowering medication were excluded from the analysis.
Patients in the “on” drug group were further filtered based on their laboratory collection date relative to their first medication order start date. Serum/plasma uric acid laboratory tests collected before the patient’s first medication order start date or within 7 days after their first medication order start date were excluded. A minimum of 7 days between medication order start date and laboratory value collection date was chosen to allow drug levels to reach steady‐state and for an effect to be seen. For patients with more than one laboratory value, only the laboratory value closest to the first medication order start date was included. In total, 273 patients met these criteria and were in the “on” drug group. Last, patients were age‐matched and sex‐matched using the MatchIt package 16 in R to be comparable in both groups, which resulted in a final sample sizes of 273 patients “on” drug and 2730 patients “off” drug (Table S1).
In order to address the possibility that underlying disease or drug class contributed to the differences seen in uric acid levels, for the therapeutic class‐specific analysis looking at immunosuppressants, patients were further assigned to subgroups. More specifically, patients in the “on cyclosporine” drug group were filtered to only include patients with a prescription to cyclosporine and exclude patients with prescriptions for any other immunosuppressants. Conversely, the “on other immunosuppressants” drug group was filtered to only include patients with at least one prescription for an immunosuppressant, but exclude patients with a prescription to cyclosporine. In addition to the inclusion/exclusion criteria above, patients with prescriptions to eltrombopag were also excluded from both groups. This resulted in 174 patients in the “on cyclosporine” drug group and 3503 patients in the “on other immunosuppressants” drug group who met the medication order inclusion/exclusion criteria and had at least one serum/plasma uric acid level reported. Patients in both groups were further filtered based on their laboratory collection date relative to their first medication order start date for cyclosporine for the “on cyclosporine” drug group and any other immunosuppressant (except cyclosporine) for the “on other immunosuppressants” drug group, respectively. The rest of the analysis was performed as described for the “on” drug group above; after age‐matching and sex‐matching the groups, we had 119 patients in the “on cyclosporine” drug group and 833 patients in the “on other immunosuppressants” drug group (Table S1).
Wilcoxon rank sum test with continuity correction was performed to evaluate if there was a significant difference in laboratory values when comparing both groups and ggplot2 was used to plot the data in R (version 3.4.0).
RESULTS
Study cohort
Healthy volunteers of European or Asian ancestry were screened for participation in this study. These ethnicities were chosen due to the high minor allele frequencies of BCRP p.Q141K in individuals of Asian (29%) and European (9%) ancestries. 17 Of the 178 subjects screened, 19 completed the study and allopurinol was well‐tolerated. Most subjects were excluded based on ABCG2 genotype, some were excluded based on HLA‐B*58:01 genotype and concomitant medications. Demographics of the study subjects by genotype can be found in Table 1. There was a significant difference in SCr concentrations between the homozygous reference (CC) and heterozygous (CA) groups (adjusted p value: 2.99 × 10−7) and CC and homozygous variant (AA) groups (adjusted p value: 1.19 × 10−6). Additionally, baseline SUA was significantly different between the CC and CA groups (adjusted p value: 5 × 10−4). If we exclude women from this comparison given the higher ratio of women to men in the CC group compared with the other groups (CA and AA), we continue to see a significant difference in SCr concentrations between the CC and CA groups (adjusted p value: 0.00338) and CC and AA groups (adjusted p value: 1.73 × 10−5), as well as a slight difference between the CA and AA groups (adjusted p value: 0.0474). In addition, excluding women from the analysis results in a greater significant difference in baseline SUA among the various genotype groups: CC and CA (adjusted p value: 5.29 × 10−13), CC and AA (adjusted p value: 0.00176), and CA and AA (adjusted p value: 0.0163).
Table 1.
Demographics and clinical characteristics of study cohort
| Demographics and clinical characteristics | Genotype | ||
|---|---|---|---|
| (0) CC | (1) CA | (2) AA | |
| No. of patients | 9 | 7 | 3 |
| Male/Female | 4/5 | 6/1 | 2/1 |
| Weight, kg | 71.5 ± 6.25 | 69.9 ± 7.31 | 73.4 ± 4.54 |
| Male, kg | 72.3 ± 8.89 | 71.7 ± 8.37 | 73.8 ± 7.82 |
| Female, kg | 70.9 ± 9.63 | 59.0 | 72.6 |
| Race | |||
| East Asian | 4 | 5 | 1 |
| South Asian | 2 | 1 | 0 |
| Filipino | 0 | 1 | 1 |
| European | 1 | 0 | 0 |
| Mixed | 2 | 0 | 1 |
| Serum creatinine, mg/dl | 0.716 ± 0.010 | 0.819 ± 0.016 a | 0.844 ± 0.023 a |
| Baseline serum uric acid, mg/dl | 4.62 ± 0.284 | 6.70 ± 0.368 b | 6.07 ± 0.133 |
Weight, serum creatinine, and baseline serum uric acid are reported as average ± standard error. All statistical tests were done using one‐way ANOVA followed by Tukey’s post hoc test for multiple test correction.
Abbreviations: AA, homozygous variant; CA, heterozygous; CC, homozygous reference.
Serum creatinine was significantly different between (0) CC and (1) CA (adjusted p value: 2.99 × 10−7) and between (0) CC and (2) AA (adjusted p value: 1.19 × 10−6).
Baseline serum uric acid was significantly different between (0) CC and (1) CA (adjusted p value: 5 × 10−4).
BCRP p.Q141K associates with a longer half‐life of oxypurinol and the concentration‐time profile of oxypurinol demonstrates enterohepatic recycling
Plasma concentration‐time profiles and the noncompartmental PK parameters were similar for allopurinol regardless of genotype (Table 2 and Figure 1a). Oxypurinol half‐life was significantly longer in patients homozygous for the p.Q141K variant compared with patients homozygous for the reference allele when using a one‐way ANOVA followed by Tukey’s post hoc test for multiple test correction (34.2 ± 12.2 h vs. 19.1 ± 1.42 h, adjusted p value: 0.047; Table 2 and Figure 1b). However, BCRP p.Q141K genotype had no statistically significant effect on any other PK parameter, such as maximum concentration achieved (Cmax), area under the curve, and time to maximum concentration (Table 2).
Table 2.
Summary of the effect of the BCRP p.Q141K variant allele on the PK parameters of allopurinol and oxypurinol computed by noncompartmental analysis
| Parameter | (0) CC | (1) CA | (2) AA |
|---|---|---|---|
| No. of patients | 9 | 7 | 3 |
| Allopurinol | |||
| Cmax, ng/ml | 1370 ± 130 | 1930 ± 617 | 1550 ± 453 |
| Tmax, h | 1.5 (0.5–2) | 1 (0.5–2) | 2 (1–2) |
| AUC0–10, μg*h/mla | 3.78 ± 0.346 | 3.47 ± 0.549 | 2.76 ± 0.637 |
| CL/F, L/h | 94.5 ± 9.73 | 97.8 ± 12.5 | 92.8 ± 26.6 |
| V/F, L | 149 ± 17.0 | 160 ± 22.3 | 148 ± 40.9 |
| t 1/2, h | 1.10 ± 0.060 | 1.14 ± 0.078 | 1.11 ± 0.025 |
| Oxypurinol | |||
| Cmax, ng/ml | 5930 ± 409 | 6440 ± 592 | 6380 ± 1350 |
| Tmax, h | 4 (1.5–10) | 4 (1.5–6) | 6 (3–6) |
| AUC0–24, μg*h/ml | 93.9 ± 8.07 | 93.4 ± 9.99 | 105 ± 20.6 |
| CL/F, L/h b | 1.89 ± 0.227 | 1.81 ± 0.276 | 1.38 ± 0.466 |
| V/F, L b | 49.9 ± 4.53 | 61.9 ± 9.58 | 56.6 ± 11.6 |
| t 1/2, h b | 19.1 ± 1.42 | 23.9 ± 1.35 | 34.2 ± 12.2 c |
All data are reported as mean ± standard error except for Tmax, which is reported as median (range).
Abbreviations: AA, homozygous variant; AUC0–10, area under the concentration‐time curve from t = 0 h to t = 10 h; AUC0–24, area under the concentration‐time curve from t = 0 h to t = 24 h; BCRP, breast cancer resistance protein; CA, heterozygous; CC, homozygous reference; CL/F, apparent clearance; Cmax, maximum plasma concentration; PK, pharmacokinetic; Tmax, time to maximum concentration; V/F, apparent volume of distribution; t 1/2, terminal half‐life.
N = 16 for AUC0–10 since three subjects had concentrations below the limit of quantification at t = 10 h.
N = 18 because t 1/2 could not be accurately estimated for one subject.
Half‐life was significantly longer in the homozygous variant group compared with the homozygous reference group when using an ANOVA comparison followed by Tukey’s post hoc test for multiple test correction (adjusted p value: 0.047).
Figure 1.
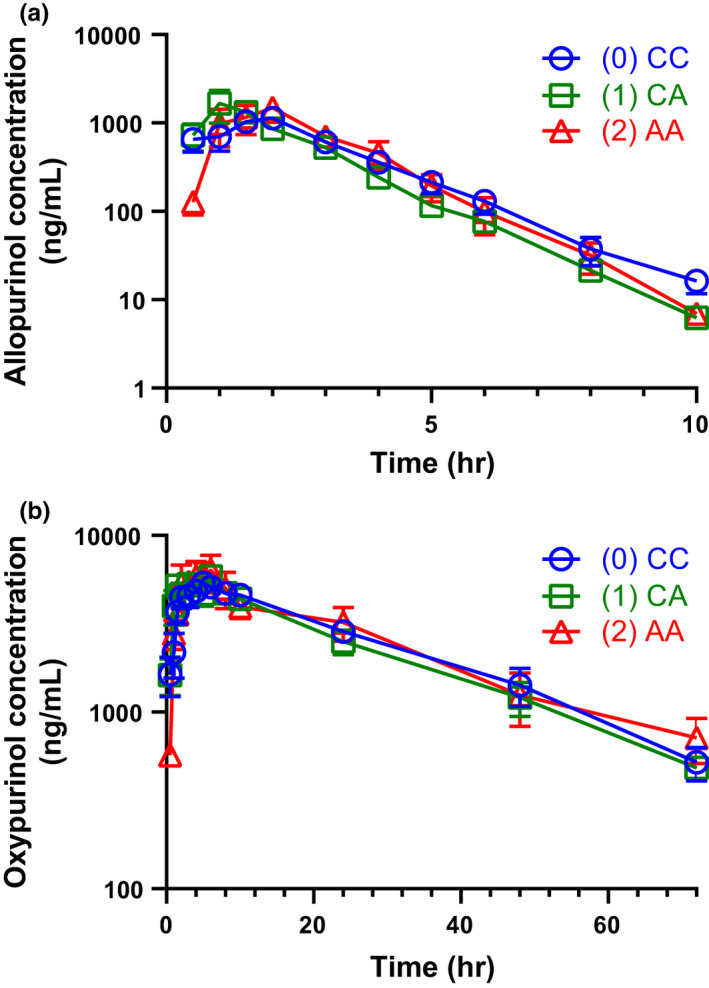
Concentration time profiles for (a) allopurinol and (b) oxypurinol after a single dose of allopurinol to healthy volunteers. Allopurinol and oxypurinol concentrations were determined following a 300 mg single oral dose of allopurinol. (0) CC, homozygous reference; (1) CA, heterozygous; (2) AA, homozygous variant. Data represent the mean ± standard error from 19 individuals of various genotypes (see Table 1); note that data were missing or below the limit of quantification at several timepoints so not all mean ± standard error are representative of all (n = 19) subjects. No error bars are present if the standard error is encompassed within the point
Additionally, multiple peaks were observed in the individual concentration‐time profiles of oxypurinol, suggesting that enterohepatic recycling may be occurring (Figure S1). Experimental studies showed that xanthine oxidase activity is present in mouse intestinal lumen (Figure S2), suggesting that the intestinal lumen is a potential site of action for oxypurinol.
PK/PD covariate model shows a significant effect of gender on oxypurinol volume of distribution and genotype and SCr on baseline SUA
A one‐compartment PK model with first order absorption was found to best fit the oxypurinol concentration‐time data. Parameter and BSV estimates (noted in parentheses) for total apparent clearance (CL/Fm), volume of distribution (V/Fm), and the formation rate constant (Kfm) were 1.74 L/h (23.6%), 57.0 L (18.7%), and 0.771 h−1(55.2%), respectively (Table 3). In the PK/PD model, BSV was estimated for baseline SUA (13.3%). The rest of parameter estimates can be found in Table 3.
Table 3.
Parameter estimates in the final oxypurinol PK/PD covariate model
| Parameter |
Estimate (%RSE) [95% CI] |
BSV (%RSE) [95% CI] |
|
|---|---|---|---|
| PK model | |||
| Kfm, 1/h |
0.771 (17.8) [0.535, 1.06] |
55.2% (38.1) [29.0, 69.6] |
|
| CL/Fm, L/h |
1.74 (7.18) [1.51, 2.00] |
23.6% (41.5) [9.61, 29.5] |
|
|
|
= 57.0 (8.05) [48.1, 65.6] |
18.7% (41.7) [7.53, 22.9] |
|
| = 0 | — | ||
|
= −0.248 (35.7) [−0.395, −0.067] |
— | ||
| PK/PD model | |||
| Emax | 1 | — | |
| C50, ng/ml |
2590 (11.6) [2170, 3330] |
— | |
|
|
= 4.61 (5.64) [4.19, 5.23] |
13.3% (61.4) [4.07, 18.6] |
|
| = 0 | — | ||
|
= 0.354 (29.3) [0.139, 0.543] |
— | ||
|
= 0.244 (40.4) [0.047, 0.452] |
— | ||
|
= 0.488 (24.6) [0.246, 0.717] |
— | ||
| Residual variability | |||
| Proportional error for PK (%CV) |
25.4 (11.1) [19.6, 30.7] |
— | |
| Proportional error for PD (%CV) |
7.67 (5.54) [6.81, 8.45] |
— | |
BSV is reported as %CV.
The additive part of the combined residual error model was fixed to 0 mg/dl (PD model) and to 0.01 ng/ml (PK model). Residual standard errors (%RSE) and 95% confidence intervals (95% CI) were obtained from the bootstrap analyses.
Abbreviations: %CV, coefficient of variation; BSV, between subject variability; C50, concentration needed to achieve 50% effect; CI, confidence interval; CL/Fm, apparent clearance of oxypurinol; Emax, maximum effect; Fm refers to the fraction of the allopurinol dose which can be converted into oxypurinol; Kfm, formation rate constant; PD, pharmacodynamic; PK, pharmacokinetic; RSE, relative standard error; SCr, serum creatinine; SUA, serum uric acid; V/Fm, apparent volume of distribution.
Gender, genotype, race, serum creatinine, and weight were tested for their effects on the PK parameters of oxypurinol as well as the effects of oxypurinol on SUA. The final PK/PD model included gender as a significant covariate on apparent V/Fm. BCRP p.Q141K genotype and SCr were also included as significant covariates for baseline SUA (Table 3). Women had a 24.8% lower V/Fm compared with men. Baseline SUA changed by 48.8% for every 1 mg/dl difference in SCr. Additionally, baseline SUA increased by 35.4% and 24.4% for the genotypes of CA or AA, respectively, as compared with reference BCRP p.Q141K (CC). Based on the results from our noncompartmental analysis where half‐life was significantly longer in patients homozygous for the BCRP p.Q141K variant, we were interested in exploring the effect of genotype on CL/Fm and V/Fm. In our model, CL/Fm also appeared to correlate with genotype; however, it was not significant and thus BCRP p.Q141K genotype was not included as a covariate for CL/Fm nor V/Fm in the final model. VPCs and GOF plots confirmed that the final PK/PD model adequately described the observed data (Figure 2, Figure S3, and Figure S4).
Figure 2.
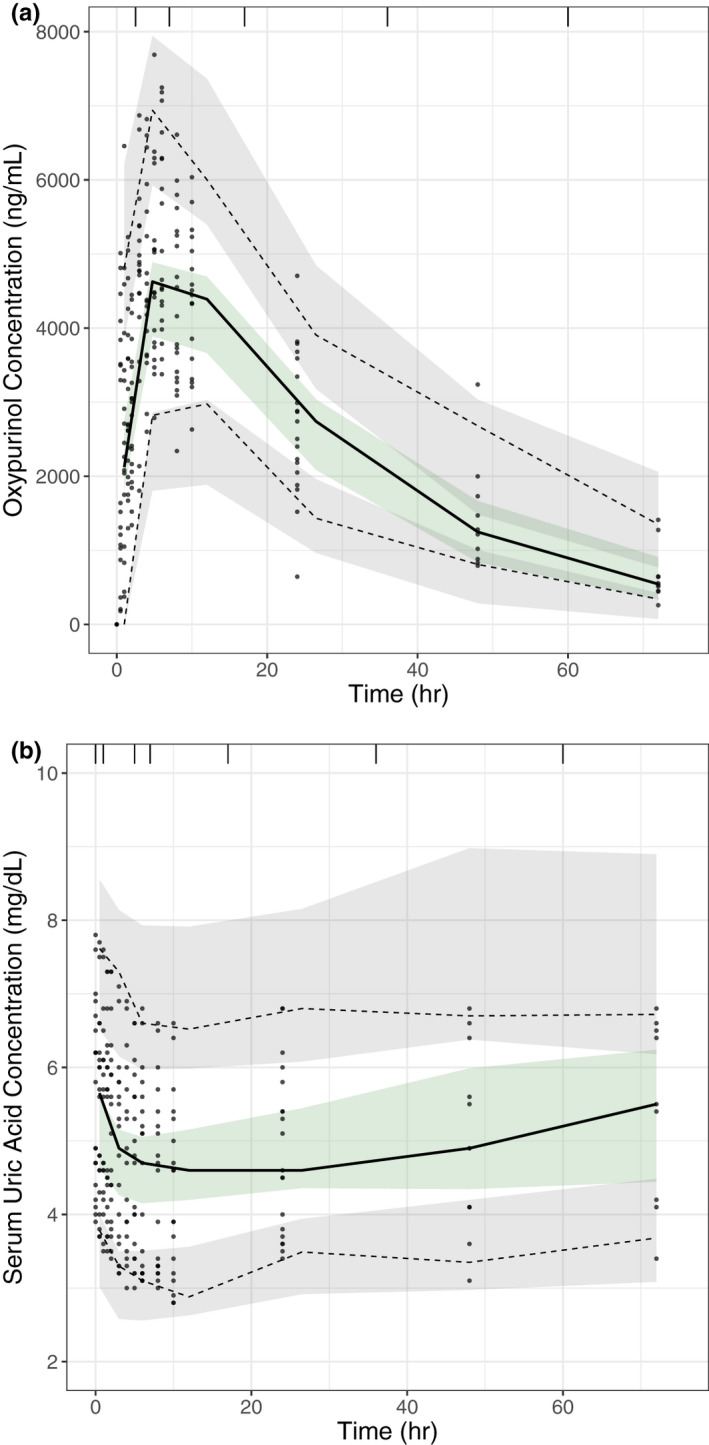
Evaluation of oxypurinol pharmacokinetic (PK) and pharmacokinetic/pharmacodynamic (PK/PD) models through visual predictive checks. PK and PK/PD model evaluation of oxypurinol concentrations and serum uric acid concentrations after single dose administration of allopurinol. Visual predictive checks show the observed data (grey dots), the median (solid line) and 5th and 95th percentiles (dashed lines) of the observed data, and the 95% confidence intervals of the model simulated data (shaded areas) for (a) plasma oxypurinol concentrations and (b) serum uric acid concentrations
Simulations were performed to visualize the role of genotype and SCr on the PD effects of oxypurinol after 30 days of once daily dosing of 300 mg of allopurinol. Simulations confirmed that individuals who have at least one variant allele or SCr levels greater than 0.78 mg/dl (the average SCr value in our dataset) have (1) higher baseline SUA levels on average and (2) are less likely to achieve and maintain the target concentration of less than 6 mg/dl given a flat dose of allopurinol (Figure 3).
Figure 3.
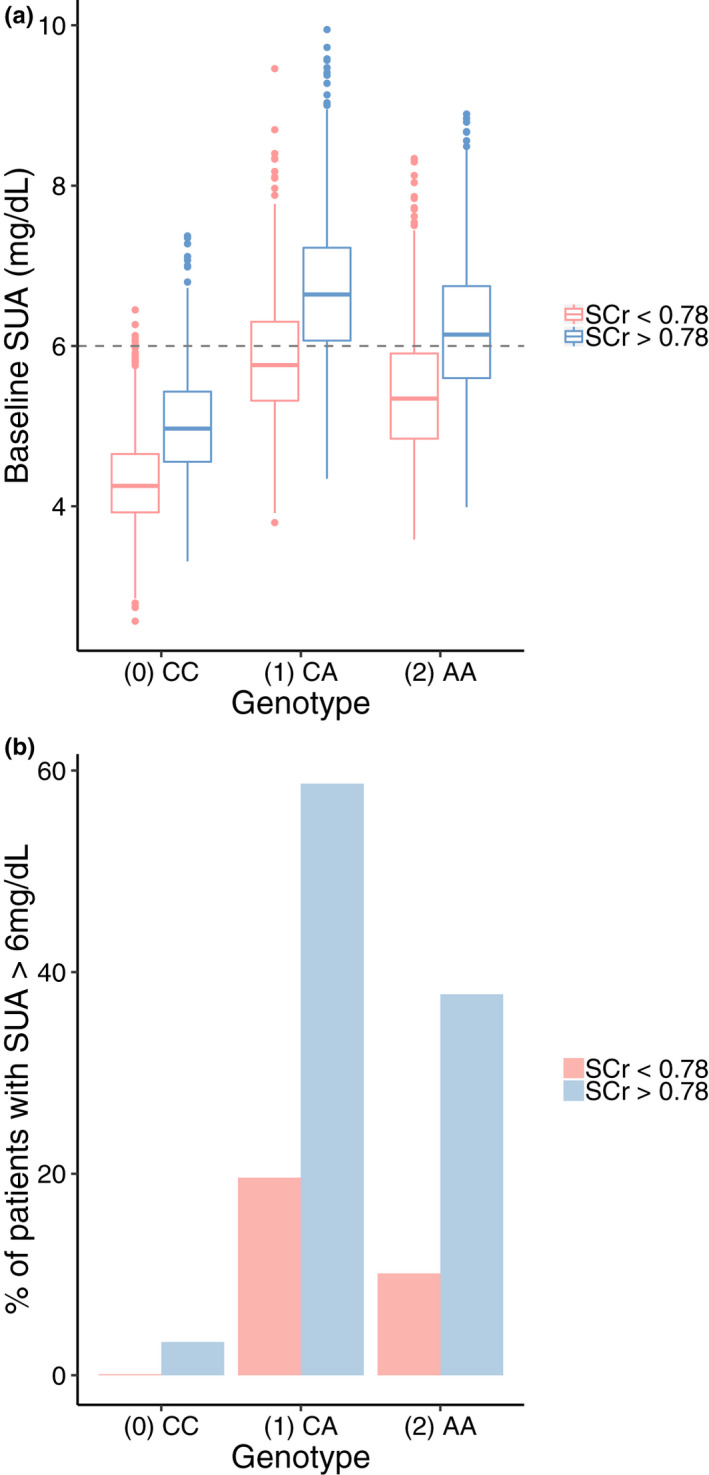
Simulated plasma concentrations of serum uric acid (SUA) at (a) baseline and (b) following allopurinol administration. Model predicted (a) baseline SUA concentrations and (b) percentage of patients with SUA levels greater than 6 mg/dl on day 31 after 30 days of once daily dosing of 300 mg of allopurinol stratified by serum creatinine (SCr) and genotype. Simulations were run using 1000 patients per genotype + SCr group (total n = 6000). Percentages reported were calculated for each of the six genotype + SCr groups. AA, homozygous variant; CA, heterozygous; CC, homozygous reference
Electronic health record analyses show clinical inhibitors of BCRP can phenocopy the p.Q141K variant
To investigate whether drugs that are clinical inhibitors of BCRP can phenocopy the effect of p.Q141K on baseline SUA, we mined the EHRs at UCSF and identified subjects both on and off BCRP inhibitors with SUA levels in the database. Specifically, we compared uric acid levels in patients prescribed cyclosporine and/or eltrombopag, both of which are identified as clinical inhibitors of BCRP by the US Food and Drug Administration (https://www.fda.gov/drugs/drug‐interactions‐labeling/drug‐development‐and‐drug‐interactions‐table‐substrates‐inhibitors‐and‐inducers), to uric acid levels in patients not prescribed either of those drugs. Based on the inclusion and exclusion criteria described in the methods, we were able to classify patients as “on” drug (i.e., prescribed cyclosporine and/or eltrombopag) or “off” drug (Figure 4a).
Figure 4.
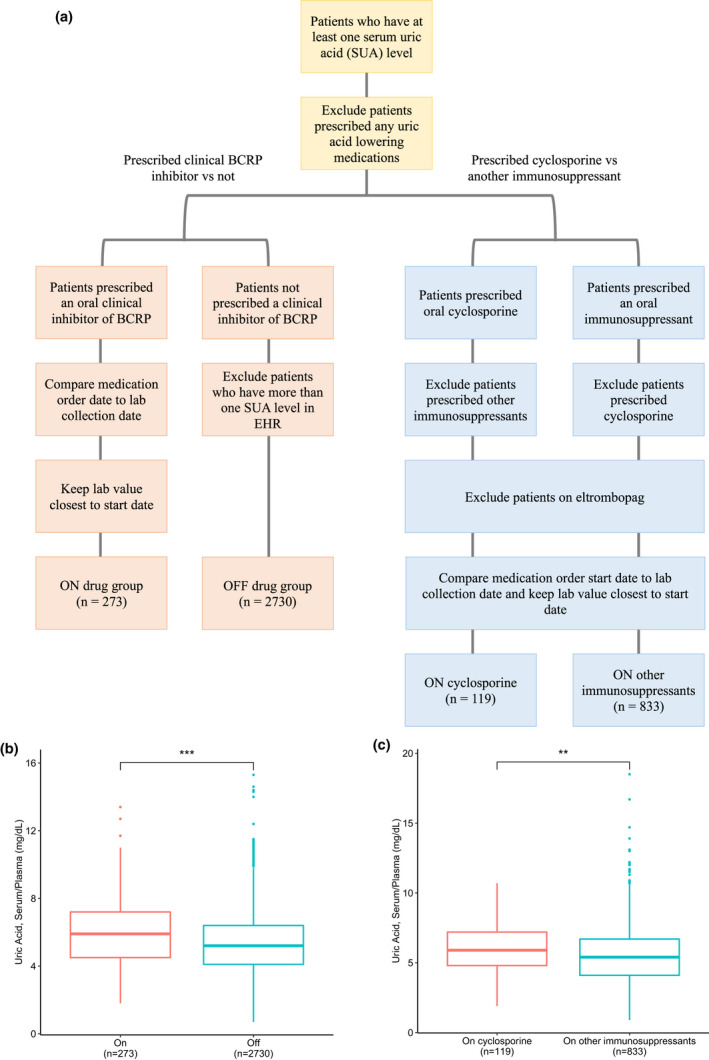
Uric acid levels in patients prescribed at least one clinical inhibitor of breast cancer resistance protein (BCRP) compared with levels in patients not prescribed a clinical inhibitor of BCRP from electronic health record (EHR) data. (a) Flow chart describing the inclusion/exclusion criteria for the analyses and (b) boxplots comparing uric acid laboratory values in patients prescribed cyclosporine and/or eltrombopag versus patients not prescribed cyclosporine and/or eltrombopag (p value: 1.74 × 10−6) and (c) patients prescribed cyclosporine (and no other immunosuppressant) versus patients prescribed other immunosuppressants (except cyclosporine; p value: 0.00462). **p ≤ 0.01, ***p ≤ 0.001, following a Wilcoxon rank sum test with continuity correction
A significant difference in uric acid levels was observed between the two groups (p value: 1.74 × 10−6; n = 273 “on” drug, n = 2730 “off” drug), with patients prescribed at least one clinical inhibitor of BCRP having higher levels compared to age‐matched and sex‐matched patients not prescribed either (5.95 mg/dl vs. 5.36 mg/dl; Figure 4b). To test the sensitivity of this analysis and selection of controls, 10 iterations were performed of randomly selected 273 age‐matched and sex‐matched patients from the “off” drug group to allow for a 1:1 ratio between the “on” drug and “off” drug groups; the “on” drug group had significantly higher uric acid levels compared with the “off” drug group in every iteration tested (Figure S5). Additional analyses with a maximum separation date of 1 year between the first medication order start date and laboratory collection date still showed a significant difference (p value = 0.00074) in uric acid levels between the “on” drug (n = 199) and “off” drug (n = 1990) groups, further demonstrating the robustness of our analysis.
In order to address the possibility that underlying disease or drug class contributed to the differences seen in uric acid levels, additional analyses were performed within the class of immunosuppressants. Uric acid levels in patients prescribed cyclosporine were compared with those prescribed other immunosuppressants (Figure 4a). Individuals prescribed cyclosporine (and no other immunosuppressants) had significantly higher uric acid levels (p value: 0.00462; n = 119 “on cyclosporine,” n = 833 “on other immunosuppressants”) compared with age‐matched and sex‐matched individuals prescribed other immunosuppressants (5.98 mg/dl vs. 5.58 mg/dl; Figure 4c).
DISCUSSION
BCRP p.Q141K has the potential to affect the PK and PD of many drugs by reducing efflux and increasing systemic levels. However, interestingly, p.Q141K has been associated with poor response to allopurinol in two genomewide association studies (GWAS) 6 , 7 and in candidate gene studies. 8 , 18 The current study provides the first investigation into the relationship between BCRP p.Q141K and allopurinol/oxypurinol PK/PD in a prospective clinical trial and BCRP inhibition and uric acid levels using real‐world data.
This study resulted in three major findings. First, we found that p.Q141K associated with a longer half‐life of oxypurinol. Second, we showed that gender was a significant covariate for oxypurinol V/Fm and higher baseline SUA levels caused by factors such as BCRP genotype and kidney function may lead to inadequate response to allopurinol. Finally, we found that clinical inhibitors of BCRP (cyclosporine and eltrombopag) associated with increased SUA levels, suggesting the potential of these drugs to cause hyperuricemia and increased risk for gout in susceptible patient populations.
One of our major findings was that the half‐life of oxypurinol was significantly longer in subjects who were homozygous for the reduced function variant compared to those who were homozygous for the reference allele of BCRP. Because half‐life is dependent on CL and V, this effect may have been driven by slight (albeit not significant) decreases in CL/F and increases in V/F in individuals homozygous for the variant allele compared with those homozygous for the reference allele (Table 2). CL of oxypurinol is determined to a large extent by kidney function. Data from BioBank Japan suggest that the reduced function allele, p.Q141K, associates with reduced kidney function (lower estimated glomerular filtration rate and thus increased SCr levels) in Asian subjects, 19 consistent with a trend toward reduced CL in the individuals homozygous for p.Q141K, which was observed in both our noncompartmental analysis and PK model. The slight increases in V/F in individuals homozygous for the variant allele may reflect increased distribution of oxypurinol into tissues in which BCRP is expressed (e.g., intestine, colon, liver, kidneys, brain, and thyroid 20 ), consistent with a reduced ability of the variant transporter to protect these tissues. Despite this finding, the overall exposure and Cmax of oxypurinol remained similar between genotype groups (Table 2). Further, the finding that oxypurinol half‐life is increased with BCRP genotype may be confounded by the limited sampling time in some subjects (24 h) compared with the oxypurinol half‐life; however, the trend remains when excluding these subjects.
To our knowledge, no other study has demonstrated enterohepatic recycling of oxypurinol and these findings need to be confirmed in additional PK studies. Preliminary studies identified xanthine oxidase activity in the contents obtained from the mouse intestinal lumen (Figure S2). This result is consistent with a previous electron microscopic study showing the presence of the enzyme in the mucous of the duodenum 21 and reports suggesting that the enzyme may be secreted into the intestinal lumen 22 , 23 or be expressed and secreted by gut microbiota. 24 Although speculative, intestinal lumen xanthine oxidase may be a target of oxypurinol, and enterohepatic recycling of oxypurinol may contribute to its overall pharmacological effects on reducing SUA. An intestinal lumen target for oxypurinol is consistent with GWAS results suggesting that individuals who harbor the reduced function variant of BCRP (who would have less enterohepatic recycling) have a poorer response to the drug. 6 , 7 , 8
Using EHRs as real‐world evidence, clinical inhibitors of BCRP associated with increased SUA levels, demonstrating that prescription drug inhibitors of BCRP may phenocopy the BCRP p.Q141K variant. Our therapeutic‐class analysis suggested that these observations were a result of inhibition of BCRP and not due to a drug class or underlying disease effect. The list of drugs used in the “on other immunosuppressants” drug group were: azathioprine, mycophenolic acid, mycophenolate mofetil, tacrolimus, and sirolimus, and the literature suggests that none of these drugs appear to interact with BCRP at clinically relevant concentrations; however, limited to no information was found about clinical inhibition of BCRP by mycophenolate mofetil, mycophenolic acid, and azathioprine, or its metabolite, 6‐mercaptopurine. 25 , 26 , 27
Simulations after 300 mg once daily dosing of allopurinol showed that subjects with higher baseline SUA due to BCRP p.Q141K genotype or SCr levels were less likely to maintain healthy (<6 mg/dl) SUA levels. Baseline SUA is an important determinant of allopurinol response, as shown in previous studies. 28 , 29 , 30 , 31 The results of this current study suggest that concomitant BCRP inhibitors may also contribute to increases in SUA levels and may contribute to hyperuricemia, and therefore allopurinol response. Subjects with higher baseline SUA for any reason may require higher doses of allopurinol to achieve and maintain healthy SUA levels. However, effect sizes observed in these simulations are based on a small sample size and need to be further investigated and validated.
There are a number of limitations to this study. First, allopurinol was dosed orally, as allopurinol for i.v. injection is only indicated for prevention of tumor lysis syndrome. 32 Oral dosing limited our ability to precisely detect effects of BCRP p.Q141K on bioavailability, and therefore on CL/F and V/F of the drug. Additionally, sample size of this trial was small, which was powered to detect large differences in oxypurinol clearance. Thus, there may be differences in oxypurinol PK parameters among genotype groups that went undetected in our study. Similarly, our model was limited due to the small sample size and study design; a larger study with a multiple dose regimen in patients diagnosed with gout is needed to better compare genotype groups, identify significant covariates, capture the multiple peaks in the concentration‐time profile, and improve our simulations. Although we attempted to include enterohepatic circulation in the model by evaluating different previously published modeling approaches, 33 this addition was not significant in terms of objective function and did not adequately capture the multiple peaks, most likely due to the limited data and large variation observed. Finally, our EHR analysis was limited by lack of data on the BCRP genotype of the patients, the indication for which the patients were prescribed the respective medications, and how long they were on each medication. As more EHR data becomes available for research purposes, we will be able to account for these variables and covariates, increase the sample size and robustness of our analysis, and potentially perform a joint GWAS and EHR analysis in the future.
The renewed interest in the treatment of gout stems from its growing prevalence and associated morbidities. Allopurinol remains the first‐line treatment, but recent GWAS have indicated that subjects harboring the BCRP p.Q141K variant may not respond as well to allopurinol. 6 , 7 , 8 Overall, our comprehensive study was able to identify covariates important in the PK and PD of oxypurinol and use real‐world data to phenocopy the p.Q141K variant using clinical inhibitors of BCRP. Additionally, this study provides evidence that individuals who harbor the reduced function variant of BCRP may have lower levels of oxypurinol in their intestine. If further studies show the intestine to be an important site of action for oxypurinol, this could help explain the poor response seen in these individuals, despite the similar systemic exposure of oxypurinol.
CONFLICT OF INTEREST
The authors report no conflicts of interest.
AUTHOR CONTRIBUTIONS
B.V., D.J.B., L.Z., M.G‐C., and K.M.G. wrote the manuscript. D.J.B., B.V., L.Z., M.G‐C., R.M.S., M.S, and K.M.G. designed the research. B.V., D.J.B., L.Z., and M.G‐C. performed the research. B.V., D.J.B., L.Z., and M.G‐C. analyzed the data. M.S., R.M.S., and K.M.G. contributed new reagents/analytical tools.
CODE AVAILABILITY
The NONMEM script for the final PK and PK/PD model is available as Supplementary Information.
Supporting information
Supplementary Material
Code S1
ACKNOWLEDGMENTS
The authors would like to acknowledge Sook Wah Yee, Celeste Eng, and Emma Hughes for helpful discussions. We would also like to thank Idit Kosti for help with extracting EHR data, Kathy Cheung for help with trial management, and Mina Azimi, Chenling Xiong, and Josefina Priotti for animal sample processing. We would like to acknowledge and thank all subjects for their participation as well as the coordinators and site personnel involved in the clinical study.
Funding information
This study was funded by NIH R01DK103729. M.S. is funded by P30AR070155.
REFERENCES
- 1. FitzGerald JD, Dalbeth N, Mikuls T, et al. American College of Rheumatology Guideline for the management of gout. Arthritis Rheumatol. 2020;72:879‐895. [DOI] [PubMed] [Google Scholar]
- 2. Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450‐2461. [DOI] [PubMed] [Google Scholar]
- 3. Wright DFB, Duffull SB, Merriman TR, Dalbeth N, Barclay ML, Stamp LK. Predicting allopurinol response in patients with gout. Br J Clin Pharmacol. 2016;81:277‐289. 10.1111/bcp.12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol. 2006;33:1646‐1650. [PubMed] [Google Scholar]
- 5. Kannangara DRW, Graham GG, Wright DFB, et al. Individualising the dose of allopurinol in patients with gout. Br J Clin Pharmacol. 2017;83:2015‐2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brackman DJ, Yee Sook W, Enogieru OJ, et al. Genome‐wide association and functional studies reveal novel pharmacological mechanisms for allopurinol. Clin Pharmacol Ther. 2019;106:623‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wen CC, Yee SW, Liang X, et al. Genome‐wide association study identifies ABCG2 (BCRP) as an allopurinol transporter and a determinant of drug response. Clin Pharmacol Ther. 2015;97:518‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wallace MC, Roberts RL, Nanavati P, et al. Association between ABCG2 rs2231142 and poor response to allopurinol: replication and meta‐analysis. Rheumatology. 2018;57:656‐660. [DOI] [PubMed] [Google Scholar]
- 9. Day RO, Graham GG, Hicks M, McLachlan AJ, Stocker SL, Williams KM. Clinical Pharmacokinetics and Pharmacodynamics of Allopurinol and Oxypurinol. Clin Pharmacokinet. 2007;46:623‐644. 10.2165/00003088-200746080-00001. [DOI] [PubMed] [Google Scholar]
- 10. Brackman DJ, Giacomini KM. Reverse translational research of ABCG2 (BCRP) in human disease and drug response. Clin Pharmacol Ther. 2018;103:233‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denney W, Duvvuri S, Buckeridge C. Simple, automatic noncompartmental analysis: the PKNCA Package. J Pharmacokinet Pharmacodyn. 2015;42:11‐107. [Google Scholar]
- 12. Li H, Limenitakis JP, Fuhrer T, et al. The outer mucus layer hosts a distinct intestinal microbial niche. Nat Commun. 2015;6:8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stocker SL, McLachlan AJ, Savic RM, et al. The pharmacokinetics of oxypurinol in people with gout. Br J Clin Pharmacol. 2012;74:477‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lindbom L, Pihlgren P, Jonsson EN, Jonsson N. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79:241‐257. [DOI] [PubMed] [Google Scholar]
- 15. Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2013;2:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ho DE, Imai K, King G, Stuart EA. MatchIt: nonparametric preprocessing for parametric causal inference. J Stat Softw. 2011;42. 10.18637/jss.v042.i08. [DOI] [Google Scholar]
- 17. Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roberts RL, Wallace MC, Phipps‐Green AJ, et al. ABCG2 loss‐of‐function polymorphism predicts poor response to allopurinol in patients with gout. Pharmacogenomics J. 2017;17:201‐203. [DOI] [PubMed] [Google Scholar]
- 19. Nagai A, Hirata M, Kamatani Y, et al. Overview of the BioBank Japan Project: study design and profile. J Epidemiol. 2017;27:S2‐S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fohner AE, Brackman DJ, Giacomini KM, Altman RB, Klein TE. PharmGKB summary: very important pharmacogene information for ABCG2. Pharmacogenet Genomics. 2017;27:420‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Den Munckhof RJ, Vreeling‐Sindelarova H, Schellens JP, Van Noorden CJ, Frederiks WM. Ultrastructural localization of xanthine oxidase activity in the digestive tract of the rat. Histochem J. 1995;27:897‐905. [PubMed] [Google Scholar]
- 22. Vettenranta K, Raivio KO. Xanthine oxidase during human fetal development. Pediatr Res. 1990;27:286‐288. [DOI] [PubMed] [Google Scholar]
- 23. Harrison R. Milk xanthine oxidase: Properties and physiological roles. Int Dairy J. 2006;16:546‐554. [Google Scholar]
- 24. Guo Z, Zhang J, Wang Z, et al. Intestinal microbiota distinguish gout patients from healthy humans. Sci Rep. 2016;6:20602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pawarode A, Shukla S, Minderman H, et al. Differential effects of the immunosuppressive agents cyclosporin A, tacrolimus and sirolimus on drug transport by multidrug resistance proteins. Cancer Chemother Pharmacol. 2007;60:179‐188. [DOI] [PubMed] [Google Scholar]
- 26. Köck K, Brouwer KLR. A perspective on efflux transport proteins in the liver. Clin Pharmacol Ther. 2012;92:599‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Wolf C, Jansen R, Yamaguchi H, et al. Contribution of the drug transporter ABCG2 (breast cancer resistance protein) to resistance against anticancer nucleosides. Mol Cancer Ther. 2008;7:3092‐3102. [DOI] [PubMed] [Google Scholar]
- 28. Köttgen A, Albrecht E, Teumer A, et al. Genome‐wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013;45:145‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuo H, Takada T, Ichida K, et al. ABCG2/BCRP dysfunction as a major cause of gout. Nucleosides Nucleotides Nucleic Acids. 2011;30:1117‐1128. [DOI] [PubMed] [Google Scholar]
- 30. Matsuo H, Yamamoto K, Nakaoka H, et al. Genome‐wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann Rheum Dis. 2016;75:652‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakayama A, Matsuo H, Takada T, et al. ABCG2 is a high‐capacity urate transporter and its genetic impairment increases serum uric acid levels in humans. Nucleosides Nucleotides Nucleic Acids. 2011;30:1091‐1097. [DOI] [PubMed] [Google Scholar]
- 32. Cairo MS, Coiffier B, Reiter A, Younes A, Panel TE. Recommendations for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578‐586. [DOI] [PubMed] [Google Scholar]
- 33. Okour M, Brundage RC. Modeling enterohepatic circulation. Curr Pharmacol Rep. 2017;3:301‐313. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Code S1