

Abstract
Consensus guidelines exist for genotype‐guided fluoropyrimidine dosing based on variation in the gene dihydropyrimidine dehydrogenase (DPYD). However, these guidelines have not been widely implemented in North America and most studies of pretreatment DPYD screening have been conducted in Europe. Given regional differences in treatment practices and rates of adverse events (AEs), we investigated the impact of pretreatment DPYD genotyping on AEs in a Canadian context. Patients referred for DPYD genotyping prior to fluoropyrimidine treatment were enrolled from December 2013 through November 2019 and followed until completion of fluoropyrimidine treatment. Patients were genotyped for DPYD c.1905+1G>A, c.2846A>T, c.1679T>G, and c.1236G>A. Genotype‐guided dosing recommendations were informed by Clinical Pharmacogenetics Implementation Consortium guidelines. The primary outcome was the proportion of patients who experienced a severe fluoropyrimidine‐related AE (grade ≥3, Common Terminology Criteria for Adverse Events version 5.0). Secondary outcomes included early severe AEs, severe AEs by toxicity category, discontinuation of fluoropyrimidine treatment due to AEs, and fluoropyrimidine‐related death. Among 1394 patients, mean (SD) age was 64 (12) years, 764 (54.8%) were men, and 47 (3.4%) were DPYD variant carriers treated with dose reduction. Eleven variant carriers (23%) and 418 (31.0%) noncarriers experienced a severe fluoropyrimidine‐related AE (p = 0.265). Six carriers (15%) and 284 noncarriers (21.1%) experienced early severe fluoropyrimidine‐related AEs (p = 0.167). DPYD variant carriers treated with genotype‐guided dosing did not experience an increased risk for severe AEs. Our data support a role for DPYD genotyping in the use of fluoropyrimidines in North America.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Dihydropyrimidine dehydrogenase (DPD) deficiency is associated with fluoropyrimidine‐related adverse events (AEs), and screening for DPD deficiency can be carried out using DPYD genotype testing of clinically relevant variants, as noted in the Clinical Pharmacogenetics Implementation Consortium guidelines.
WHAT QUESTION DID THIS STUDY ADDRESS?
Given the paucity of data relating to pretreatment use of DPYD genotyping in North America, this Canadian study adds new insights to the clinical impact of DPYD testing.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
DPYD genotype‐guided dosing can ameliorate fluoropyrimidine‐related AE risk for patients treated with fluoropyrimidine dose and regimens prescribed in North America.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Although the European Medicines Agency supports DPD deficiency screening, this study suggests that similar efforts should be undertaken in North America.
INTRODUCTION
Five‐fluorouracil (5‐FU) and capecitabine are fluoropyrimidines used in the treatment of solid tumours. 1 , 2 , 3 , 4 Unfortunately, ~30% of patients experience severe fluoropyrimidine‐related toxicity. 5 , 6 Dihydropyrimidine dehydrogenase (DPD, gene name DPYD) is the rate‐limiting enzyme in fluoropyrimidine catabolism. 7 DPD deficiency increases the risk of fluoropyrimidine‐related toxicity, 8 and there are heritable DPYD variants associated with decreased enzyme function and thereby DPD deficiency. 9 Meta‐analyses have narrowed the list of clinically relevant genetic variants allowing the implementation of genotype‐guided dosing. 10 , 11 , 12
In 2013, the Clinical Pharmacogenetics Implementation Consortium (CPIC) published a consensus guideline detailing fluoropyrimidine dosing recommendations for 3 DPYD variants associated with reduced enzymatic activity: DPYD c.1905+1G>A (*2A, rs39818290), c.2846A>T (rs67376798), and c.1679T>G (*13, rs55886062). 13 For heterozygous carriers of an individual variant, a 50% dose reduction was recommended, whereas avoidance of fluoropyrimidines was recommended for homozygous or compound heterozygous variant carriers. A fourth intronic variant, DPYD c.1129‐5923C>G (rs75017182, in linkage disequilibriumwith DPYD c.1236G>A [rs56038477]) was added to the guideline in 2017. 9 These recommendations were also refined based on enzymatic activity scores (AS). 14 The AS of each patient is the sum of the individual alleles where each allele is assigned a score of 0 to 1 based on functional characterization. The AS of clinically relevant alleles are 0 for DPYD c.1905+1G>A or c.1679T>G and 0.5 for DPYD c.2846A>T or c.1129‐5923C>G. A 25% to 50% dose reduction was recommended for intermediate metabolizers with an AS of 1.5 and a 50% dose reduction was recommended for an AS of 1 and avoidance for an AS of 0 to 0.5. In 2018, results from Henricks et al. 15 led to further updates of the CPIC guidelines to recommend a 50% dose reduction for AS of either 1 or 1.5. 16 In addition, following a well‐tolerated initial dose reduction, the CPIC encourages cautious dose escalation and with concurrent therapeutic drug monitoring if available. Of note, the CPIC guidelines provide reference for patients with available genotype data and do not comment on the necessity of preemptively determining the DPYD genotype.
In addition to the CPIC guidelines for response to known DPYD variants, Dutch and French initiatives have published guidelines that explicitly recommend DPD deficiency screening prior to fluoropyrimidine therapy. 17 , 18 Despite this, adoption of pretreatment DPYD genotyping in Canada has been limited and currently is widely accessible only in Quebec. Given the abundance of data linking complete DPD deficiency to severe toxicity, a randomized controlled trial of pretreatment DPD deficiency screening was considered to be inappropriate for our center. The only two‐arm comparative study of DPD deficiency screening was terminated prematurely due to the fluoropyrimidine‐related death of a DPD‐deficient patient in the control arm. 19 However, two prospective DPYD single arm genotype‐guided studies were completed in the Netherlands, the first examined the impact of one variant (c.1905+1G>A), 20 and the second assessed four variants (c.1905+1G>A, c.2846A>T, c.1679T>G, and c.1236G>A). 15 These studies demonstrated that genotype‐guided dosing reduces the risk of adverse events (AEs) for DPYD variant carriers in a European population compared to the historical rate in DPYD variant carriers receiving the standard of care.
In contrast, the impact of pretreatment DPYD genotype‐guided fluoropyrimidine dosing in North America is unpublished. There is an important distinction between results from a European population and the potential results in a North American population. Work by Haller et al. has identified regional variation in fluoropyrimidine‐related AEs between the United States and Europe. 21 Therefore, there is a need for regional data to support regional implementation. Here, we conducted a study to determine the impact of pretreatment DPYD genotype‐guided dosing on patient safety at a tertiary care center in London, Ontario, Canada. We hypothesized that DPYD variant carriers who received a genotype‐guided dosing would have no greater risk of fluoropyrimidine‐related AEs as compared with noncarriers.
PATIENTS AND METHODS
Study Sample
We conducted a single‐center retrospective study of patients referred to the Personalized Medicine clinic at London Health Sciences Centre (LHSC), London, Ontario, Canada, for DPYD genotype testing between December 1, 2013, and November 30, 2019. The study was approved by the Institutional Review Board at Western University and all patients provided written informed consent. Of 1945 patients referred for testing, 1845 were tested prior to fluoropyrimidine treatment. At initiation, the study was based on the 2013 CPIC guidelines 13 ; however, in response to the 2017 update, 9 the DPYD c.1236G>A variant was added to the testing panel in May 2018. Consequently, 41 c.1236G>A carriers identified retrospectively were removed from the study as they did not receive appropriate genotype‐guided dosing. Two compound heterozygous carriers were identified among the genotype‐guided patients and the treating oncologists were advised to select an alternative therapy. There were 1394 patients who initiated treatment through LHSC prior to December 1, 2019, that were included in the genotype‐guided study (Figure 1, baseline characteristics are summarized in Table 1). Prior chemotherapy, radiation therapy, concurrent antineoplastic therapies, and other concomitant medications were allowed. Baseline characteristics for patients lost to follow‐up are shown in Table S1.
Figure 1.
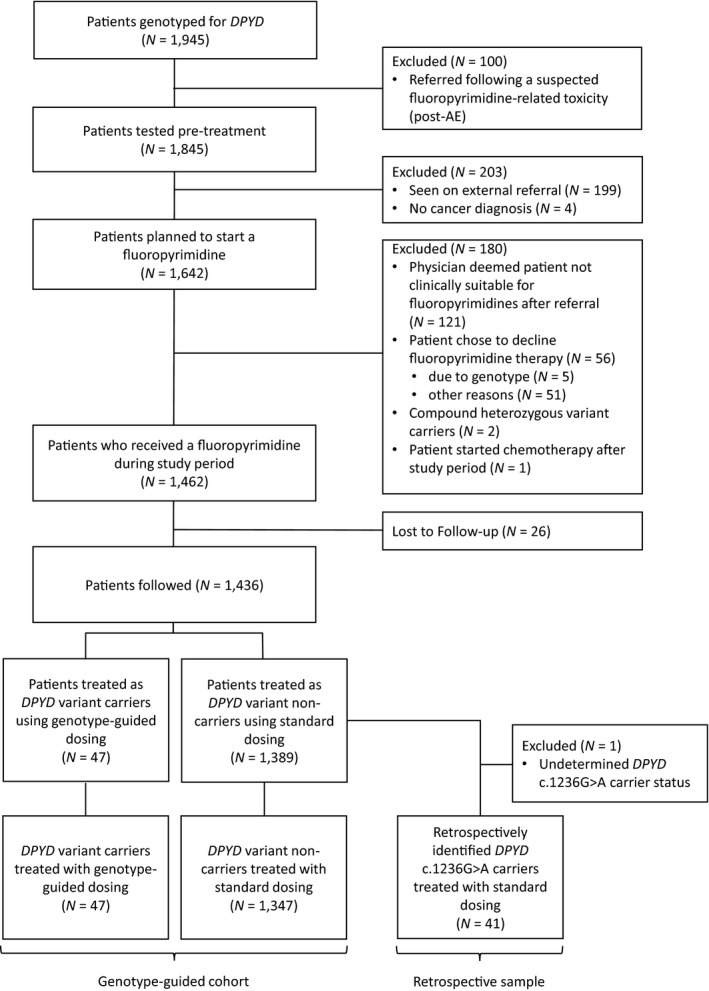
Flow diagram illustrating the study cohort. AE, adverse event; DPYD, dihydropyrimidine dehydrogenase
Table 1.
Baseline patient characteristics
| Characteristic | Genotype‐guided cohort | Retrospective sample | |
|---|---|---|---|
| Noncarrier (N = 1347) | Carrier (N = 47) | c.1236G>A carrier (N = 41) | |
| Sex, N (%) | |||
| Female | 605 (44.9) | 25 (53) | 13 (31) |
| Male | 742 (55.1) | 22 (47) | 28 (68) |
| Race, N (%) | |||
| White | 1267 (94) | 45 (96) | 40 (98) |
| Other a | 32 (2.4) | 1 (2) | 1 (2) |
| Unknown b | 48 (3.6) | 1 (2) | 0 (0) |
| Age, mean (SD), years | 64 (12) | 62 (13) | 66 (10.4) |
| Body surface area, mean (SD), m2 | 1.9 (0.3) | 1.9 (0.2) | 1.94 (0.27) |
| Tumor site, N (%) | |||
| Colorectal | 779 (57.8) | 25 (53) | 21 (51) |
| Gastric and esophagus | 189 (14.0) | 7 (15) | 5 (12) |
| Pancreas | 106 (7.9) | 6 (13) | 2 (5) |
| Breast | 89 (6.6) | 3 (6) | 1 (2) |
| Anus | 48 (3.6) | 1 (2) | 3 (7) |
| Head and neck | 27 (2.0) | 2 (4) | 3 (7) |
| Other c | 109 (8.1) | 3 (6) | 6 (15) |
| Regimen, N (%) | |||
| Capecitabine with radiation | 277 (20.6) | 11 (23) | 11 (27) |
| Capecitabine monotherapy d | 229 (17.0) | 7 (15) | 8 (20) |
| Capecitabine with oxaliplatin | 130 (9.7) | 2 (4) | 2 (5) |
| Capecitabine with other agents e | 68 (5.0) | 3 (6) | 1 (2) |
| FOLFOX d | 228 (16.9) | 8 (17) | 9 (22) |
| FOLFIRI/FOLFIRINOX | 135 (10.0) | 8 (17) | 0 (0) |
| 5‐FU with cisplatin/carboplatin | 128 (9.5) | 4 (9) | 3 (7) |
| 5‐FU with other agents f | 152 (11.3) | 4 (9) | 7 (17) |
| DPYD genotype, N (%) | |||
| Wild‐type | 1347 (100) | 0 (0) | 0 (0) |
| c.2846A>T heterozygous | 0 (0) | 19 (40) | 0 (0) |
| c.1905+1G>A heterozygous | 0 (0) | 9 (19) | 0 (0) |
| c.1679T>G heterozygous | 0 (0) | 1 (2) | 0 (0) |
| c.1236G>A heterozygous | 0 (0) | 18 (38) | 41 (100) |
Abbreviation: 5‐FU, 5‐fluorouracil.
Other includes Black, Asian, and Indigenous.
Due to self‐declaration of race not all patients opted to provide this information and it remains unknown.
Other included appendix and small bowel, genitourinary, hepatobiliary, and primary site unknown.
Including with and without biologic agents.
Including gemcitabine, lapatinib, temozolomide, docetaxel, epirubicin, and mitomycin + radiation.
Including Degramount, FEC‐D, and FLOT regimens, in addition to mitomycin + radiation.
DPYD genotype testing and dosing recommendations
Whole blood samples were collected from each patient and DNA was extracted using the MagNA Pure Compact Instrument (Roche). DNA was assessed on a ViiA 7 real‐time polymerase chain reaction system (ThermoFisher Scientific) using TaqMan allelic discrimination assays (ThermoFisher Scientific) for DPYD c.1905+1G>A (assay ID: C__30633851_20), c.2846A>T (assay ID: C__27530948_10), c.1679T>G (assay ID: C__11985548_10), and c.1236G>A (assay ID: C__25596099_30). Variant c.1236G>A is known to be in strong linkage disequilibrium with c.1129‐5923C>G and was used as a proxy for genotyping, which is in alignment with the CPIC guidelines.
Results and dosing recommendations were provided to the referring physicians within the patients’ electronic health records (EHRs). Recommendations were as follows: for noncarriers, dose as per standard of care; for simple heterozygous carriers, apply a 50% initial dose reduction, and consider attempting dose escalation in subsequent cycles pending patient tolerance. A 25% to 50% initial dose reduction was recommended for heterozygous carriers of c.1236G>A upon its addition to the testing panel, with the same additional recommendation to attempt dose escalation based on patient tolerability. Avoidance of fluoropyrimidines in homozygous or compound heterozygous variant carriers was recommended throughout the study, recommendations are summarized in Table 2. Final treatment decisions were at the discretion of the treating oncologist.
Table 2.
Dose Recommendations used in the study
| Variant Status | Genotype | AS a | Recommendation |
|---|---|---|---|
| Noncarrier | ‐/‐ | 2 | Standard dosing |
| Simple heterozygous carriers | ‐/c.1236G>A b | 1.5 | 25%–50% Dose reduction |
| ‐/c.2846G>A | 1.5 | 50% Dose reduction | |
| ‐/c.1905+1G>A | 1 | ||
| ‐/c.1679T>G | 1 | ||
| Compound heterozygous carriers | c.1236G>A/c.2846A>T c | 1 | Avoid fluoropyrimidines |
| c.1236G>A/c.1905+1G>A | 0.5 | ||
| c.1236G>A/c.1679T>G | 0.5 | ||
| c.2846A>T/c.1905+1G>A | 0.5 | ||
| c.2846A>T/c.1679T>G | 0.5 | ||
| c.1905+1G>A/c.1679T>G | 0 | ||
| Homozygous carriers | c.1236G>A/c.1236G>A d | 1 | |
| c.2846A>T/c.2846A>T d | 1 | ||
| c.1905+1G>A/c.1905+1G>A | 0 | ||
| c.1679T>G/c.1679T>G | 0 |
Abbreviations and Symbols: AS, activity score; ‐, negative for tested variants.
The predicted AS, assuming nontested variants are functional with an AS of 1 per allele.
The c.1236G>A was added to the testing panel in 2018 as a proxy for haplotype‐B3 and the causative variant DPYD c.1129‐5923C>G.
Despite an AS of 1, we recommend avoiding fluoropyrimidines in c.1236G>A/c.2846A>T patients, however, no patients with this genotype were detected.
Despite an AS of 1, we recommend avoiding fluoropyrimidines in c.1236G>A/c.1236G>A or c.2846A>T/c.2846A>T patients, however, no patients with this genotype were detected.
Data collection
Treatment data, including regimen, dose, and radiation use, were collected from LHSC pharmacy records. Clinical variables and toxicity data were obtained by standardized review of the patients’ EHRs by trained study personnel, each record was reviewed independently by two study members. Toxicity data were recorded from clinic notes, admission records, discharge summaries, and emergency department reports. Severe AEs included grade greater than or equal to 3 toxicities according to the National Cancer Institutes’ Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. 22 Only those AEs determined to be possibly, probably, or definitely related to the fluoropyrimidine components were included in the outcome, following the standard definitions proposed in the National Institutes of Health protocol template for phase II/III trials. 23 Based on these principles, the definitions require the AE to occur within 30 days of fluoropyrimidine administration, be pharmacologically plausible, and not be attributable to another component of the regimen. The effect of removing and reinstating the fluoropyrimidine were also considered when these challenges occurred. Based on the literature, the major toxicity categories considered were gastrointestinal (including primarily: diarrhea, oral mucositis, and nausea/vomiting), myelosuppression (primarily neutropenia/febrile neutropenia, as well as thrombocytopenia, and unexplained anemia), cardiac (sudden onset cardiac toxicity during fluoropyrimidine administration), and Palmar‐Plantar Erythrodysesthesia (Hand‐Foot Syndrome). The remaining AEs, in which fluoropyrimidines were likely contributors, were grouped under the other heading. The AEs reported by the initial reviewers and attribution of causality was reviewed by a medical oncology fellow under the supervision of a practicing medical oncologist. Conflicts in the records were reviewed by the initial coders and the reviewing medical oncologist. Patients were followed for their entire treatment period and until toxicity resolved.
Outcomes
The primary outcome was severe (grade ≥3, CTCAE version 5.0) fluoropyrimidine‐related AEs. We included a secondary outcome of early fluoropyrimidine‐related AEs during the first two cycles of treatment. Secondary outcomes further included fluoropyrimidine‐related AEs by toxicity category, proportion of patients discontinuing fluoropyrimidines due to fluoropyrimidine‐related AEs, and fluoropyrimidine‐related deaths.
Statistical methods
The primary outcome was compared between DPYD variant carriers and noncarriers using a χ2 test. Other dichotomous outcomes were compared using χ2 or Fisher’s Exact test as appropriate. Fisher’s Exact tests were used when cell values in contingency tables were less than or equal to 5. A test for noninferiority between AEs in the variant carriers and noncarriers was performed using a two‐one sided test of equivalence. The smallest effect size of interest (SESOI) was determined using the lower bound for the 95% confidence interval (CI) of the risk for the c.1236G>A variant carriers in the literature multiplied by the event rate in noncarriers in this study. The c.1236G>A demonstrated the lowest increased risk and using this value to set the SESOI was considered a conservative approach. Unadjusted relative risk was used to show the risk of grade greater than or equal to 3 AE in our genotype‐guided study and within the literature. Unadjusted relative risks are reported due to the low number of events among variant carriers, and for consistency with previous genotype‐guided studies. A multivariable logistic regression determined the adjusted odds ratios and is available within the supplementary data. A Wilcoxon Mann‐Whitney U test was used to compare the number of cycles administered between variant carriers and noncarriers. Descriptive statistics are shown using number (percentage), mean (SD), and median (interquartile range [IQR]) as applicable. Reported p values are for two‐sided tests, with p < 0.05 considered significant. All analyses were performed using R (version 4.0.2; R Foundation Inc.; http://cran.r‐project.org/). In addition, the package “tidyverse” was used for data processing and both “epiR” and “TOSTER” were used for analysis, the script used for analysis is available in the supplementary materials.
RESULTS
Study population
Among the 1394 patients provided genotype‐guided dosing, the mean (SD) age was 64 (12) years and 764 (54.8%) were men. The most common primary tumor site was colorectal (804, 57.7%). Overall fluoropyrimidine use was distributed between capecitabine (727, 52.2%) and 5‐FU (667, 47.8%). Forty‐seven patients (3.4%) were heterozygous carriers for one of DPYD c.2846A>T (19, 1.4%), c.1236G>A (18, 1.3%), c.1905+1G>A (9, 0.6%), or c.1679T>G (1, <0.1%). The retrospectively identified c.1236G>A carriers did not appear to differ from the primary study, with the most common primary tumor site being colorectal (21, 51%), and an approximately equal use of capecitabine (23, 56%), and 5‐FU (18, 44%). However, the retrospective sample contained more men (28/41, 68%) than women. The baseline characteristics are summarized in Table 1.
Physician compliance with dose recommendations
We confirmed that variant carriers were treated according to the dose recommendations provided to the treating oncologist. The mean initial dose intensity was 52% (18) of ideal for variant carriers and 87.4% (15.2) for noncarriers (Table 3). Variant carriers received a median (IQR) of 6 (2–7) cycles of fluoropyrimidine treatment, and noncarriers received a median of 4 (2–6) cycles. We also assessed the mean dose intensity throughout the treatment period and found that variant carriers received a mean dose intensity over the total treatment period of 55% (15), whereas mean intensity for noncarriers was 84.2% (14.7).
Table 3.
Severe fluoropyrimidine‐related adverse events during total treatment period
| Genotype‐guided cohort | Retrospective sample | |||||||
|---|---|---|---|---|---|---|---|---|
| Noncarrier (N = 1347) | Carrier (N = 47) | P value a | c.1905+1G>A (N = 9) | c.2846A>T (N = 19) | c.1679T>G (N = 1) | c.1236G>A (N = 18) | c.1236 G>A (N = 41) | |
| Initial dose intensity, mean (SD) | 87.4 (15.2) | 52 (18) | NA | 47 (16) | 47 (21) | 43 (NA) | 59 (13) | 85 (17) |
| Dose intensity, mean (SD) | 84.2 (14.7) | 55 (13) | NA | 46 (8) | 55 (15) | 50 (NA) | 59 (12) | 85 (17) |
| Treatment cycles, median (IQR) | 4 (2–6) | 6 (2–7) | 0.201 | 6 (2–8) | 6 (4–8) | 6 (NA) | 4 (2–6) | 2 (2–4) |
| Total severe AEs b (all cycles), N (%) | N (%) | N (%) | N (%) | |||||
| Global c | 418 (31.0) | 11 (23) | 0.265 | 3 (33) | 5 (26) | 0 (0) | 3 (17) | 14 (34) |
| Gastrointestinal | 167 (12.4) | 6 (12) | 0.940 | 2 (22) | 2 (11) | 0 (0) | 2 (11) | 7 (17) |
| Myelosuppression | 157 (11.7) | 6 (12) | 0.816 | 2 (22) | 2 (11) | 0 (0) | 2 (11) | 2 (5) |
| Cardiac | 33 (2.4) | 0 (0) | 0.625 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| HFS | 35 (2.6) | 1 (2) | >0.99 | 0 (0) | 1 (5) | 0 (0) | 0 (0) | 2 (5) |
| Other d | 113 (8.4) | 2 (4) | 0.425 | 1 (11) | 1 (5) | 0 (0) | 0 (0) | 5 (12) |
| AE‐related death e | 10 (0.7) | 0 (0) | >0.99 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Discontinued treatment f | 232 (17.2) | 10 (21) | 0.437 | 2 (22) | 3 (16) | 0 (0) | 5 (28) | 7 (17) |
Abbreviations: AEs, adverse events; HFS, hand‐foot syndrome; IQR, interquartile range; NA, not applicable.
P value for treatment cycles was calculated based on Wilcoxon‐Mann‐Whitney test. P values for fluoropyrimidine‐related AEs calculated using the following tests: Global, Gastrointestinal, Myelosuppression, and Discontinued Treatment utilized χ2 tests; Cardiac, HFS, Other, and AE‐related Death utilized Fisher’s Exact Test.
Grade ≥3 by Common Terminology Criteria for Adverse Events version 5.0.
Global includes all fluoropyrimidine‐related AEs grade ≥3 and fluoropyrimidine‐related deaths. This does not include discontinuation.
Other grade ≥3 AEs included: fatigue, infections, neurotoxicities, and laboratory abnormalities.
At least one fluoropyrimidine‐related AE contributed significantly to death.
Patients discontinuing treatment with fluoropyrimidines due to a fluoropyrimidine‐related AE of any grade.
Toxicity outcomes
There were no significant differences in the primary or secondary toxicity outcomes between genotype‐guided variant carriers and noncarriers. We observed that 23% (11/47) of variant carriers, and 31.0% (418/1347) of noncarriers experienced severe fluoropyrimidine‐related AEs during their total treatment periods (p = 0.265; Table 3). We next examined severe fluoropyrimidine‐related AEs that occurred during the early cycles (1–2) of fluoropyrimidine treatment. We found that 13% (6/47) of genotype‐guided variant carriers compared to 21.4% (284/1347) of genotype‐guided noncarriers experienced an early fluoropyrimidine‐related AE (p = 0.167; Table 4). Secondary analyses of the major AE categories, proportion discontinuing fluoropyrimidines due to AEs and fluoropyrimidine‐related deaths, during the total treatment period or the first two cycles, did not show any significant differences between genotype‐guided variant carriers and noncarriers. Additionally, we performed noninferiority testing comparing the risk for global severe fluoropyrimidine‐related AEs between carriers and noncarriers both during the total treatment and limited to the early cycles (Figure 2). In both early and total treatment periods, the CIs included no difference but did not cross the noninferiority margin. Therefore, we conclude that genotype‐guided variant carriers do not experience increased risk of fluoropyrimidine‐related AEs compared with noncarriers receiving the standard of care dosing practices. We determined the unadjusted relative risk (RR) of grade greater than or equal to 3 fluoropyrimidine‐related AEs in our genotype‐guided variant carriers to allow for comparison to literature values (Table 5). 11 , 15 We report unadjusted RR values due to the small number of genotype‐guided variant carriers in our cohort and the literature. We obtained historical values for RR of fluoropyrimidine‐related AEs without genotype‐guidance from a meta‐analysis by Meulendijks et al. 11 In our cohort, genotype‐guided variant carriers were not at a significantly elevated risk for severe fluoropyrimidine‐related AEs compared with noncarriers. Indeed, with the recommended 50% dose reduction the RR was 1.08 (95% CI: 0.43–2.74) for c.1905+1G>A carriers, and 0.85 (95% CI: 0.40–1.82) for c.2846A>T carriers. With the recommended 25% to 50% dose reduction recommendations, the RR for genotype‐guided c.1236G>A carriers was 0.54 (95% CI: 0.19–1.52). Finally, the single c.1679T>G carrier in our genotype‐guided cohort was treated with a 50% dose reduction and did not suffer any fluoropyrimidine‐related AEs during treatment. 11 Additionally, we performed a secondary calculation of multivariable logistic regression adjusting for age, sex, regimen, and initial intensity of therapy. We do not include this in the primary report due to the small sample size of variants and the potential for the introduction of bias during adjustment. However, we note that there were no significant differences from the unadjusted predictions and these results can be found in Table S2.
Table 4.
Early severe fluoropyrimidine‐related AEs
| Genotype‐guided cohort | Retrospective sample | |||||||
|---|---|---|---|---|---|---|---|---|
| Noncarrier (N = 1347) | Carrier (N = 47) | P value a | c.1905+1G>A (N = 9) | c.2846A>T (N = 19) | c.1679T>G (N = 1) | c.1236G>A (N = 18) | c.1236 G>A (N = 41) | |
|
Early severe AEs b (cycles 1–2), N (%) |
N (%) | N (%) | N (%) | |||||
| Global c | 284 (21.1) | 6 (13) | 0.167 | 2 (22) | 3 (16) | 0 (0) | 1 (5) | 10 (24) |
| Gastrointestinal | 131 (9.7) | 3 (6) | 0.616 | 1 (11) | 1 (5) | 0 (0) | 1 (5) | 5 (12) |
| Myelosuppression | 102 (7.6) | 5 (11) | 0.401 | 2 (22) | 2 (11) | 0 (0) | 1 (5) | 1 (2) |
| Cardiac | 26 (1.9) | 0 (0) | >0.99 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| HFS | 13 (1.0) | 0 (0) | >0.99 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (5) |
| Other d | 68 (5.0) | 2 (4) | >0.99 | 1 (11) | 1 (5) | 0 (0) | 0 (0) | 3 (7) |
| AE‐related death e | 8 (0.6) | 0 (0) | >0.99 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Discontinued treatment f | 137 (10.2) | 5 (11) | 0.808 | 2 (22) | 1 (5) | 0 (0) | 2 (11) | 4 (10) |
Abbreviations: AEs, adverse events; HFS, hand‐foot syndrome.
P values for fluoropyrimidine‐related AEs calculated using the following tests: Global utilized χ2 test; Gastrointestinal, Myelosuppression, Cardiac, HFS, Other, AE‐related death, and Discontinued Treatment utilized Fisher’s Exact Test.
Grade ≥3 by Common Terminology Criteria for Adverse Events version 5.0.
Global includes all fluoropyrimidine‐related AEs grade ≥3 and fluoropyrimidine‐related deaths. This does not include discontinuation.
Other AEs included: fatigue, infections, neurotoxicities, and laboratory abnormalities.
At least one fluoropyrimidine‐related AE contributed significantly to death.
Patients discontinuing treatment with fluoropyrimidines due to a fluoropyrimidine‐related AE of any grade.
Figure 2.
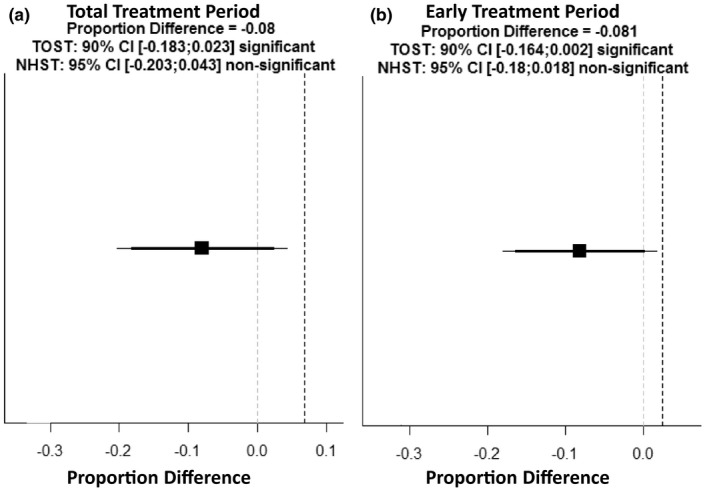
Plotting results of noninferiority comparison for global severe fluoropyrimidine‐related AEs between genotype‐guided variant carriers and noncarriers. Difference is variant carriers minus noncarriers, less than zero genotype‐guided variant carriers are at less risk than standard of care noncarriers. The first panel compares proportion of severe AEs during total treatment period (a), the inferiority bound is 6.82%, the genotype‐guided variant carriers do not experience increased risk of severe AEs in the total treatment period. The second panel compares proportion of severe AEs during early treatment period (b), the inferiority bound is 2.52%, the genotype‐guided variant carriers do not experience increased risk of severe AEs in the early treatment period. AE, adverse event; CI, confidence interval
Table 5.
Unadjusted relative risk of severe fluoropyrimidine‐related adverse events
| DPYD variant |
Genotype‐guided dosing current cohort a RR (95% CI) d |
Patients treated without genotype‐guided dosing b RR (95% CI) d |
Genotype‐guided dosing literature cohort c RR (95% CI) d |
|---|---|---|---|
| c.1905+1G>A | 1.08 (0.43–2.74) | 2.87 (2.14–3.86) | 1.31 (0.63–2.72) |
| c.2846A>T | 0.85 (0.40–1.82) | 3.11 (2.25–4.28) | 2.00 (1.19–3.34) |
| c.1679T>G | NA e | 4.30 (2.10–8.80) | NA e |
| c.1236G>A | 0.54 (0.19–1.52) | 1.72 (1.22–2.42) | 1.69 (1.18–2.42) |
Abbreviations: CI, confidence interval; NA, not applicable; RR, relative risk.
Our genotype‐guided cohort: 50% dose reduction recommended for carriers of c.1905+1G>A, c.2846A>T, and c.1679T>G; 25% −50% dose reduction for carriers of c.1236G>A.
Meulendijks et al. historical cohort derived from a meta‐analysis 11 : standard of care dosing with adjustment due to tolerability resulting in the assumption that given no genotype was known the dose intensity was equivalent between DPYD variant carriers and noncarriers.
Henricks et al. genotype guided cohort 16 : 50% dose reduction recommended for carriers of c.1905+1G>A or c.1679T>G; 25% dose reduction for carriers of c.2856A>T or c.1236G>A. Followed by dose escalation pending patient tolerance.
Unadjusted RRs with 95% CIs are discussed due to small sample size of variant carriers in genotype‐guided cohorts. Risks are calculated compared with noncarriers of the individual variant of interest.
Only one c.1679T>G carrier was detected in each genotype‐guided cohort. In both cohorts, the carrier was treated with 50% dose reduction and did not suffer a fluoropyrimidine‐related adverse event.
Retrospectively identified DPYD c.1236G>A carriers
DPYD c.1236G>A carriers identified retrospectively in May 2018 (N = 41) were removed from the genotype‐guided cohort as they were treated as DPYD variant noncarriers. We predicted that these c.1236G>A carriers would experience an increased risk of fluoropyrimidine‐related AEs given they were treated with standard dosing. However, c.1236G>A carriers treated with standard dosing did not experience an elevated toxicity profile (Table 3). In brief, 34% (14/41) of the retrospectively identified c.1236G>A carriers experienced a severe fluoropyrimidine‐related AE during the total treatment period and 24% (10/41) experienced an early severe fluoropyrimidine‐related AE (Table 4). In summary, compared to the genotype‐guided cohort, the unadjusted relative risk of global severe fluoropyrimidine‐related AEs was 1.09 (0.71–1.68).
DISCUSSION
We report the impact of pretreatment DPYD genotype‐guided fluoropyrimidine dosing on AEs in a Canadian hospital assessed through retrospective follow‐up of the Personalized Medicine Clinic. We show that when treated with genotype‐guided dosing for DPYD c.1905+1G>A, c.1679T>G, c.2846A>T, or c.1236G>A, the proportion of variant carrying patients who experienced severe fluoropyrimidine‐related AEs was not statistically different from noncarriers. We found that a 50% dose reduction for DPYD c.1905+1G>A and c.2846A>T carriers ameliorated the severe fluoropyrimidine‐related AE risk compared to the historical RR for carriers treated with full dose (Table 5). Previously, Henricks et al. reported that a 25% initial dose reduction in carriers of DPYD c.2846A>T did not eliminate the elevated risk of severe fluoropyrimidine‐related AEs. 15 Together these findings suggest that an initial 50% dose reduction is an appropriate dosing strategy for carriers of DPYD c.1905+1G>A and c.2846A>T, consistent with the current CPIC guidelines. 9
The Personalized Medicine Clinic attempted to provide DPYD genotype‐guided dosing in alignment with the best available evidence. Indeed, the genotyping for DPYD c.1236G>A as a proxy for variant c.1129‐5923C>G starting in 2018 reflects the latest CPIC guideline recommendations that note the association of c.1129‐5923C>G with severe fluoropyrimidine‐related AEs. 11 , 24 In order to account for this in this analysis, we carried out retrospective genotyping for DPYD c.1236G>A for patients who had been enrolled prior to inclusion of this variant as part of the DPYD test panel. We hypothesized that our patients who were DPYD c.1236G>A carriers treated with standard dosing would exhibit an increased risk of fluoropyrimidine‐related AEs in alignment with previous meta‐analysis data, as cited in the CPIC guidelines. 11 , 14 However, the retrospectively identified DPYD c.1236G>A carriers in our study did not demonstrate an increased risk. In the meta‐analysis by Meulendijks et al. that demonstrated an increased risk associated with DPYD c.1236G>A, however, the included studies consisted of only European populations (N = 4261). 11 Subsequently to the meta‐analysis publication, a large association study of American patients with colorectal cancer (N = 1953) demonstrated no significant association between DPYD c.1129‐5923C>G and fluoropyrimidine‐related AEs (RR 1.27, 95% CI: 0.97–1.67) in their population. 25 However, in the American study, they also confirmed that the proxy variant was in complete linkage disequilibrium with the causal variant. Lee et al. did demonstrate a trend toward an association and significance in a secondary outcome associating the c.1129‐5923C>G variant with neutropenia. 25 Given the difference between these previous findings and the known regional difference between the United States and European populations, we suggest the lack of significant association in our c.1236G>A carriers may reflect this difference. However, this difference was not proven and may be due to the limited sample size of retrospective c.1236G>A carriers in this study. The CPIC currently supports a 50% dose reduction for DPYD c.1236G>A carriers, followed by dose escalation if the patient tolerates the reduced dose. More evidence is needed to elucidate the extent of the potential regional effect on carriers of this variant. In the meantime, we continue to support the CPIC recommendations for DPYD c.1236G>A carriers. Given the uncertainty, therapeutic drug monitoring may be useful to limit AEs during dose escalations. 26
Limitations
The first major limitation of our study design is the experimental design. A robust two‐arm comparative study directly comparing genotype‐guided dosing to standard of care therapy would have provided stronger evidence to support these findings. However, a two‐arm comparative study was deemed inappropriate given the body of evidence associating DPYD variation and fluoropyrimidine‐related AEs prior to initiating the program at the Personalized Medicine Clinic. The retrospective collection of AE outcomes also limits the design. However, as listed in the Methods sections, systems were in place to limit the bias of this data collection and the pragmatic nature was necessary given limitations of the clinic at the time of study initiation. As well, our study design lacks disease progression or survival outcomes. However, it has previously been shown that c.1905+1G>A carriers treated with a 50% starting dose reduction achieved the same fluoropyrimidine exposure as noncarriers with standard dosing. 20 Additionally, a retrospective survival analysis showed no difference in survival outcomes between variant carriers receiving genotype‐guided dosing and noncarriers receiving standard dosing. 27 These data suggest that the DPYD variant carriers treated with a dose reduction achieve the same systemic exposure and therapeutic outcomes. The four variants tested in this study have been validated in studies predominated by White people of European descent, as was our study population. Additional DPYD variants may play an important role in other patient populations (e.g., DPYD c.557A>G in people of African descent). 28 Further research in other patient populations is needed to validate the utility of DPYD genotype‐guided dosing in more diverse populations. Finally, this study used DPYD genotype testing as a pretreatment screening method for DPD deficiency, however, we did not assess other methods of detecting DPD deficiency in this patient population.
CONCLUSION
Health Canada and the US Food and Drug Administration include warnings that DPD‐deficient patients are at an increased risk of severe AEs on fluoropyrimidine product labels. 1 , 2 , 3 However, to date, neither agency has recommended any pretreatment screening methods despite consensus guidelines from expert groups in Europe. 17 , 18 In March of 2019, the French Medicines Agency triggered a formal review of preemptive DPD deficiency screening by the European Medicines Agency (EMA), and, in April 2020, the EMA issued a recommendation for DPD deficiency testing prior to initiation of fluoropyrimidines. Our data support equivalent efforts to study and implement DPD deficiency screening through DPYD genotype testing be undertaken within North America.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
T.J.W., U.T.S., and R.B.K. wrote the manuscript. W.A.T., U.I.S., and R.B.K. designed the research. T.J.W., B.L.P., W.A.T., R.M.L., J.L., S.N., V.P., D.K., J.M., V.S., S.S., Y‐H.C., S.W., E.W., U.I.S., and R.B.K. performed the research. T.J.W., B.L.P., S.M., J.L., V.P., D.K., S.S., and Y‐H.C. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Cam Ross and Yinyin Liao for excellent technical assistance. The authors would also like to thank the support staff of London Health Sciences Centre, London Regional Cancer Centre, and Lawson Health Research Institute for their continued support of ongoing projects. We also thank the many physicians who referred patients for participation in this study. Finally, we extend our gratitude to the patients and their supportive family members that participated in this study.
Funding
This study was supported by the Wolfe Medical Research Chair in Pharmacogenomics (to R.B.K.) and the Ontario Research Fund – Research Excellence RE08‐063 (to R.B.K.).
REFERENCES
- 1. Hoffman‐La Roche Ltd. Xeloda (Capecitabine) Product Monograph. (2019). <https://pdf.hres.ca/dpd_pm/00051991.PDF>.
- 2. Hoffman‐La Rocher Ltd . Xeloda (Capecitabine) Package Insert. (2019). <https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/020896s037lbl.pdf>.
- 3. Sandoz Canada Inc. Fluorouracil Injection Product Monograph. (2012). <https://pdf.hres.ca/dpd_pm/00016156.PDF>.
- 4. Spectrum Pharmaceuticals , Fluorouracil Package Insert. (2016). <https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/012209s040lbl.pdf>.
- 5. Mikhail SE, Sun JF, Marshall JL. Safety of capecitabine: a review. Expert Opin Drug Saf. 2010;9:831‐841. [DOI] [PubMed] [Google Scholar]
- 6. Lee AM, Shi Q, Pavey E, et al. DPYD variants as predictors of 5‐fluorouracil toxicity in adjuvant colon cancer treatment (NCCTG N0147). J Natl Cancer Inst. 2014;106:dju298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heggie GD, Sommadossi JP, Cross DS, Huster WJ, Diasio RB. Clinical pharmacokinetics of 5‐fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res. 1987;47:2203‐2206. [PubMed] [Google Scholar]
- 8. Diasio RB, Beavers TL, Carpenter JT. Familial deficiency of dihydropyrimidine dehydrogenase. Biochemical basis for familial pyrimidinemia and severe 5‐fluorouracil‐induced toxicity. J Clin Invest. 1988;81:47‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amstutz U, Henricks LM, Offer SM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103:210‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosmarin D, Palles C, Church D, et al. Genetic markers of toxicity from capecitabine and other fluorouracil‐based regimens: investigation in the QUASAR2 study, systematic review, and meta‐analysis. J Clin Oncol. 2014;32:1031‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meulendijks D, Henricks LM, Sonke GS, et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine‐associated toxicity: a systematic review and meta‐analysis of individual patient data. Lancet Oncol. 2015;16:1639‐1650. [DOI] [PubMed] [Google Scholar]
- 12. Terrazzino S, Cargnin S, Re MD, et al. DPYD IVS14+1G>A and 2846A>T genotyping for the prediction of severe fluoropyrimidine‐related toxicity: a meta‐analysis. Pharmacogenomics 2013;14:1255‐1272. [DOI] [PubMed] [Google Scholar]
- 13. Caudle KE, Thorn CF, Klein TE, et al. Clinical pharmacogenetics implementation consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clin Pharmacol Ther. 2013;94:640‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henricks LM, Lunenburg CATC, Meulendijks D, et al. Translating DPYD genotype into DPD phenotype: using the DPYD gene activity score. Pharmacogenomics. 2015;16:1‐10. [DOI] [PubMed] [Google Scholar]
- 15. Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype‐guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19:1459‐1467. [DOI] [PubMed] [Google Scholar]
- 16. CPIC .CPIC® Guideline for Fluoropyrimidines and DPYD. (2018). Revised November 2018 <https://cpicpgx.org/guidelines/guideline‐for‐fluoropyrimidines‐and‐dpyd/>.
- 17. Loriot M‐A, Ciccolini J, Thomas F, et al. Dépistage du déficit en dihydropyrimidine deshydrogénase (DPD) et sécurisation des chimiothérapies à base de fluoropyrimidines: mise au point et recommandations nationales du GPCO‐Unicancer et du RNPGx. B Cancer. 2018;105:397‐407. [DOI] [PubMed] [Google Scholar]
- 18. Lunenburg CATC, van der Wouden CH, Nijenhuis M, et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene–drug interaction of DPYD and fluoropyrimidines. Eur J Hum Genet. 2019;28(4):508‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boisdron‐Celle M, Capitain O, Faroux R, et al. Prevention of 5‐fluorouracil‐induced early severe toxicity by pre‐therapeutic dihydropyrimidine dehydrogenase deficiency screening: assessment of a multiparametric approach. Semin Oncol. 2017;44:13‐23. [DOI] [PubMed] [Google Scholar]
- 20. Deenen MJ, Meulendijks D, Cats A, et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J Clin Oncol. 2015;34:227‐234. [DOI] [PubMed] [Google Scholar]
- 21. Haller DG, Cassidy J, Clarke SJ, et al. Potential regional differences for the tolerability profiles of fluoropyrimidines. J Clin Oncol. 2008;26:2118‐2123. [DOI] [PubMed] [Google Scholar]
- 22. National Institutes of Health; National Cancer Institute . Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. Revised November 2017. <https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf>.
- 23. National Institutes of Health Office of Science Policy . NIH‐FDA Phase 2 and 3 IND/IDE Clinical Trial Protocol Template vol 0.1 Published February 2016. <https://osp.od.nih.gov/wp‐content/uploads/2014/01/Protocol_Template_05Feb2016_508.pdf>.
- 24. Nie Q, Shrestha S, Tapper EE, et al. Quantitative contribution of rs75017182 to dihydropyrimidine dehydrogenase mRNA splicing and enzyme activity. Clin Pharmacol Ther. 2017;102:662‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee AM, Shi Q, Alberts SR, et al. Association between DPYD c.1129‐5923 C>G/hapB3 and severe toxicity to 5‐fluorouracil‐based chemotherapy in stage III colon cancer patients. Pharmacogenet Genom. 2016;26:133‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beumer JH, Chu E, Allegra C, et al. Therapeutic drug monitoring in oncology: international association of therapeutic drug monitoring and clinical toxicology recommendations for 5‐fluorouracil therapy. Clin Pharmacol Ther. 2018;105:598‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Henricks LM, van Merendonk LN, Meulendijks D, et al. Effectiveness and safety of reduced‐dose fluoropyrimidine therapy in patients carrying the DPYD*2A variant: a matched pair analysis. Int J Cancer. 2019;144:2347‐2354. [DOI] [PubMed] [Google Scholar]
- 28. Offer SM, Lee AM, Mattison LK, et al. A DPYD Variant (Y186C) in individuals of African ancestry is associated with reduced DPD enzyme activity. Clin Pharmacol Ther. 2013;94:158‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material