

Abstract
Estimating early exposure of drugs used for the treatment of emergent conditions is challenging because blood sampling to measure concentrations is difficult. The objective of this work was to evaluate predictive performance of two early concentrations and prior pharmacokinetic (PK) information for estimating early exposure. The performance of a modeling approach was compared with a noncompartmental analysis (NCA). A simulation study was performed using literature‐based models for phenytoin (PHT), levetiracetam (LEV), and valproic acid (VPA). These models were used to simulate rich concentration‐time profiles from 0 to 2 h. Profiles without residual unexplained variability (RUV) were used to obtain the true partial area under the curve (pAUC) until 2 h after the start of drug infusion. From the profiles with the RUV, two concentrations per patient were randomly selected. These concentrations were analyzed under a population model to obtain individual population PK (PopPK) pAUCs. The NCA pAUCs were calculated using a linear trapezoidal rule. Percent prediction errors (PPEs) for the PopPK pAUCs and NCA pAUCs were calculated. A PPE within ±20% of the true value was considered a success and the number of successes was obtained for 100 simulated datasets. For PHT, LEV, and VPA, respectively, the median value of the success statistics obtained using the PopPK approach of 81%, 92%, and 88% were significantly higher than the 72%, 80%, and 67% using the NCA approach (p < 0.05; Mann–Whitney U test). This study provides a means by which early exposure can be estimated with good precision from two concentrations and a PopPK approach. It can be applied to other settings in which early exposures are of interest.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Conducting pharmacokinetic (PK) studies with intensive sampling in emergent conditions like status epilepticus is challenging. Hence, getting a reliable estimate of early drug exposure to correlate with treatment response in order to develop exposure‐response relationships is difficult.
WHAT QUESTION DID THIS STUDY ADDRESS?
The results obtained in this simulation study support the notion that a two‐sample approach can be used to estimate early drug exposure. The population PK (PopPK) approach was found to be superior in estimating early exposure as compared with the standard noncompartmental analysis (NCA) approach.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This PK simulation study shows that using prior information based on the PopPKs of the drug and just two plasma concentration measures per patient in the first 2 h post drug administration, it is possible to adequately estimate early drug exposure. The study not only provides an alternative method to estimate early exposure but also demonstrates that it is superior to the standard NCA approach.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This study provides a means by which early exposure can be estimated with good precision using limited sampling. This approach can be applied to any setting in which early exposures are of interest and these exposures can be related to outcomes of interest in clinical and translational pharmacology.
INTRODUCTION
Treatments used for the management of emergent conditions are usually administered intravenously to rapidly achieve therapeutic drug concentrations at the site of action as it is these early concentrations that will drive the response to treatment. Plasma concentrations are often used as a surrogate to reflect the concentrations at the site of action. Although extensive blood sampling to measure drug exposure is considered as the reference standard for pharmacokinetic (PK) modeling using traditional methods like noncompartmental analysis (NCA), rich blood sampling is challenging when conducting studies in emergent conditions and in young children. One solution to this problem is to use sparse sampling (1–4 samples) at opportunistic schedules. 1 Population PK (PopPK) modeling is better suited to utilize sparse PK samples than traditional approaches, such as NCA or individual compartmental modeling, because information from each individual can be utilized, even if it is a single concentration, in the estimation of PopPK parameters. Further, models can include estimates of between‐subject variability, allowing for the prediction of an individual’s early exposures.
This challenge of estimating early drug exposures was encountered during the conduct of the Established Status Epilepticus Treatment Trial (ESETT)—a comparative‐effectiveness study of fosphenytoin (FOS), levetiracetam (LEV), and valproic acid (VPA) in patients with benzodiazepine‐refractory status epilepticus (SE). 2 , 3 The study drugs were administered as a 10‐minute i.v. infusion in patients who continued to experience convulsive seizures despite receiving an adequate dose of benzodiazepines. The primary outcome of ESETT was clinical cessation of seizures with improved responsiveness at 60 min after the start of study drug infusion without the need for additional antiseizure medications. The objective of an ancillary PK study was to measure early exposure of the study drugs (within the first 2 h post‐administration) and relate it to the treatment response. Two PK samples were chosen to measure early drug concentrations, the first within the time window of 20 and 50 min (W1) and the second between 60 and 120 min (W2) following the start of the 10‐minute drug infusion.
Using ESETT as a motivating example, we were interested in testing if using two blood samples, like those collected in the specified time‐windows in ESETT, would give a reliable estimate of early drug exposure. Testing this in a real‐world setting was not reasonable because extensive early blood sampling is not feasible during the treatment of an emergent condition like convulsive SE, as the therapy includes stabilizing the patient, initiating electrocardiogram and electroencephalographic monitoring, treating the underlying cause of seizures, and attempting i.v. access to administer pharmacotherapy quickly. 4 As an alternative, we chose to conduct a PK simulation study. The metric of exposure used was partial area under the drug concentration‐time curve (pAUC) until 2 h after the start of drug infusion. The objective of this work was to evaluate the predictive performance of using two concentration measures and prior information about the drug for estimating early drug exposure, using a PopPK modeling approach. Further, the predictive performance of the PopPK approach for estimating early drug exposure was compared with the standard NCA approach. The drugs used in ESETT (FOS, LEV, and VPA) were chosen as example drugs for this simulation study.
METHODS
Published PopPK models used for this work
Literature‐based PopPK models for FOS (prodrug of phenytoin [PHT] 5 ), LEV, and VPA were used for this simulation work. 6 , 7 , 8 The models were selected after a comprehensive review of published PopPK models for i.v. administration that utilized intense sampling in healthy volunteers or patients who were not on chronic therapy of that drug. For the FOS/PHT model, data from 24 healthy adults, and 7 adult and 20 pediatric patients that included those with SE, acute repetitive seizures, or neurosurgical patients in whom FOS was administered to prevent seizures were used. For the LEV model, data from 11 adult patients treated with i.v. LEV for SE (10 with convulsive and 1 with nonconvulsive SE) were used 7 ; whereas for VPA, peak and trough concentrations after i.v. administration in 102 newly diagnosed, treatment‐naïve patients with epilepsy were used. 8
The value of residual unexplained variability (RUV) was not reported for the LEV model. 7 For the FOS/PHT model, an RUV value (σ2 exponential error 0.00148 and σ2 additive error 0.317) was reported, 6 which was much lower than other FOS/PHT PK models. 9 , 10 , 11 To reflect the unexplained variability typically seen in a clinical study, particularly in an emergency department, a proportional RUV with 20% coefficient of variation (CV) modeled as shown in Equation 1 was used for the LEV and FOS/PHT models.
| (1) |
where Cij , is the measured plasma concentration for subject i at time j, Pred ij is the corresponding model predicted concentration for subject i at time j, and ε is residual error term distributed normally with mean 0 and variance of σ 2. All models included allometric scaling using a median weight of 75 kg and an exponent of 0.75 for all clearance terms (total body clearance and intercompartmental clearance) and 1 for all volume terms (central and peripheral).
Doses and weights used for simulations
Table 1 provides the model structure and parameter values used for this work. The doses and body weights used came from ESETT. The doses used, 20 mg/kg for FOS, 40 mg/kg for VPA, and 60 mg/kg for LEV, were weight‐based to a maximum of 75 kg and capped thereafter. 12 The proportion of adults:children in the simulated patients was 60:40. Adult weights were sampled from a normal distribution with mean 75 kg and SD 19.3 kg, whereas weights for children were sampled from normal distribution with mean 20 kg and SD 7.3 kg. The lowest weight of a child enrolled in ESETT was 9 kg. Hence, the lower end of the simulated weights was capped at 9 kg.
TABLE 1.
Details of the PopPK models used
| PHT | LEV | VPA | |
|---|---|---|---|
| Reference | Tanaka et al. 5 | Uges et al. 6 | Park et al. 7 |
| Population |
24 healthy volunteers +14 adult patients +33 pediatric patients Mean age 19.6 (±15.6) Mein weight 43.8 (±21.3) |
≥18 years (mean age 64 years; mean weight 64 kg) | ≥16 years (mean age 45 years; mean weight 60 kg) |
| PK Model | Two‐compartment | Two‐compartment | One‐compartment |
| CL (L/HR/60 KG) | 1.61 (44% CV) | 2.86 (30.9% CV) | 0.849 (32% CV) |
| V (L/60 KG) | 20.8 (40.1% CV) | 27.0 (18.7% CV) | 15.1 (18% CV) |
| V(peripheral) (L/60 KG) | 26 (20.7% CV) | – | – |
| Q | 53 (52.1% CV) | – | – |
| K12 (/HR) | – | 0.24 (50% CV) | – |
| K21 (/HR) | – | 0.70 (31.4% CV) | – |
| RUV reported |
3.85% CV (exponential) 0.56 SD (additive) |
Not reported | 26.7% CV (Proportional) |
| RUV used | 20% CV (proportional) | 20% CV (proportional) | 26.7% CV (Proportional) |
| Dose | 20 mg/kg for ≤75 kg; 1500 mg for >75 kg | 60 mg/kg for ≤75 kg; 4500 mg for >75 kg | 40 mg/kg for ≤75 kg; 3000 mg for >75 kg |
Abbreviations: CL, clearance; CV, coefficient of variation; LEV, levetiracetam; PHT, phenytoin; PK, pharmacokinetic; PopPK, population pharmacokinetics; RUV, residual unexplained variability; SD, standard deviation; V, volume; VPA, valproic acid.
Simulation and modeling
The metric of early exposure chosen for this work was pAUC 0–2 h after the start of drug infusion. Given the relatively long terminal half‐life of LEV (6–8 h 13 ), PHT (12–29 h 14 ), and VPA (16 h 15 ), we assumed that the early exposure of these drugs would be mainly driven by the volume of distribution.
Figure 1 shows a schematic of the methodology used. The steps used were as follows:
FIGURE 1.
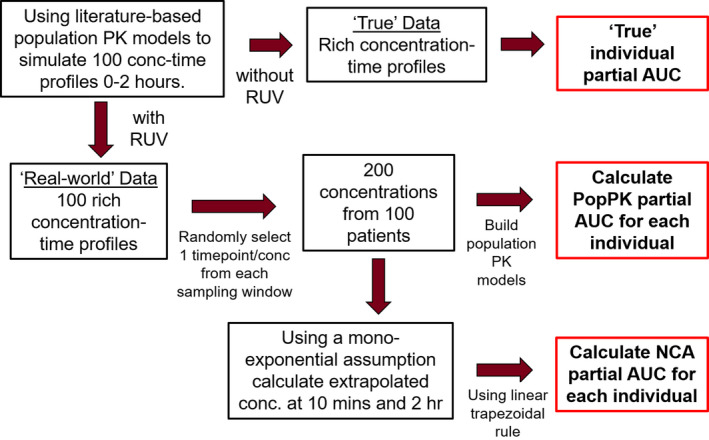
Schematic of the methodology used. AUC, area under the concentration‐time curve from 0 to 2 h after the start of study drug infusion; NCA, non‐compartmental analysis; PK, pharmacokinetic; RUV, residual unexplained variability
Step 1: Simulating concentration time profiles with and without the RUV
Using the selected PopPK models, rich concentration‐time profiles (from 0 to 2 h after the start of study‐drug infusion) including the RUV (to generate random‐noise corrupted concentrations) and without the RUV (true concentrations) were simulated for 100 individuals for PHT (active form of FOS), LEV, and VPA. Simulated profiles were generated using R (version 3.6.1), RStudio (version 1.2.5042) and mrgsolve package.
Step 2: Calculation of true pAUC
The profiles without the RUV were used to calculate the “true” pAUC for each individual by integrating the concentrations in the central compartment over 2 h after the start of study drug infusion. This true pAUC was used as a reference for comparison.
Step 3: pAUC calculation using a PopPK analysis
Using the rich concentration‐time profiles with the RUV, one data point in W1 from among 5 possible data points at 24, 30, 36, 42, and 48 min and another in W2 from among 10 possible data points at 66, 72, 78, 84, 90, 96, 102, 108, 114, and 120 min, were randomly selected for each patient to generate a sparse sampled dataset of 200 concentrations from 100 simulated patients (R version 3.6.1, RStudio version 1.2.5042). These data were analyzed under a PopPK model (NONMEM version 7.3; ICON Development Solutions) using the first order conditional estimation method (ADVAN13). Due to the sparseness of data, the population‐level clearance, peripheral volume of distribution, and intercompartmental clearance terms were fixed to literature values with the between subject variability set to zero. The population‐level central volume of distribution was estimated along with its variability. The RUV was also estimated. This was called the PopPK approach and the individual 2‐h pAUCs called the “PopPK pAUCs” were calculated by integration of the concentration in the central compartment from time zero to time 2 h.
Step 4: pAUC estimation using NCA
As a comparator for the PopPK approach, pAUC estimates were also generated using NCA. Using the two data points per patient in the sparse sampled dataset of 100 simulated patients and a mono‐exponential assumption, concentrations at 10 min after the start of study drug infusion were calculated using back‐extrapolation. Similarly, concentrations at 2 h were calculated using forward‐extrapolation. Using a linear trapezoidal rule (PKPDmisc package, R version 3.6.1, RStudio version 1.2.5042), the “NCA pAUC” from zero to 2 h for each simulated patient was then calculated with these 4 data points. This was called the NCA approach.
Step 5: Evaluation of predictive performance
The PopPK pAUC and NCA pAUC were compared with the true pAUC. The percent prediction error (PPE) was calculated as a test statistic and shown in Equation 2.
| (2) |
where sparse pAUC is PopPK pAUC or NCA pAUC.
It is not critical that a predicted pAUC exactly match the true pAUC and we set a threshold for an acceptable prediction. Success in estimating early exposure for each simulated patient had a value of one if the PPE for pAUC was within ±20%, otherwise it was zero.
Step 6: Generating a distribution of the success statistic across 100 different datasets
Steps 1–5 were repeated 100 times with 100 different datasets obtained using different seed values. The number of simulated individuals with success value of 1 (i.e., individuals with PPE within ±20%) per dataset was obtained and a distribution of the success statistic was generated for PHT, LEV, and VPA for both the PopPK and NCA approaches.
Step 7: Comparing the predictive performance of PopPK and NCA
As the success statistic obtained in step six for the PopPK and NCA approaches may not be normally distributed, a two‐sample t‐test may not be appropriate. Hence, its nonparametric counterpart, the Mann–Whitney U test, was used to test if the distribution of the success statistic was significantly different between PopPK and NCA approaches.
Plots were generated using R (version 3.6.1), RStudio (version 1.2.5042), and ggplot2 package.
RESULTS
Distribution of the success statistic
Figure 2, Figure 3, and Figure 4 shows the distribution of the success statistic for PHT, LEV, and VPA, respectively, across 100 datasets obtained using the NCA approach (dashed bars) and the PopPK approach (solid bars). Median (5th and 95th percentile) value of the success statistic for PHT was 72% (65% and 79%) for the NCA approach and 81% (73% and 88%) for the PopPK, for LEV it was 80% (73% and 87%) for the NCA approach and 92% (87% and 96%) for the PopPK approach, and for VPA it was 67% (58% and 73%) for the NCA approach and 88% (82% and 93%) for the PopPK approach.
FIGURE 2.
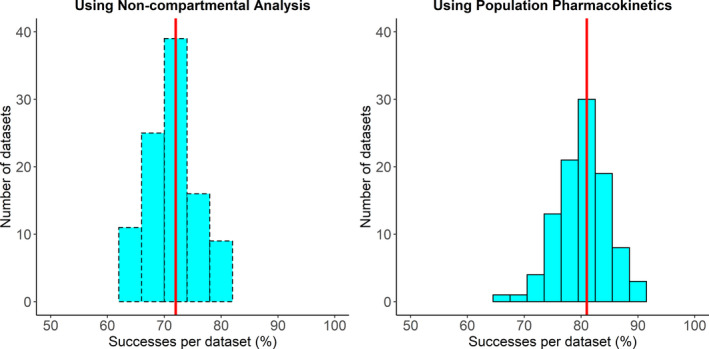
Distribution of percent of successes per dataset for phenytoin using noncompartmental analysis (left) and population pharmacokinetic analysis (right). The red line indicates the median percent of successes per dataset
FIGURE 3.
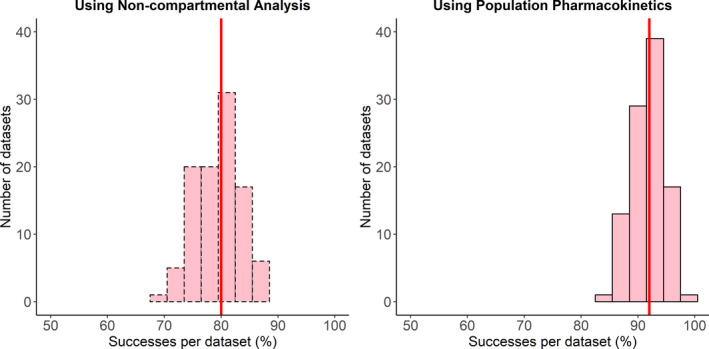
Distribution of percent of successes per dataset for levetiracetam using noncompartmental analysis (left) and population pharmacokinetic analysis (right). The red line indicates the median percent of successes per dataset
FIGURE 4.
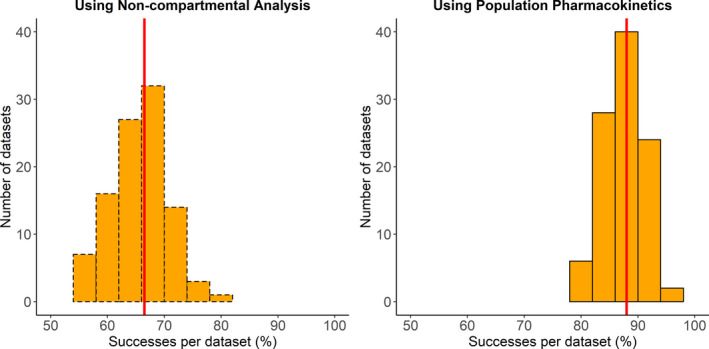
Distribution of percent of successes per dataset for valproic acid using noncompartmental analysis (left) and population pharmacokinetic analysis (right). The red line indicates the median percent of successes per dataset
Comparing the predictive performance of PopPK and NCA
Table 2 shows the results of the Mann–Whitney U test comparing the distribution of success statistic between the PopPK and NCA approaches. The difference in the location of the median for the success statistic distributions obtained using the two approaches was highly significant (p < 2.2e‐16) for all three drugs and showed that the PopPK approach performed significantly better than a standard NCA approach.
TABLE 2.
Results of Mann–Whitney U test comparing distribution of success statistic between the PopPK approach and NCA approach
| Drug | W‐statistic | Difference in location of the median of success statistic between PopPK and NCA (95% confidence interval) | Associated p value |
|---|---|---|---|
| PHT | 792 | 9 (7–10) | <2.2e−16 |
| LEV | 51.5 | 12 (11–13) | <2.2e−16 |
| VPA | 2 | 22 (21–23) | <2.2e−16 |
Abbreviations: LEV, levetiracetam; NCA, noncompartmental analysis; PHT, phenytoin; PopPK, population pharmacokinetics; VPA, valproic acid.
DISCUSSION
Our results from this PK simulation study using FOS, LEV, and VPA as example drugs show that using data from two blood samples collected from each patient over the first 2 h after acute drug administration and prior drug information, it is possible to estimate early drug exposure reasonably well using the PopPK approach. Further, a significantly higher number of simulated patients had their pAUC estimate closer to the true value (within ±20% PPE) with the PopPK approach compared with the standard NCA approach. These results demonstrate the superiority of the PopPK approach over the standard NCA approach to estimate early drug exposure under these conditions.
Although much of the literature on PK sampling limitations is focused on pediatric studies, 16 , 17 , 18 similar challenges are also encountered in emergent conditions. This is further limited by the potential need to measure early drug exposure. Under these conditions, it is often impractical to collect multiple blood samples without interfering with patient care, and initial samples may be delayed by the need to obtain informed consent. 18 Hence, wider sampling windows and flexibility in timing is essential. Based on a PubMed search, we found no published reports focusing on estimation of early drug exposures using sparse (less than 3) samples in emergent conditions. There are several reports of PopPK models developed using concentration‐time data collected within 48 h after acute administration of rescue therapies. 19 , 20 , 21 Most of these models rely on more extensive blood sampling and did not use prior information about the drugs. Our approach is novel and focused on estimating early drug exposure with reasonable precision using very sparse data (2 concentrations per patient) in the first 2 h post drug administration and a PopPK modeling approach.
Often, a single blood sample drawn per patient is used as the measure of drug exposure. 22 , 23 , 24 , 25 A single sample represents exposure at one instant in time and can be highly variable; thus this is likely to be imprecise. Further, a rapidly changing concentration‐time profile will increase this imprecision. In this simulation study, two concentration measures within the first 2 h post drug administration and a PopPK approach was utilized to estimate early drug exposures. As an alternative, we also tested a standard NCA approach, which has fewer assumptions. 26 Whereas NCA assumes that there is no error in drug concentrations, a PopPK approach imposes a pharmaco‐statistical model on the data, and the error in the form of RUV is estimated during the model fitting process. This could explain why the pAUC estimates generated using the PopPK approach were closer to the true value than those generated by the NCA approach.
Our approach is driven by prior information available for FOS/PHT, LEV, and VPA. Although the RUV value used for the VPA model was the same as that reported in the published model, 8 a proportional RUV with 20% CV was used for FOS and LEV models to reflect the variability seen in clinical settings. Whereas it is possible to reduce analytical error, several other sources of errors, including but not limited to incorrect time record of sample collection, dosing errors like administration of incorrect dose volume, time of dose administration, etc., that occur in a clinical setting, may contribute to the RUV. The probabilities of these errors are likely to be increased in emergency conditions.
Our analysis has several limitations. It was assumed that the subjects in the simulation study were similar to the literature population and that the exact times of dosing and sampling are known. Concentrations are changing rapidly during early exposures and may be sensitive to model misspecification, which was not incorporated into this analysis. The same structural model was used for the simulations and for fitting the sparse dataset using the PopPK approach. Additionally, optimal sampling design (e.g., POPED) was not utilized as it is difficult to control the exact time of sampling in an emergency department setting.
Using a PopPK modeling analysis with three antiseizure medications as example drugs, we demonstrate that early drug exposures can be estimated adequately utilizing prior information on the drug and sparse concentration data collected early after drug administration. The PopPK approach was found to be significantly better at estimating early drug exposure as compared with a standard NCA approach. This approach can be used in other emergent conditions like convulsive SE, or in other situations where early drug exposure is of interest. These results encourage further investigation of pharmacometric models of drug exposure, which can then be related to factors affecting outcomes in difficult emergent settings.
CONFLICT OF INTEREST
All authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
A.G.S., V.I., R.C.B., L.D.C., J.M.C., and J.C.C. wrote the manuscript. A.G.S., V.I., R.C.B., L.D.C., J.M.C., J.C.C., J.K., R.S., and J.J.E. designed the research. A.G.S., V.I., R.C.B., and L.D.C performed the research. A.G.S. analyzed the data. V.I., R.C.B., and L.D.C contributed new reagents/analytical tools.
Supporting information
Supporting information
ACKNOWLEDGMENTS
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke, National Institutes of Health, or the United States Government.
Previous presentation: This work has been presented as an abstract at the American Conference on Pharmacometrics (ACoP10) held in Orlando, Florida, in October 2019.
Funding information
Research reported in this publication was supported by National Institutes of Health, National Institutes of Neurological Disorders and Stroke under Award R01NS099653 (Clinical trials.gov identifier NCT01960075).
REFERENCES
- 1. Jonsson EN, Wade JR, Karlsson MO. Comparison of some practical sampling strategies for population pharmacokinetic studies. J Pharmacokinet Biopharm. 1996;24(2):245‐263. [DOI] [PubMed] [Google Scholar]
- 2. Kapur J, Elm J, Chamberlain JM, et al. Randomized trial of three anticonvulsant medications for status epilepticus. N Engl J Med. 2019;381(22):2103‐2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chamberlain JM, Kapur J, Shinnar S, et al. Efficacy of levetiracetam, fosphenytoin, and valproate for established status epilepticus by age group (ESETT): a double‐blind, responsive‐adaptive, randomised controlled trial. Lancet. 2020;395(10231):1217‐1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glauser T, Shinnar S, Gloss D, et al. Evidence‐based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American epilepsy society. Epilepsy Curr. 2016;16(1):48‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fischer JH, Patel TV, Fischer PA. Fosphenytoin: clinical pharmacokinetics and comparative advantages in the acute treatment of seizures. Clin Pharmacokinet. 2003;42(1):33‐58. [DOI] [PubMed] [Google Scholar]
- 6. Tanaka J, Kasai H, Shimizu K, Shimasaki S, Kumagai Y. Population pharmacokinetics of phenytoin after intravenous administration of fosphenytoin sodium in pediatric patients, adult patients, and healthy volunteers. Eur J Clin Pharmacol. 2013;69(3):489‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uges JWF, Van Huizen MD, Engelsman J, et al. Safety and pharmacokinetics of intravenous levetiracetam infusion as add‐on in status epilepticus. Epilepsia. 2009;50(3):415‐421. [DOI] [PubMed] [Google Scholar]
- 8. Park HM, Kang SS, Lee YB, et al. Population pharmacokinetics of intravenous valproic acid in Korean patients. J Clin Pharm Ther. 2002;27(6):419‐425. [DOI] [PubMed] [Google Scholar]
- 9. Odani A, Hashimoto Y, Takayanagi K, et al. Population pharmacokinetics of phenytoin in Japanese patients with epilepsy: analysis with dose‐dependent clearance model. Biol Pharm Bull. 1996;19(3):444‐448. [DOI] [PubMed] [Google Scholar]
- 10. Ahn JE, Cloyd JC, Brundage RC, et al. Phenytoin half‐life and clearance during maintenance therapy in adults and elderly patients with epilepsy. Neurology. 2008;71(1):38‐43. [DOI] [PubMed] [Google Scholar]
- 11. Moffett BS, Weingarten MM, Schmees LR, Galati M, Erklauer J, Riviello JJ. Fosphenytoin population pharmacokinetics in the acutely ill pediatric population. Pediatr Crit Care Med. 2018;19(8):748‐754. [DOI] [PubMed] [Google Scholar]
- 12. Sathe AG, Elm JJ, Cloyd JC, et al. The association of patient weight and dose of fosphenytoin, levetiracetam and valproic acid with treatment success in status epilepticus. Epilepsia. 2020;61:e66‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. KEPPRA . KEPPRA®(levetiracetam injection for intravenous use)[package insert]. Smyrna, GA: UCB Inc.; 2008:1‐16. [Google Scholar]
- 14. CEREBYX . CEREBYX®(fosphenytoin sodium injection)[package insert]. New York, NY: Pfizer Injectables; 2015:1‐22. [Google Scholar]
- 15. DEPACON . DEPACON® (valproate sodium for intravenous injection)[package insert]. Lake Forest, IL: Hospira, Inc.; 2017:1‐38. [Google Scholar]
- 16. Laughon MM, Benjamin DK, Capparelli EV, et al. Innovative clinical trial design for pediatric therapeutics. Expert Rev Clin Pharmacol. 2011;4(5):643‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Howie SRC. Blood sample volumes in child health research: review of safe limits. Bull World Health Organ. 2011;89(1):46‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barker CIS, Standing JF, Kelly LE, et al. Pharmacokinetic studies in children: recommendations for practice and research. Arch Dis Child. 2018;103(7):695‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilmshurst JM, Van Der Walt JS, Ackermann S, Karlsson MO, Blockman M. Rescue therapy with high‐dose oral phenobarbitone loading for refractory status epilepticus. J Paediatr Child Health. 2010;46(1–2):17‐22. [DOI] [PubMed] [Google Scholar]
- 20. Gonzalez D, Chamberlain JM, Guptill JT, et al. Population pharmacokinetics and exploratory pharmacodynamics of lorazepam in pediatric status epilepticus. Clin Pharmacokinet. 2017;56(8):941‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ku LC, Hornik CP, Beechinor RJ, et al. Population pharmacokinetics and exploratory exposure‐response relationships of diazepam in children treated for status epilepticus. CPT Pharmacometrics Syst Pharmacol. 2018;7(11):718‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wheless JW, Clarke D, Hovinga CA, et al. Rapid infusion of a loading dose of intravenous levetiracetam with minimal dilution: a safety study. J Child Neurol. 2009;24(8):946‐951. [DOI] [PubMed] [Google Scholar]
- 23. Piper JD, Hawcutt DB, Verghese GK, Spinty S, Newland P, Appleton R. Phenytoin dosing and serum concentrations in paediatric patients requiring 20 mg/kg intravenous loading. Arch Dis Child. 2014;99(6):585‐586. [DOI] [PubMed] [Google Scholar]
- 24. Selioutski O, Grzesik K, Vasilyeva ON, et al. Evaluation of phenytoin serum levels following a loading dose in the acute hospital setting. Seizure. 2017;2017(52):199‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perrenoud M, André P, Buclin T, et al. Levetiracetam circulating concentrations and response in status epilepticus. Epilepsy Behav. 2018;2018(88):61‐65. [DOI] [PubMed] [Google Scholar]
- 26. Gabrielsson J, Weiner D. Non‐compartmental analysis. Meth Mol Biol. 2012;377‐389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information