

Abstract
Nephrogenic diabetes insipidus (NDI) is rarely considered against more common differentials such as diabetes mellitus in patients presenting with polydipsia and polyuria. Hypokalemia and hypercalcemia are known to induce NDI, but not much is known about hypomagnesemia. Hypokalemia refractory to therapy should prompt consideration of hypomagnesemia.
Keywords: Aquaporin‐2, arginine vasopressin, diabetes mellitus, hypokalemia, hypomagnesemia, nephrogenic diabetes insipidus
1. INTRODUCTION
Nephrogenic diabetes insipidus (NDI) is a significantly rarer condition than diabetes mellitus (DM). Both forms of diabetes (DI and DM) can be difficult to distinguish if they were to occur simultaneously as both present similarly, with polydipsia and polyuria. NDI results from the failure of the kidney to concentrate urine due to the insensitivity of the distal nephron to respond to antidiuretic hormone (ADH), also known as arginine vasopressin (AVP). This leads to polyuria of more than 3L in 24 hours with a urinary osmolality of less than 300 mOsm/kg H2O and specific gravity of less than 1.005, causing an increase in plasma osmolality in response to raised serum sodium and urea. 1 , 2 NDI can be hereditary or an acquired disorder. Acquired NDI is commonly caused by drugs such as lithium or metabolic imbalances, such as hypokalemia and hypercalcemia. 3 Refractory hypokalemia management has been attributed to hypomagnesemia. The co‐administration of magnesium and potassium is essential for correcting persistent hypokalemia. 4 Here, we describe a case of a type 1 diabetic who acquired NDI due to hypokalemia and hypomagnesemia.
2. CASE REPORT
A 27 year old previously healthy male was referred to a hospital with a two‐week history of vomiting, high‐grade fever, and abdominal pain, which was diagnosed as appendicitis. On admission to the referral facility, he was diagnosed with a new presentation of type 1 diabetes mellitus. Postappendectomy, he was referred to our facility for further management. On arrival, he was semi‐conscious, barely responsive to pain, and had labored breathing. He also had electrolyte imbalance with severe hypokalemia of 1.6 mmol/L and an elevated random blood glucose level.
Physical examination revealed a well‐nourished albeit severely dehydrated male weighing about 53kg. He was febrile and had Kussmaul breathing. His vitals were as follows: BP = 151/69 mm Hg, HR = 132 beats per minute, RR = 32 breaths per minute, SPO2 = 96% on room air, temperature =38.9℃, and random blood glucose =16.6 mmol/L. A bedside ultrasound revealed a collapsed inferior vena cava (IVC). His initial point‐of‐care venous blood gas showed metabolic acidosis (pH =7.20, PCO2 = 21.5 mmHg, HCO3 = 10.1 mmol/L) with severe hypokalemia of 2.2 mmol/L (3.5–5.0 mmol/L), hypernatremia of 167 mmol/L (135–145 mmol/L), and hyperchloremia of 125 mmol/L (98–109 mmol/L). Serial venous blood gases and electrolytes were performed every 3 to 6 hours (Table 1). Urine analysis was unremarkable including the absence of urinary ketones.
TABLE 1.
Blood gas and electrolytes from admission to the following 3 days.
| Initial | 3hrs | 6hrs | 12hrs | 24hrs | 48hrs | 72hrs | |
|---|---|---|---|---|---|---|---|
| pH | 7.20 | 7.26 | 7.38 | 7.37 | 7.52 | 7.50 | 7.44 |
| PCO2 | 21.5 | 36.3 | 29.4 | 46 | 41.6 | 45.1 | 38.8 |
| HCO3 − | 10.1 | 15.7 | 17.6 | 26.4 | 33.4 | 34.9 | 26 |
| Na+ | 167 | 164 | 160 | 159 | 155 | 151 | 146 |
| K+ | 2.2 | 1.69 | 2.2 | 1.8 | 1.94 | 3.17 | 3.01 |
| Cl− | 125 | 117 | 119 | 115 | 115 | 108 | 105 |
Central venous access was obtained in the right subclavian vein, and a 4‐L bolus of Ringers lactate was administered followed by a free water deficit maintenance using 5% dextrose water admixed with potassium chloride (KCl) 40 – 80 mmol which ran at 125 ml/h (KCl rate of 10 – 20 mmol/h). Simultaneously, a fast‐acting insulin infusion at 0.05 U/kg/h (2.5 U/h) was also administered. Post‐operative broad‐spectrum parenteral antibiotics were initiated, and the patient was admitted to the ICU.
After 24 hours of admission, we noted the patient had a urinary output of 9 L of dilute urine with a negative fluid balance of 3 L. His serum potassium was persistently low despite receiving a continuous KCl infusion for over 24 hours. At this point, his serum potassium level was 1.94 mmol/L. Serum magnesium levels were ordered to rule out probable cause of refractory hypokalemia, which revealed hypomagnesemia of 0.50 mmol/L (0.66 −1.25 mmol/L). All other blood investigations including brain and abdominal CT scan were unremarkable except for the ECG which showed diffuse U‐wave morphology (Figure 1) correlating with hypokalemia and/or hypomagnesemia. Urinary specific gravity was found to be low with a value of 1.005. Plasma levels of antidiuretic hormone (ADH) and urine osmolality were not available at our center.
FIGURE 1.
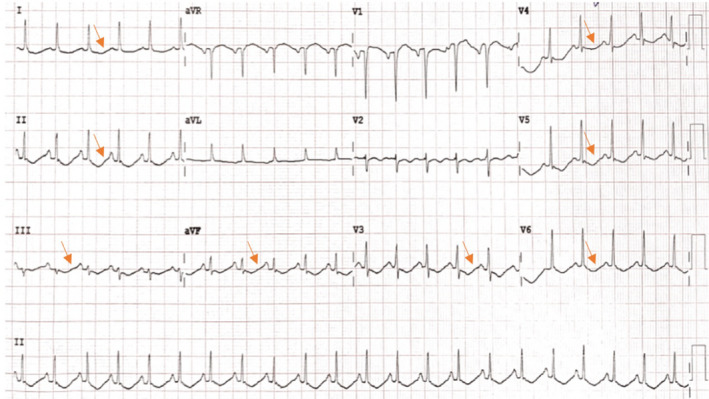
Diffuse U waves on ECG correlating with hypokalemia and hypomagnesemia.
Based on these findings, a diagnosis of nephrogenic diabetes insipidus (NDI) was made secondary to hypomagnesemia induced severe hypokalemia. The treatment was modified by the addition of 4g of magnesium sulfate along with 40 mmol of KCl in 5% dextrose water (500 ml bottle) which ran at a rate of 125 ml/h, along with the same insulin rate of 0.05 U/kg/h (2.5 U/h). This treatment plan continued for 48 hours until his electrolytes normalized. In addition, oral bendroflumethiazide 5 mg twice a day, indomethacin 50 mg thrice a day, eplerenone 25 mg twice a day, and magnesium trisilicate 250 mg thrice a day were added to his treatment.
Improvement in potassium and magnesium levels were seen on the 3rd day postadmission with significant ECG improvement with the disappearance of U waves (Figure 2). A decrease in the exaggerated amount of urine output was also observed with an improvement in the patient's hydration status. On the 8th day in the ICU, the patient had significant clinical improvement with normal serum electrolytes and an adequate positive fluid balance (input of 3.5 L and output of 2.9 L in 24 h). Thereafter, he was transferred from the ICU to the general ward to continue with the management and follow‐up. Further follow‐up as an outpatient postdischarge has yielded normal electrolyte results.
FIGURE 2.
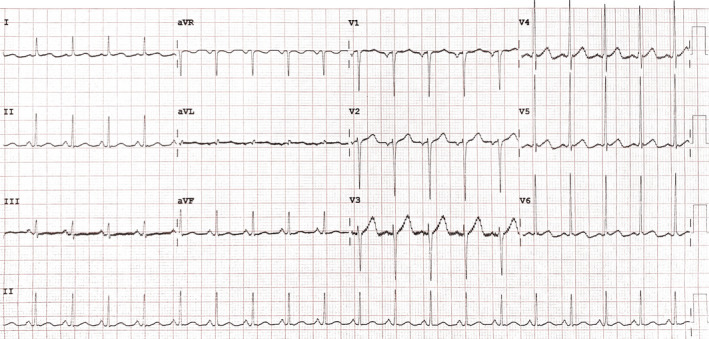
Two days on potassium chloride with magnesium sulfate infusion.
3. DISCUSSION
Determining whether diabetes insipidus is central or nephrogenic is challenging in resource limited set‐ups especially if a patient has pre‐existing diabetes mellitus. We were able to diagnose NDI based on patient's clinical presentation of excessive polyuria and severe hypokalemia and hypomagnesemia and started the management on the second day with the resources available to us. Confirmatory tests like urine osmolality and plasma levels of arginine vasopressin (AVP) were not available, so the diagnosis was made based on the available diagnostic tools and clinical judgment.
The ability of kidneys to concentrate urine is regulated through water balancing activities mediated via a complex AVP/AVP V2 receptor/Aquaporin‐2 axis. Aquaporin‐2 (AQP2) are the principal water channel cells that regulate water permeability at the collecting duct epithelium mediated chiefly via AVP effects. 5 Both hereditary and acquired causes of NDI lead to decreased in the AQP2 expressions and AVP action, thus causing similar consequences of free urinary water loss and increased plasma osmolality. Acquired NDI is more frequently encountered than the hereditary form, usually being drug‐induced or due to electrolyte disturbance. Lithium is not the only culprit, and there are a number of drugs (antibiotics, antifungals, and antineoplastic) responsible in decreasing AQP2 expression and targeting, hence causing acquired NDI. 6
In respect to metabolic causes of NDI, hypokalemia, and hypercalcemia have been identified from animal studies. It has been observed that hypokalemia and hypercalcemia lead to autophagic degradation of AQP2 protein abundance in the inner medullary collecting ducts that impair urine concentrating ability. Within their studies, it was observed that correcting hypokalemia and hypercalcemia ameliorates autophagic activities on AQP2 reversing NDI within 24 – 48 hours. 7 , 8 In this patient, hypokalemia induced NDI was refractory to therapy despite high doses of parenteral potassium supplementation until a coexistence of hypomagnesemia was sought. We corrected the hypokalemia and hypomagnesemia concurrently, which eventually led to significant clinical improvement in correlation with ECG U‐wave disappearance (Figure 2).
Magnesium and potassium are cations which are predominantly intracellular. This distribution is what maintains the stability of the membrane potential and decreases cell excitability. 9 Deficiency of magnesium usually goes unrecognized thus exacerbating potassium wasting through the impairment of Na‐K‐ATPase activity. 4 Though hypomagnesemia has been associated directly toward hypokalemia, our knowledge is limited in identifying whether the former electrolyte has any obvious physiological impact in directly causing NDI.
4. CONCLUSION
This was a rare clinical occurrence that we observed and treated in our practice. It is unknown whether similar occurrences have been taking place in our setting that are going unnoticed and being mismanaged due to its rarity and a delay in establishing diagnoses. Our emphasis is to always maintain a high level of suspicion in similar clinical presentations to ensure NDI does not go unrecognized. We also hope that this will spark further physiological studies to take place regarding electrolyte anomalies inducing NDI.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
Ahmad Mwinyi gathered the data and wrote the original draft. Esmail Sangey and Kishan Chudasama involved in reviewing and editing of the manuscript. Esmail Sangey involved in supervision and submission of the manuscript. All took care of the patient and have approved the final version of the manuscript.
ETHICAL APPROVAL
Informed and written consent was obtained from the patient to publish in this case report.
ACKNOWLEDGMENTS
We thank all Emergency and ICU department team of Shree Hindu Mandal Hospital for their involvement in the intensive treatment and care of this patient. Published with written consent of the patient.
Sangey E, Chudasama K, Mwinyi A. The combined effect of hypomagnesemia and hypokalemia inducing nephrogenic diabetes insipidus in a patient with type 1 diabetes mellitus. Clin Case Rep. 2021;9:e04564. 10.1002/ccr3.4564
1. DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in Authorea at http://doi.org/10.22541/au.161669000.08417124/v1
REFERENCES
- 1. Velásquez‐Jones L, Medeiros‐Domingo M. Nephrogenic diabetes insipidus. Bol Méd Hosp Infant México Engl Ed. 2014;71(6):332‐338. [DOI] [PubMed] [Google Scholar]
- 2. Kalra S, Zargar AH, Jain SM, et al. Diabetes insipidus: The other diabetes. Indian J Endocrinol Metab. 2016;20(1):9‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khanna A. Acquired nephrogenic diabetes insipidus. Semin Nephrol. 2006;26(3):244‐248. [DOI] [PubMed] [Google Scholar]
- 4. Huang C‐L, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. 2007;18(10):2649‐2652. [DOI] [PubMed] [Google Scholar]
- 5. Radin MJ, Yu M‐J, Stoedkilde L, et al. Aquaporin‐2 regulation in health and disease. Vet Clin Pathol Am Soc Vet Clin Pathol. 2012;41(4):455‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moeller HB, Rittig S, Fenton RA. Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment. Endocr Rev. 2013;34(2):278‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khositseth S, Uawithya P, Somparn P, et al. Autophagic degradation of aquaporin‐2 is an early event in hypokalemia‐induced nephrogenic diabetes insipidus. Sci Rep. 2015;5(1). 10.1038/srep18311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khositseth S, Charngkaew K, Boonkrai C, et al. Hypercalcemia induces targeted autophagic degradation of aquaporin‐2 at the onset of nephrogenic diabetes insipidus. Kidney Int. 2017;91(5):1070‐1087. [DOI] [PubMed] [Google Scholar]
- 9. Chakraborti S, Chakraborti T, Mandal M, Mandal A, Das S, Ghosh S. Protective role of magnesium in cardiovascular diseases: a review. Mol Cell Biochem. 2002;238(1–2):163‐179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are openly available in Authorea at http://doi.org/10.22541/au.161669000.08417124/v1