

INTRODUCTION
N‐nitrosodimethylamine (NDMA), a probable human carcinogen, was discovered decades ago, although elevated levels in several widely used drugs were detected only recently. Several medications that are now known to contain NDMA have remained available, whereas ranitidine has been completely withdrawn from the market. In the following paper, we discuss the overview of events that led to the withdrawal of ranitidine and compare evidence and subsequent regulatory response of other medications also containing NDMA.
Our prior commentary detailed what is known about N‐nitrosodimethylamine (NDMA), initial US Food and Drug Administration (FDA) evaluation of affected ranitidine products, and limits in existing data to assess best practices for NDMA detection in drug products. 1 Briefly, NDMA is considered a probable carcinogen by the International Agency for Research on Cancer, 2 given that several animal studies have demonstrated adverse effects with chronic exposure to NDMA, including tumors of the liver, lungs, and stomach. NDMA metabolism in the liver results in the formation of methyldiazonium, a molecule that readily chelates to DNA and other macromolecules with cytochrome P450 families CYP2E and CYP2A believed to be key mediators in the formation of this metabolite. In vitro metabolism data from human liver samples suggests that formation of the methyldiazonium metabolite is comparable to that of laboratory animals, and thus NDMA may also be a procarcinogen in humans. Although human exposure to N‐nitrosamine is a common occurrence from numerous sources, including manufacturing byproducts, food and beverages, and even drinking water, several medications have recently been reported to contain NDMA in quantities greater than the acceptable daily intake (ADI) limit set by the FDA.
NDMA presence above the ADI in marketed drugs was first reported in 2018 when an American manufacturer of valsartan, an angiotensin II receptor blocker (ARB) frequently prescribed to treat hypertension and heart failure, 3 alerted the FDA that NDMA had been detected. The FDA confirmed levels ranging from below the ADI of 0.096 μg up to 20.19 μg per tablet 4 (Table 1). Batches of ARBs losartan and irbesartan were also affected, resulting in nearly 40 recalls, although unaffected lots remained on the market. The impurity was thought to be linked to manufacturing changes in 2014, wherein sodium nitrite integral to synthesis of many ARBs containing a tetrazole ring, reacted with amines resulting in formation of NDMA. Neither regulators nor industry had anticipated increased risk of NDMA formation in ARBs, thus testing for NDMA was not regular practice.
Table 1.
Comparison of medications known to contain NDMA
| Medication | Highest NDMA level, µg, per dose | Maximum daily dosing, mg | Cause of NDMA | FDA response |
|---|---|---|---|---|
| Ranitidine | 0.86 per 300 mg | 300 | Undetermined but increases with application of heat | Complete market withdrawal |
| Nizatidine | 0.02 per 150 mg | 300 | Undetermined | Voluntary recalls of affected products |
| Irbesartan | Unpublished | 300 | Manufacturing process | Voluntary recalls of affected products |
| Losartan | Unpublished | 100 | Manufacturing process | Voluntary recalls of affected products |
| Valsartan | 20.19 per 320 mg | 320 | Manufacturing process | Voluntary recalls of affected products |
| Metformin a | 0.19 per 500 mg | 2000 | Undetermined | Voluntary recalls of affected products |
Comparison of NDMA levels of affected medications and cause of impurity as reported by the FDA as well as agency response. 4 , 6 , 8 Typical maximum daily dosing of oral products is reported.
Abbreviations: FDA, US Food and Drug Administration; NDMA, N‐nitrosodimethylamine.
Extended‐release formation.
In September 2019, a new petition alerted the FDA to elevated NDMA levels in ranitidine, 5 an H2‐receptor antagonist (H2RA), prompting a voluntary recall. Although the FDA reported NDMA concentrations ninefold higher than the ADI, the levels were much lower than those reported by the petitioner possibly due to different laboratory practices and higher temperatures used in detection methods, prompting the FDA to communicate specific testing guidance to manufacturers. Although the FDA continues to determine root causes, the agency has confirmed NDMA formation in ranitidine is not purely secondary to manufacturing processes, 6 unlike affected ARBs. Suppositions about the exact mechanism remain, and the petitioner has highlighted the potential for instability of ranitidine, which contains both a nitrite and a dimethylamine group. 5 Other nitrites introduced into the gastrointestinal tract have also been considered possible biological sources reacting to form NDMA in ranitidine, but the FDA strongly rejects this theory, as the agency’s simulated gastric and intestinal fluid testing results did not yield elevated levels.
Several countries have reported NDMA above the ADI limit in nizatidine products, an H2RA structurally similar to ranitidine. The Korea Ministry of Food and Drug Safety suspended manufacturing and prescription of 13 nizatidine products following detection of NDMA at or above the same threshold used as the FDA. 7 Initial testing of four nizatidine products conducted by the FDA were found to be below the ADI. Voluntary recalls of nizatidine in the United States have been initiated, however, the levels in the recalled products have not been published. The FDA has not publicly announced plans for further situational testing of NDMA in nizatidine, such as shelf stability or exposure to heat.
Recently, NDMA was found in international metformin products, prompting FDA evaluation. Initial FDA testing of metformin, the medication commonly used in the treatment for type 2 diabetes mellitus, revealed either nondetectable or reassuring levels. 8 In March 2020, the same petitioner that reported NDMA in ranitidine released analysis of 38 metformin batches. Sixteen of the batches from 11 companies exceeded ADI limits when common daily treatment dose was factored, and variability in NDMA levels among batches even within a single company was described. 9 By May, the agency announced voluntary recalls of metformin extended‐release formulations. Inconsistency between lots was confirmed, but the FDA reported levels were again generally lower than recorded by the petitioner. The agency stated the source of NDMA remains unknown and has not been detected in immediate release metformin or the API. The petitioner posits the NDMA may be primarily due to contamination during manufacturing as opposed to instability of the molecule. Voluntary recalls have continued with unaffected lots remaining on the market.
With ongoing evaluation of the potentially affected drugs, in April 2020, the FDA requested immediate withdrawal of all ranitidine products, citing that NDMA levels could increase when stored at high temperatures, such as might occur during distribution, handling, or even normal storage conditions. The European Medicines Agency shortly thereafter, recommended suspending all ranitidine products in the European Union. Informing the decision was another petition to the agency claiming ranitidine has limited shelf‐life stability, and progressively accumulates NDMA at elevated temperatures (Figure 1). 10 Evaluations determined that NDMA levels could be acceptable at the manufacturing site but were at risk of increasing by the time of consumption. The highest NDMA value remained well under the maximum amount found in valsartan doses, but the small study set of ranitidine products continued to produce increasing levels of NDMA through termination of the experiment at 14 days.
Figure 1.
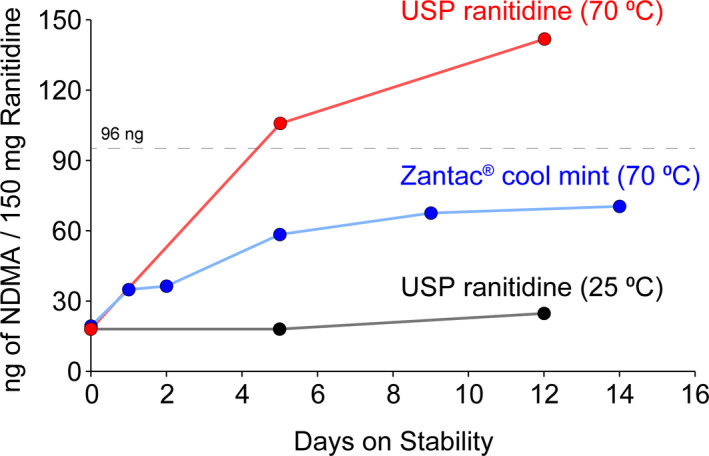
Preliminary ranitidine stability study as reported by Emery Pharma. Preliminary stability study with United States Pharmacopeial Convention (USP) ranitidine standard at 25°C, USP ranitidine standard at 70°C, and ranitidine (Zantac 150 Cool Mint), at 70°C. N‐nitrosodimethylamine (NDMA) analysis performed via liquid chromatography tandem mass spectrometry (vide infra), and quantified based on a calibration curve generated using an NDMA reference standard. Dotted line approximately indicates the daily intake limit of 96 ng for NDMA. Used by permission of Emery Pharma. 10 The generic of "Zantac" is ranitidine
The FDA recommended market withdrawal of ranitidine reiterates the need to tailor response to each affected medication’s specific characteristics, not solely basing actions on highest potential NDMA levels. Estimated treatment time‐courses, benefits of medication continuation versus risks of exposure, availability of alternative medication options, source of impurity, assumed duration for which medications have been affected, and known risk of carcinogenicity are considered. Treatment with metformin or ARBs is typically chronic, whereas H2RA treatment courses are generally recommended to be short term. Risks of untreated chronic hypertension and hyperglycemia are well‐established, so a higher hypothetical risk of cancer secondary to NDMA might be tolerated, and studies suggest metformin use may be associated with statistically significant reductions in the risk of cancer and cancer‐related mortality. 11 Risks associated with inadequate treatment of gastroesophageal reflux disease include esophagitis, esophageal stricture, esophageal ulceration, Barrett’s esophagus, esophageal dysplasia, and esophageal adenocarcinoma, but there are well‐known alternative treatment options, such as other structurally dissimilar H2RAs famotidine and cimetidine and proton pump inhibitors. When the source of NDMA is considered among the affected drugs, the regulatory actions taken toward ranitidine clarifies the more extensive actions. With proper oversight and testing, an aberrant manufacturing process might be remedied, but if the source is due to an inherently unstable molecule, such as has been proposed for ranitidine, then proper manufacturing protocols may not mitigate the risks of NDMA formation. Additionally, changes in the manufacturing of ARBs argues for a more recent presence of NDMA, whereas ranitidine may always have had the potential to develop NDMA.
Epidemiological studies designed to inform true risk due to long‐term NDMA exposure from specific medications are lacking. Although studies evaluating long‐term valsartan risk are not available, a secondary analysis of a Danish cohort revealed no increase in short‐term cancer risk for users of likely affected products. 12 With regard to ranitidine, after about 4 decades of usage, and millions of doses prescribed and purchased over the counter, a clear adverse signal with regard to malignancy has not been observed. The few studies evaluating risk of cancer from ranitidine are underpowered, focus on few specific cancers, lack appropriate case definitions, and/or appropriate control populations as well as long‐term follow‐up, and were uninformed about potential NDMA presence. A large database of millions of H2RA users is being investigated with early results reporting no increased risk of various cancers in users of ranitidine compared with structurally dissimilar famotidine. 13 However, early indication is that the dataset does not differentiate long‐term versus short‐term use. Population‐based adverse event reviews and cancer outcomes using pharmacy databases contained within hospital systems, health maintenance organizations, preferred provider organizations, and managed care plans are critically needed to provide epidemiological evidence to support or disprove an association of increased cancer risk of affected medications.
Additional examination of sources of NDMA production is imperative, and manufacturers and regulators should have a high index of suspicion given NDMA may result from production processes, storage processes, and possibly from endogenous processes. The establishment of uniform analytic methodologies leading to reproducible results under a robust number of conditions is essential. True exposure to NDMA in vivo following ranitidine use would be informative particularly should other drugs be concerning for similar unstable structures. Further analysis of NDMA ADI limits is warranted, particularly considering higher ADIs for treatment courses traditionally lasting less than 70 years of daily use. With current knowledge gaps, it is unclear if possible preventative and corrective actions may allow the return of ranitidine to the market. Considerations could include requiring temperature indicator labels, temperature‐controlled shipping and storage, labels warning about the risk of carcinogenic byproducts at high temperatures, dispersion by prescription only, addition of reducing agents, and recommendations on maximum length of treatment course.
Although the FDA continues to develop proactive industry guidance to be implemented so that all drugs can have a standardized NDMA risk assessment, continued transparency is vital especially given discrepancies in NDMA levels among laboratories. Drafting a singular regulatory response to medications affected by NDMA is unlikely given the unique characteristics of each affected medication. Ranitidine’s potential for elevated NDMA levels during storage required its withdrawal from the market, and, in the meantime, we will have to patiently await long‐term epidemiological studies.
CONFLICT OF INTEREST
All authors declared no competing interests for this work.
Funding information
No funding was received for this work.
REFERENCES
- 1. Wagner JA, Colombo JM. Medicine and media: the ranitidine debate. Clin Transl Sci. 2020;13:649‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. International Agency for Research on Cancer (IARC) . Overall evaluations of carcinogenicity: an updating of IARC monographs volumes 1–42. IARC Monogr Eval Carcinog Risks Hum. 1987; suppl. 7:1‐440. [PubMed] [Google Scholar]
- 3. Agency for Healthcare Research and Quality . Total purchases in thousands by prescribed drug, United States, 2010‐2017. Medical Expenditure Panel Survey. https://www.meps.ahrq.gov/mepsweb/. Accessed October 6, 2020.
- 4. Laboratory analysis of valsartan products. FDA statement. 2019. https://www.fda.gov/drugs/drug‐safety‐and‐availability/laboratory‐analysis‐valsartan‐products. Accessed June 1, 2020.
- 5. Light D, Kucera KR. Valisure citizen petition on ranitidine. Valisure, LLC. 2019. https://www.valisure.com/wp‐content/uploads/Valisure‐Ranitidine‐FDA‐Citizen‐Petitionv4.12.pdf. Accessed November 26, 2019.
- 6. Media briefing on FDA’s request for the removal of all ranitidine products (Zantac) from the market. FDA transcript. 2020. https://www.fda.gov/media/136672/download. Accessed June 1, 2020.
- 7. Yun‐mi K. Regulator detects NDMA also in nizatidine drugs. Korea Biomedical Review. 2019. http://www.koreabiomed.com/news/articleView.html?idxno=6861. Accessed June 1, 2020.
- 8. Woodcock J. Statement from Janet Woodcock, M.D., director of FDA’s Center for Drug Evaluation and Research, on impurities found in diabetes drugs outside the U.S. FDA Statement. 2019. https://www.fda.gov/news‐events/press‐announcements/statement‐janet‐woodcock‐md‐director‐fdas‐center‐drug‐evaluation‐and‐research‐impurities‐found. Accessed June 1, 2020.
- 9. Light D, Kucera K, Wu Q. Re. Valisure citizen petition on metformin. Valisure, LLC. 2020. https://www.valisure.com/wp‐content/uploads/Valisure‐FDA‐Citizen‐Petition‐on‐Metformin‐v3.9.pdf#:~:text=Re:%20Valisure%20Citizen%20Petition%20on%20Metformin%20.%20Dear,Petition%20(“Petition”)%20pursuant%20to%20Sections%20301(21%20U.S.C.%20§. Accessed June 1, 2020.
- 10. Najafi RR. Re: Emery Pharma citizen petition. Emery Pharma. 2020. https://emerypharma.com/wp‐content/uploads/2020/01/EP‐Ranitidine‐FDA‐Citizen‐Petition‐v21‐January‐2‐2020.pdf. Accessed June 1, 2020.
- 11. Franciosi M, Lucisano G, Lapice E, Strippoli GFM, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One. 2013;8:e71583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pottegård A, Kristensen KB, Ernst MT, Johansen NB, Quartarolo P, Hallas J. Use of N‐nitrosodimethylamine (NDMA) contaminated valsartan products and risk of cancer: Danish Nationwide Cohort Study. BMJ. 2018;362:k3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mohy‐ud‐din N, Mohyuddin GR, Syed A, Desai SM, Thakka S. Risk of cancer with use of ranitidine: results of a cohort study of 65 million US adults. Poster accepted for presentation at Digestive Disease Week 2020 Convention, May 2–5, 2020. Chicago, IL, USA.