

Abstract
Vincristine (VCR) is one of the most widely prescribed medications for treating solid tumors and acute lymphoblastic leukemia (ALL) in children and adults. However, its major dose‐limiting toxicity is peripheral neuropathy that can disrupt curative therapy. Peripheral neuropathy can also persist into adulthood, compromising quality of life of childhood cancer survivors. Reducing VCR‐induced neurotoxicity without compromising its anticancer effects would be ideal. Here, we show that low expression of NHP2L1 is associated with increased sensitivity of primary leukemia cells to VCR, and that concomitant administration of VCR with inhibitors of NHP2L1 increases VCR cytotoxicity in leukemia cells, prolongs survival of ALL xenograft mice, but decreases VCR effects on human‐induced pluripotent stem cell‐derived neurons and mitigates neurotoxicity in mice. These findings offer a strategy for increasing VCR’s antileukemic effects while reducing peripheral neuropathy in patients treated with this widely prescribed medication.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Vincristine (VCR) is a widely prescribed drug, but its use is limited by its main side effect, neurotoxicity. There are currently no strategies to mitigate VCR neurotoxicity without altering its antileukemic effects.
WHAT QUESTION DID THIS STUDY ADDRESS?
How to improve VCR efficacy while reducing its main side effect, neurotoxicity?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The present study shows for the first time the possibility of reduced VCR ‐induced neurotoxicity while improving VCR anti‐leukemia effect by using small molecules.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The current translational study could permit a safer and more efficient use of VCR.
INTRODUCTION
The success of drug therapy for many diseases is often compromised by adverse effects of medications used in the clinic. 1 , 2 This is frequently true for cancer chemotherapy, which typically has a narrow therapeutic index, damaging normal tissues at doses required for anticancer effects. Vincristine (VCR) is a widely used anticancer drug that has a very narrow therapeutic index, with a dose‐limiting toxicity of peripheral neuropathy, characterized by neuropathic pain, and sensory and motor dysfunction, 3 , 4 that can persist for decades after completing therapy, compromising the quality of life of cancer survivors. 5 There are currently no effective strategies for reducing VCR‐induced neurotoxicity while retaining its therapeutic efficacy. Therefore, our current work focused on elucidating strategies to improve the therapeutic index of VCR (i.e., increase antileukemic effects relative to neurotoxicity). We found that low expression of NHP2L1 in primary acute lymphoblastic leukemia (ALL) cells was associated with greater sensitivity to VCR, suggesting that inhibition of NHP2L1 could increase VCR’s antileukemic effects. Because ALL cells undergo mitosis whereas neurons are postmitotic, 6 , 7 we hypothesized that small molecule inhibitors of NHP2L1, 8 which is a component of the spliceosome complex 9 , 10 , 11 involved in mitosis, 12 might increase the sensitivity of leukemia cells to VCR without altering the sensitivity of neurons, thus enhancing VCR’s therapeutic index. Here, we report that dipyridamole (DIP), an inhibitor of NHP2L1, increases the sensitivity of leukemia cells to VCR and prolongs survival of ALL xenograft mice, while protecting both human‐induced pluripotent stem cell (hiPSC)‐derived neurons and mice from VCR ‐induced neurotoxicity.
METHODS
Patients
We studied pediatric patients (aged ≤21 years) with newly diagnosed ALL who were enrolled on St. Jude Total Therapy XV and XVI (Memphis, TN, USA), and used publicly available data from the Ninth ALL Dutch Childhood Oncology Group (DCOG) protocol at Erasmus Medical Center, Sophia Children’s Hospital (Rotterdam, The Netherlands), or treatment protocol 92 or 97 of the German Cooperative Study Group for Childhood Acute Lymphoblastic Leukemia (COALL; Hamburg, Germany; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE649; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE648). Informed consent was obtained from patients, their guardians, or both before enrollment. Leukemia cells were isolated by applying a Ficoll‐Hypaque gradient to bone marrow aspirates obtained at diagnosis. Genome‐wide mRNA expression in ALL cells was determined using the Affymetrix U133 microarray. Ex vivo drug sensitivity of primary ALL cells was determined using a modification of the MTT ((3‐[4,5‐dimethylthiazol‐2‐yl]‐2, 5‐diphenyl‐tetrazoliumbromide) assay, as we have described previously. 13 For the genome‐wide interrogation of mitotic genes, there were 572 genes with a mitotic phenotype, 12 395 genes were represented on the Affy HGU133A expression array. The association between the expression of these genes and VCR lethal concentration 50% (LC50) was first tested in a cohort of 92 patients in the St. Jude cohort, which led to the identification of 12 genes associated with VCR LC50 with a p value less than 0.05. Of these 12 genes, only NHP2L1 expression was significantly associated with VCR LC50 in the DCOG/COALL cohort used as validation cohort. Sensitive and resistant cells were defined the same way as previously described with sensitive cells as having a VCR LC50 of the cells of 0.391 μg/ml (0.474 µM) or less, and resistant cells as having an LC50 greater or equal to 1.758 μg/ml (2.131 µM). 13
Cell culture
The human T‐lineage leukemia cell line CCRF‐CEM was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The human pre‐B leukemia cell line NALM‐6 was obtained from the DSMZ‐German Collection of Microorganisms and Cell Cultures GmbH (Braunschweig, Germany). The cells were authenticated using Short Tandem Repeat profiling. 14 The cells tested negative for mycoplasma (MycoAlert mycoplasma detection kit; Lonza). Cells were cultured in RPMI‐1640 medium containing 2 mM glutamine and 10% (vol/vol) fetal bovine serum at 37°C with 5% CO2.
Humani PSC‐derived neurons (Peri.4 U) having greater than 90% purity and expressing βIII‐tubulin, MAP‐2, peripherin, and vGLUT2 were purchased from Ncardia BV (Gosselies, Belgium). All batches of iPSC‐derived neurons were tested for sterility, viability, purity, and morphology by Ncardia BV. Neurons were maintained according to the manufacturers’ protocol. Three independent batches of Peri.4 U neurons were used, including LOT#D059_PNN, LOT#424D_v1, and LOT#428D_v2 M.
Animals
All animal studies were approved by the St. Jude Animal Care and Use Committee. Male C57BL/6 J mice (The Jackson Laboratory) at 8 weeks of age were used. The mice had free access to food and water. The mice were maintained under 12 h light‐dark cycle.
Reagents
Dipyridamole (Sigma‐Aldrich and LKT Labs), VCR, scopoletin, tazarotene, dihydroxyflavone (Sigma‐Aldrich), quetiapine fumarate (LKT Labs and Selleck Chemicals), hymecromone, esculin (Selleck Chemicals, Houston, TX, USA), chir 99021 (Cayman Chemical), harmaline, fisetin (Tokyo Chemical Industry, Thermo Fisher Scientific, USA), aurintricarboxylic acid (Calbiochem, Millipore Sigma), piperacillin sodium (Santa Cruz Biotechnology), and olmesartan medoxomil (Ark Pharm).
NHP2L1 gene knockdown
CEM cells were infected with MISSION lentiviral transduction particles (Sigma‐Aldrich) produced from a library of sequence‐verified shRNAs targeting human NHP2L1 transcripts. The following sequence of shRNA was used to knock down the NHP2L1 gene: 5′‐ CCGGCCTGAGGTTGTGTATCATATTCTCGAGAATATGATACACAACCTCAGGTTTTTG −3′ (TRCN0000074798); 5’CCGGCAATCCATTCAGCAGTCCATTCTCGAGAATGGACTGCTGAATGGATTGTTTTTG‐3′ (TRCN0000074801). Nontarget shRNA control particles (SHC002 V; Sigma‐Aldrich) were also used. Individual cell clones were isolated in medium containing puromycin. Gene knocked down was assessed by Western blot. HiPSC‐derived neurons, Peri.4 U underwent siRNA transfection using Accell human NHP2L1 SMARTpool siRNA and a nontargeting siRNA pool as the negative control (Dharmacon). Quantitative reverse transcription polymerase chain reaction was used to measure the level of NHP2L1 expression (SNU13, Hs03025442_s1) compared with the housekeeping control Beta‐2‐microglobulin (4326319E) (both from Thermo Fisher Scientific).
Western blot analysis
Lysates from the CEM or NALM6 cell lines were separated by electrophoresis on an SDS‐polyacrylamide gel. The proteins were then electroblotted onto a Hybond‐P PVDF membrane. Protein expression was analyzed using the primary antibodies anti‐GAPDH (Santa Cruz Biotechnology #sc‐20357) and anti‐NHP2L1 (Abcam #ab95958). Horseradish peroxidase–conjugated secondary antibodies were purchased from Agilent (Santa Clara). Phospho‐histone H3 (Ser10) clone 3H10 (Millipore #05‐806); Beta tubulin (Sigma‐Aldrich#C4585) Histone H3 (proteintech#17168‐1‐AP) were used. Sororin was provided by Dr. Jan‐Michael Peters from the Research Institute of Molecular Pathology, Vienna, Austria.
High content imaging of neuronal morphological characteristics
After drug treatment, neurons were stained with 5 µg/ml Hoechsts 33342 (Sigma Aldrich) and 10 µg/ml Calcein AM (Molecular Probes; ThermoFisher Scientific) for 10 min at room temperature. Imaging was performed at 10× magnification using an ImageXpress Micro imaging device (Molecular Devices, LLC) at the University of Chicago Cellular Screening Center. The MetaXpress software Neurite Outgrowth Application Module was utilized to collect individual cell measurements of mean/median/maximum process length, total neurite outgrowth (the sum of the length of all processes), number of processes, number of branches, cell body area, mean outgrowth intensity, straightness, and cell number. A minimum of 1000 cells per condition were imaged in each of the five independent experiments.
Gene expression in the iPSC‐derived neurons Peri. 4 U
Total RNA from iPSC‐derived neurons Peri. 4 U was extracted and prepared as previously described. 15 Samples were evaluated on the Affymetrix GeneChip Human Transcriptome (HTA) 2.0 Array (Affymetrix) and data generated at the University of Chicago Genomics Core. The raw data were processed using the Affymetrix Power Tools (APT version 1.16) and the HTA 2.0 array annotation library (release 1.0). Background subtraction, normalization, and probe‐set summarization were performed using the APT software. Briefly, gene‐level data were background corrected and normalized using the Robust Multi‐array Average method on perfectly matched probes through median polish. 16 The summarized gene‐level data were log2 transformed for downstream analysis.
Cell viability assay
HiPSC‐derived neurons cell viability was assessed at 72 h postdrug treatment by the Cell Titer‐Glo 2.0 assay (Promega), which measures ATP.
Ex vivo drug sensitivity (MTT) assay
Ex vivo drug sensitivity of CEM and NALM6 cell lines was determined using a modification of the MTT ((3‐[4,5‐dimethylthiazol‐2‐yl]‐2, 5‐diphenyl‐tetrazoliumbromide) assay, as described previously. 13 Briefly, cells in mid‐log phase were exposed to serial dilutions of the different drugs and analyzed after 3 days of incubation. The concentration of drug required for 50% cell kill (LC50) was used as the measure of relative sensitivity to each compound.
Synergy studies: Response surface modeling
A modification of the response surface modeling approach 17 , 18 , 19 implemented in MatLab (version R2019a, Mathworks) was used to determine changes in the response of two drugs given in combination from an additive response. A drug combination was considered either synergistic or antagonistic if α, the interaction term describing the change in response relative to the additive model, was either positive or negative, respectively. We quantified the effects of VCR in the presences of a fixed concentration of the second drug as the ratio of the estimated additive half‐maximal effective concentration (EC50) to estimated actual EC50 of VCR and the fixed concentration of the second drug.
Intracellular concentration of VCR and DIP in leukemia cell lines and in hiPSC‐derived neurons
After treatment with VCR and DIP, the cells were harvested, washed twice with Hank’s Balanced Salt Solution (HBSS; Sigma Aldrich), pelleted, and stored at −80°C. The cells were later resuspended in HBSS, sonicated to fully lyse the cells, and after centrifugation the supernatant was collected. Quantitation of analytes was carried out with a Waters ACQUITY separation system (Waters, Milford, MA, USA) and Xevo TQ triple‐quadrupole system (Beverly, MA, USA). Separation was achieved on a Waters ACQUITY BEHC18 column (1.7 µm, 50 × 2.1 mm) using a column heater operating at 40°C with Waters ACQUITY in‐line filter. Autosampler temperature was maintained at 15°C ± 5°C. The gradient mobile phase was composed of acetonitrile (B) and 15 mM ammonium acetate in 0.02% formic acid water (A). The flow rate was 0.6 ml/min and the separation was completed within 6 min. The instrument was equipped with an electrospray interface, and was controlled by Masslynx version 4.1 software (Waters). The analysis was performed in MRM mode: m/z 505.46 greater than 429.27 for DIP, m/z 525.6 greater than 449.3 for DIP_d3; m/z 825.36 greater than 807.39 for VCR, and m/z 828.56 greater than 810.57 for VCR _d3. The tandem mass spectrometry conditions were as follows: capillary voltage: 0.5 kV; source temperature: 150°C; desolvation temperature: 600°C; cone gas flow:0 l/h; and desolvation gas flow: 1000 l/h.
Vincristine intracellular concentration modeling
Vincristine intracellular (VCRintracellular) concentrations with respect to VCR and DIP extracellular concentration and incubation time were modeled with the following multivariate Hill function:
For each of the two extracellular concentrations of VCR [2 nM (1.65 µg/L) and 40 nM (33 µg/L)] the dependent variables Vmax , KM , n, ΔVmax , and ΔKM were estimated relative to the two independent variables DIP: extracellular concentration of DIP (0 µM, 2.5 µM [1.265 mg/L], 5 µM [2.525 mg/L], 10 µM [5.05 mg/L) and the incubation time t (1, 6, 12, and 24 hours). If the parameters ΔVmax or ΔKM were significantly different from zero then the effect of DIP concentration significantly altered the steady‐state intracellular concentration of VCR.
DIP intracellular concentration modeling
Dipyridamole intracellular (DIPintracellular) concentration with respect to VCR and DIP extracellular concentration and incubation time were modeled with the following multivariate linear function:
For each of the three extracellular concentrations of DIP (0 µM, 2.5 µM [1.265 mg/L], 5 µM [2.525 mg/L], 10 µM [5.05 mg/L]) the dependent variables b, m, Δb, and Δm were estimated relative to the two independent variables VCR: extracellular concentration of VCR (0, 2 nM [1.65 µg/L], and 40 nM [33 µg/L]) and the incubation time t (1, 6, 12, and 24 h). If the parameters Δb or Δm were significantly different from zero, then the effect of VCR concentration significantly altered the steady‐state intracellular concentration of DIP.
In vivo pharmacokinetic studies
Methods for the in vivo pharmacokinetic (PK) studies performed in mice are provided in the Supplementary Methods.
Mouse neurotoxicity assessment: Mechanical allodynia
Mice were acclimated before testing. The mice were treated, from day 1 to day 10, intraperitoneally with VCR or saline and by oral gavage with DIP or the vehicle of DIP (1% methylcellulose and 1% Tween 80, pH adjusted to 2.0 with HCl). Mechanical allodynia was evaluated in mice using von Frey filaments (0.16, 0.4, 0.6, 1, and 2 g) prior to drug treatment, at days 7 and 10 of drug administration, and at day 14 (4 days after discontinuation of treatment). Each mouse was tested 10 times in increasing order of filament force. The data obtained from three different replicate experiments comprise both cross‐sectional and time‐series measurements of responses to stimulus under multiple conditions. The withdraw threshold (g) was determined as the minimal force filament for which a response was obtained at least 5 times out of the 10 stimulations.
Statistical analyses
Student’s t‐test was used to compare the means between two different experimental conditions.
Analyses were done using SAS, version 9.4 (Cary, NC), R (The R Project http://www.r‐project.org/), and SigmaPlot 11.0 (Systat Software).
Statistical analyses of longitudinal in vivo data in mice accounted for possible between‐subject heterogeneity and within‐subject correlation. The longitudinal mean response was modeled by a mixed effect linear model, with treatment group, filament level, baseline response, and batch as fixed effects and a random effect to account for intra‐animal longitudinal correlation with compound symmetry working covariance structure. The primary effect of interest was a comparison of neuropathy events among the treatment groups. To visually depict the cross‐sectional and cumulative proportion of mice positive for neuropathy (NP+) in each treatment group, we dichotomized the response (NP+ vs. negative for neuropathy [NP‐]) using a threshold of the 95th percentile of the percentage of response of the von Frey measurement at the baseline in all treatments and filament levels pooled. Any measurement above or equal to this threshold was defined as NP+. The 95th percentile of the von Frey measurements in all treatments pooled together was 90, thus any von Frey measurement exceeding 90 defined a positive finding for NP+. Neuropathy was assessed at days 7, 10, and 14, with the day of the first measurement greater than or equal to 90 defining the time to the first NP+ event in the cumulative proportion analysis and figure. In the mouse ALL xenograft survival study, all mice reached the study end point by the time of data freeze, therefore no observed survival time was censored. Survival time distributions were compared using exact Wilcoxon rank sum test between the different treatment groups.
RESULTS
NHP2L1 expression and VCR LC50 in primary leukemia cells
In a genome‐wide interrogation of genes whose mRNA expression was significantly related to the sensitivity of primary ALL cells to VCR (LC50), we identified NHP2L1 expression as the top mitosis‐related 12 gene significantly associated with VCR LC50 in two different patient cohorts (Table S1). Specifically, lower NHP2L1 mRNA expression in leukemia cells isolated from diagnostic bone marrow aspirates of children with newly diagnosed ALL was significantly associated with increased sensitivity to VCR in B cell and T cell leukemia in two patient cohorts (Figure 1a,b).
FIGURE 1.
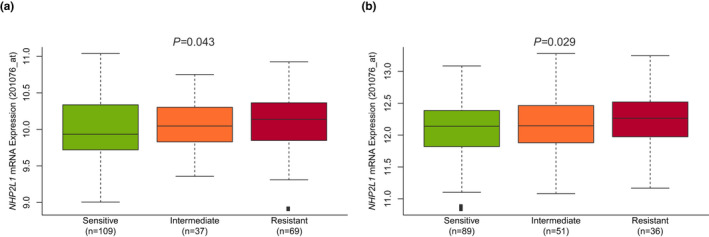
NHP2L1 expression and sensitivity of primary B leukemic cells to vincristine (VCR). NHP2L1 mRNA expression was determined by Affymetrix gene expression array. MTT was used to determine the sensitivity of primary cells to VCR. The association between log2 expression of NHP2L1 and VCR sensitivity was assessed in two cohorts of patients with acute lymphoblastic leukemia: (a) St. Jude Children’s Research Hospital and (b) Dutch Childhood Oncology Group (DCOG)/Cooperative Study Group for Childhood Acute Lymphoblastic Leukemia (COALL) cohorts. Based on previously published criteria24 sensitive cells were defined as having a VCR concentration lethal to 50% of the cells (LC50) of 0.391 μg/ml (0.474 µM) or less, and resistant cells were defined as having an LC50 of 1.758 μg/ml (2.131 µM) or more. The horizontal line inside each box depicts the median, the upper and lower limits of the box are the 75th and 25th percentiles, respectively, and the vertical bars above and below each box indicate the maximum and minimum values, respectively. The p values were determined by linear regression comparing NHP2L1 gene expression and leukemia cell sensitivity to VCR
NHP2L1 knockdown and VCR LC50 in leukemia cells
To determine whether a low level of NHP2L1 expression increased leukemia cell sensitivity to VCR, we reduced the expression of NHP2L1 by infecting two human leukemia cells (the T‐lineage cell line CCRF‐CEM and the B‐lineage NALM6 cell lines) with shRNA against NHP2L1. As shown in Figure 2a–c, the knockdown of NHP2L1 using two different shRNA clones resulted in a significant reduction in NHP2L1 protein and significantly increased sensitivity of human CCRF‐CEM and NALM6 ALL cells to VCR (Figure 2b–d), consistent with the association of lower NHP2L1 expression with greater VCR sensitivity (low LC50) in primary leukemia cells isolated from patients with newly diagnosed ALL (Figure 1).
FIGURE 2.
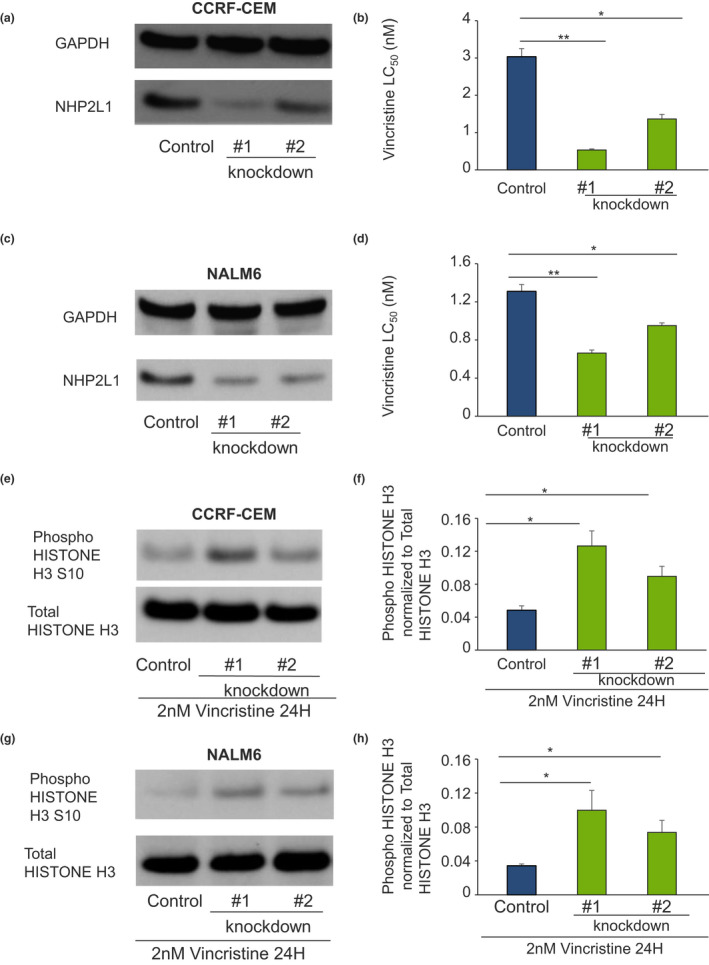
Knockdown of NHP2L1 and sensitivity to vincristine (VCR) and mitotic arrest in leukemic cell lines. Human leukemia cells infected with two different shRNAs against NHP2L1 show a decrease in NHP2L1 protein levels in CCRF‐CEM (a) and in NALM6 (c). After knockdown of NHP2L1, cells were treated for 72 h in the presence of increasing concentrations of VCR. VCR sensitivity (lethal concentration 50% [LC50]) Cell viability determined by MTT in CCRF‐CEM (b) and in NALM6 (d) are represented. Error bars represent SDs of three replicate experiments. Student’s t‐test was used to calculate p values. *p < 0.05, **p < 0.01. Human leukemia cells (CCRF‐CEM and NALM6) were infected with two different shRNAs (#1 and #2) against NHP2L1 and treated with vincristine for 24 h. Representative blots of phosphorylated Serine 10 in Histone H3 protein and of total Histone H3 after knockdown of NHP2L1 and 24‐h treatment with VCR were shown in CEM (e) and NALM6 cells (g). Phospho Histone H3 protein levels were quantified by densitometry, normalized to total Histone H3 signal and expressed in arbitrary units for the CEM cells (f) and for the NALM6 cells (h). Values are means ± SD of three independent experiments. Student’s t‐test was used to calculate p values. *p < 0.05, **p < 0.01
NHP2L1 knockdown and VCR‐induced accumulation of mitotic cells
To assess the mechanisms by which NHP2L1 influences VCR‐induced cell death through mitotic arrest, we assessed the effects of VCR on HISTONE H3 phosphorylation at serine 10 (Phospho‐HISTONE H3 [Ser10]), a mitosis marker, 20 in CEM and in NALM6 cells. We documented increased VCR‐induced HISTONE H3 phosphorylation (Ser10) after NHP2L1 knockdown (Figure 2e–h), indicating that knockdown of NHP2L1 sensitizes cells to the microtubule inhibitor VCR in part by inducing mitotic arrest.
Small molecule inhibitors of NHP2L1‐U4 interaction synergize with VCR in leukemic cells
NHP2L1’s known function is via interaction with the spliceosome component U4 RNA. We developed an assay based on time‐resolved FRET to detect the interaction between NHP2L1 and U4 5’‐stem loop and used this to conduct a high‐throughput screen for small molecules inhibiting this interaction. 8 Given the greater VCR sensitivity of primary ALL cells with lower NHP2L1 expression, we hypothesized that inhibitors of the NHP2L1‐U4 interaction would potentiate the effects of VCR in ALL cells. We further assessed the effects of several small molecule hits in this analysis (scopoletin, quetiapine fumarate, hymecromone, harmaline, fisetin, esculin, dihydroxyflavone, chir 99021, aurintricarboxylic acid, tazarotene, DIP, piperacillin sodium, and olmesartan medoxomil) on VCR sensitivity in human leukemic cells. Synergistic effects between these small molecules and VCR were assessed by the response surface modeling method 17 , 18 , 19 (based on α). This revealed that DIP, tazarotene, and quetiapine fumarate synergize with VCR cytotoxicity in human leukemia CEM cells (Figure S1a–m). We also validated these results in a second human leukemia cell line, NALM6, with DIP providing the highest level of synergy (Figure S1n–r and Table 1).
TABLE 1.
Estimate of drug‐drug interaction between vincristine and second drug (dipyridamole, tazarotene, and quetiapine fumarate) in CEM and NALM6 ALL cell‐lines
| Drug name | CEM α | CEM p value | NALM6 α | NALM6 p value |
|---|---|---|---|---|
| Dipyridamole | 11.6 | 2.45 × 10−9 | 4.45 | 6.9 × 10−25 |
| Tazarotene | 0.945 | 9.66 × 10−7 | 1.42 | 1.43 × 10−11 |
| Quetiapine fumarate | 0.787 | 0.0235 | 2.33 | 1.73 × 10−7 |
Values of α significantly greater than zero indicate synergy and values significantly less than zero indicate antagonism. The p value indicates the significance of the interaction term (α) relative to additive (0) and was determined using a t‐test.
Abbreviation: ALL, acute lymphoblastic leukemia.
Effects of these small molecules on VCR‐induced neurotoxicity in human iPSC‐derived neurons
To assess the effects of these small molecules on VCR‐induced neurotoxicity, we used neurons derived from human iPSCs. These hiPSC‐derived neurons were previously validated as a model to evaluate chemotherapy‐induced neurotoxicity, 21 , 22 including VCR‐induced neurotoxicity. 23 In contrast to ALL cells, we documented significantly greater resistance of hiPSC‐derived neurons to VCR in the presence of these small molecules (Table 2). This protection against VCR‐induced toxicity in neurons was evidenced by the antagonistic effects of these small molecules as measured by neurite outgrowth, branches, processes, and cytotoxicity compared with VCR alone (Table 2), whereas knockdown of NHP2L1 in hiPSC‐derived neurons did not have any effect on these morphologic phenotypes (Figure S2). Overall, our results show that DIP, quetiapine fumarate, and tazarotene synergized with VCR in leukemia cells but mitigated VCR effects on neurons (Figure S3). Because DIP showed the strongest degree of synergy with VCR in leukemia cells and protection against VCR toxicity in iPSC‐derived neurons, as documented for hiPSC‐derived neurons outgrowth and ability to expand in branches and processes (Figure 3a,b and Figure S4), DIP was chosen for further experiments.
TABLE 2.
Estimate of drug‐drug interaction between vincristine and second drug (dipyridamole, tazarotene, and quetiapine fumarate) in hiPSC‐derived neurons
| Drug name | Branches | Processes | Outgrowth | Viability | ||||
|---|---|---|---|---|---|---|---|---|
| α | p value | α | p value | α | p value | α | p value | |
| Dipyridamole | −3.43 | 2.8 × 10−12 | −1.75 | 5.07 × 10−9 | −1.98 | 5.69 × 10−8 | −3.7 | 4.83 × 10−11 |
| Tazarotene | −1.16 | 0.032 | −1.59 | 2.05 × 10− 8 | −1.63 | 2.9 × 10−7 | −0.72 | 0.25 |
| Quetiapine fumarate | −2.29 | 2.22 × 10−7 | −1.25 | 1.47 × 10−4 | −1.67 | 6.32 × 10−6 | −0.007 | 0.99 |
Values of α significantly greater than zero indicate synergy and values significantly less than zero indicate antagonism. The p value indicates the significance of the interaction term (α) relative to additive (0) and was determined using a t‐test.
Abbreviation: hiPSC, human‐induced pluripotent stem cell.
FIGURE 3.
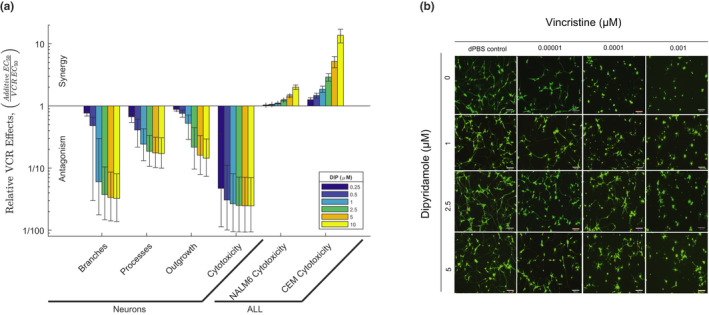
Effects of small molecule inhibitor of NHP2L1 U4 interaction on neurons and leukemic cells treated with vincristine (VCR). (a) VCR effects relative to dipyridamole (DIP) on neurons and two acute lymphoblastic leukemia (ALL) cell lines (CEM and NALM6). The y‐axis represents the ratio of the additive half‐maximal effective concentration (EC50) to the actual EC50 for the combination of VCR and DIP. Each bar represents a fixed concentration of DIP (0.25 µM [0.125 mg/L], 0.5 µM [0.25 mg/L], 1 µM [0.5 mg/L], 2.5 µM [1.265 mg/L], 5 µM [2.525 mg/L], and 10 µM [5.05 mg/L]). The whiskers are the 95% confidence interval of the measure. A ratio of one indicates the combination is additive, whereas a ratio greater or less than one indicates that the combination is either synergistic or antagonistic, respectively. (b) Representative images of human‐induced pluripotent stem cell (hiPSC) neurons (Peri.4 U) 72 h after treatment with increasing concentrations of VCR with or without increasing concentrations of DIP. Images were taken from the ImageXpress Micro imaging microscope at 10× magnification
Mechanisms by which DIP alters VCR effects in ALL cells and in neurons
To understand the mechanisms by which DIP influences VCR‐induced cell death, we assessed the effect of VCR on Histone H3 phosphorylation in the presence of increasing concentration of DIP in CEM and in NALM6 leukemia cells. DIP induced a dose‐dependent increase in phospho‐histone H3 (Ser10) levels in CEM (Figure 4a,b) and in NALM6 cells (Figure 4c,d), consistent with the increased accumulation of mitotic cells prior to VCR‐induced cell death. The primary function of NHP2L1 through its interaction with U4 involves the splicing of SORORIN pre‐mRNA. Disruption of this interaction compromises SORORIN splicing and decreases the abundance of mature SORORIN protein. 24 We documented a dose‐dependent effect of DIP on SORORIN protein levels, with the highest concentration of DIP (10 µM [5.05 mg/L]) producing the lowest level of SORORIN protein in CEM (Figure 4e,f) and in NALM6 cells (Figure 4g,h), consistent with DIP exerting its effects via interference with NHP2L1/U4 interaction.
FIGURE 4.
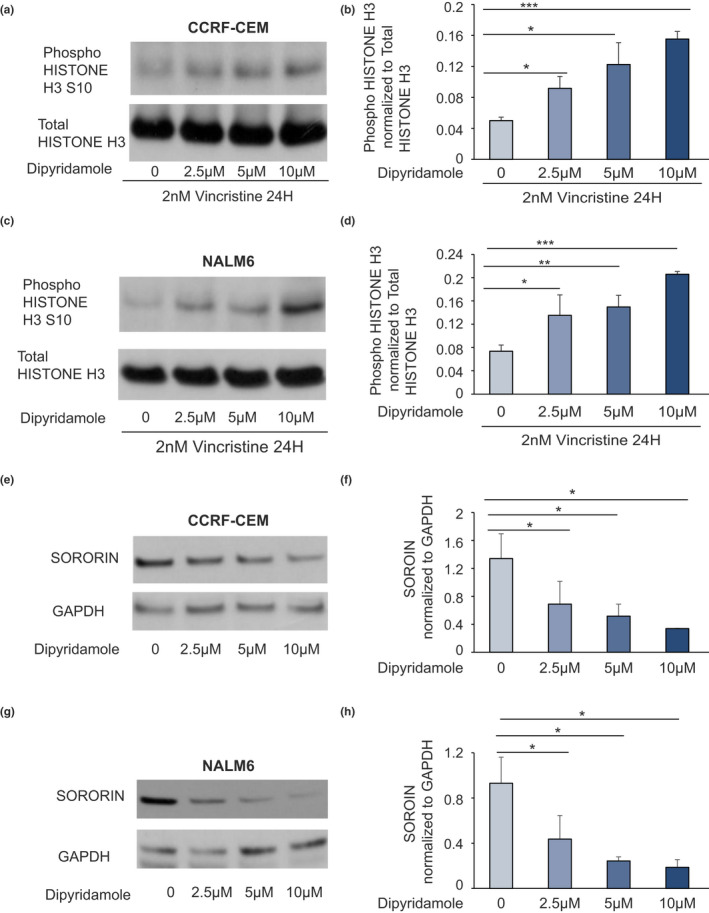
Effects of dipyridamole (DIP) on vincristine (VCR)‐induced mitotic arrest and on SORORIN protein level. Human leukemia cells (CCRF‐CEM and NALM6) were treated with vincristine (2 nM [1.65 µg/L]) for 24 h alone or with increasing concentrations of DIP. Representative blots of phosphorylated Serine 10 in Histone H3 protein and of total Histone H3 after 24‐h treatment with VCR and increasing concentrations of DIP for human acute lymphoblastic leukemia cell lines CEM (a) and NALM6 (c) are shown. Phospho Serine 10 Histone H3 protein levels were quantified by densitometry, normalized to total Histone H3 signal and expressed in arbitrary units for the CEM cells (b) and for the NALM6 cells (d). Values are means ± SD of three independent experiments. Student’s t‐test was used to calculate p values. *p < 0.05, **p < 0.01, ***p < 0.001. G2 synchronized cells were treated with increasing concentration of DIP, and expression of SORORIN was document by Western blot in CEM (e) and NALM6 (g) cells. The level of SORORIN was normalized to the level of GAPDH and the ratio was plotted for the CEM (f) and the NALM6 (h) cells. Values are means ±SD of three independent experiments. Student’s t‐test was used to calculate p values. *p < 0.05, **p < 0.01, ***p < 0.001
Because DIP is a known inhibitor of the ABCB1 efflux transporter, we assessed the effects of DIP on the intracellular concentration of VCR and vice versa. There was a significant increase in the intracellular concentration of VCR with increasing concentrations of DIP in CEM (Figure 5a) and in NALM6 leukemia cells (Figure S5a), indicating that DIP can also affect sensitivity of leukemia cells by increasing the intracellular concentration of VCR. In contrast, increasing concentrations of VCR did not significantly increase the intracellular concentration of DIP in CEM (Figure 5b) or in NALM6 cells (Figure S5b). We also assessed the intracellular concentrations of DIP and VCR in hiPSC‐derived neurons, documenting that DIP did not increase VCR intracellular concentrations (Figure 5c,d), but instead trended toward lower VCR intracellular concentrations in the presence of DIP in hiPSC‐derived neurons. Gene expression analyses indicated low expression of ABCB1 (relative to other transporters), the main efflux transporter of VCR, in hiPSC‐derived neurons but not in the leukemia cells (Tables S2 and S3).
FIGURE 5.
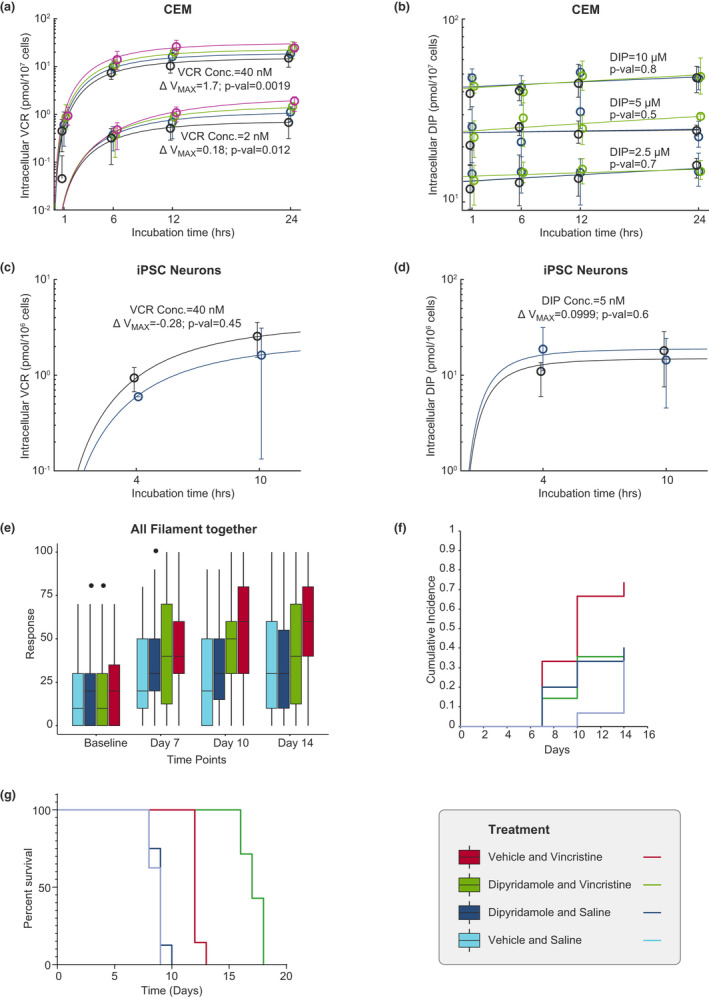
Intracellular concentration of vincristine (VCR) or dipyridamole (DIP) in CEM human leukemia cell line, or human‐induced pluripotent stem cell (hiPSC)‐derived neurons relative to incubation time (hours) and VCR neurotoxicity in mice. Intracellular concentration of VCR was determined in (a) CEM acute lymphoblastic leukemia (ALL) cells after exposure of the cells to 2 nM (1.65 µg/L) or 40 nM (33 µg/L) of VCR with or without increasing concentrations of DIP: 0 µM in black, 2.5 µM (1.265 mg/L) in blue, 5 µM (2.525 mg/L) in green, and 10 µM (5.05 mg/L) in magenta. The circles and whiskers represent the median and range of the data. The maximal rate of metabolism (ΔVmax ) represents the change in Vmax due to the presence of DIP. The p values test whether the change in Vmax (ΔVmax ) due to the presence of DIP differs from 0 using the t‐test (a). Intracellular concentration of DIP was determined in (b) CEM after treating leukemia cells with 2.5 µM (1.265 mg/L), 5 µM (2.525 mg/L), or 10 µM (5.05 mg/L) of DIP with or without increasing concentrations of VCR: 0 nM in black, 2 nM (1.65 µg/L) in blue, and 40 nM (33 µg/L) in green. The circles and whiskers represent the median and range of the data. The p values test whether the change in the slope due to the presence of VCR is different from 0 using the t‐test (b). Intracellular levels of DIP were not significantly related to extracellular VCR concentrations. Intracellular concentration of VCR was determined in human‐induced pluripotent stem cell (hiPSC)‐derived neurons after treatment of the cells with 40 nM (33 µg/L) of VCR with or without 5 µM (2.525 mg/L) of DIP: 0 µM in black, and 5 µM (2.525 mg/L) in blue (c). The p values test if ∆Vmax differs due to the presence or absence of dipyridamole using the t‐test (c). The circles and whiskers represent the median and range of the data. The ΔVmax represents the change in Vmax due to the presence of DIP. Intracellular concentration of DIP was determined in hiPSC‐derived neurons after treating the cells with 5 µM of DIP with or without 40 nM (33 µg/L) of VCR: 0 nM in black, and 40 nM (33 µg/L) in blue. The circles and whiskers represent the median and range of the data (d). The p values test whether ∆Vmax due to the presence of vincristine is different from 0 using the t‐test (d). (e) Paw withdrawal responses expressed as percentage were determined using von Frey filaments at baseline, day 7, day 10 and day 14 in mice receiving one of the following treatments: saline and vehicle of DIP; saline and DIP; VCR and vehicle of DIP; and VCR and DIP. (f) Cross‐sectional and cumulative proportion (CPN) of mice positive for neurotoxicity in each treatment group is depicted. Positive neurotoxicity was defined as values higher than 95th percentile of the von Frey measurements. (g) NSG mice were inoculated with human CEM leukemia cells. After engraftment, the mice were treated once a week with saline and vehicle (controls), VCR, DIP, or the two drugs together. Percentage survival was determined for each group of mice. Mice receiving VCR and vehicle showed a significantly prolonged survival compared with controls (saline + vehicle as well as saline + dipyridamole; p value <0.001). The addition of DIP to VCR significantly prolonged the survival of mice compared with mice treated with only VCR and vehicle (p value <0.001)
In vivo PK studies
PK studies verified that mice had clinically relevant plasma drug concentrations with doses of VCR 0.1 mg/kg intraperitoneal and DIP 200 mg/kg p.o. These studies documented total plasma concentrations of VCR and DIP of up to 10 ng/ml (12.1 nM) and 5000 ng/ml (9.9 µM), respectively. Clinically, studies have reported plasma concentrations up to 31.2 ng/ml (37.8 nM) for VCR 25 and 6000 ng/ml (11.9 µM) for DIP. 26 There was no significant difference in VCR systemic clearance in mice treated with VCR and DIP versus VCR alone (4.88 L/hr/kg vs. 5.96 L/hr/kg, respectively, p = 0.153; Figure S6a). Likewise, concomitant VCR administration did not significantly affect the apparent oral clearance or plasma exposure of oral DIP in mice (p = 0.950; Figure S6b). Overall, these data indicate that concomitant DIP 200 mg/kg p.o. daily does not affect VCR clearance or plasma exposure in mice.
In vivo effects of DIP on VCR‐induced peripheral neuropathy and on VCR’s antileukemic effects in a mouse xenograft model
To determine whether DIP alters VCR‐induced peripheral neuropathy in mice, we used von Frey filaments. 27 We compared the percentage paw withdrawal response as a measure of neurotoxicity in mice among four different treatment groups of mice, using a mixed effect model with baseline response, filament strength, and measurement timepoint as covariates in the analysis. We observed a significant increase in paw withdrawal responses of mice receiving VCR alone. Importantly, the administration of DIP with VCR significantly lowered this response (Figure 5e, Figure S6c–f, and Table S4), indicating in vivo mitigation of the neurotoxic effects of VCR. These results were confirmed by a significantly higher cumulative incidence of cutaneous pain sensitivity (Figure 5f) and a significant decrease in mechanical sensitivity threshold in mice receiving VCR alone compared with mice treated with VCR and DIP (Figure S6g), consistent with neuroprotective effects of DIP observed in the hiPSC‐derived neurons. Using NSG mice inoculated with human CEM leukemia cells, we documented that DIP significantly prolonged the survival of mice treated with VCR (Figure 5g) consistent with the synergistic effects of DIP and VCR seen in leukemia cells ex vivo.
DISCUSSION
The sensitivity of primary leukemia cells to antileukemic agents, measured in ex vivo assays as LC50, is a good predictor of in vivo drug response in patients with ALL. 13 , 28 , 29 In a genome‐wide interrogation study based on the sensitivity of primary ALL cells to VCR, we discovered that low expression of NHP2L1 is associated with increased sensitivity to VCR and this was validated by manipulating NHP2L1 in human ALL cell lines. VCR exerts its antileukemic effects by interfering with microtubule formation and mitotic spindle dynamics, 30 , 31 , 32 thereby inducing mitotic arrest and apoptosis. 33 Low NHP2L1 is also associated with mitotic arrest, 24 consistent with low expression of NHP2L1 sensitizing ALL cells to VCR, as both increase accumulation of mitotic cells. Previous studies have shown synergistic effects between antimitotic drugs, such as VCR and proteins involved in mitosis. 34 , 35 , 36 We previously identified US Food and Drug Administration (FDA)‐approved small molecules that are inhibitors of NHP2L1 function, and here we documented synergy between VCR and several of these NHP2L1 inhibitors, including tazarotene, quetiapine fumarate, and dipyridamole. Tazarotene is an acetylenic retinoid used for the topical treatment of acne and psoriasis. 37 Quetiapine fumarate is an antipsychotic used in the treatment of depression, schizophrenia, and bipolar disorder. 38 Dipyridamole inhibits platelet aggregation and thrombus formation and is widely used clinically as an antiplatelet drug with no major side effects. 39
We further documented that DIP decreases the cellular level of SORORIN, a protein that requires normal spliceosome (NHP2L1) function to maintain adequate cellular levels to regulate sister chromatid cohesion during mitosis. 40 Mitotic arrest could be caused by either defects in mitotic spindle dynamics caused by VCR and (or) defects in sister chromatid cohesion following reduction of SORORIN protein level. 24 , 41 , 42 We also observed increased intracellular concentrations of VCR in leukemia cells when ALL cells were treated concomitantly with DIP. VCR is transported out of leukemia cells mainly by the multidrug efflux pump P‐glycoprotein (P‐gp; encoded by ABCB1). 43 , 44 DIP is a well‐known inhibitor of P‐gp 45 and DIP has been shown to increase the intracellular concentration of P‐gp substrates 45 , 46 , 47 consistent with the increased level of VCR in leukemia cells treated with DIP. DIP did not increase the intracellular concentration of VCR in hiPSC neurons, perhaps due to the low constitutive expression of ABCB1 in hiPSC‐derived neurons. We have not assessed the impact of differences in plasma proteins, which is unlikely to be substantial given the modest (around 75%) plasma protein binding of VCR. 48
Low expression or knockdown of NHP2L1 increased sensitivity of leukemic cells to VCR, whereas knockdown of NHP2L1 did not have any effect on VCR‐mediated toxicity in iPSC‐derived neurons. The differing effect of NHP2L1 on ALL cells versus hiPSC‐derived neurons is likely also because neurons are post‐mitotic cells and NHP2L1 acts primarily on mitotic cells. Thus, there are at least two potential reasons why DIP has different effects on VCR in leukemia cells versus hiPSC‐derived neurons: (1) DIP increases intracellular VCR by inhibiting the efflux transporter (ABCB1) in ALL cells but not in iPSC‐derived neurons, and (2) the inhibition of NHP2L1 has different consequences in cells undergoing mitosis (ALL) versus post‐mitotic cells, such as neurons. We cannot exclude the possibility of other mechanisms by which DIP differentially alters VCR sensitivity in leukemia cells and hiPSC‐derived neurons, as it is known to have several additional properties (anti‐oxidant and anti‐inflammatory). 49 , 50
We have previously identified an inherited variant in CEP72, which encodes a protein essential for microtubule formation that increases the risk and severity of VCR‐induced acute neuropathy, 23 providing a strategy for identifying patients at highest risk of this common toxicity. Our present discovery of small molecules capable of increasing the sensitivity of ALL cells to VCR and prolonging survival of mice bearing human leukemia cells, whereas simultaneously protecting neurons from VCR‐induced neurotoxicity constitutes another step toward improving the efficacy while mitigating the neurotoxicity of VCR.
In conclusion, we have shown that DIP, a small molecule inhibitor of NHP2L1, synergizes with VCR in leukemic cells while mitigating VCR‐induced neurotoxicity, opening the possibility of improving the therapeutic index of this widely prescribed anticancer agent, which will require a prospective clinical trial to validate in patients.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
B.D., W.E.E., C.W., M.E.D., J.C.P., D.E., W.L., W.Y., Y.F., D.P., C.C., S.M.D., W.Z., E.J.B., K.R.C., S.W.P., L.L., B.B.R., R.J.A., J.A.B., D.C.F., L.J.J., K.K.N., T.C., S.S.Z., S.J., C.H.P., M.V.R. wrote the manuscript; B.D. and W.E.E. designed the research. B.D., C.W., D.E., W.L., S.M.D., and I.I. performed the research. B.D., C.W., J.C.P., D.E., W.Y., Y.F., D.P., C.C., B.B.F., and L.J.J. analyzed the data. B.D., W.E.E., M.E.D., M.V.R., C.H.P., S.J., R.P., M.L.D.B., S.S.Z., and T.C. supervised the research.
Supporting information
Fig S1‐S6
Supplementary Material
Tables S1‐S4
ACKNOWLEDGMENTS
The authors thank Dr. Jan‐Michael Peters (Research Institute of Molecular Pathology, Vienna) for providing SORORIN antibody. We thank Siamac Sahely, John Stukenborg, Hannah Williams, Sean Savage, Michael Anderson, Chandra Savage, Kerry Heath, Jack Carpenter, Cara Goodrum, Madoka Inoue, Jennifer McCommon, Clemothy Bell and Yingzhe Wang, for their technical assistance with experiments. We thank Jerry Harris, Elizabeth Stevens, and Joshua Stokes for preparation of the figures. We thank the Shared Research Resources of the St. Jude Comprehensive Cancer Center for imaging and gene‐expression profiling, for pharmacokinetics, for preclinical pharmacokinetics, and the St. Jude Children’s Research Hospital Animal Resource Center for husbandry and the Cellular Screening Center and the Genomics Cores at the University of Chicago for use of the ImageXpress and gene array processing, respectively.
Funding information
This work was supported, in part, by National Institutes of Health (NIH) Grants R37 CA36401 (W.E.E.); U01 GM92666 (W.E.E.); R21 CA222764 (M.E.D.), R01 MH097742 (S.S.Z.), R01 DC012833 (S.S.Z.), St. Jude Comprehensive Cancer Center Support Grant CA21765 from the National Cancer Institute; University of Chicago Comprehensive Cancer Center Women’s Board (M.E.D.), University of Chicago Cancer Center Support Grant CA014599, and by ALSAC.
REFERENCES
- 1. Calcabrini C, Maffei F, Turrini E, Fimognari C. Sulforaphane potentiates anticancer effects of doxorubicin and cisplatin and mitigates their toxic effects. Front Pharmacol. 2020;11:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alam M, Yadav RK, Minj E, Tiwari A, Mehan S. Exploring molecular approaches in Amyotrophic lateral sclerosis: drug targets from clinical and pre‐clinical findings [published online ahead of print April 27, 2020). Curr Molec Pharmacol. https://doi.org/ 10.2174/1566524020666200427214356. [DOI] [PubMed] [Google Scholar]
- 3. Bradley WG, Lassman LP, Pearce GW, Walton JN. The neuromyopathy of vincristine in man. Clinical, electrophysiological and pathological studies. J Neurol Sci. 1970;10:107‐131. [DOI] [PubMed] [Google Scholar]
- 4. Gidding CEM, Meeuwsen‐de Boer GJ, Koopmans P, Uges DRA, Kamps WA, de Graaf SSN. Vincristine pharmacokinetics after repetitive dosing in children. Cancer Chemother. Pharmacol. 1999;44:203‐209. [DOI] [PubMed] [Google Scholar]
- 5. Bjornard KL, Gilchrist LS, Inaba H, et al. Peripheral neuropathy in children and adolescents treated for cancer. Lancet Child Adolesc Health. 2018;2:744‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luserna G, di Rorà A, Martinelli G, Simonetti G. The balance between mitotic death and mitotic slippage in acute leukemia: a new therapeutic window? J Hematol Oncol. 2019;12:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Iwata R, Casimir P, Vanderhaeghen P. Mitochondrial dynamics in postmitotic cells regulate neurogenesis. Science (New York, NY). 2020;369:858‐862. [DOI] [PubMed] [Google Scholar]
- 8. Diouf B, Lin W, Goktug A, et al. Alteration of RNA splicing by small‐molecule inhibitors of the interaction between NHP2L1 and U4. SLAS Discov Adv Life Sci R & D. 2018;23:164‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu S, Li P, Dybkov O, et al. Binding of the human Prp31 Nop domain to a composite RNA‐protein platform in U4 snRNP. Science. 2007;316:115–120. [DOI] [PubMed] [Google Scholar]
- 10. Falb M, Amata I, Gabel F, Simon B, Carlomagno T. Structure of the K‐turn U4 RNA: a combined NMR and SANS study. Nucleic Acids Res. 2010;38:6274‐6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vidovic I, Nottrott S, Hartmuth K, Lührmann R, Ficner R. Crystal structure of the spliceosomal 15.5kD protein bound to a U4 snRNA fragment. Mol. Cell. 2000;6:1331‐1342. [DOI] [PubMed] [Google Scholar]
- 12. Neumann B, Walter T, Hériché J‐K, et al. Phenotypic profiling of the human genome by time‐lapse microscopy reveals cell division genes. Nature. 2010;464:721‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holleman A, Cheok MH, den Boer ML, et al. Gene‐expression patterns in drug‐resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med. 2004;351:533‐542. [DOI] [PubMed] [Google Scholar]
- 14. Stabley DL, Holbrook J, Harris AW, et al. Establishing a reference dataset for the authentication of spinal muscular atrophy cell lines using STR profiling and digital PCR. Neuromusc Dis. 2017;27:439‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morrison G, Liu C, Wing C, Delaney SM, Zhang W, Dolan ME. Evaluation of inter‐batch differences in stem‐cell derived neurons. Stem Cell Res. 2016;16:140‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Irizarry RA. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England). 2003;4:249‐264. [DOI] [PubMed] [Google Scholar]
- 17. Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995;47:331‐385. [PubMed] [Google Scholar]
- 18. Minto CF, Schnider TW, Short TG, Gregg KM, Gentilini A, Shafer SL. Response surface model for anesthetic drug interactions. Anesthesiology. 2000;92:1603‐1616. [DOI] [PubMed] [Google Scholar]
- 19. Jonker DM, Visser SA, van der Graaf PH, Voskuyl RA, Danhof M. Towards a mechanism‐based analysis of pharmacodynamic drug‐drug interactions in vivo. Pharmacol Ther. 2005;106:1‐18. [DOI] [PubMed] [Google Scholar]
- 20. Abdelfattah N, Rajamanickam S, Panneerdoss S, et al. MiR‐584‐5p potentiates vincristine and radiation response by inducing spindle defects and DNA damage in medulloblastoma. Nat Commun. 2018;9:4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wheeler HE, Wing C, Delaney SM, Komatsu M, Dolan ME. Modeling chemotherapeutic neurotoxicity with human induced pluripotent stem cell‐derived neuronal cells. PLoS One. 2015;10:e0118020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wing C, Komatsu M, Delaney SM, Krause M, Wheeler HE, Dolan ME. Application of stem cell derived neuronal cells to evaluate neurotoxic chemotherapy. Stem Cell Res. 2017;22:79‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diouf B, Crews KR, Lew G, et al. Association of an inherited genetic variant with vincristine‐related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA. 2015;313:815‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sundaramoorthy S, Vázquez‐Novelle MD, Lekomtsev S, Howell M, Petronczki M. Functional genomics identifies a requirement of pre‐mRNA splicing factors for sister chromatid cohesion. EMBO J. 2014;33:2623‐2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moore AS, Norris R, Price G, et al. Vincristine pharmacodynamics and pharmacogenetics in children with cancer: a limited‐sampling, population modelling approach. J Paediatr Child Health. 2011;47:875‐882. [DOI] [PubMed] [Google Scholar]
- 26. Willson JK, Fischer PH, Tutsch K, et al. Phase I clinical trial of a combination of dipyridamole and acivicin based upon inhibition of nucleoside salvage. Can. Res. 1988;48:5585‐5590. [PubMed] [Google Scholar]
- 27. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55‐63. [DOI] [PubMed] [Google Scholar]
- 28. Hongo T, Yajima S, Sakurai M, Horikoshi Y, Hanada R. In vitro drug sensitivity testing can predict induction failure and early relapse of childhood acute lymphoblastic leukemia. Blood. 1997;89:2959‐2965. [PubMed] [Google Scholar]
- 29. Kaspers GJL, Veerman AJP, Pieters R, et al. In vitro cellular drug resistance and prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Blood. 1997;90:2723‐2729. [PubMed] [Google Scholar]
- 30. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253‐265. [DOI] [PubMed] [Google Scholar]
- 31. Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA. 1993;90:9552‐9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Toh HC, Sun L, Koh CH, Aw SE. Vinorelbine induces apoptosis and caspase‐3 (CPP32) expression in leukemia and lymphoma cells: a comparison with vincristine. Leukemia Lymphoma. 1998;31:195‐208. [DOI] [PubMed] [Google Scholar]
- 33. Blajeski AL, Phan VA, Kottke TJ, Kaufmann SH. G(1) and G(2) cell‐cycle arrest following microtubule depolymerization in human breast cancer cells. J Clin Investig. 2002;110:91‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hugle M, Belz K, Fulda S. Identification of synthetic lethality of PLK1 inhibition and microtubule‐destabilizing drugs. Cell Death Differ. 2015;22:1946‐1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ikezoe T, Yang J, Nishioka C, et al. A novel treatment strategy targeting polo‐like kinase 1 in hematological malignancies. Leukemia. 2009;23:1564‐1576. [DOI] [PubMed] [Google Scholar]
- 36. Tannous BA, Kerami M, Van der Stoop PM, et al. Effects of the selective MPS1 inhibitor MPS1‐IN‐3 on glioblastoma sensitivity to antimitotic drugs. J. Natl Cancer Inst. 2013;105:1322‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tang‐Liu DD, Matsumoto RM, Usansky JI. Clinical pharmacokinetics and drug metabolism of tazarotene: a novel topical treatment for acne and psoriasis. Clin Pharmacokinet. 1999;37:273‐287. [DOI] [PubMed] [Google Scholar]
- 38. Chang K, DelBello M, Garrett A, et al. Neurofunctional correlates of response to quetiapine in adolescents with bipolar depression. J Child Adolesc Psychopharmacol. 2018;28:379‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gresele P, Momi S, Falcinelli E. Anti‐platelet therapy: phosphodiesterase inhibitors. Br J Clin Pharmacol. 2011;72:634‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Valcárcel J, Malumbres M. Splicing together sister chromatids. The EMBO J. 2014;33:2601‐2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oka Y, Varmark H, Vitting‐Seerup K, et al. UBL5 is essential for pre‐mRNA splicing and sister chromatid cohesion in human cells. EMBO Rep. 2014;15:956‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Watrin E, Demidova M, Watrin T, Hu Z, Prigent C. Sororin pre‐mRNA splicing is required for proper sister chromatid cohesion in human cells. EMBO Rep. 2014;15:948‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Safa AR, Glover CJ, Meyers MB, Biedler JL, Felsted RL. Vinblastine photoaffinity labeling of a high molecular weight surface membrane glycoprotein specific for multidrug‐resistant cells. J Biol Chem. 1986;261:6137‐6140. [PubMed] [Google Scholar]
- 44. Cornwell MM, Safa AR, Felsted RL, Gottesman MM, Pastan I. Membrane vesicles from multidrug‐resistant human cancer cells contain a specific 150‐ to 170‐kDa protein detected by photoaffinity labeling. Proc Natl Acad Sci USA. 1986;83:3847‐3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verstuyft C. Dipyridamole enhances digoxin bioavailability via P‐glycoprotein inhibition. Clin Pharmacol Ther. 2003;73:51‐60. [DOI] [PubMed] [Google Scholar]
- 46. Hirose M, Takeda E, Ninomiya T, Kuroda Y, Miyao M. Synergistic inhibitory effects of dipyridamole and vincristine on the growth of human leukaemia and lymphoma cell lines. Br J Cancer. 1987;56:413‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shalinsky DR, Slovak ML, Howell SB. Modulation of vinblastine sensitivity by dipyridamole in multidrug resistant fibrosarcoma cells lacking mdr1 expression. Br J Cancer. 1991;64:705‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Donigian DW, Owellen RJ. Interaction of vinblastine, vincristine and colchicine with serum proteins. Biochem Pharmacol. 1973;22:2113‐2119. [DOI] [PubMed] [Google Scholar]
- 49. Ciacciarelli M, Zerbinati C, Violi F, Iuliano L. Dipyridamole: a drug with unrecognized antioxidant activity. Curr Top Med Chem. 2015;15:822‐829. [DOI] [PubMed] [Google Scholar]
- 50. Balakumar P, Nyo YH, Renushia R, et al. Classical and pleiotropic actions of dipyridamole: not enough light to illuminate the dark tunnel? Pharmacol Res. 2014;87:144‐150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S6
Supplementary Material
Tables S1‐S4