

Abstract
Apomorphine is an on‐demand treatment of “OFF” episodes in patients with Parkinson’s disease (PD). A joint parent‐metabolite population pharmacokinetic (PK) model characterized apomorphine and apomorphine‐sulfate following administration of apomorphine sublingual film (APL) and two formulations of subcutaneous apomorphine. Overall, 2485 samples from 87 healthy subjects and 71 patients with PD and “OFF” episodes were analyzed using nonlinear mixed‐effects modeling. Apomorphine PK was adequately described by a two‐compartment model with first‐order transit absorption via both routes of administration and first‐order metabolism to apomorphine‐sulfate with one‐compartment disposition and first‐order elimination. Bioavailability of apomorphine sublingual film was ~ 18% relative to subcutaneous apomorphine. Among covariates tested, only body weight had a large effect on apomorphine exposure (maximum plasma concentration and area under the concentration–time curve [AUC0–∞]), with greater weight resulting in lower exposure. Model‐predicted apomorphine exposure was similar between apomorphine sublingual film 30 mg and subcutaneous apomorphine 5 mg (median AUC0–24, 66.7 ng•h/mL, geometric mean ratio of 0.99; 90% confidence interval [CI], 0.96−1.03) and was comparable between apomorphine sublingual film 35 mg and subcutaneous apomorphine 6 mg (median AUC0–24, 75.4 and 80.0 ng•h/mL, respectively; geometric mean ratio of 0.94; 90% CI, 0.90−0.97) administered every 2 h for a maximum of 5 doses per day. In a typical patient with PD, predicted apomorphine exposure increased with increasing doses of apomorphine sublingual film; however, the increase was less than dose proportional. Similar apomorphine exposure was predicted in patients with mild renal impairment versus normal renal function. PK properties of apomorphine sublingual film support its administration for a wide range of patients with PD and “OFF” episodes, regardless of demographic and clinical characteristics.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Pharmacokinetic (PK) properties of apomorphine sublingual film (APL‐130277; APL), an on‐demand treatment for “OFF” episodes in patients with Parkinson’s disease (PD), have not been previously described comprehensively.
WHAT QUESTION DID THIS STUDY ADDRESS?
A population PK model of apomorphine and its inactive metabolite, apomorphine sulfate, was developed to characterize PK properties of APL and two formulations of subcutaneous apomorphine.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Compared with subcutaneous apomorphine, APL has a relative bioavailability of ~ 18%. Increase in apomorphine exposure (peak plasma concentration and area under the concentration–time curve [AUC0–∞]) was less than dose proportional with no obvious difference in elimination half‐life across the dose range studied. Model‐predicted apomorphine exposure at higher doses of APL (30–35 mg) was comparable to subcutaneous apomorphine (5–6 mg) across a range of patient characteristics tested as covariates, including mild renal impairment; greater body weight resulted in lower exposure.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Sublingual apomorphine PK properties support administration of APL as an on‐demand treatment for a wide range of patients with PD and “OFF” episodes.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder worldwide. 1 Advancing age is the greatest risk factor for this chronic and progressive disease 1 ; therefore, cases of PD in the United States continue to rise and are expected to reach ~ 1.2 million by 2030. 2 PD is characterized by a loss of dopaminergic neurons in the brain, and the use of carbidopa/levodopa, which crosses the blood–brain barrier where it is converted to dopamine, is the mainstay of therapy. 1 , 3 , 4 However, after 10 years of treatment with carbidopa/levodopa, nearly all patients develop motor fluctuations 3 , 5 , 6 including “OFF” episodes, defined as periods in which treated symptoms of PD re‐emerge or worsen. 3 Fluctuations in blood levodopa levels during carbidopa/levodopa treatment can result in reduced brain bioavailability, particularly in patients with chronic PD, and likely contribute to “OFF” episodes. 4
Apomorphine is a nonergoline dopamine agonist with strong affinity for the D1 and D2 receptors. 7 , 8 Subcutaneous apomorphine (APOKYN®; US WorldMeds, LLC, Louisville, KY, available in the United States, and APO‐go®; Britannia Pharmaceuticals, Reading, Berkshire, UK, available in Europe) are available as on‐demand treatments of “OFF” episodes 9 , 10 and have been shown to be highly effective in this context. 7 , 8 , 11 , 12 However, use of subcutaneous apomorphine has been limited for various reasons, including complexity of assembly, injection‐site reactions, and systemic adverse events, such as nausea/vomiting and dyskinesias. 12 , 13 , 14
Orally administered apomorphine has limited clinical utility due to extensive first‐pass hepatic metabolism and poor gut absorption. 15 , 16 The bioavailability of oral apomorphine is less than 4% in patients with PD 17 ; therefore, different routes of administration, including sublingual administration, have been investigated as alternatives for apomorphine delivery. 18 The sublingual tissues have a rich blood supply that can support rapid absorption and onset of action. 15
To address the aforementioned limitations, apomorphine sublingual film (KYNMOBI®; Sunovion Pharmaceuticals Inc., Marlborough, MA, USA) was developed and is now approved as an on‐demand treatment of “OFF” episodes for patients with PD. 19 Apomorphine sublingual film (APL) consists of two layers: (1) a drug layer, designed for stability, rapid diffusion, and maximal bioavailability, and (2) a buffer layer, designed to neutralize acid generation and enhance drug permeability. 15 In a double‐blind, placebo‐controlled, randomized phase III trial, patients who received apomorphine sublingual film at doses of 10–35 mg experienced significantly greater improvement in motor function at 30 min postdose at week 12, as assessed by part III (Motor Examination) of the Movement Disorder Society Unified Parkinson’s Disease Rating Scale, compared with those who received placebo (−11.1 vs. −3.5, respectively; least squares mean difference, −7.6, p = 0.0002), and the drug was generally safe and well‐tolerated. 20
We developed a population pharmacokinetic (PopPK) model to characterize the PK properties of apomorphine and its major inactive metabolite, apomorphine sulfate, after single and multiple doses of apomorphine sublingual film and subcutaneous apomorphine.
METHODS
Data source
Pooled PK data were obtained from nine clinical studies (5 phase I, 3 phase II, and 1 phase III) of apomorphine sublingual film (Table S1). The studies that contributed to the PK data used for this analysis were approved by institutional review boards at each study site, and written informed consent was obtained from all individuals before study initiation. Each study was conducted in accordance with the International Council for Harmonization Guideline for Good Clinical Practice and the Declaration of Helsinki. Two commercially available preparations of subcutaneous apomorphine were used in studies CTH‐103, CTH‐200, and CTH‐203: APOKYN 9 (US WorldMeds, LLC; available in the United States) and APO‐go 10 (Britannia Pharmaceuticals; available in Europe).
Data analysis platform
PK analyses were performed using nonlinear mixed‐effects modeling methodology as implemented in NONMEM software (version 7.3; ICON Development Solutions), and data manipulations, postprocessing, and graphical analysis were performed using R (version 3.1.2 or higher; R Foundation for Statistical Computing). The first‐order conditional estimation with eta‐epsilon interaction (FOCEI) method in NONMEM was used during all stages of model development.
Model development
Model development followed these general sequential steps: exploratory graphical data analyses; base model development (structural and residual error); covariate model development; model optimization and finalization; and model evaluation and/or validation.
Base model development
Initially, a one‐compartment model with first‐order absorption and first‐order elimination was used to characterize the apomorphine concentration‐time profile following apomorphine sublingual film and apomorphine subcutaneous injection administration. If multiple distribution phases were observed, the one‐compartment model was replaced starting with a two‐compartment model. The initial model was parameterized in terms of clearance (CL/F) and distribution volume (V/F) parameters (e.g., ADVAN2 or ADVAN4 subroutine in PREDPP in NONMEM software).
Following identification of the compartmental model for apomorphine, apomorphine‐sulfate metabolite data were incorporated with a single disposition compartment with all apomorphine elimination occurring via conversion to the sulfate metabolite. Additionally, to account for the higher observed apomorphine‐sulfate concentrations following sublingual administration, a direct absorption pathway for apomorphine‐sulfate was incorporated, representing oral absorption of the swallowed portion of the sublingual dose (F5 ). Bioavailability via the sublingual route of administration (F 1) was parameterized as the product of the fraction of the sublingual dose not swallowed, and thus available for absorption via this route, and the estimated bioavailability relative to the subcutaneous route. Bioavailability via the subcutaneous route (F 4) was assumed to be 100%.
Standard goodness‐of‐fit plots were used to assess potential lack‐of‐fit for the initial model. Based on the goodness‐of‐fit diagnostics, additional model complexities, such as additional distribution compartments, alternate absorption pathways (e.g., transit absorption compartment), or nonlinearities in key model parameters were considered. Transit absorption compartments were evaluated separately for each route of administration (e.g., sublingual and subcutaneous) in order to adequately describe the absorption profile. In addition, pertinent covariates (e.g., dose) were evaluated at the base model stage to stabilize the base model and to facilitate easier identification of the full model in subsequent steps. Specifically, dose‐dependencies in bioavailability were evaluated if indicated based on exploratory assessment of observed peak concentrations versus dose. Dose‐dependencies were described using power models as the reference approach with saturable functions (e.g., maximal effect [Emax]) used in the event power models inadequately described the observed data. The FOCEI method in NONMEM was utilized in the model development.
Random effect model development
Interindividual variability (IIV) of the PK parameters was incorporated using a log normal random effects model of the form:
| (1) |
where θi is the individual value of the parameter (e.g., CL/F) for the ith patient, θ TV is the typical value model parameter, and patient specific random variable (ηi ) denotes the IIV accounting for the ith individual’s deviation from the typical value. The ηi’s were assumed to have a normal distribution with a zero mean and variance ω 2. The approximate percent coefficient of variation (%CV) was reported as:
| (2) |
The multivariate vector of interindividual random effects (across parameters within each individual) has variance‐covariance matrix Ω IIV . A diagonal Ω was estimated initially with off‐diagonal elements considered, as needed.
Residual variability, a composite measure of assay error, dose/sample time collection errors, model misspecification, and any other unexplained variability within a patient, was initially modeled using the log‐transformed error model:
| (3) |
where Yij denoted the observed concentration for the ith individual at time tj , Cij denoted the corresponding predicted concentration based on the PK model, and ε ij denoted the intra‐individual (residual) random effect, which is assumed to have a normal distribution with a zero mean and variance σ 2.
A complete battery of diagnostic plots was generated to evaluate the adequacy of the base model fit. Plots of population weighted residuals (WRES) versus TIME, individual weighted residuals (IWRES) versus TIME, and conditional weighted residuals (CWRES) versus TIME were evaluated for random scatter around the zero line. The residual (WRES, IWRES, and CWRES) plots were also used to identify potential outliers. The model resulting from the previous steps is considered the base PK model.
Covariate model development
Clinical judgment and mechanistic plausibility were used to determine which covariates were tested and on which parameters. Table 1 provides a list of the covariates that were considered for the PopPK model. Several intrinsic and extrinsic patient factors were planned for evaluation in the covariate model development. Typical baseline demographic characteristics, including body weight, sex, and patient status (patients with PD vs. healthy subjects), were evaluated on CL/F and V/F. Additional covariates included on CL/F were creatinine clearance, biomarkers of hepatic function, and concomitant medications. Because age and race were highly correlated with patient status, they were not tested as covariates as planned a priori. Although some degree of correlation exists between other covariates (e.g., body weight and patient status; and body weight and sex), this was judged to be modest and these covariates were both tested on model parameters.
TABLE 1.
Covariates evaluated in the population pharmacokinetic analysis
| Parameter | Covariates |
|---|---|
| Absorption rate parameters such as k a | Position under tongue (drug strip facing up or down), contact time for sublingual administration, concomitant antiemetic use, formulation, a population (healthy subject vs. patient with PD and “OFF” episodes), age, and race |
| F | Concomitant antiemetic use, formulation, b population (healthy subject vs patient with PD and “OFF” episodes), age, and race |
| CL/F | Baseline body weight, sex, race, creatinine clearance, aspartate aminotransferase, alanine aminotransferase, albumin, and total bilirubin |
| V/F | Baseline body weight, sex, and race |
Abbreviations: CL/F, apparent drug clearance; F, bioavailability; k a, absorption rate constant; PD, Parkinson’s disease; V/F, apparent volume of distribution.
Two formulations of apomorphine sublingual film, an exploratory formulation (K) and the commercial formulation (O), were used in the studies that contributed to the population pharmacokinetic model.
Two formulations of subcutaneous apomorphine, APOKYN and APO‐go, were used in certain studies that contributed to the population pharmacokinetic model.
The relationship between continuous covariates and the typical value of PK parameters was described using power models:
| (4) |
where θ REF and θx are the fixed‐effect parameters and x REF is a reference value of the covariate xij . In the context of this analysis, the reference values represent the approximate median of the population. The relationship between categorical covariates (xij ) and the typical value of PK parameters was modeled as:
| (5) |
or as:
| (6) |
where θ REF and θx are fixed‐effect parameters and xij is the indicator variable, which is equal to one or zero dependent on the category of the covariates. In Equation 5, the lower bound values for θx were constrained to be negative, such that PK parameters will always be positive. In the parameterization of Equation 6, θx is the log of the fractional change in the typical value for a specific categorical covariate. This parameterization was selected to prevent negative PK parameter values in future simulation activities.
All covariates were initially included simultaneously in a full model. Due to overparameterization, the full model procedure was unsuccessful and a stepwise forward selection procedure was used to identify a stable working full model. The covariate‐parameter relationship, which had the largest decrease in objective function value (OFV) and met the inclusion criteria of change in OFV (δOFV) greater than 3.8 (p < 0.05) was included into the working full model. The stepwise forward selection process was repeated until all covariate‐parameter relationships met the inclusion criteria. Once a working full model was identified, a complete battery of diagnostic plots was generated. Plots of the individual random effect values versus covariate values were generated in order to identify any covariate effects that have not been properly accounted for. In addition, box‐plots of the η values versus dose and study were generated to evaluate dose invariance and adequacy of pooling studies for this analysis, respectively.
Final model development
The final PopPK model was developed using the backward elimination procedure. The working full model was reduced to the final parsimonious model by a stepwise backward elimination procedure. The covariate‐parameter relationship that had the lowest change in OFV and did not meet the inclusion criteria of δOFV less than 10.8 (p < 0.001) was eliminated and the stepwise backward elimination procedure was repeated until all covariate‐parameters met the inclusion criteria.
Assessment of model predictive performance (validation)
The predictive performance of the final PopPK model was evaluated using visual predictive checks. Model parameters were fixed to their estimated values from the final model run and used to simulate 1000 data sets of plasma concentrations replicating the design, population, dose regimens, sample sizes, and observed covariate distributions from the observed data set. The full joint covariate distribution in the observed data set was maintained for these simulations by using all of the observed data as the input for simulations (e.g., all healthy subjects and patients with PD were included). The observed 5th, 50th, and 95th percentiles of apomorphine and/or apomorphine sulfate concentration were stratified by time and compared with the 5th and 95th percentiles (90% confidence interval [CI]) of the 1000 simulated summary measures at corresponding percentiles (5th, 50th, and 90th) of the simulated data and plotted to provide a visual assessment of the predictive performance of the PopPK model.
Simulation
The final PopPK model was then used to simulate and predict apomorphine and apomorphine sulfate exposures under maximal dosing conditions for apomorphine sublingual film (30 or 35 mg every 2 h for a maximum of 5 doses per day) and/or subcutaneous apomorphine (5 or 6 mg every 2 h for a maximum of 5 doses per day), which were not observed in clinical trials. Covariate information from patients with PD in the data set was used to create a simulation data set of 1000 patients with PD by nonparametric bootstrapping of complete covariate vectors to preserve correlation between covariates. Parameter estimates from the final PopPK model were fixed to the estimated values and used to generate individual PK parameter estimates for each of the 1000 virtual patients with PD and to generate 1000 24‐h concentration–time profiles for apomorphine and apomorphine sulfate under each dosing condition. Median (50th percentile) and 90% prediction interval (PI; 5th and 95th percentiles) were calculated for maximum plasma concentration (Cmax) and area under the concentration–time curve from time 0 to 24 hours (AUC0–24). Additionally, geometric mean ratios (90% CI) of AUC0–24 were calculated for 30 mg sublingual apomorphine relative to 5 mg subcutaneous apomorphine and 35 mg sublingual apomorphine relative to 6 mg subcutaneous apomorphine to facilitate comparison of overall exposure following these dosing regimens.
RESULTS
Model population
The final PopPK model included data from 158 individuals (87 healthy subjects and 71 patients with PD and “OFF” episodes) with 2485 measurable PK samples of apomorphine, including 101 individuals (39 healthy subjects and 62 patients with PD and “OFF” episodes) with 1182 measurable PK samples of apomorphine sulfate, a major inactive metabolite of apomorphine (Table S1). Compared with healthy subjects, patients with PD and “OFF” episodes were older (mean age, 65.0 vs. 27.2 years, respectively), had greater body weight (mean, 80.0 vs. 65.8 kg, respectively), were predominantly White (94%) as compared with healthy subjects who were predominantly Asian (98%), and had slightly lower creatinine clearance levels (mean, 94 vs. 114 mL/min, respectively; Table 2). Among the doses evaluated, nearly half of the healthy subjects received 15 mg of apomorphine sublingual film (48%), whereas 24%, 13%, 28%, and 14% of patients with PD and “OFF” episodes received 10 mg, 15 mg, 20 mg, and 25 mg of apomorphine sublingual film, respectively (Table 2).
TABLE 2.
Baseline characteristics of healthy subjects and patients with PD in the population pharmacokinetic data set by study
| Characteristic | Healthy subjects (N = 87) | Patients with PD and “OFF” episodes (N = 71) |
|---|---|---|
| Age, years, mean (range) | 27.2 (21.0, 50.0) | 65.0 (45.6, 88.1) |
| Sex, n (%) | ||
| Male | 65 (74.7) | 49 (69.0) |
| Female | 22 (25.3) | 22 (31.0) |
| Body weight, kg, mean (range) | 65.8 (50.3–87.8) | 80.0 (50.7–146.1) |
| Race, n (%) | ||
| White | 0 | 67 (94.4) |
| Black | 0 | 3 (4.2) |
| Asian | 85 (97.7) | 0 |
| Other | 2 (2.3) | 1 (1.4) |
| Dose of apomorphine, mg, n (%) a | ||
| Subcutaneous apomorphine | ||
| 2 | 32 (24.4) | 0 |
| 3 | 12 (9.2) | 3 (3.0) |
| 4 | 0 | 2 (2.0) |
| 5 | 0 | 3 (3.0) |
| Sublingual apomorphine | ||
| 10 | 13 (9.9) | 24 (24.0) |
| 15 | 63 (48.1) | 13 (13.0) |
| 20 | 0 | 28 (28.0) |
| 25 | 11 (8.4) | 14 (14.0) |
| 30 | 0 | 7 (7.0) |
| 35 | 0 | 5 (5.0) |
| 50 | 0 | 1 (1.0) |
| Formulation of apomorphine sublingual film, n (%) a | ||
| K | 48 (48.5) | 9 (12.7) |
| O | 51 (51.5) | 62 (87.3) |
| Albumin, g/dL mean (range) | 4.5 (3.5, 5.3) | 4.4 (3.8, 4.9) |
| Alanine aminotransferase, IU/L, mean (range) | 23 (8, 50) | 20 (3, 64) |
| Aspartate aminotransferase, IU/L, mean (range) | 22 (12, 42) | 21 (10, 60) |
| Bilirubin, mg/dL mean (range) | 0.73 (0.28, 2.28) | 0.56 (0.20, 1.40) |
| Creatinine clearance, mL/min, mean (range) | 114 (86, 155) | 94 (52, 213) |
Abbreviations: K, exploratory formulation; O, commercial formulation; PD, Parkinson’s disease.
Doses of subcutaneous apomorphine: 2–5 mg; doses of apomorphine sublingual film: 10–35 mg, 50 mg. Percentages are based on a larger number of total individuals than listed because some individuals received more than one dosage strength and/or formulation of apomorphine sublingual film in certain studies.
PopPK analysis
Base model
The base PopPK model simultaneously fitted apomorphine and apomorphine sulfate concentration–time profiles using a two‐compartment model for apomorphine and a one‐compartment model for apomorphine sulfate, with first‐order transit absorption and first‐order elimination for both analytes. The transit absorption models characterized the slight delay in apomorphine absorption via both sublingual and subcutaneous administration. A proportional residual error model was used for both apomorphine and apomorphine sulfate. The base model included four structural covariates: (1) effect of apomorphine sublingual film dose on sublingual apomorphine bioavailability (F 1); (2) effect of apomorphine sublingual film contact time under the tongue on sublingual administration absorption rate constant (k a); (3) effect of body weight on apomorphine volume of distribution (V 2/F); and (4) effect of study CTH‐103 on V 2/F. Study CTH‐103 as a covariate was added to the base model to account for the very high peak concentrations, particularly following subcutaneous administration, observed in this study. Key model parameters (e.g., CL/F, V 2/F, effect of female sex on the volume of distribution for apomorphine sulfate [V 3/F]) differed by less than 15% with outlier observations excluded; therefore, these samples were not considered influential and were retained in the analysis.
Covariate model
Of the nine covariates included in the working full model following the stepwise forward addition procedure, five were found to be statistically significant in backward elimination and were retained in the final PopPK model, including the four structural covariates incorporated at the base model development stage. The effect of patient status (healthy subject vs. patient with PD and “OFF” episodes), subcutaneous apomorphine dose, subcutaneous apomorphine formulation, and sublingual apomorphine formulation (e.g., pre‐commercial formulation K vs. commercial formulation O) were not statistically significant covariates.
Final model
The concentration‐time profiles of apomorphine and apomorphine sulfate following sublingual and subcutaneous administration were adequately described in the final PopPK model by simultaneous two‐ and one‐compartment models, respectively, with first‐order transit absorption input and first‐order elimination (Figure 1). Parameter estimates for the final PopPK model are provided in Table 3. Mean (95% CI) typical apomorphine apparent systemic clearance and apparent central volume of distribution were 81 (41, 120) L/h and 438 (366, 510) L, respectively. Bioavailability of apomorphine sublingual film was estimated to be ~ 18% (fraction not swallowed and available for sublingual absorption × fraction absorbed relative to subcutaneous injection) relative to subcutaneous apomorphine for both healthy subjects and patients with PD and “OFF” episodes. Covariate effects of sublingual apomorphine dose on F 1, contact time of sublingual administration on k a, and body weight on V 2/F represent the power to which the ratio of the covariate value to the reference value (20 mg, 2 min, and 69.3 kg, respectively) is raised. Covariate effects for study CTH‐103 on V 2/F and female sex on V 3/F represent the proportional shift from the reference condition. The magnitude of the IIV identified for the rate constant for metabolism of apomorphine to apomorphine sulfate (k23), V 2/F, V 3/F, k a for sublingual administration, and F 1 ranged from 35.7 to 41.7 %CV. The residual unexplained variabilities for apomorphine and apomorphine sulfate were 44% and 58%, respectively.
FIGURE 1.
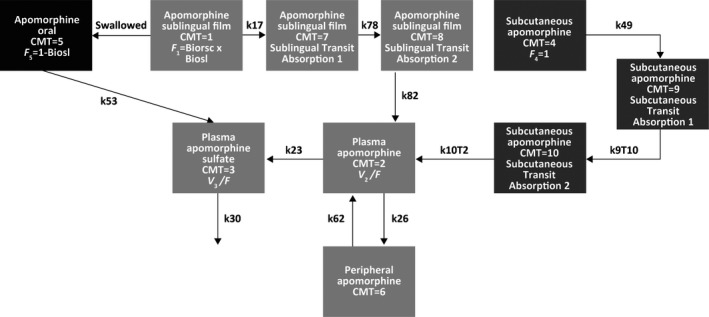
The final pharmacokinetic model was a two‐compartment model for apomorphine (CMT = 2 for plasma and CMT = 6 for peripheral) and a one‐compartment model for apomorphine sulfate (CMT = 3). Sublingual administration of apomorphine (CMT = 1) included two transit absorption compartments (CMT = 7 and CMT = 8), and subcutaneous administration of apomorphine (CMT = 4) included two transit compartments (CMT = 9 and CMT = 10). The fraction of the apomorphine sublingual film dose that was swallowed was modeled as a first‐order absorption process (k53) of apomorphine sulfate reaching the systemic circulation from the gastrointestinal tract (CMT = 5). The first‐order absorption rate constants for apomorphine sublingual film transit absorption were k17, k78, and k82, where k17 = k78 = k82. The first‐order absorption rate constants for subcutaneous apomorphine were k49, k9T10, and k10T2, where k49 = k9T10 = k10T2. The parameter k23 represented the first‐order metabolism of apomorphine to apomorphine sulfate, and k30 was the first‐order elimination of apomorphine sulfate. Finally, k26 was the distribution rate constant for apomorphine moving from the central to tissue compartments, and k62 was the distribution rate constant for apomorphine moving from the tissue to central compartments. Biorsc, bioavailability of apomorphine sublingual film relative to subcutaneous apomorphine; Biosl, fraction of apomorphine sublingual film dose not swallowed; CMT, compartment; F 1, relative bioavailability of apomorphine sublingual film; F 4, relative bioavailability of subcutaneous apomorphine; F 5, fraction of apomorphine sublingual film dose swallowed and absorbed as metabolite; V 2/F, volume of distribution for apomorphine; V 3/F, volume of distribution for apomorphine sulfate.
TABLE 3.
Parameter estimates for the final population pharmacokinetic model
| Parameter | Estimate | ASE | % RSE | 95% CI | Units |
|---|---|---|---|---|---|
| CL/F | 80.7 | 20.2 | 25.0 | (41.1–120) | L/h |
| V 2/F | 438 | 36.7 | 8.39 | (366–510) | L |
| V 3/F | 1.42 | 0.49 | 34.2 | (0.468–2.38) | L |
| k a for sublingual administration | 6.58 | 1.06 | 16.2 | (4.49–8.67) | h–1 |
| k a for subcutaneous administration | 17.6 | 1.48 | 8.40 | (14.7–20.5) | h–1 |
| k30 | 1.28 | 0.494 | 38.5 | (0.32–2.25) | h–1 |
| k a for apomorphine sulfate absorption from the gastrointestinal tract | 0.205 | 0.180 | 87.9 | (−0.15 to 0.55) | h–1 |
| Fraction not swallowed and available for sublingual absorption | 0.910 | 0.0533 | 5.86 | (0.805–1.01) | |
| Fraction absorbed relative to subcutaneous administration | 0.202 | 0.0258 | 12.8 | (0.152–0.253) | |
| k26 | 0.613 | 0.0859 | 14.0 | (0.445–0.781) | h–1 |
| k62 | 0.00480 | 0.000762 | 15.9 | (0.00330–0.00630) | h–1 |
| Dose of sublingual apomorphine on F 1 | −0.206 | 0.339 | 165 | (−0.870 to 0.459) | |
| Contact time under the tongue for sublingual film on k a for sublingual administration | –0.194 | 0.119 | 61.5 | (–0.428 to –0.0400) | |
| Body weight on V 2/F | 1.53 | 0.379 | 24.8 | (0.788–2.27) | |
| Study CTH−103 on V 2/F | −0.555 | 0.0476 | 8.59 | (–0.648 to −0.462) | |
| Female sex on V 3/F | –0.310 | 0.0663 | 21.4 | (–0.440 to –0.180) | |
| Residual variability | |||||
| Apomorphine | 44.3 | 1.45 | 3.26 | (41.5–47.2) | % |
| Apomorphine sulfate | 58.0 | 2.21 | 3.81 | (53.6–62.3) | % |
| Interindividual variability | |||||
| k23 | 35.9 | (16.6–47.9) | %CV | ||
| V 2/F | 41.7 | (27.5–52.1) | %CV | ||
| V 3/F | 40.3 | (27.9–49.7) | %CV | ||
| k a for sublingual administration | 35.7 | (24.7–41.1) | %CV | ||
| F 1 | 38.3 | (27.2–46.7) | %CV | ||
Abbreviations: % RSE, percent relative standard error; %CV, percent coefficient of variation; ASE, asymptotic standard error; CI, confidence interval; CL/F, apparent apomorphine clearance; F 1, bioavailability of apomorphine sublingual film; k23, first‐order rate constant for the metabolism of apomorphine to apomorphine sulfate and equal to CL/V 2; k26, distribution rate constant for apomorphine moving from central to tissue compartment; k30, elimination rate constant for apomorphine sulfate; k62, distribution rate constant for apomorphine moving from tissue to central compartment; k a, first‐order absorption rate constant; V 2/F, apparent volume of distribution for apomorphine; V 3/F, apparent volume of distribution for apomorphine sulfate.
Model evaluation
Inspection of goodness‐of‐fit and concordance plots suggested that the final PopPK model adequately described the plasma apomorphine and apomorphine sulfate concentration–time profiles after sublingual and subcutaneous apomorphine administration (Figures S1–S3). In addition, the predicted apomorphine and apomorphine sulfate concentration–time profiles adequately described the observed apomorphine and apomorphine sulfate exposure for all studies, formulations, and doses in a visual predictive check, which confirmed the predictive performance of the final PopPK model (Figure S2).
Influence of covariates on model‐predicted apomorphine exposure
The impact of covariates included in the final PopPK model on apomorphine exposure was investigated for a typical patient with PD (i.e., 65‐year‐old White man with a body weight of 78 kg who received a 20‐mg dose of apomorphine sublingual film with the film strip positioned drug side down, a contact time under the tongue of 3 min, and who did not receive concomitant antiemetic medication). Body weight was the only covariate with a large magnitude of effect on apomorphine exposure (Cmax) and AUC from time 0 to infinity (AUC0–∞). Greater body weight resulted in lower exposure to apomorphine. For example, Cmax and AUC0–∞ were 2.62 ng/mL and 6.77 ng•h/mL, respectively, for an individual who weighed 114 kg compared with 4.48 ng/mL and 10.60 ng•h/mL, respectively, for an individual who weighed 78 kg. Contact time under the tongue and sex were associated with a negligible change in Cmax and AUC0–∞ (Figure 2). Findings were similar for apomorphine sulfate exposure, except that women also had higher Cmax values than men.
FIGURE 2.
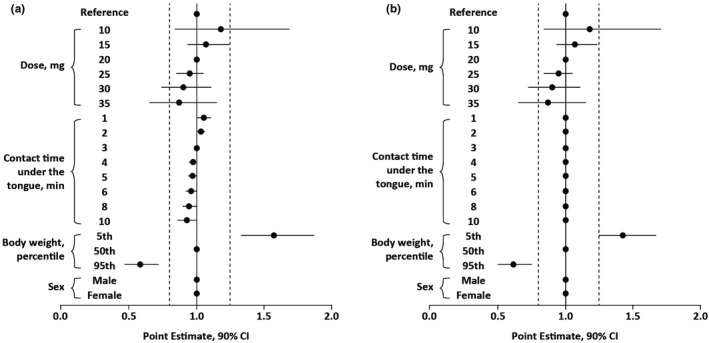
Influence of dose covariates on apomorphine (a) Cmax and (b) AUC0–∞ after administration of apomorphine sublingual film based on a typical patient with PD (i.e., 65‐year‐old White man with a body weight of 78 kg who received a 20‐mg dose of apomorphine sublingual film with the film strip positioned drug side down, a contact time under the tongue of 3 min, and who did not receive concomitant antiemetic medication). AUC0–∞, area under the concentration–time curve from time 0 to infinity; CI, confidence interval; Cmax, maximum plasma concentration; PD, Parkinson's disease.
The relationship between apomorphine dosage and exposure was also investigated. In healthy subjects, apomorphine exposure measured by Cmax increased with increasing doses of apomorphine sublingual film (Figure S4a). The increase was nearly dose proportional over the sublingual dose range of 10 to 25 mg evaluated in healthy subjects. Over the wider dose range of 10 to 50 mg administered to patients with PD and “OFF” episodes, the relationship between Cmax and the apomorphine sublingual film dose was less than proportional and tended to plateau at higher doses (Figure S4b). Power models were used to explore dose proportionality of Cmax in the observed data, with the slope estimate of the regression of the logarithm of Cmax and the logarithm of dose representing the power of the dose–exposure relationship. 21 The power estimate was not significantly different from unity (1.01; 95% CI, 0.37−1.65) over the subcutaneous apomorphine dose range of 2−5 mg; however, it was less than dose proportional over the sublingual apomorphine dose range of 10−50 mg (0.44; 95% CI, 0.13−0.75). Correspondingly, dose was included as a covariate of sublingual apomorphine bioavailability in the model using a power function. Relative to a 20‐mg dose of apomorphine sublingual film, dose‐normalized apomorphine Cmax was 18% higher with a 10‐mg dose and 9% lower with a 30‐mg dose.
The final PopPK model was also used to simulate and predict exposures of apomorphine and apomorphine sulfate after administration of 30‐mg and 35‐mg doses of apomorphine sublingual film and was compared with model‐predicted exposures at 5‐mg and 6‐mg doses of subcutaneous apomorphine, administered every 2 h for a maximum of 5 doses per day (Figure 3). Median (90% PI) predicted Cmax for apomorphine was ~ 9 ng/mL (3.24−21.5 ng/mL) with 30‐mg apomorphine sublingual film and ~ 10 ng/mL (3.66–24.3 ng/mL) with the 35‐mg dose. Median (90% PI) predicted Cmax for apomorphine was ~ 10 ng/mL (4.89−20.9) with 5‐mg subcutaneous apomorphine and ~ 12 ng/mL (5.86–25.0 ng/mL) with the 6‐mg dose. Subcutaneous administration of apomorphine is predicted to result in apomorphine sulfate Cmax concentrations that are ~ 35% of sublingual administration at both of these comparative doses. Median predicted AUC0–24 was 66.7 ng•h/mL for both apomorphine sublingual film (30 mg) and subcutaneous apomorphine (5 mg) administered every 2 h for a maximum of 5 doses per day, corresponding to a geometric mean ratio of 0.99 (90% CI, 0.96−1.03). At the highest doses, median predicted AUC0–24 was 75.4 ng•h/mL for apomorphine sublingual film (35 mg) and 80.0 ng•h/mL for subcutaneous apomorphine (6 mg), corresponding to a geometric mean ratio of 0.94 (90% CI, 0.90−0.97).
FIGURE 3.
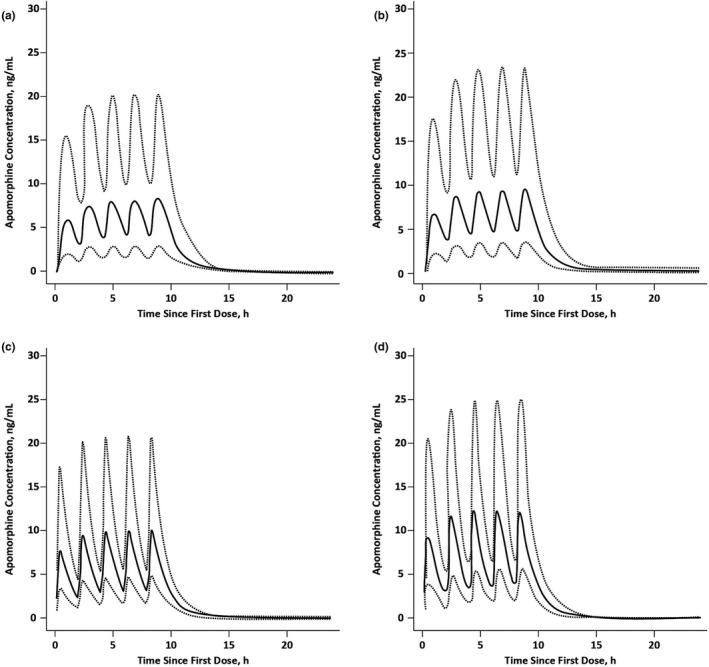
Predicted apomorphine exposure after administration of (a) apomorphine sublingual film 30 mg, (b) apomorphine sublingual film 35 mg, (c) subcutaneous apomorphine 5 mg, or (d) subcutaneous apomorphine 6 mg administered every 2 h for a total of 5 doses per day. Solid lines are median; dotted lines are 90% prediction interval.
Creatinine clearance was not found to be a statistically significant covariate of apomorphine PK in covariate analysis. In order to further describe any potential impact of renal impairment on exposure, we described the individual predicted apomorphine exposure in patients with PD and “OFF” episodes stratified by renal impairment category. Among the study population, 26 patients with PD were classified as having mild renal impairment (creatinine clearance ≥50 and <80 mL/min) and 45 patients had normal renal function (creatinine clearance ≥80 mL/min). After administration of apomorphine sublingual film (10 mg), individual predictions indicated that patients with PD and mild renal impairment had similar predicted apomorphine exposure compared with those with normal renal function (Table 4). Findings were similar for apomorphine sulfate exposure.
TABLE 4.
Apomorphine exposure parameters for patients with PD in the population pharmacokinetic data set classified as having normal a or mildly impaired b renal function
| Patients with PD and normal renal function a | Patients with PD and mild renal impairment b | |
|---|---|---|
| Cmax, ng/mL c , d | ||
| Mean | 2.74 | 2.97 |
| Median | 2.45 | 2.07 |
| Minimum, maximum | 1.00, 6.08 | 0.69, 8.79 |
| AUC0–∞, ng·h/mL c , d | ||
| Mean | 6.08 | 6.56 |
| Median | 5.34 | 5.31 |
| Minimum, maximum | 2.66, 14.2 | 2.18, 20.4 |
Abbreviations: AUC0–∞, area under the concentration‐time curve from time 0 to infinity; Cmax, maximum plasma concentration; PD, Parkinson’s disease.
Creatinine clearance ≥80 mL/min.
Creatinine clearance ≥50 and <80 mL/min.
Individual predicted value after a single 10‐mg dose of apomorphine sublingual film.
Between‐group differences in mean Cmax (p = 0.60) and AUC (p = 0.59) were not statistically significant based on a two‐sample t‐test performed with a two‐sided alpha of 0.05.
DISCUSSION
This is the first comprehensive report of the PK properties of this new apomorphine drug product, apomorphine sublingual film, from a large number of PK samples; previous reports referred to earlier sublingual formulations. 16 In this analysis, a PopPK model was successfully developed that describes the PK of apomorphine and apomorphine sulfate after sublingual and subcutaneous administration to 158 individuals across 9 clinical studies. Apomorphine PK was adequately described by a two‐compartment model, and apomorphine sulfate PK was described by a one‐compartment model. Absorption of apomorphine after sublingual and subcutaneous administration was modeled using transit absorption models to characterize the slight delay in apomorphine absorption.
The bioavailability of apomorphine sublingual film was ~ 18% relative to subcutaneously administered apomorphine. These results are consistent with previous findings from a standalone comparative bioavailability study in 8 patients with PD. 22 Although apomorphine is believed to show linear kinetics with dose‐independent clearance, systemic exposure following sublingual administration tends to increase in a less than dose proportional manner. Compared with a 10‐mg dose of apomorphine sublingual film, the relative bioavailability of a 30‐mg dose of apomorphine sublingual film is ~ 80%. The lack of dose proportionality observed in apomorphine exposure appeared to be more pronounced in patients with PD (and may be attributed to extrinsic factors, such as sublingual film contact time under the tongue, dry mouth, etc.) compared with healthy subjects; it is to be noted that the dose range of apomorphine sublingual film studied in healthy subjects was limited due to tolerability (10–25 mg) compared with that in patients with PD (10–50 mg). In clinical practice, individual dose titration of apomorphine sublingual film is expected to accommodate and allow for dose optimization and proper dose selection.
IIV for k23, V 2/F, V 3/F, k a for sublingual administration, and F 1 ranged from 35.7 to 41.7 %CV. A proportional error model was used to describe the residual unexplained variabilities in the PK of apomorphine and its inactive metabolite (apomorphine sulfate), and the magnitude ranged from 44% to 58%.
Five covariates were found to be statistically significant and were included in the final PopPK model, and their impact on apomorphine exposure was investigated in a typical patient with PD. There was no significant difference in the PK of apomorphine between healthy subjects and patients with PD and “OFF” episodes based on the covariate model analysis. Body weight was the only covariate with a large magnitude of effect on apomorphine exposure (Cmax and AUC0–∞), with greater weight resulting in lower exposure. The covariates of contact time under the tongue and sex resulted in a negligible change in exposure that is not expected to be clinically meaningful.
Simulations using the final PopPK model predicted similar median apomorphine exposure (AUC) with apomorphine sublingual film 30 mg administered every 2 h for a maximum of 5 doses per day compared with apomorphine 5 mg given subcutaneously on the same schedule. Median predicted apomorphine exposure was comparable with the highest doses of apomorphine sublingual film (35 mg) and subcutaneous apomorphine (6 mg) administered on this schedule (AUC0–24 of 75.4 and 80.0 ng•h/mL, respectively). Simulations also indicated that patients with mild renal impairment had similar apomorphine exposure to patients with normal renal function. Patients with moderate renal impairment were not specifically evaluated in the current analysis. Of note, in other PK studies, administration of apomorphine sublingual film did not result in differences in apomorphine exposure between patients with mild renal impairment and patients with normal renal function. 19 In contrast, administration of subcutaneous apomorphine resulted in increased AUC0–∞ and Cmax by ~ 16% and 50%, respectively, in patients with moderate renal impairment compared with healthy subjects. 9
This PopPK analysis has some limitations. The model was based on a modest sample of clinical study participants (87 healthy subjects and 71 patients with PD and “OFF” episodes). Doses for each individual were determined by titration to an effective and tolerable dose, rather than by random assignment (see Table S1) 19 ; this may introduce bias because persons with faster clearance may titrate to higher doses. Additionally, limited PK samples were obtained between 6 and 12 h postdose and there was a lack of PK data beyond 12 h to better inform the model. Finally, healthy subjects and patients with PD represent distinct subpopulations in this analysis, and the observed dose‐concentration relationship appears approximately dose‐proportional among healthy subjects and less than dose‐proportional among patients with PD. The model‐predicted relationship between dose and bioavailability is predicted to be less than dose‐proportional but may reflect other intrinsic (e.g., dry mouth) and extrinsic (e.g., inaccuracies in dosing diaries) factors that impact absorption or model estimates. Furthermore, patient population may not have been detected as a significant covariate due to collinearity with other covariate values (e.g., body weight) or the high variability among patients with PD.
The PK properties of apomorphine sublingual film described in this comprehensive PopPK analysis support its administration as an on‐demand treatment for patients with PD and “OFF” episodes. Drug exposure with apomorphine sublingual film is similar across a range of patient characteristics tested as covariates, which should facilitate treatment across a wide range of patients. Although apomorphine exposure was found to be lower with greater body weight, apparent clearance of apomorphine with apomorphine sublingual film is not influenced by weight. 19 In addition, the comparable predicted apomorphine exposure with maximal dosing of sublingual (30 or 35 mg every 2 h for a maximum of 5 doses per day) and subcutaneous (5 or 6 mg every 2 h for a maximum of 5 doses per day) administration is expected to further support the administration of apomorphine sublingual film across the full dose range.
CONFLICT OF INTEREST
F.A., Y.‐Y.C., G.G., and B.N. are employees of Sunovion Pharmaceuticals Inc. (Fort Lee, NJ, and Marlborough, MA, USA). R.L.C. and S.C. were contracted by Sunovion Pharmaceuticals Inc. D.B. was an employee of Sunovion Pharmaceuticals Inc. when the study was conducted and is now a consultant for Sunovion Pharmaceuticals Inc.
AUTHOR CONTRIBUTIONS
F.A., R.L.C., Y.‐Y.C., S.C., G.G., D.B., and B.N. wrote the manuscript. F.A., Y.‐Y.C., and G.G. designed the research. F.A., R.L.C., Y.‐Y.C., S.C., and G.G. performed the research and analyzed the data.
Supporting information
Fig S1‐S4
Table S1
ACKNOWLEDGMENTS
Medical writing and editorial assistance were provided by Payal N. Gandhi, PhD, of The Lockwood Group (Stamford, CT, USA) and Lisa Baker, PhD, on behalf of The Lockwood Group. This assistance was supported by funding from Sunovion Pharmaceuticals Inc. (Marlborough, MA, USA).
Funding information
This analysis and the associated clinical studies were supported by funding from Sunovion Pharmaceuticals Inc. (Marlborough, MA, USA).
REFERENCES
- 1. Mhyre TR, Boyd JT, Hamill RW, Maguire‐Zeiss KA. Parkinson's disease. Subcell Biochem. 2012;65:389‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marras C, Beck JC, Bower JH, et al. Prevalence of Parkinson's disease across North America. NPJ Parkinsons Dis. 2018;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olanow CW, Stocchi F. Levodopa: a new look at an old friend. Mov Disord. 2018;33:859‐866. [DOI] [PubMed] [Google Scholar]
- 4. Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease. Neurology. 2009;72(suppl 4):S1‐S136. [DOI] [PubMed] [Google Scholar]
- 5. Ahlskog JE, Muenter MD. Frequency of levodopa‐related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16:448‐458. [DOI] [PubMed] [Google Scholar]
- 6. Thanvi BR, Lo TC. Long term motor complications of levodopa: clinical features, mechanisms, and management strategies. Postgrad Med J. 2004;80:452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dewey RB Jr, Hutton JT, LeWitt PA, Factor SA. A randomized, double‐blind, placebo‐controlled trial of subcutaneously injected apomorphine for parkinsonian off‐state events. Arch Neurol. 2001;58:1385‐1392. [DOI] [PubMed] [Google Scholar]
- 8. Boyle A, Ondo W. Role of apomorphine in the treatment of Parkinson's disease. CNS Drugs. 2015;29:83‐89. [DOI] [PubMed] [Google Scholar]
- 9. APOKYN . APOKYN® [US package insert]. Louisville, KY: US WorldMeds; 2020. [Google Scholar]
- 10. APO‐go . APO‐go® [summary of product characteristics]. Reading, Berkshire, UK: Britannia Pharmaceuticals; 2020. [Google Scholar]
- 11. Hughes AJ, Bishop S, Kleedorfer B, et al. Subcutaneous apomorphine in Parkinson's disease: response to chronic administration for up to five years. Mov Disord. 1993;8:165‐170. [DOI] [PubMed] [Google Scholar]
- 12. Manson AJ, Turner K, Lees AJ. Apomorphine monotherapy in the treatment of refractory motor complications of Parkinson's disease: long‐term follow‐up study of 64 patients. Mov Disord. 2002;17:1235‐1241. [DOI] [PubMed] [Google Scholar]
- 13. Bhidayasiri R, Chaudhuri KR, LeWitt P, Martin A, Boonpang K, van Laar T. Effective delivery of apomorphine in the management of Parkinson disease: practical considerations for clinicians and Parkinson nurses. Clin Neuropharmacol. 2015;38:89‐103. [DOI] [PubMed] [Google Scholar]
- 14. Bowron A. Practical considerations in the use of apomorphine injectable. Neurology. 2004;62(suppl 4):S32‐S36. [DOI] [PubMed] [Google Scholar]
- 15. Bilbault T, Taylor S, Walker R, Grundy SL, Pappert EJ, Agro A. Buccal mucosal irritation studies of sublingual apomorphine film (APL‐130277) in Syrian golden hamsters. Ther Deliv. 2016;7:611‐618. [DOI] [PubMed] [Google Scholar]
- 16. Argiolas A, Hedlund H. The pharmacology and clinical pharmacokinetics of apomorphine SL. BJU Int. 2001;88(suppl 3):18‐21. [DOI] [PubMed] [Google Scholar]
- 17. Gancher ST, Nutt JG, Woodward WR. Absorption of apomorphine by various routes in parkinsonism. Mov Disord. 1991;6:212‐216. [DOI] [PubMed] [Google Scholar]
- 18. Borkar N, Mu H, Holm R. Challenges and trends in apomorphine drug delivery systems for the treatment of Parkinson’s disease. Asian J Pharm Sci. 2018;13:507‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. KYNMOBI® . KYNMOBI® [US package insert]. Marlborough, MA: Sunovion Pharmaceuticals Inc.; 2020. [Google Scholar]
- 20. Olanow CW, Factor SA, Espay AJ, et al. Apomorphine sublingual film for off episodes in Parkinson's disease: a randomised, double‐blind, placebo‐controlled phase 3 study. Lancet Neurol. 2020;19:135‐144. [DOI] [PubMed] [Google Scholar]
- 21. Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17:1278‐1283. [DOI] [PubMed] [Google Scholar]
- 22. Agbo F, Isaacson S, Ellenbogen A, et al. Comparative bioavailability of apomorphine sublingual film and subcutaneous apomorphine formulations in patients with Parkinson’s disease and “OFF” episodes: preliminary results [abstract EPO1165]. Eur J Neurol. 2019;26:445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4
Table S1