

Abstract
Background & Aim
Abundantly expressed in the metabolically active cells including hepatocytes, N-nicotinamide methyltransferase (NNMT) catalyzes S-adenosylmethionine (SAM)-dependent methylation/degradation of nicotinamide, the predominant precursor for intracellular nicotinamide adenine dinucleotide (NAD+) regeneration via the salvage pathway. The enzyme is emerging to play an important role in regulating metabolism, however, the exact regulatory mechanism(s) underlying NNMT expression remains unclear and its potential implication in alcoholic liver disease (ALD) is completely unknown.
Methods
Both traditional Lieber-De Carli and the NIAAA mouse model of ALD were employed. A small scale of chemical screening and ChIP assay were performed. NNMT inhibition was achieved via both genetic (adenoviral shRNA delivery) and pharmacological approach.
Results
Chronic alcohol consumption induces liver endoplasmic reticulum (ER) stress and upregulates hepatic NNMT expression. ER stress inducers upregulated NNMT expression in both AML-12 hepatocytes and mice. PERK-ATF4 pathway activation is the main contributor in ER stress-mediated NNMT upregulation in the liver. Alcohol consumption fails to upregulate NNMT in liver-specific ATF4 knockout mice. Both adenoviral NNMT knockdown and NNMT inhibitor administration prevents fatty liver development induced by chronic alcohol feeding, which is associated with the downregulation of an array of genes involved in de novo lipogenesis pathway, including Srebf1, Acaca, Acacb and Fasn. Further investigations revealed that NNMT inhibition-induced lipogenic pathway activation was independent of its NAD+-enhancing action; however, incremental cellular NAD+ contents due to NNMT inhibition was associated with marked liver AMPK activation.
Conclusions
ER stress, in specific, the PERK-ATF4 pathway activation, is mechanistically involved in hepatic NNMT upregulation in response to chronic alcohol exposure and NNMT overexpression in the liver plays an important role in the pathogenesis of ALD.
Keywords: ALD, NNMT, ATF4, ER stress, lipogenesis
Graphical Abstract
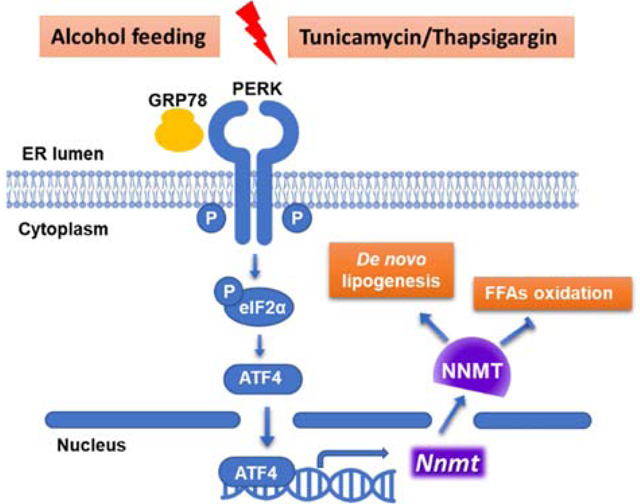
Exact pathogenesis of alcoholic fatty liver disease remains to be clarified. In this study, we have identified ER stress-induced upregulation of liver nicotinamide methyltransferase (NNMT), the enzyme catalyzing nicotinamide degradation using s-adenosylmethionine as a methyl donor, to be a pathological regulator in alcoholic fatty liver development. NNMT inhibition protects against fatty liver development resulted from chronic alcohol consumption, which is associated with suppressed de novo lipogenic activity and enhanced AMPK activation. Thus, our data suggest that NNMT may be a potential therapeutic target for the treatment of alcoholic liver disease.
Lay summary
In this study, we have identified ER stress-induced upregulation of liver nicotinamide methyltransferase (NNMT), the enzyme catalyzing nicotinamide degradation using s-adenosylmethionine as a methyl donor, to be a pathological regulator in alcoholic fatty liver development. NNMT inhibition protects against fatty liver development resulted from chronic alcohol consumption, which is associated with suppressed de novo lipogenic activity and enhanced AMPK activation. Thus, our data suggest that NNMT may be a potential therapeutic target for the treatment of alcoholic liver disease.
Introduction
Alcoholic liver disease (ALD) ranks among the major causes of all liver diseases, affecting millions of patients worldwide. The pathological process of the disease development encloses early steatosis, steatohepatitis, with some individuals ultimately progressing to fibrosis and liver failure.1 Despite well-characterized disease progression and improved understanding on its pathogenesis, the exact mechanism(s) underpinning its pathogenesis remain unclear and there are currently no accepted therapies available to prevent or cure the disease. Therefore, a better understanding of the mechanism(s) and risk factors that mediate the initiation and progression of this disease is essential and urgently required for the development of rational targeted therapies to prevent or treat it clinically.
N-nicotinamide methyltransferase (NNMT) is a methyltransferase, catalyzing S-adenosylmethionine (SAM)-dependent methylation/degradation of nicotinamide (an amide of vitamin B3/niacin), a predominant precursor for cellular NAD+ synthesis via salvage pathway.2,3 Abundantly expressed in the metabolically active tissues, such as the liver and adipose tissue, NNMT is emerging to be a critical player in the regulation of nutrients metabolism and energy homeostasis. Whole-body NNMT overexpression leads to reduced hepatic NAD+ and SAM levels in mice.3 Importantly, the NNMT-overexpressing mice manifest worsened hepatic steatosis in the setting of chronic high-fat diet feeding, which is associated with increased expression of the genes invovled in fatty acid uptake and decreased VLDL secretion.3 Moreover, adipose tissue NNMT expression levels are positively associated with obesity and insulin resistance in both experimental and clinical settings.4,5 Accordantly, NNMT knockdown via either antisense oligonucleotides or chemical inhibitor protects against diet-induced obesity in mice.5,6 Genetic NNMT deficiency improves insulin sensitivity in diet-induced obesity in male mice7 and overexpression of NNMT is associated with reduced cellular methylation potential.3,8 The central location of NNMT in both SAM- and NAD+-dependent metabolic and signaling pathways suggest a critical role of this enzyme in ALD pathogenesis in that both are well-documented to be attributable to the pathogenesis of ALD.9–12 Indeed, the supplementation with exogenous SAM,12 nicotinamide,3,10 or nicotinic acid13 has all been reported to protect against alcohol-induced liver damage in mice. Nevertheless, whether and how chronic alcohol exposure affects hepatic NNMT expression, as well as whether and how altered hepatic NNMT contributes to ALD development remain to be completely unanswered questions.
A disruption of the balance between the unfolded proteins and the ER folding machinery inside the ER lumen leads to the accumulation of unfolded proteins, a condition coined as ER stress.14 The ER stress instigates the unfolded protein response (UPR) to restore ER homeostasis via attenuating protein translation, increasing protein folding, and promoting degradation of unfolded proteins.15 UPR comprises three canonical signaling pathways including: inositol-requiring enzyme 1-α (IRE1α), RNA-dependent protein kinase-like ER kinase (PERK), activating transcription factor 6 (ATF6). IRE1α has both endoribonuclease activity and kinase activity, whose activation leads to generation of transcription factor X-box binding protein 1 (XBP1s) and c-Jun N-terminal kinase (JNK) activation, respectively. PERK activation selectively induces activating transcription factor 4 (ATF4) via phosphorylating/activating eukaryotic translation initiation factor 2α (eIF2α). ATF6 functions as a transcription factor to activate expression of ER chaperones, such as 78 kDa glucose-regulated protein (GRP78).15–17
The pathogenic association between ER stress and ALD has been documented both experimentally and clinically,18–20 however, exact mechanism(s) with respect to how ER stress contributes to fatty liver in response to chronic alcohol exposure remain ambiguous. In this study, we demonstrate for the first time that chronic alcohol exposure upregulates hepatic NNMT expression via the PERK-ATF4 pathway activation of UPR in the liver. We further show that NNMT knockdown via either adenoviral delivery of NNMT shRNA or chemical inhibitor administration is protective against alcoholic fatty liver development. Finally, our data unravel that the preventive effect of NNMT inhibition on fatty liver development is associated with both de novo lipogenesis pathway suppression and AMPK activation.
Materials and methods
Animal studies
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago (Chicago, IL) and consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Both the NIAAA model21 and the tradition model of ALD22 were employed. NNMT knockdown was achieved via retro-orbital injection of either Ad-GFP-U6-m-NNMT-shRNA or corresponding control vector (Vector Biolabs, Malvern, PA). The liver-specific ATF4 knockout mice (ALKO) were generated by crossing ATF4flox/flox (f/f) mice (generously provided by Dr. Christopher M. Adams from University of Iowa) with albumin-Cre mice (Jackson Laboratory, Bar Harbor, ME). (see supplementary information for details).
Cell culture studies
Both AML12 hepatocytes, a non-transformed mouse hepatocyte cell line, and HepG2 cells, a human hepatoma cell line, were obtained from American Type Culture Collection (ATCC, Manassas, VA). (see supplementary information for details).
Total RNA isolation and quantitative real-time PCR
Total RNA isolation, generation of cDNA, and real-time quantitative PCR (q-PCR) were conducted as previously reported.23 Primer sequences used are listed in Table 1. The comparative threshold cycle (or 2 Ct) method was used to calculate relative mRNA expression levels.
TABLE 1.
PRIMERS IN USE
| PRIMER | SEQUENCES (5’ – 3’) |
|---|---|
| Mouse-Nnmt-F | CTTTGGGTCCAGACACTGTGCA |
| Mouse-Nnmt-R | CCAGAGCCAATGTCAATCAGGAG |
| Mouse-Atf4-F | AACCTCATGGGTTCTCCAGCGA |
| Mouse-Atf4-R | CTCCAACATCCAATCTGTCCCG |
| Mouse-Srebf1-F | CGACTACATCCGCTTCTTGCAG |
| Mouse-Srebf1-R | CCTCCATAGACACATCTGTGCC |
| Mouse-Srebf2-F | AGAAAGAGCGGTGGAGTCCTTG |
| Mouse-Srebf2-R | GAACTGCTGGAGAATGGTGAGG |
| Mouse-Fasn-F | CACAGTGCTCAAAGGACATGCC |
| Mouse-Fasn-R | CACCAGGTGTAGTGCCTTCCTC |
| Mouse-Acaca-F | GTTCTGTTGGACAACGCCTTCAC |
| Mouse-Acaca-R | GGAGTCACAGAAGCAGCCCATT |
| Mouse-Acacb-F | AGAAGCGAGCACTGCAAGGTTG |
| Mouse-Acacb-R | GGAAGATGGACTCCACCTGGTT |
| Mouse-Gapdh-F | CATCACTGCCACCCAGAAGACTG |
| Mouse-Gapdh-R | ATGCCAGTGAGCTTCCCGTTCAG |
| Human-NNMT-F | GTTTGGTTCTAGGCACTCTGCAG |
| Human-NNMT-R | AGAGCCGATGTCAATCAGCAGG |
| Human-GAPDH-F | GTCTCCTCTGACTTCAACAGCG |
| Human-GAPDH-R | ACCACCCTGTTGCTGTAGCCAA |
Chromatin immunoprecipitation analysis (ChIP)
ChIP analysis was performed according to the instruction of Pierce Agarose ChIP Kit (Pierce Biotechnology, Rockford, USA). AML12 cells were seeded in 6-well plate before treated with either DMSO or thapsigargin for 6 hours. 2×106 cells were fixed by adding 16% formaldehyde directly into the culture media, which was diluted to 1% finally. After 10 min fixing in room temperature, the media was added glycine to stop fixing. The upstream 2000bp region of the NNMT transcription start site was selected as the gene promoter region. The ATF4 binding site was predicted in the JASPAR database (http://jaspar.genereg.net/). The purified, immunoprecipitated DNA was analyzed by qPCR. The primers used in ChIP qPCR were as following:
Forward: 5’-ACGGTTATGTCCCTGGTTCT-3’;
Reverse: 5’-CCATATGCGCATCAAACTGG-3’.
Statistical analysis
All data are expressed as mean ± SEM. The sample size was calculated using size power analysis methods (GraphPad StatMate) for a priori determination based on the standard deviations of previous experiments. The minimal sample size for each group as 5 animals was determined after calculation. Statistical analysis was performed using a Student’s t test or one-way ANOVA analysis for the comparison of 2 groups and 3 or more groups, respectively. GraphPad Prism software (version 6.0) was used to calculate statistical significance. Statistical tests were used as described in the Figure legends and statistical significance was indicated as follows: *p <0.05; **p <0.01; ***p <0.001; ****p <0.0001; ns, not significant. For further details regarding the materials and methods used, please refer to the CTAT table.
Results
ER stress upregulates NNMT expression in both hepatocytes and the livers
NNMT catalyzes SAM-dependent methylation/degradation of nicotinamide (Fig. 1A). The effect of ER stress on NNMT expression in hepatocytes was directly examined in cultured AML12 cells, a non-transformed mouse hepatocyte cell line. Two well-established and mechanistically distinct chemical ER stress inducers, thapsigargin and tunicamycin, were employed to induce ER stress in AML12 cells. While thapsigargin induces UPR/ER stress via raising cytosolic Ca2+ level,24 tunicamycin induces UPR/ER stress via inhibiting N-linked glycosylation, a posttranslational modification essential for proper protein folding.25 The expression of NNMT at both mRNA and protein level was determined by real-time PCR and Western blotting, respectively. Both thapsigargin and tunicamycin increased NNMT gene expression robustly, which can be detected at as early as 3 hours and peaked at 6 hours (Fig. 1B). In response to thapsigargin, a strong unfolded protein response (UPR) was observed, which was evidenced by a robust induction of GRP78 protein and ATF4, signature indicators of UPR induction (Fig. 1C). Correspondingly, a markedly enhanced NNMT protein abundance was detected (Fig. 1C). The effect of ER stress on NNMT expression was also examined in HepG2 cells, a human hepatoma cell line and the similar impact was observed (Fig. 1D), although the basal level of NNMT gene expression was much lower in HepG2 cells in comparison to that in AML12 cells (Fig. 1E). To corroborate our in vitro observations, animal experiments were subsequently conducted. To induce ER stress, male C57 BL/6N mice (10-wk old) were administrated with either vehicle or tunicamycin via intraperitoneal injection. ER stress induction and hepatic NNMT expression were determined 16 hours later. Tunicamycin administration resulted in a significant and conspicuous increment of NNMT expression at both mRNA and protein level (Fig. 1F & G), which was concomitant with a notable induction of UPR in the liver, evidenced by a markedly enhanced GRP78 protein expression upon tunicamycin administration (Fig. 1G).
Figure 1.
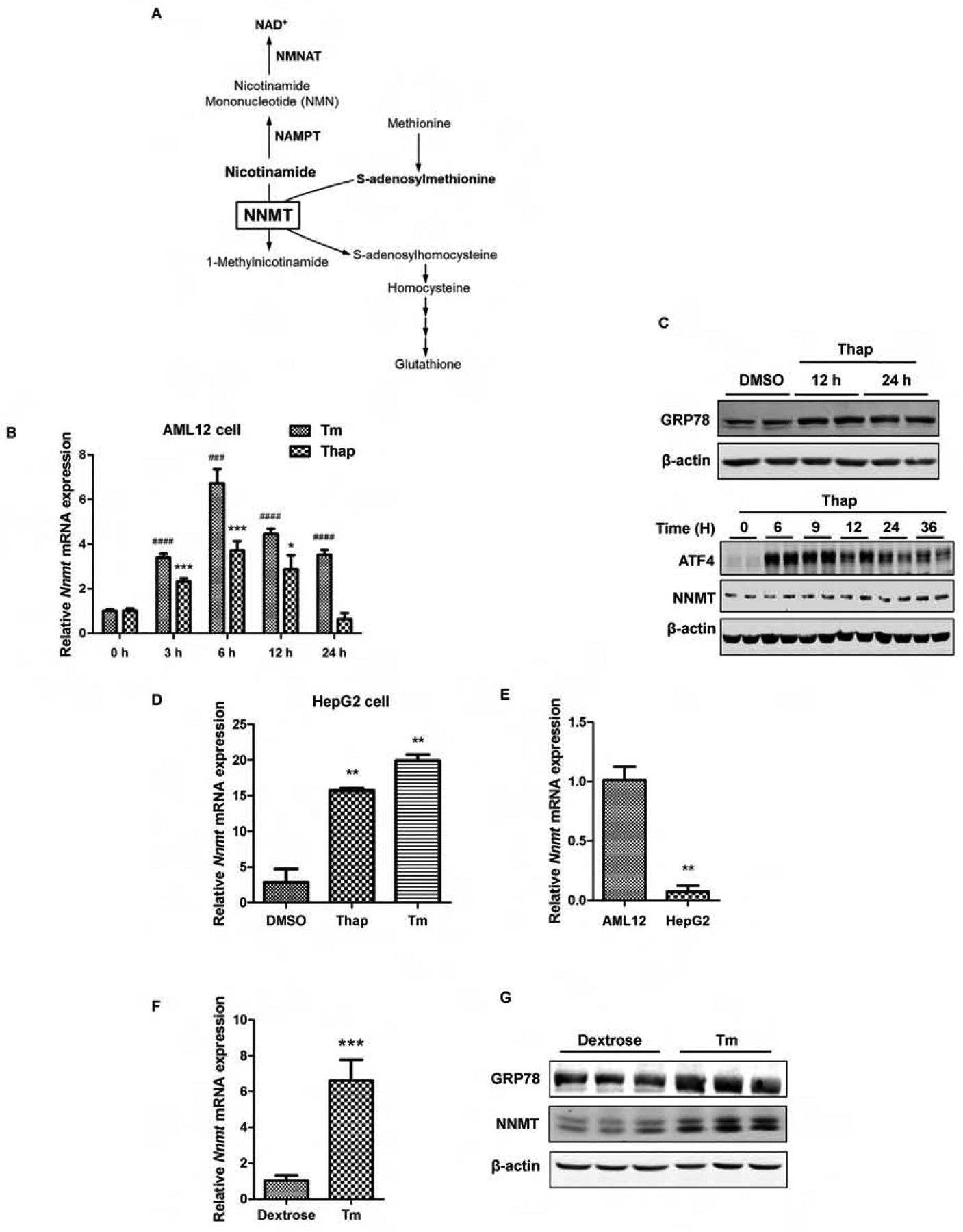
ER stress upregulates NNMT expression. (A) Schematic illustration of simplified NNMT-regulated metabolic pathways. NAMPT: Nicotinamide Phosphoribosyltransferase; NMNAT: nicotinamide mononucleotide adenylyltransferase. (B) Time-course effects of ER stress inducers on NNMT gene expression. AML12 cells were treated with either thapsigargin (Thap, 2 μM) or tunicamycin (Tm, 5 μM) for indicated time periods. NNMT mRNA expression was detected by q-PCR. (C) Time course changes of GRP78, ATF4, and NNMT protein abundance. AML12 hepatocytes were treated with thapsigargin (Thap, 2 μM) for indicated time periods. GRP78, ATF4, and NNMT protein expressions were detected via Western-blotting. (D & E) ER stress upregulates NNMT expression in HepG2 cells. HepG2 cells were treated with either thapsigargin (Thap, 2 μM) or tunicamycin (Tm, 5 μM) for 6 hours and NNMT gene expression measured by q-PCR. (F & G) Tunicamycin administration upregulates NNMT expression at both mRNA and protein level. Ten-week old male C57BL/6N mice were injected with tunicamycin (2 mg/kg BW) or isovolumic vehicle (150 mM dextrose) intraperitoneally. Hepatic NNMT gene expression and protein abundance were determined. Data are expressed as mean ± SEM. Student’s t test was used for statistical evaluation (**p < 0.01, ***, ###p < 0.001, ####p < 0.0001 versus corresponding control).
The PERK-ATF4 branch of UPR contributes to ER stress-triggered NNMT upregulation
The UPR consists of three ER transmembrane protein sensors: inositol requiring enzyme 1α (IRE1α), pancreatic ER kinase (PERK), and activating transcription factor 6 (ATF6).15,17 To determine the potential mechanistic involvement of these three canonical UPR branches in ER stress inducer-elicited NNMT upregulation, the specific inhibitors for IRE1α (STF083010 for its endonuclease activity and SP600125 for its kinase activity), PERK (GSK2606414), and ATF6 (Ceapin-A7) were applied to pretreat AML12 cells for 2 hours before ER stress induction with either thapsigargin or tunicamycin. NNMT gene expression was measured by real-time q-PCR. As shown in Fig. 2A, whereas neither IRE1α nor ATF6 inhibitor affected NNMT upregulation upon thapsigargin exposure, PERK inhibitor almost completely ablated thapsigargin-elicited NNMT overexpression. Similarly, PERK inhibitor also ameliorated tunicamycin-induced NNMT overexpression. Notably, PERK activator alone led to a significant NNMT upregulation (Fig. 2B), suggesting that this branch of UPR plays a mechanistic role in regulating ER stress-induced NNMT upregulation. To confirm this notion, we transfected AML12 cells with siRNA for ATF4, a downstream transcription factor in response to PERK activation, for overnight, followed by tunicamycin exposure. As shown in Fig. 2C & D, ATF4 siRNA transfection resulted an approximate 60% reduction of ATF4 expression. Importantly, ATF4 siRNA knockdown dramatically blunted NNMT upregulation in response to either thapsigargin or tunicamycin stimulation, supporting that the activation of the PERK-ATF4 branch of UPR is mechanistically attributed to ER stress-induced NNMT upregulation. To further consolidate this notion, the ChIP assay was performed by selecting the 2000 bp upstream region in front of NNMT transcription start site as prediction area of ATF4 binding site. ChIP-qPCR confirmed that ATF4 is able to bind with NNMT promoter region to regulate its gene expression directly (Fig. 2E).
Figure 2.
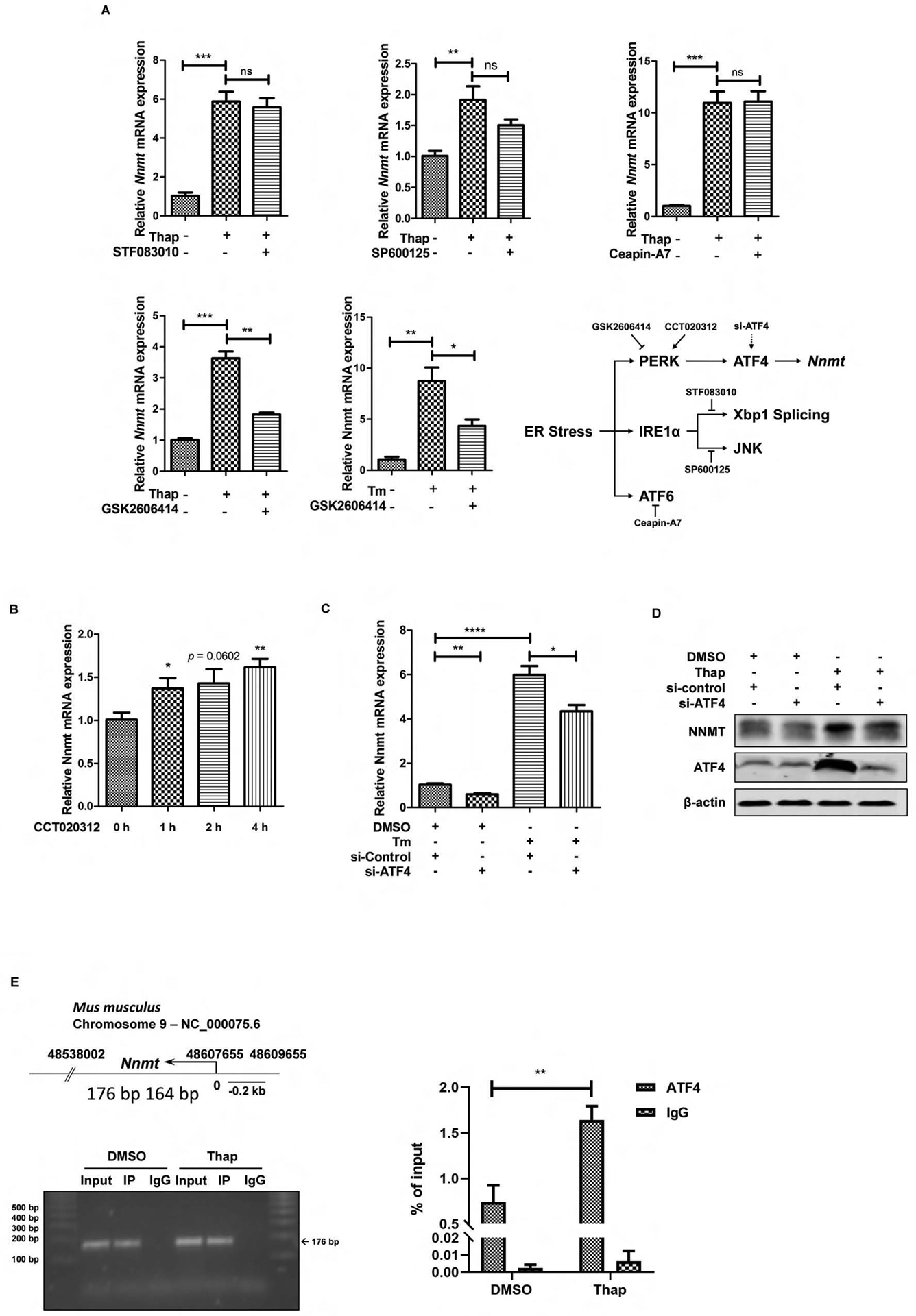
PERK-ATF4 pathway contributes to ER stress-triggered NNMT upregulation. (A) The chemical screening study identifies PERK inhibitor blunts NNMT upregulation upon challenging with ER-stress inducers. AML12 cells were treated with either thapsigargin (Thap, 2 μM) or tunicamycin (Tm, 5 μM) for 6 hours with or without a two-hour pretreatment with IRE1α inhibitor (STF083010 at 40 μM for its endoribonuclease activity and SP600125 at 30 μM for its kinase activity), PERK inhibitor (GSK2606414, 150 nM), or ATF6 inhibitor (Ceapin-A7, 10 μM). NNMT gene expression was detected by q-PCR. (B) Chemical activator of PERK upregulates NNMT gene expression. AML12 cells were treated with PERK activator (CCT020312, 30 μM) for 0–4 h and NNMT gene expression was detected by q-PCR. (C & D) ATF4 siRNA knockdown alleviates ER stress-triggered NNMT upregulation. AML12 cells were transfected with either scrambled siRNA or ATF4 siRNA for overnight, followed by tunicamycin exposure. NNMT mRNA expression and protein abundance were determined at 6 hours. Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus corresponding control. Differences between 2 groups were determined using Student’s t test. (E) Effect of thapsigargin on ATF4 recruitment to the NNMT promoter. The numbers (176–164bp) indicate nucleotide position of a putative ATF4 binding site within 2000 bp upstream of transcription starting site. AML12 cells were treated with/without thapsigargin (2 μM) for 6 hours and ATF4 recruitment was determined by ChIP assay. The associated DNA was quantified by real-time qPCR with primer pairs specific for the putative ATF4 binding site . Values are normalized by input DNA. Results are expressed as the mean ± SEM fold induction vs basal levels. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus corresponding control. Differences between 2 groups were determined using Student’s t test.
NNMT knockdown protects against ER stress-induced fat accumulation in the liver
ER stress induction with tunicamycin administration is associated with the development of hepatic steatosis.26 To determine the pathophysiologic significance of NNMT upregulation in ER stress-induced fatty liver development, we conducted NNMT knockdown experiment via retro-orbital delivery of either empty vector or NNMT shRNA-containing adenovirus. Adenoviral NNMT shRNA delivery significantly reduced NNMT gene expression in the liver (~55% reduction), while NNMT expression in adipose tissue (both epididymal fat and inguinal fat) was not significantly affected (Fig. 3A). Tunicamycin was administered via intraperitoneal injection at a dose of 2 mg/kg BW and liver samples harvested 16 hours later. As shown in Fig. 3B–D, adenoviral knockdown of NNMT ameliorated tunicamycin-induced fatty liver development, as made evident by both biochemical assay and histological/morphological examination.
Figure 3.
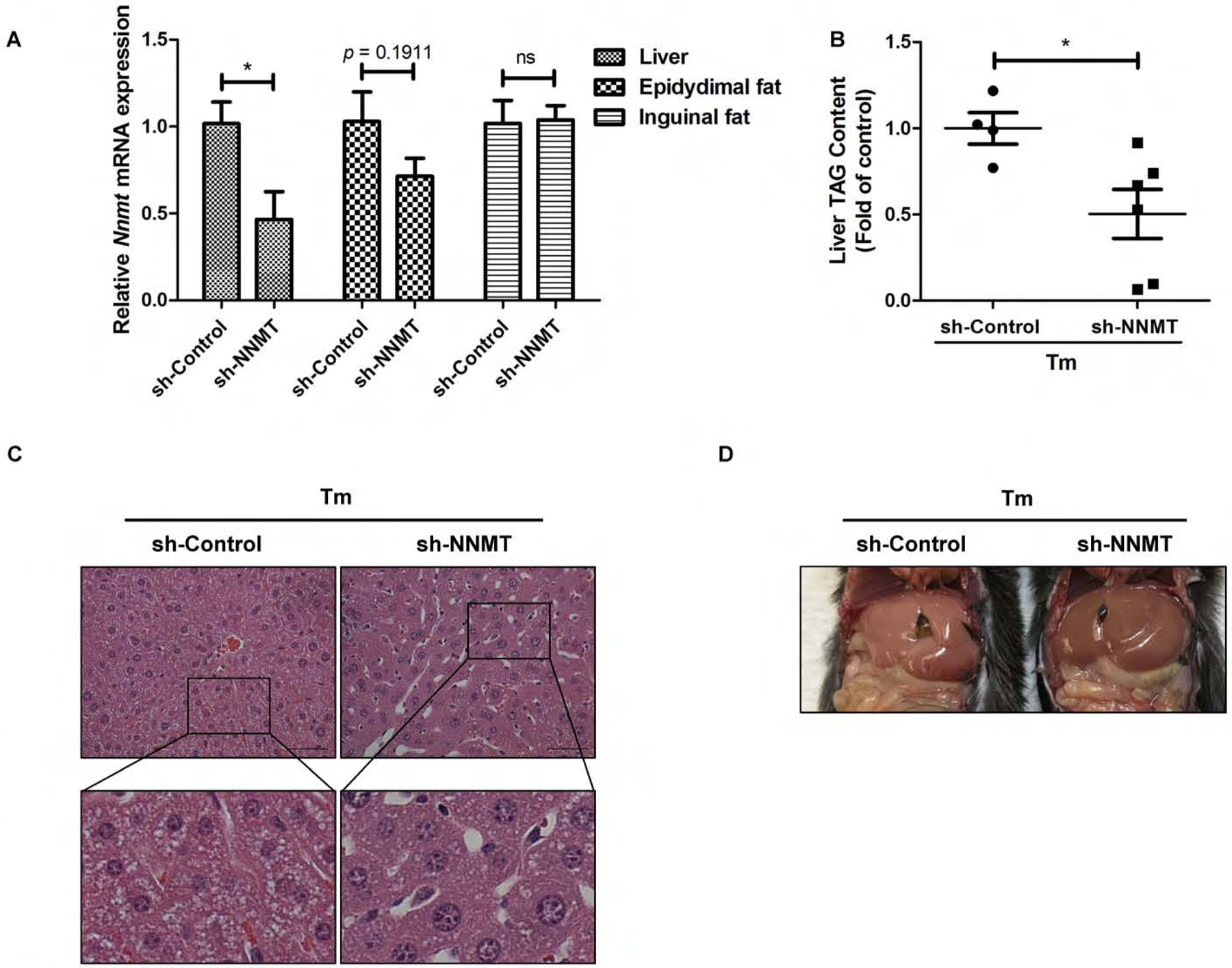
NNMT shRNA knockdown protects against ER stress-induced fat accumulation in the liver. Ten-week old male C57BL/6N mice were retro-orbitally injected with either sh-control or sh-NNMT. (A) NNMT gene expression in the liver and adipose tissues after adenoviral delivery of NNMT shRNA. (B) shRNA knockdown of NNMT ameliorates hepatic fat accumulation in a mouse ER stress model. Ten-week old male C57BL/6N mice were retro-orbitally injected with either sh-control or sh-NNMT, followed by tunicamycin administration (2 mg/kg BW, ip) for 16 hours. Liver TAG contents were measured. (C & D) Histological (H&E staining) and morphological observation. Data are expressed as mean ± SEM (n = 5–8). Student’s t test was used for statistical evaluation (*p < 0.05 versus corresponding control).
Chronic alcohol consumption upregulates NNMT expression in the liver
Chronic alcohol consumption is associated with hepatic ER stress.16,19,27 The fact that hepatic ER stress upregulates NNMT expression prompted us to posit that chronic alcohol consumption is associated with NNMT upregulation in the liver. To test our hypothesis, we examined liver NNMT expression using two well-established and widely-employed mouse models of ALD: 1. traditional Lieber-De Carli diet feeding model (5-week alcohol-containing liquid diet feeding, hereafter referred to as “the tradition model”); and 2. chronic plus binge ethanol feeding model (10-day feeding with alcohol-containing liquid diet plus one gavage, hereafter referred to as “the NIAAA model”). As shown in Fig. 4A & B, a robust increase of NNMT expression was observed in both mouse models of ALD when compared to their corresponding isocaloric pair-fed counterparts. NNMT catalyzes methylation of nicotinamide to generate 1-methylnicotinamide (1-MNA), therefore, intracellular 1-MNA concentration serves as an indicator of NNMT functionality. The LC-MS/MS analysis of liver 1-MNA levels showed that the alcohol-induced upregulation of NNMT expression was concomitant with its functional enhancement, evidenced by significantly increased liver tissue 1-MNA levels in alcohol-fed animals in comparison to their pair-fed counterparts (Fig. 4C). Furthermore, ethanol-induced NNMT upregulation was also observed in AML12 cells. As shown in Fig. 4D, the inclusion of ethanol (100 and 200 mM) in the culture media led to NNMT upregulation, which peaked at 48 hours post-ethanol addition.
Figure 4.
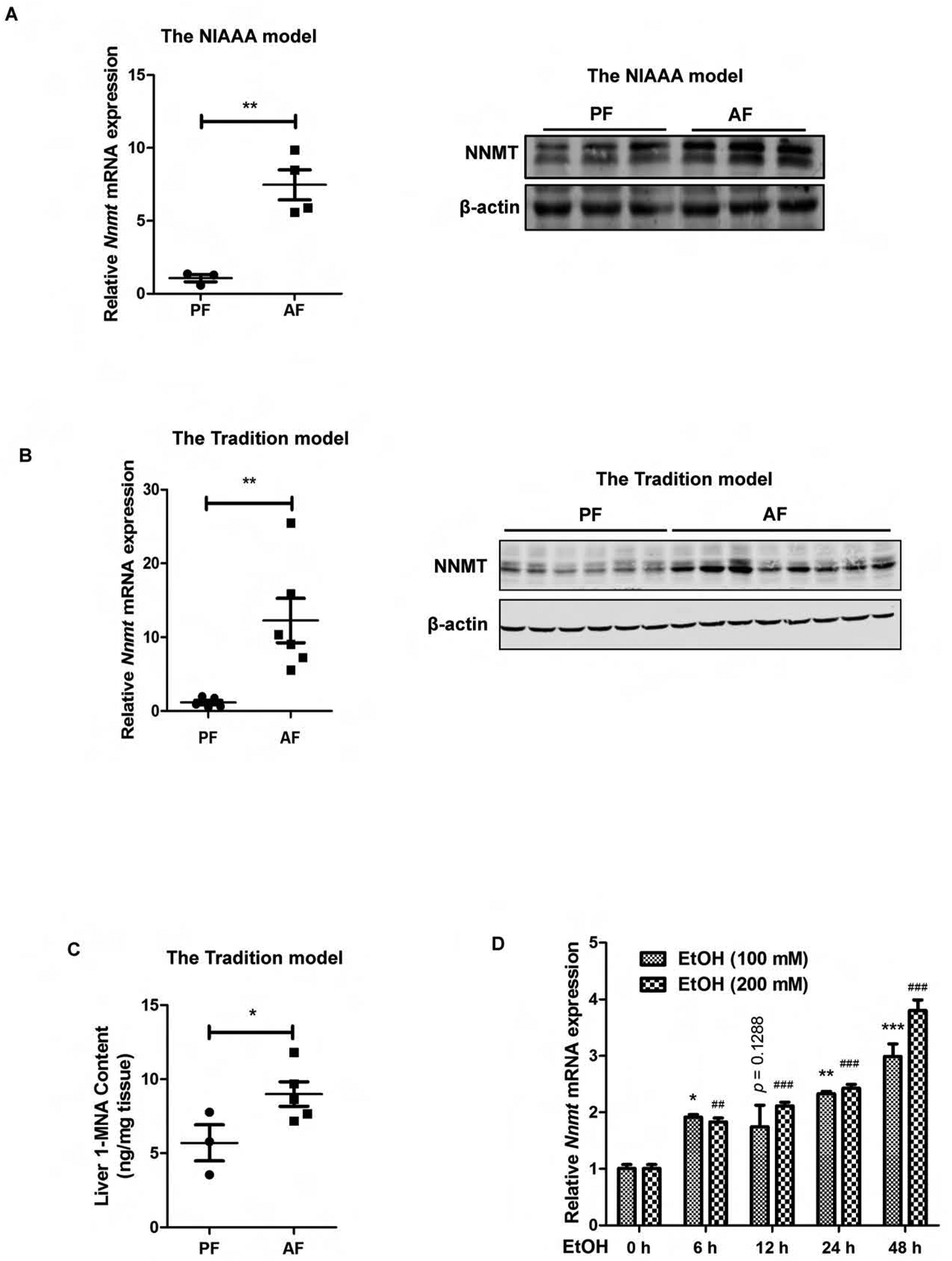
Chronic alcohol consumption upregulates hepatic NNMT expression. (A) NNMT expression in the NIAAA model. (B) NNMT expression in the traditional model. (C) Hepatic 1-MNA contents in the NIAAA ALD model. (D) Ethanol exposure upregulates NNMT gene expression in AML12 cells. AML12 were cultured with media containing ethanol (100 and 200 mM, respectively) for indicated time periods. NNMT gene expression was detected by q-PCR. Data are expressed as mean ± SEM (n =5–8). Differences between 2 groups were determined using Student’s t test (*p < 0.05, **p < 0.01, ***, ###p < 0.001 versus control).
The PERK-ATF4 pathway activation contributes to alcohol-induced hepatic NNMT overexpression
Chronic alcohol consumption induces ER stress in the liver.16,19,20 Based on the evidence that PERK-ATF4 pathway activation plays a mechanistic role in ER stress-induced NNMT upregulation (Fig. 2), we examined PERK activation status and ATF4 protein abundance in the NIAAA mouse model of ALD. The activated PERK was observed in alcohol-fed mice, which was evidenced by increased p-eIF2α protein abundance, the direct downstream target of PERK activation (Fig. 5A). Moreover, ATF4 protein abundance in both whole liver tissue lysate and nuclear lysate was significantly increased in alcohol-fed mice when compared with their isocaloric counterparts (Fig. 5B). Similarly, a significantly increased hepatic ATF4 protein expression in response to alcohol feeding was detected when the assay was conducted using the mice from the tradition model of ALD (Fig. 5C). To determine whether ATF4 activation contributes to alcohol-induced hepatic NNMT upregulation, the liver-specific ATF4 knockout mice and their age-and gender-matched control animals were exposed to either isocaloric pair-fed control diet (PF) or Lieber-De Carli alcohol diet28 for 5 weeks (the tradition ALD model) and liver NNMT expression was determined. As shown in Fig. 5D & E, a significant elevation of hepatic NNMT expression at both mRNA and protein level was observed in control animals (f/f), which is consistent with our previous observation (Fig. 4); however, in the liver-specific ATF4 knockout mice (ALKO), alcohol-induced NNMT upregulation was profoundly compromised. These data altogether suggest that the PERK-ATF4 pathway activation contributes to hepatic NNMT upregulation in the setting of chronic alcohol consumption.
Figure 5.
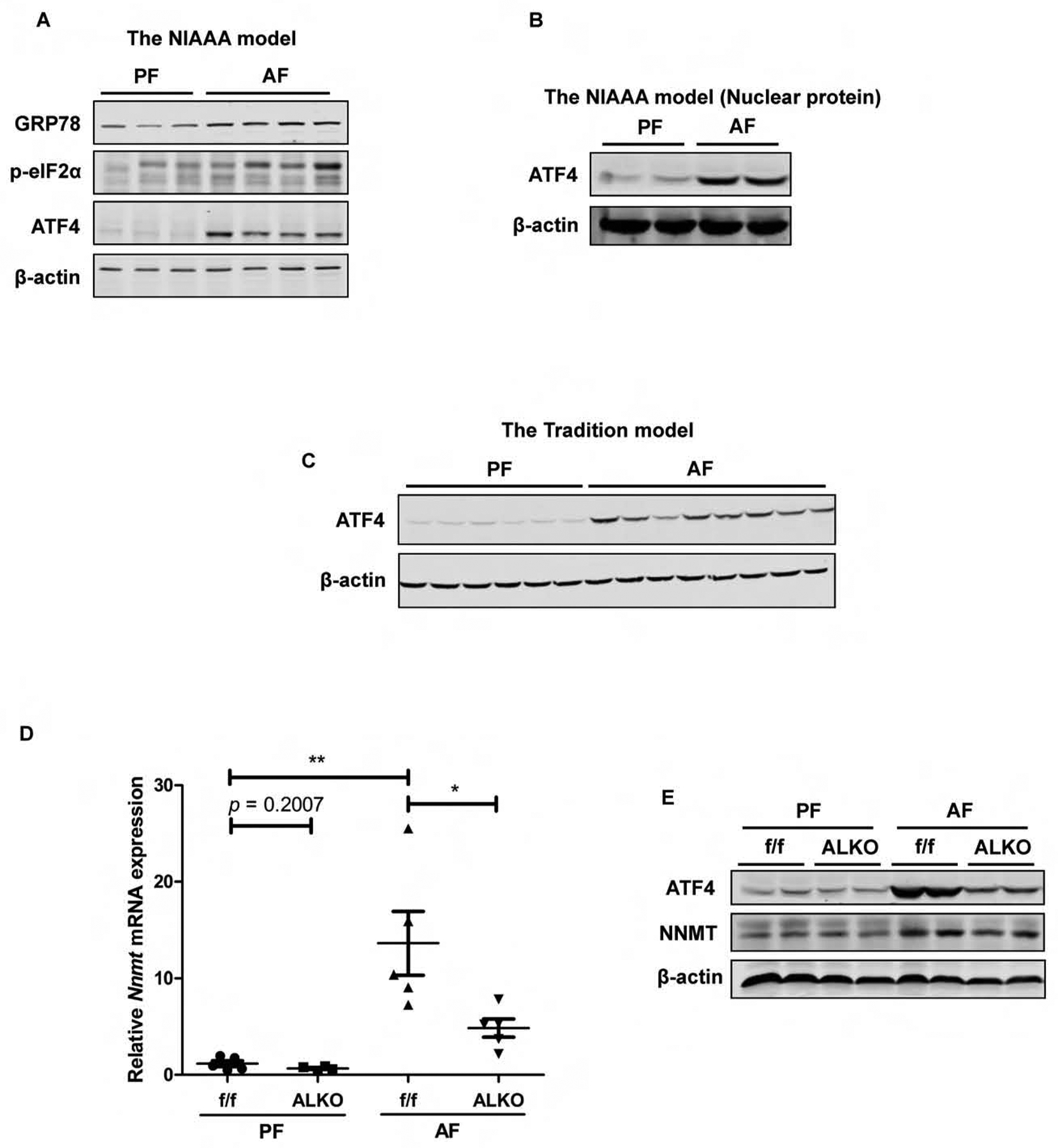
The PERK-ATF4 pathway activation contributes to hepatic NNMT upregulation in the setting of chronic alcohol consumption. (A) Chronic alcohol exposure activates hepatic PERK-ATF4 pathway in the NIAAA model of ALD. (B) Chronic alcohol exposure increases nuclear ATF4 protein abundance in in the NIAAA model of ALD. (C) Chronic alcohol exposure increases hepatic ATF4 protein abundance in the tradition model of ALD. (D & E) The liver-specific ATF4 knockout (ALKO) mice manifests blunted NNMT upregulation in response to chronic alcohol feeding in the tradition model of ALD. Data are expressed as mean ± SEM (n = 5–8). Differences between 2 groups were determined using Student’s t test (*p < 0.05, **p < 0.01 versus ATF4flox/flox (f/f)/PF mice).
NNMT inhibition protects against alcohol-induced fatty liver development
The potential pathological role of hepatic NNMT overexpression in ALD was subsequently determined using the NIAAA model via either adenoviral knockdown of NNMT expression or NNMT inhibitor (JBSNF-000088) administration (ip injection). Retro-orbital injection of Ad-GFP-U6-m-NNMT-shRNA resulted in a significant downregulation of NNMT expression in the liver (Fig. 6A). As shown in Fig. 6B–D, neither alcohol feeding nor NNMT knockdown affected plasma TAG, total cholesterol, and glucose concentrations. Chronic alcohol exposure resulted in a significant increase of liver fat contents in control animals with empty vector delivery, as confirmed by both biochemical measurement and histological examination; however, the adenoviral knockdown of NNMT expression ameliorated fatty liver development in response to alcohol exposure (Fig. 6E & F). The similar effects were observed when NNMT inhibitor was administered (Fig. 6 G & H). A TG-reducing action of NNMT inhibitor in AML12 cells was also observed (Fig. 6I). These data altogether suggest that NNMT overexpression contributes to alcohol-induced fatty liver development.
Figure 6.
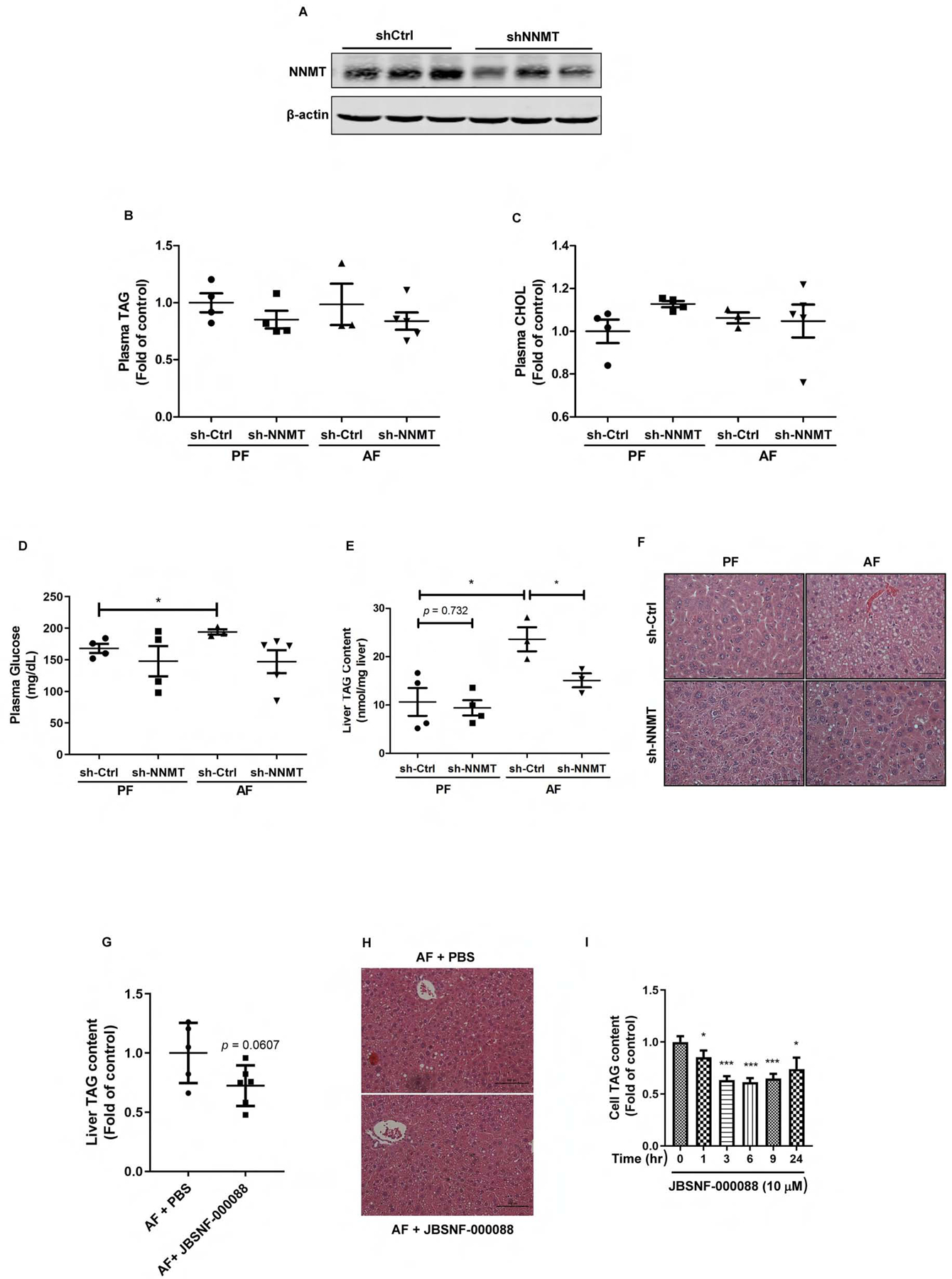
NNMT inhibition protects against alcoholic fatty liver development. Ten-week old male C57BL/6N mice were retro-orbitally injected with either Ad-GFP-U6-m-NNMT-shRNA or corresponding control vector, followed by the NIAAA model regimen. (A) Hepatic NNMT protein abundance; (B) Plasma TAG levels, (C) Plasma total cholesterol levels; (D) Plasma glucose levels; (E) The liver TAG levels. (F) H&E staining of liver tissues. Ten-week old male C57BL/6N mice were injected (ip) with either PBS or JBSNF-00008, a chemical inhibitor of NNMT, twice a day in the NIAAA model regimen. (G) The liver TAG levels; (H) H&E staining of liver tissues. AML12 cells were treated with JBSNF-00008 (10 μM), a chemical inhibitor of NNMT, for indicated time durations. (I) Intracellular TAG levels were measured. Data are expressed as mean ± SEM (n = 5–8). Differences between groups were determined using one-way ANOVA analysis (*p < 0.05 versus control vector/PF mice).
Neither alcohol nor NNMT knockdown affects adipose tissue lipolysis in the NIAAA model of ALD
Both enhanced adipose tissue lipolytic response and hepatic de novo lipogenesis pathway activation was reported to contribute to hepatic steatosis in the setting of chronic alcohol consumption.29–34 To investigate mechanism(s) underlying NNMT knockdown-conferred protection against alcoholic fatty liver development, we first examined the effect of NNMT knockdown on adipose tissue lipolysis in the NIAAA model through the measurement of circulatory glycerol and free fatty acids levels. Comparable plasma glycerol and free fatty acids levels were observed between control (empty vector-injected) and NNMT knockdown mice no matter whether ethanol was included in the diet or not (Fig. 7A & B). Moreover, in control mice injected with empty vector, alcohol-feeding had no effect on two adipose tissue lipolysis parameters when compared with isocaloric pair-fed animals (Fig. 7A & B), suggesting that adipose tissue lipolysis plays a minimal role in the development of fatty liver in the NIAAA model of ALD.
Figure 7.
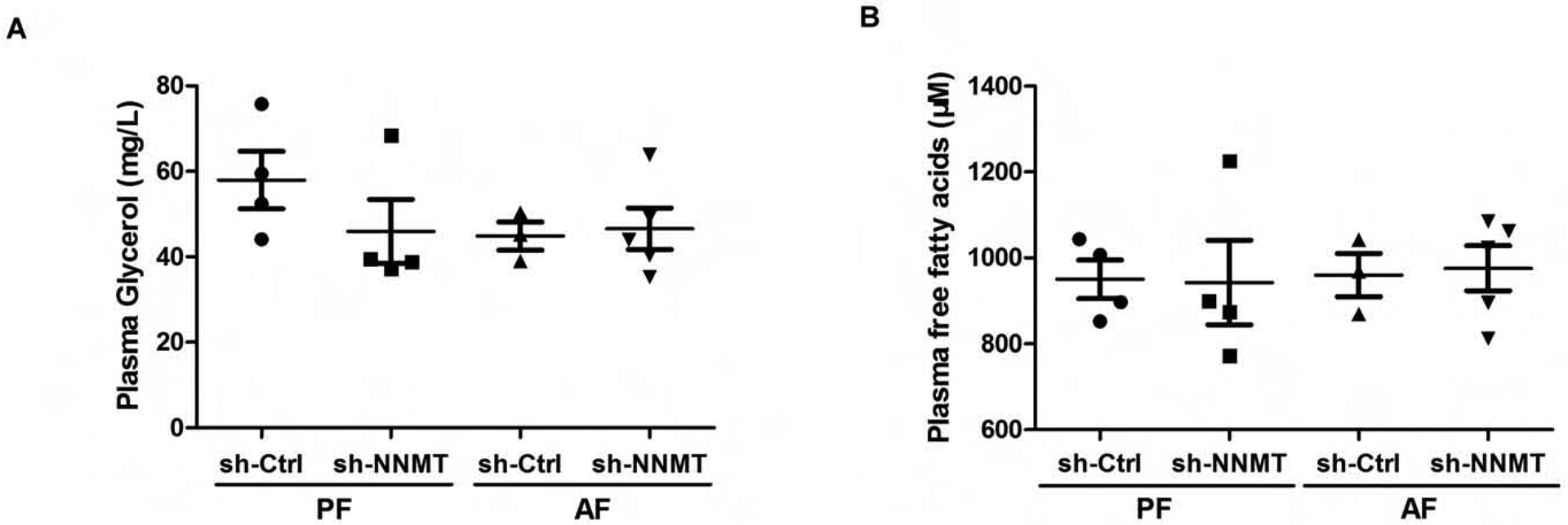
Neither alcohol consumption nor NNMT adenoviral knockdown influences adipose tissue lipolysis. Ten-week old male C57BL/6N mice were retro-orbitally injected with either Ad-GFP-U6-m-NNMT-shRNA or corresponding control vector, followed by the NIAAA model regimen. (A) The plasma glycerol levels. (B) Plasma free fatty acids levels. Data are expressed as mean ± SEM (n = 5–8). Differences between groups were determined using one-way ANOVA analysis (*p < 0.05 versus control vector/PF mice).
NNMT inhibition suppresses hepatic de novo lipogenic activity
We next examined whether the de novo lipogenesis pathway in the liver could be affected by NNMT inhibition. To test this, an array of genes involved in the de novo lipogenesis pathway were measured using liver tissues from the NIAAA model of ALD. As shown in Fig. 8A & B, the livers of alcohol-fed mice with NNMT inhibition, through both genetical and pharmacological approach, manifested a significant reduction of several key genes participated in the de novo lipogenic pathway when comparing with these in alcohol-fed control mice. The similar phenomena were observed when we retrospectively examined the effects of NNMT shRNA knockdown on de novo lipogenic genes in the tunicamycin-treated mice livers (Fig. 8C). To further substantiate these in vivo observations, we performed in vitro cell culture experiments to directly determine the effects of NNMT siRNA knockdown on de novo lipogenesis and on ethanol-induced intracellular TAG accumulation. AML12 cells were transfected with either scrambled siRNA (si-Ctrl) or NNMT siRNA (si-NNMT) for overnight, followed by the exposure with/without ethanol (200 mM)-containing media for another 12 hours. The effects of NNMT knockdown on de novo lipogenesis gene expression as well as on ethanol-induced TAG accumulation were examined. As shown in Fig. 8D, siRNA knockdown of NNMT led to a significant reduction of several key genes involved in the de novo lipogenesis pathway. Importantly, NNMT siRNA knockdown attenuated ethanol-elicited intracellular TAG accumulation (Fig. 8E). Similarly, we also observed that NNMT siRNA knockdown ameliorated intracellular TAG accumulation induced by oleic acid exposure (Fig. 8F).
Figure 8.
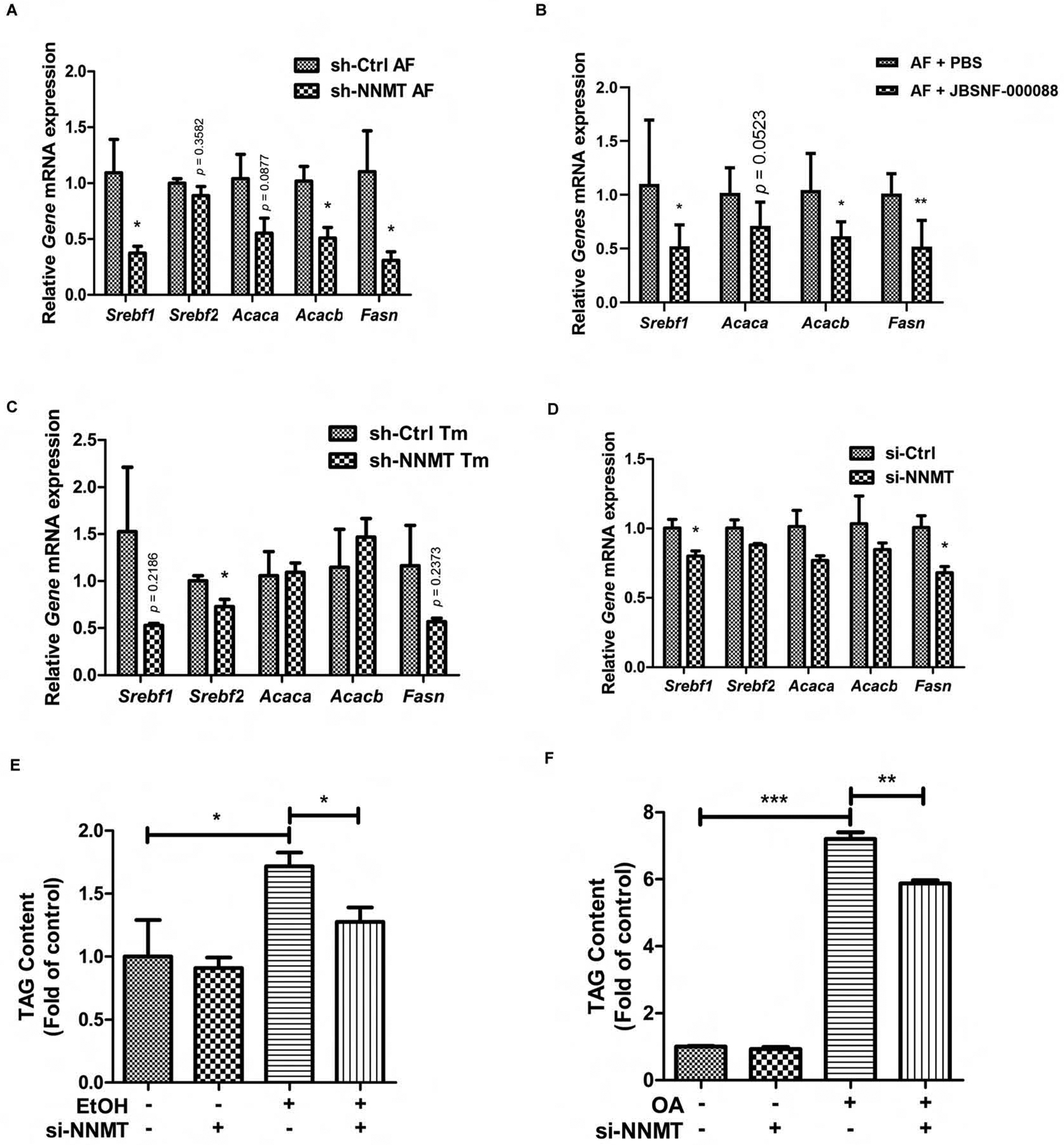
NNMT inhibition, via both genetic and pharmacological approaches, inhibits de novo lipogenesis in hepatocytes and the liver. Ten-week old male C57BL/6N mice were either retro-orbitally injected with Ad-GFP-U6-m-NNMT-shRNA or injected with JBSNF-00008 twice a day, followed by either the NIAAA model regimen or acute ER stress induction with tunicamycin (Tm) injection. (A & B) Hepatic expression of genes involved in de novo lipogenesis pathway in the NIAAA model of ALD. (C) Hepatic expression of genes involved in de novo lipogenesis pathway in acute ER stress mice model. AML12 hepatocytes were transfected with either si-control or si-NNMT for overnight, followed by either ethanol (200 mM) or oleic acid (OA, 0.4 mM) exposure for 12 hours. (D) Expression of genes invovled in de novo lipogenesis pathway in response to ethanol exposure. (E) Intracellular TAG concentrations in response to ethanol exposure. (F) Intracellular TAG concentrations in response to oleic acid exposure. Data are expressed as mean ± SEM. Differences between 2 groups were determined using Student’s t test (*p < 0.05, **p < 0.01, ***p < 0.001 versus corresponding control).
The inhibitory effect of NNMT inhibition on de novo lipogenic gene expressions is NAD+-independent
Given the critical involvement of NNMT in maintaining cellular nicotinamide and NAD+ homeostasis, it is conceivable to postulate that NNMT inhibition may suppress de novo lipogenesis pathway via activating the NAD+-Sirt1 pathway. To test this hypothesis, we first examined the effect of NNMT inhibition on hepatic NAD+ concentrations. NNMT inhibition via both genetic and pharmacological approach led to an increment of hepatic NAD+ contents (Fig. 9 A & B), whereas NNMT overexpression in AML12 cells was associated with declined intracellular NAD+ levels (Fig. 9C). In attempt to determine whether incremental hepatic NAD+ levels is attributable to the observed suppressive effect of NNMT inhibition on de novo lipogenic genes via upregulating/activating Sirt1, exogenous nicotinamide riboside (NR), a potent NAD+ -enhancing agent, was injected (ip) to male C57 BL/6N mice. The liver samples were harvested 24 hours later. NR administration resulted in a significant reduction of liver TG contents (Fig. 9D), which was associated increased hepatic NAD+ levels (Fig. 9E & F). Unexpectedly, despite a significant increase of cellular NAD+ levels, neither Sirt1 expression (at both mRNA and protein level) nor its activities were altered by NR administration (Fig. 9G & H). Similarly, NR administration had no effects on lipogenic genes in comparison to these in control animals (Fig. 9I). Intriguingly, a robust increase of p-AMPK protein abundance was observed in response to NR administration (Fig. 9J). To substantiate this observation, we treated AML12 cells with NR directly and examined AMPK activation status via Western blotting for p-AMPK. NR treatment elevated intracellular NAD contents (Fig. 9K). Whereas a dose-dependent effect of NR on AMPK activation was observed (Fig. 9L), NR treatment failed to alter Sirt1 expression. Furthermore, the time-course effect of NNMT inhibitor on AMPK activation was detected in AML12 hepatocytes (Fig. 9M).
Figure 9.
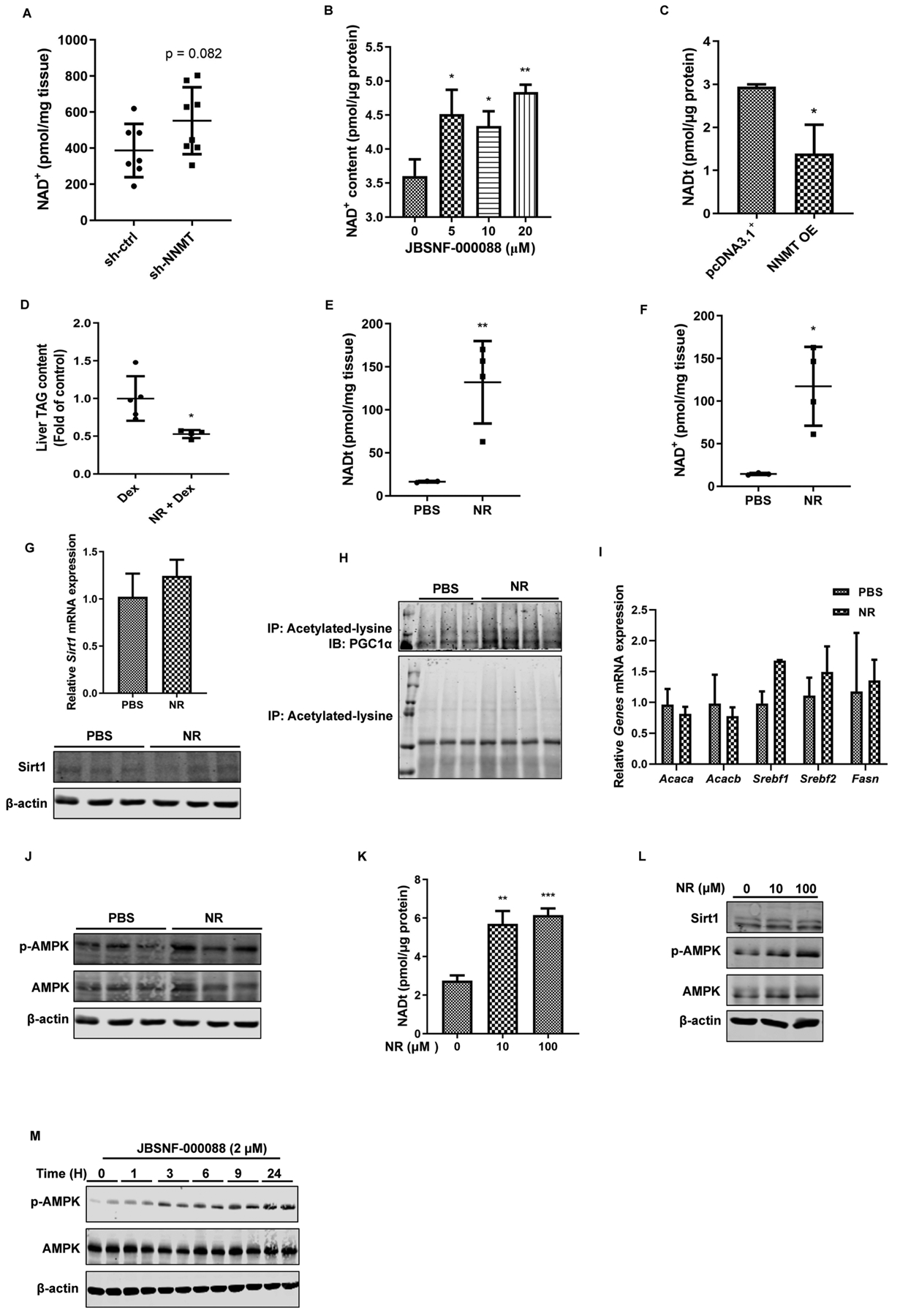
The inhibitory effect of NNMT inhibition on de novo lipogenic gene expressions is NAD+- independent. Ten-week old male C57BL/6N mice were retro-orbitally injected with either Ad-GFP-U6-m-NNMT-shRNA or control shRNA, followed by either the NIAAA model regimen. Hepatic NAD+ levels were measured (A). (B) AML12 cells were treated with JBSNF-00008 at indicated doses for 16 hours. Intracellular NAD+ levels were measured. (C) NNMT overexpression in AML12 hepatocytes resulted in declined intracellular NAD+ contents. Ten-week old male C57BL/6N mice received a single dose of 400 mg/kg BW nicotinamide ribose (NR) through ip injection. After 24 hours, the mice were sacrificed, followed by collection of the plasma and liver samples. (D) Liver TAG contents. (E & F) Hepatic total NAD and NAD+ levels. (G) Hepatic Sirt1 expression at both mRNA and protein levels. (H) Hepatic PGC1α acetylation status. (I) Liver lipogenic gene expressions. (J) Liver p-AMPK and total AMPK protein abundance. AML12 cells were treated with either NR or JBSNF-00008 for indicated doses and time durations. (K) Effects of NR on intracellular total NAD contents. (L) Effects of NR on Sirt1 protein expression and AMPK activation. (M) Time-course effects of JBSNF-00008 on AMPK activation. Data are expressed as mean ± SEM. Differences between 2 groups were determined using Student’s t test (*p < 0.05, **p < 0.01, ***p < 0.001 versus corresponding control).
Discussion
In the present study, we provide initial evidence that hepatic NNMT upregulation contributes to alcoholic fatty liver development. We demonstrate that alcohol consumption increases hepatic NNMT expression and activity via ER stress induction and the PERK-ATF4 pathway plays a mechanistic role in both ER stress- and alcohol-mediated NNMT upregulation. NNMT inhibition ameliorates alcoholic fatty liver development, which is associated with incremental cellular NAD+ concentrations and de novo lipogenesis pathway inhibition. Intriguingly, the inhibitory effects of NNMT inhibition on de novo lipogenic gene expressions appear to be NAD+-independent.
NNMT catalyzes SAM-dependent methylation/degradation of nicotinamide (Fig. 1A) and is emerging to be a novel metabolic regulator.29,30 Although several signal pathways and transcription factors, including STAT3 and HNF-1β, have been reported to regulate NNMT gene expression,31,32 the exact regulatory mechanism(s) remains to be ambiguous. ER stress plays a central role in a variety of metabolic disorders, including alcoholic fatty liver diseases.33,34 We found in this study: 1. ER stress upregulates hepatic NNMT expression; 2. PERK positively controls NNMT expression; 3. ATF4 knockdown prevents ER stress-triggered NNMT upregulation; 4. the liver-specific ATF4 knockout mice express lower basal hepatic NNMT expression levels than their littermates; 5. ATF4 binds to NNMT promoter region to stimulate its transcription. To the best of our knowledge, we are the first to report that ER stress upregulates hepatic NNMT expression via the PERK-ATF4 pathway activation.
In line with many previous reports,19,27,35 we showed that alcohol-induced fatty liver development is associated with a profound ER stress induction in the liver. As expected, a robust hepatic NNMT upregulation was observed in two well-established ALD mouse models. ER stress contributes to alcoholic fatty liver disease;36 however, the exact mechanisms remain to be completely clarified. All three canonical pathways of UPR have been documented to contribute to the development of fatty liver.37–39 Whereas the PERK-ATF4 branch activation led to TAG accumulation via upregulating de novo lipogenesis,40 ATF4 deficiency ameliorated both age-related and diet-induced fatty liver development.28,41 To gain insight into the pathophysiological role of NNMT upregulation in ER stress-associated fatty liver development, we investigated the effect of NNMT adenoviral knockdown on fat accumulation in both ER stress mouse model (tunicamycin administration) and the NIAAA mouse model of ALD. Our results clearly showed that NNMT knockdown attenuated hepatic fat accumulation induced by both tunicamycin and alcohol, suggesting that hepatic NNMT upregulation contributes to the pathogenesis of alcoholic fatty liver development. This notion was further consolidated by the observation that JBSNF-000088, a chemical inhibitor of NNMT, protected against alcoholic fatty liver development in the NIAAA mouse model of ALD.
Incremental adipose tissue fatty acids release and liver de novo lipogenesis activation represent important pathological factors implicating in ALD pathogenesis.42,43 Chronic alcohol consumption enhanced adipose tissue lipolysis and the amelioration of which prevented mice from developing fatty liver.44,45 Although NNMT is also abundantly expressed in adipose tissue, our data didn’t support that the beneficial effects of NNMT inhibition on ALD are derived from its impact on adipose tissue lipolysis in that: 1. unlike the liver, adenoviral delivery of NNMT shRNA via retro-orbital injection only minimally decreased NNMT expression in adipose tissue; 2. no obviously enhanced adipose tissue lipolysis in response to alcohol feeding was observed in the NIAAA model of ALD; 3. hepatic NNMT knockdown did not affect adipose tissue lipolytic response in the setting of alcohol feeding. Instead, we uncovered that hepatic NNMT inhibition was associated with the downregulation of an array of genes involved in the de novo lipogenesis pathway. This observation was further confirmed in cell culture experiments in which we demonstrated that NNMT siRNA knockdown in AML12 cells prevented ethanol-induced fat accumulation, concomitant with downregulation of several genes in the de novo lipogenesis pathway, suggesting that the inhibition of hepatic de novo lipogenesis contributes to the ameliorative effect of NNMT downregulation on fatty liver development.
As illustrated in Fig. 1A, NNMT locates at the crossroad of methionine/SAM and nicotinamide/NAD+ metabolism. Therefore, altered NNMT expression could potentially have an impact on cellular concentration of NAD+, whose biosynthesis utilizes nicotinamide as a predominant precursor, and/or cellular methylation status, potentially resulting from depleted cellular SAM contents. Intracellular NAD+ is a physiological activator of SIRT1, which serves as a suppressor of SREBP-1c via deacetylation reactions.46 Supplementation of exogenous NAD+ enhancers, such as nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR), prevented fatty liver development via activating SIRT1, at least partially involving suppressed SREBP-1c activation.10,47 While, as expected, our data confirmed that NNMT is a negative regulator of cellular NAD+ contents, our studies using nicotinamide riboside (NR) in both mice and hepatocytes did not support the notion that cellular NAD+ elevation is obligatory to induce Sirt1 expression and activation. Intriguingly, we unraveled that the incremental cellular NAD+ levels were associated with a robust AMPK activation. Therefore, it is conceivable to conclude that both inhibitory de novo lipogenesis and enhanced fatty acids oxidation due to AMPK activation may account for the beneficial effects of NNMT inhibition in ALD.
NNMT overexpression is associated with a reduced intracellular methylation status.3,29 Methylation is critical for the synthesis of phosphatidylcholine, a major membrane component. A recent study elegantly demonstrated that a reduced cellular SAM concentration is associated with elevated SREBP-1-dependent de novo lipogenesis pathway activation and lipid droplet accumulation in C. elegans, mouse liver, and human cells via a feedback mechanism whereby transcriptionally active SREBP-1 is controlled by cellular PC levels.48 The NAD+ - independent de novo lipogenesis pathway suppression by NNMT inhibition suggests that an alteration in SAM-regulated methylation reactions may be invovled in this process. Further research is warranted to delineate and clarify the precise contribution of this pathway in the beneficial effect of downregulating NNMT on the prevention of alcoholic fatty liver disease.
In conclusion, in this study we provide original experimental evidence that the PERK-ATF4 pathway activation plays a mechanistic role in alcohol-induced hepatic NNMT overactivation and hepatic NNMT upregulation plays a pathological role in the development of alcoholic fatty liver diseases. Given that ER stress plays a critical role in many types of metabolic disorders and NNMT is abundantly expressed in a variety of metabolically active cell types, our findings underscores the potential mechanistic implication of ER stress-triggered NNMT overexpression in these pathological conditions. Our results suggest that targeting NNMT can be a potential therapeutic choice for the treatment of ALD as well as other metabolic disorders, with ER stress being the principal pathomechanism.
Supplementary Material
Highlights.
Chronic alcohol consumption upregulates NNMT expression and activity in the liver.
The activation of the PERK-ATF4 pathway of UPR contributes to alcohol-induced hepatic NNMT upregulation.
Adenoviral knockdown of NNMT protects against fatty liver development in response to both ER stress and chronic alcohol exposure.
NNMT inhibition enhances cellular NAD+ levels.
NNMT knockdown suppresses de novo lipogenesis pathway in hepatocytes and the liver.
Acknowledgments:
We thank Christopher M. Adams (Department of Internal Medicine and Molecular Physiology and Biophysics and Fraternal Order of Eagles Diabetes Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa) for kindly providing the ATF4flox/flox mice.
Financial support statement:
This work was in part funded by US NIH Grants NIAAA R21AA025363 (to Z. S.) and NIAAA R01AA026603 (to Z. S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: No
References:
- [1].Osna NA, Donohue TM Jr., Kharbanda KK. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res 2017;38:147–161. [PMC free article] [PubMed] [Google Scholar]
- [2].Pissios P Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol Metab 2017;28:340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Komatsu M, Kanda T, Urai H, Kurokochi A, Kitahama R, Shigaki S, et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD (+) metabolism. Sci Rep 2018;8:8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kannt A, Pfenninger A, Teichert L, Tonjes A, Dietrich A, Schon MR, et al. Association of nicotinamide-N-methyltransferase mRNA expression in human adipose tissue and the plasma concentration of its product, 1-methylnicotinamide, with insulin resistance. Diabetologia 2015;58:799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014;508:258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Neelakantan H, Vance V, Wetzel MD, Wang HL, McHardy SF, Finnerty CC, et al. Selective and membrane-permeable small molecule inhibitors of nicotinamide N-methyltransferase reverse high fat diet-induced obesity in mice. Biochem Pharmacol 2018;147:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brachs S, Polack J, Brachs M, Jahn-Hofmann K, Elvert R, Pfenninger A, et al. Genetic Nicotinamide N-Methyltransferase (Nnmt) Deficiency in Male Mice Improves Insulin Sensitivity in Diet-Induced Obesity but Does Not Affect Glucose Tolerance. Diabetes 2019;68:527–542. [DOI] [PubMed] [Google Scholar]
- [8].Ulanovskaya OA, Zuhl AM, Cravatt BF. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol 2013;9:300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 2008;295:G833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang S, Wan T, Ye M, Qiu Y, Pei L, Jiang R, et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1alpha/mitochondrial biosynthesis pathway. Redox Biol 2018;17:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mukhopadhyay P, Horvath B, Rajesh M, Varga ZV, Gariani K, Ryu D, et al. PARP inhibition protects against alcoholic and non-alcoholic steatohepatitis. J Hepatol 2017;66:589–600. [DOI] [PubMed] [Google Scholar]
- [12].Esfandiari F, You M, Villanueva JA, Wong DH, French SW, Halsted CH. S-adenosylmethionine attenuates hepatic lipid synthesis in micropigs fed ethanol with a folate-deficient diet. Alcohol Clin Exp Res 2007;31:1231–1239. [DOI] [PubMed] [Google Scholar]
- [13].Li Q, Xie G, Zhang W, Zhong W, Sun X, Tan X, et al. Dietary nicotinic acid supplementation ameliorates chronic alcohol-induced fatty liver in rats. Alcohol Clin Exp Res 2014;38:1982–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007;8:519–529. [DOI] [PubMed] [Google Scholar]
- [15].Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081–1086. [DOI] [PubMed] [Google Scholar]
- [16].Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol 2011;54:795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol 2012;22:R622–626. [DOI] [PubMed] [Google Scholar]
- [18].Kaplowitz N, Than TA, Shinohara M, Ji C. Endoplasmic reticulum stress and liver injury. Semin Liver Dis 2007;27:367–377. [DOI] [PubMed] [Google Scholar]
- [19].Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003;124:1488–1499. [DOI] [PubMed] [Google Scholar]
- [20].Longato L, Ripp K, Setshedi M, Dostalek M, Akhlaghi F, Branda M, et al. Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxid Med Cell Longev 2012;2012:479348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang Z, Dou X, Li S, Zhang X, Sun X, Zhou Z, et al. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology 2014;59:1381–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang X, Wang Z, Li J, Gu D, Li S, Shen C, et al. Increased 4-hydroxynonenal formation contributes to obesity-related lipolytic activation in adipocytes. PLoS One 2013;8:e70663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A 1990;87:2466–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Parodi AJ. Role of N-oligosaccharide endoplasmic reticulum processing reactions in glycoprotein folding and degradation. Biochem J 2000;348 Pt 1:1–13. [PMC free article] [PubMed] [Google Scholar]
- [26].Jo H, Choe SS, Shin KC, Jang H, Lee JH, Seong JK, et al. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 2013;57:1366–1377. [DOI] [PubMed] [Google Scholar]
- [27].Ji C Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J Gastroenterol Hepatol 2008;23 Suppl 1:S16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Seo J, Fortuno ES 3rd, Suh JM, Stenesen D, Tang W, Parks EJ, et al. Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes 2009;58:2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Eckert MA, Coscia F, Chryplewicz A, Chang JW, Hernandez KM, Pan S, et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 2019;569:723–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hong S, Moreno-Navarrete JM, Wei X, Kikukawa Y, Tzameli I, Prasad D, et al. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat Med 2015;21:887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tomida M, Ohtake H, Yokota T, Kobayashi Y, Kurosumi M. Stat3 up-regulates expression of nicotinamide N-methyltransferase in human cancer cells. J Cancer Res Clin Oncol 2008;134:551–559. [DOI] [PubMed] [Google Scholar]
- [32].Xu J, Capezzone M, Xu X, Hershman JM. Activation of nicotinamide N-methyltransferase gene promoter by hepatocyte nuclear factor-1beta in human papillary thyroid cancer cells. Mol Endocrinol 2005;19:527–539. [DOI] [PubMed] [Google Scholar]
- [33].Cao SS, Kaufman RJ. Targeting endoplasmic reticulum stress in metabolic disease. Expert Opin Ther Targets 2013;17:437–448. [DOI] [PubMed] [Google Scholar]
- [34].Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 2017;13:477–491. [DOI] [PubMed] [Google Scholar]
- [35].Ji C Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem Res Int 2012;2012:216450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lee JS, Zheng Z, Mendez R, Ha SW, Xie Y, Zhang K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol Lett 2012;211:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zheng Z, Zhang C, Zhang K. Role of unfolded protein response in lipogenesis. World J Hepatol 2010;2:203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].DeZwaan-McCabe D, Sheldon RD, Gorecki MC, Guo DF, Gansemer ER, Kaufman RJ, et al. ER Stress Inhibits Liver Fatty Acid Oxidation while Unmitigated Stress Leads to Anorexia-Induced Lipolysis and Both Liver and Kidney Steatosis. Cell Rep 2017;19:1794–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008;320:1492–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lauressergues E, Bert E, Duriez P, Hum D, Majd Z, Staels B, et al. Does endoplasmic reticulum stress participate in APD-induced hepatic metabolic dysregulation? Neuropharmacology 2012;62:784–796. [DOI] [PubMed] [Google Scholar]
- [41].Xiao G, Zhang T, Yu S, Lee S, Calabuig-Navarro V, Yamauchi J, et al. ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J Biol Chem 2013;288:25350–25361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Steiner JL, Lang CH. Alcohol, Adipose Tissue and Lipid Dysregulation. Biomolecules 2017;7. [Google Scholar]
- [43].You M, Arteel GE. Effect of ethanol on lipid metabolism. J Hepatol 2019;70:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dou X, Xia Y, Chen J, Qian Y, Li S, Zhang X, et al. Rectification of impaired adipose tissue methylation status and lipolytic response contributes to hepatoprotective effect of betaine in a mouse model of alcoholic liver disease. Br J Pharmacol 2014;171:4073–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhou Z, Wang L, Song Z, Saari JT, McClain CJ, Kang YJ. Zinc supplementation prevents alcoholic liver injury in mice through attenuation of oxidative stress. Am J Pathol 2005;166:1681–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M, et al. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem 2010;285:33959–33970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Uddin GM, Youngson NA, Sinclair DA, Morris MJ. Head to Head Comparison of Short-Term Treatment with the NAD(+) Precursor Nicotinamide Mononucleotide (NMN) and 6 Weeks of Exercise in Obese Female Mice. Front Pharmacol 2016;7:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Walker AK, Jacobs RL, Watts JL, Rottiers V, Jiang K, Finnegan DM, et al. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 2011;147:840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.