

Abstract
As apex predators, pinnipeds are considered to be useful bioindicators of marine and coastal environments. Endemic to a small archipelago in the South Pacific, the Juan Fernandez fur seal (JFFS) is one of the less‐studied members of the pinniped family Otariidae. This study aimed to characterize the fecal microbiome of the JFFS for the first time, to establish a baseline for future studies of host–microbial–environment interactions and monitoring programs. During two consecutive reproductive seasons, 57 fecal samples were collected from seven different JFFS colonies within the Juan Fernandez Archipelago, Chile. Bacterial composition and abundance were characterized by sequencing the V4 region of the 16S rRNA gene. The overall microbiome composition was dominated by five phyla: Firmicutes (40% ±24), Fusobacteria (30% ±17), Bacteroidetes (22% ±10), Proteobacteria (6% ±4), and Actinobacteria (2% ±3). Alpha diversity was higher in Tierras Blancas. However, location was not found to be a dominant driver of microbial composition. Interestingly, the strongest signal in the data was a negative association between the genera Peptoclostridium and Fusobacterium, which explained 29.7% of the total microbial composition variability between samples. The genus Peptoclostridium has not been reported in other pinniped studies, and its role here is unclear, with interpretation challenging due to a lack of information regarding microbiome functionality in marine mammals. As a first insight into the JFFS fecal microbiome, these results contribute towards our understanding of the natural microbial diversity and composition in free‐ranging pinnipeds.
Keywords: Arctophoca philippii, marine mammals, microbiome, pinnipeds, scatology
Two distinct microbial patterns were identified in Juan Fernandez fur seal fecal samples. We suspect that sex, age, and especially diet are most likely to be influencing the observed patterns.
![]()
1. INTRODUCTION
Marine environments are complex and interconnected systems subject to various environmental impacts. Pollution, climate change, disruption of the food network, and pathogen dissemination are a few examples of problems currently affecting ocean integrity and function (Halpern et al., 2019). Integrated approaches at the macro‐ and micro‐ecological levels are needed to properly understand and manage environmental threats in these kinds of complex systems. Identification and investigation of potential environmental sentinel species such as marine mammals can provide a better understanding of the deterioration or improvement of ocean health (Bossart, 2011; Hazen et al., 2019). However, to effectively use wild populations as sentinels, it is first necessary to establish a baseline.
In the last couple of decades, the study of the microbiome in wild populations has increased, due to the profound impact of host–microbial interactions on host physiology and the growing affordability of sequencing technology (Redford et al., 2012; Trevelline et al., 2019). The gastrointestinal tract, especially the colon, is recognized as one of the largest microbial reservoirs (O’Hara & Shanahan, 2006). This microbial community fulfills essential functions in digestion, metabolic activity, and immunity, and differences in species composition and abundance can therefore provide much information about the host organism. For example, following its initial acquisition during birth and lactation, the microbiome is constantly modified by factors such as age, sex, and diet (Ley et al., 2008a, 2008b; Nicholson et al., 2012). Similar factors shaping the gut microbiome in terrestrial mammals influence that of marine mammals (Nelson et al., 2013; Pacheco‐Sandoval et al., 2019; Smith et al., 2013; Stoffel et al., 2020). However, studies have also shown substantial differences between marine and terrestrial mammal gut microbiomes, even when these two groups share a similar diet (e.g., herbivore, carnivore) (Bik et al., 2016; Nelson et al., 2013). Thus, even though research into the microbiome of terrestrial mammals is at a relatively advanced stage, this information cannot be easily extrapolated to marine mammals whose microbiomes remain poorly understood particularly, those in non‐captive, natural populations. Consistent characterization of the core microbiome of these populations is therefore required as a fundamental baseline before we can attempt to understand its functions, roles, interactions, and possible uses (Shade & Handelsman, 2012).
The fecal microbiome has been characterized for eight pinniped species inhabiting the southern hemisphere, including three out of the eleven members forming the subfamily Arctocephalinae (fur seals): Arctocephalus pusillus doriferus (Smith et al., 2013), Arctocephoca australis, and Arctophoca tropicalis (Medeiros et al., 2016). Also part of the Arctocephalinae subfamily is the Juan Fernandez fur seal (Arctophoca philippii philippii) (JFFS) which is endemic to the Juan Fernandez Archipelago, a group of islands located in the middle of the Pacific Ocean 600 km away from the Chilean continental coast (Figure 1). The archipelago is a hotspot for biodiversity with a high number of endemic marine species, including the JFFS (Aguayo et al., 1971; Friedlander et al., 2016; Pompa et al., 2011). These fur seals are the only native mammals in the archipelago and, like other pinnipeds, occupy upper trophic levels in the marine food web (Ochoa Acuña & Francis, 1995; Trites, 2019). Their feeding behavior, lifespan, fat storage, and their amphibian lifestyle, which links marine and coastal environments, are some of the characteristics that make this species a great candidate to act as a marine bioindicator. However, despite showing a significant population recovery since the late 1960s and becoming an icon for local tourism, little is known about this species.
FIGURE 1.

Juan Fernandez fur seal (Arctophoca philippii philippii)
This study aimed to characterize the JFFS fecal microbiome for the first time, as a baseline for understanding the host–microbial interactions in this species. To investigate, we performed sequencing of the 16S rRNA gene, a highly conserved region of the bacterial genome, which provides a reliable overview of bacterial community composition.
2. METHODS
2.1. Sample collection
Fecal samples were collected from seven reproductive colonies of Juan Fernandez fur seals situated throughout the Juan Fernandez archipelago, Chile (coordinates: 33°38′29″S 78°50′28″W) (Figure 2). Six of the seven colonies included in this study were located on Robinson Crusoe Island: El Arenal (EA) (n = 9), Bahia El Padre (BP) (n = 23), Piedra Carvajal (PC) (n = 1), Punta Trueno (PT) (n = 1), Tierras Blancas (TB) (n = 12), and Vaquería (V) (n = 1). One colony was located on Santa Clara Island (SC). Samples were collected during two consecutive reproductive seasons (2017 and 2018), which took place between mid‐January to the end of February. Collection of samples took place before noon to limit sun exposure. The samples were collected based on consistency and color to reduce the variability caused by the delay between the defecation and collection. A disposable wooden spatula was used to expose the center of the feces to avoid collecting material in direct contact with the surrounding elements. Using a sterile Copan FLOQSwab®, a sample from the core of the feces was placed into RNAlater® (Sigma‐Aldrich) (Blekhman et al., 2016; Vlčková et al., 2012). No animal was observed defecating. Thus, it was not possible to distinguish sex or age at the time of sample collection. We used visual cues and GPS location to decrease the risk of collection from the same individual. Samples were stored at −20°C within 32 h post collection for 1–2 months until arrival in the laboratory, where they were transferred to −80°C until further analysis.
FIGURE 2.
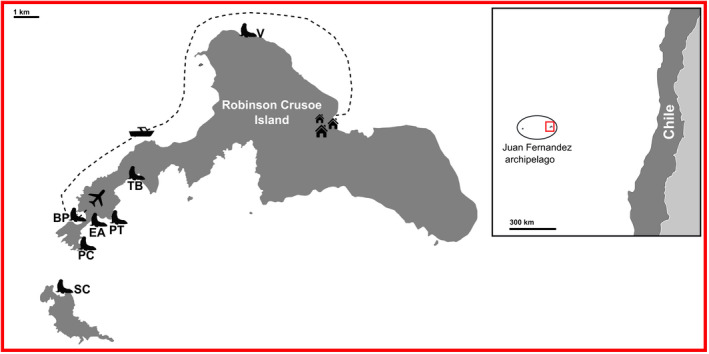
Simplified map of Robinson Crusoe and Santa Clara Islands. The plane indicates the airfield and the dotted line the access route from the airfield to San Juan Bautista Village (the only settlement on the island). Fur seal icons show the sampling locations. El Arenal (EA) (n = 9), Bahia El Padre (BP) (n = 23), Piedra Carvajal (PC) (n = 1), Punta Trueno (PT) (n = 1), Santa Clara (SC) (n = 12), Tierras Blancas (TB) (n = 10), and Vaqueria (V) (n = 1). 57 samples in total
2.2. DNA extraction and sequencing
Samples were processed as soon as possible after collection (2017 and 2018, respectively). Due to the possible batch effect introduced by processing samples in different years, comparisons between years of the collection will not be explored in this study.
Samples were thawed on ice and centrifuged at 10,000 g for 15 min to pellet the sample out of RNAlater®. Genomic DNA was extracted from each pelleted sample (approximately 180 micrograms) using the MO BIO PowerSoil DNA Isolation kit (QIAGEN) according to the manufacturer's instructions. Isolated DNA was quantified on a Qubit fluorometer (Invitrogen).
The bacterial 16S rRNA gene was polymerase chain reaction (PCR) amplified targeting a 250 bp region covering the V4 variable region. PCR amplification, barcode tagging, and library preparation were performed according to Kozich et al. (2013). Libraries were constructed using the TrueSeq DNA kit and sequenced on a MiSeqTM platform (Illumina®). The read length target changed between the two sampling years. Sequencing was performed using the v2 chemistry producing 2 × 250 bp paired‐end reads in the 2017 samples while the 2018 sequences were 2× 150 bp paired‐end reads.
2.3. Sequence data analysis and taxonomic classification
Raw sequence quality was manually assessed with FastQC v. 0.11.5 (Simon Andrews, 2010). All 57 samples contained reads of consistent length (respective to the sequencing year), and the average read quality score was above 30. A drop in base quality was observed at the ends of reads (4–5 and 8–10). Demultiplexed raw sequences were imported into QIIME2‐2019.10 (Bolyen et al., 2019) where quality control, de‐replication, read truncation, and paired read merging were performed using the DADA2 (Divisive Amplicon Denoising Algorithm) qiime2 plugin (Callahan et al., 2016). Instead of generating operational taxonomic units (OTUs) by clustering sequences based on similarity, the final output of DADA2 is a table with exact sequence variants also known as amplicon sequence variants (ASVs), which are generated by modeling and correcting Illumina sequencing errors. This step was carried out separately according to the year of collection. However, to normalize between datasets, the 250 bp reads produced from 2017 samples were truncated so that the paired reads matched the length of the paired reads from 2018 samples. To confirm consistency in paired read lengths between the two years, representative sequences generated from both years were aligned in Geneious Prime® 2020.0.5 (https://www.geneious.com) by Multiple Alignment using the Fast Fourier Transform (MAFFT) plug‐in with default settings and then assessed by eye (Katoh & Standley, 2013).
Next, a mid‐point rooted, approximately‐maximum‐likelihood phylogenetic tree for diversity analysis was generated using the qiime2 phylogeny plug‐in which uses MAFFT and the FastTree program (Price et al., 2010). Finally, taxonomies were assigned to the ASVs using a 16S‐V4‐specific classifier trained against the Silva132 database clustered at 99% sequence similarity (Quast et al., 2013).
2.4. Data processing and statistical analysis
Statistical analysis was performed in duplicate, once using all available data and again with data corresponding to the core microbiome only. The core microbiome was defined here as all the ASVs present in at least 50 percent of the samples.
Data processing and statistical analysis were carried out in R version 3.6.0 (R Core Team, 2019). To prepare the data by identifying unassigned ASVs and removing contaminants and samples with an insufficient depth of sampling prior to analysis, multiple filtering steps were applied to the data using the phyloseq package (McMurdie & Holmes, 2013). (1) Unassigned ASVs at the Kingdom level were manually inspected with the Basic Local Alignment Search Tool (BLAST) before filtering based on both BLAST results (those with non‐bacterial matches) and prevalence (ambiguous taxonomy at the phylum level with a prevalence of 1 and total abundance less than 5 reads) (Altschul et al., 1990). (2) Based on the rarefaction curve (Figure A1), three samples were identified as having an insufficient depth of sampling and were therefore removed from the statistical analysis. A threshold of 13,980 reads was used as a cut‐off. Removed samples were identified as 17JFFS16 (BP, 4463 counts), 17JFFS23 (TB, 2602 counts), and 17JFFS23 (EA, 2042 counts). (3) Possible contamination signals were also removed by running a correlation analysis and comparing clusters with a list of previously identified reagent contaminants (Salter et al., 2014). (4) Finally, the data were rarefied using the same threshold used for filtering samples (Table A1) (McKnight et al., 2019).
The overall microbiota composition was characterized by summing the non‐normalized read counts and obtaining the relative abundance at different taxonomic levels.
2.4.1. Alpha diversity
Estimates of within‐sample diversity (alpha diversity) were calculated using the phyloseq package. Three indices were included: a richness estimator, which estimates the total number of species in each sample (Chao1), and two different diversity estimators (Shannon‐Weiner and Simpson index). The latter two approaches consider richness and abundance. However, the effect of richness and rare species strongly impact the Shannon‐Weiner index, whereas the Simpson index is mainly influenced by evenness and common species.
Non‐rarefied data were used to explore the alpha diversity. To compare locations, a one‐way analysis of variance test (ANOVA) or a non‐parametric Kruskal–Wallis test was performed for each estimate. ANOVA assumptions were tested by visualization of the data and statistical testing. A Shapiro–Wilk test was used to confirm normality and Levene's test for heteroscedasticity. When exploring Shannon‐Weiner and Simpson indices, sample 18JFFS23 (SC) was identified as an outlier (standard residual >3) and was removed for these indices only. Finally, data visualization suggested samples collected from TB differed from the other locations; thus, a posthoc analysis was performed with Dunnett's or the non‐parametric Dunn's test to compare each location to TB. Samples from PC, PT, and V were not included in the location comparison due to their limited sample size (n = 1).
2.4.2. Beta diversity
To investigate variation between samples (beta diversity) two different distances were calculated using the rarefied full as well as the core datasets. Bray‐Curtis dissimilarity distance was used to look at the differences between samples based on the ASVs abundances. Weighted UniFraq distance was used to explore the phylogenetic divergence between ASVs by also taking into account the abundance of these (with an emphasis on dominant ASVs). Respective distance matrices were visualized using principal coordinate analysis plots (PCoA).
To further explore the clustering of samples (Cluster 1 versus Cluster 2) observed in the Bray‐Curtis PCoA, a permutational multivariate ANOVA (PERMANOVA) was computed with 999 permutations to test for statistically significant differences between the clusters. Finally, a Similarity Percentage breakdown analysis (SIMPER) was performed between the clusters to identify the genera that most contributed to the difference between clusters. Genera that highly contributed to dissimilarities between groups were further explored with the non‐parametric Mann‐Whitney U test.
Spearman's rank correlation coefficient (ρ) was used to explore any possible associations between the different taxa and also between the first two components of the Bray‐Curtis ordination analysis. Correlations were visualized in a correlation matrix plot, and only those significantly and strongly correlated (Rho (ρ) ≥ |0.6|) were explored further. For this method, only the core microbiome dataset was used at the genus level.
3. RESULTS
Following the removal of low‐quality sequences and merging the 2017 and 2018 datasets, a total of 2,074,038 paired reads, grouped into 595 ASVs were imported into R studio for statistical analysis. A total of 54 samples, with 2,062,763 sequences clustered into 558 ASVs remained after the filtering steps (Table A1). Three samples were removed from the analysis due to rarefaction analysis indicating the insufficient depth of sequencing. The rarefied dataset ended up with 518 ASVs and a total of 754,974 reads.
3.1. Composition of the Juan Fernandez fur seal fecal microbiome
A total of 10 bacterial phyla were detected in the feces of the JFFSs. From the total ASV counts Firmicutes (41.9%), Fusobacteria (28.2%), Bacteroidetes (22.1%), Proteobacteria (5.5%), and Actinobacteria (1.5%) dominated the bacterial composition. The total ASV counts from individual samples were very similar to the average relative abundance: Firmicutes (40% ±24), Fusobacteria (30% ±17), Bacteroidetes (22% ±10), Proteobacteria (6% ±4), and Actinobacteria (2% ±3) (Table A2). Eighty‐two bacterial families could be assigned, of which 14 had a relative abundance ≥1% of the total ASV count. Five bacterial families accounted for 78.5% of all read counts Fusobacteriaceae (28.2%) belonging to the phylum Fusobacteria, Bacteroidaceae (15.5%) from the phylum Bacteroidetes, and Ruminococcaceae (15.0%), Lachnospiraceae (10.4%), and Peptostreptococcaceae (9.4%) from the phylum Firmicutes (Figure 3a,b) (Table A3). Forty‐six ASVs were present in at least 50% of the samples (Table A4). While fourteen ASVs were present in >90% of samples, only three ASVs were present in all the samples, all of which were assigned to the genus Fusobacterium (14.9%, 6.5%, and 3.7% of the total reads respectively) (Table 1).
FIGURE 3.
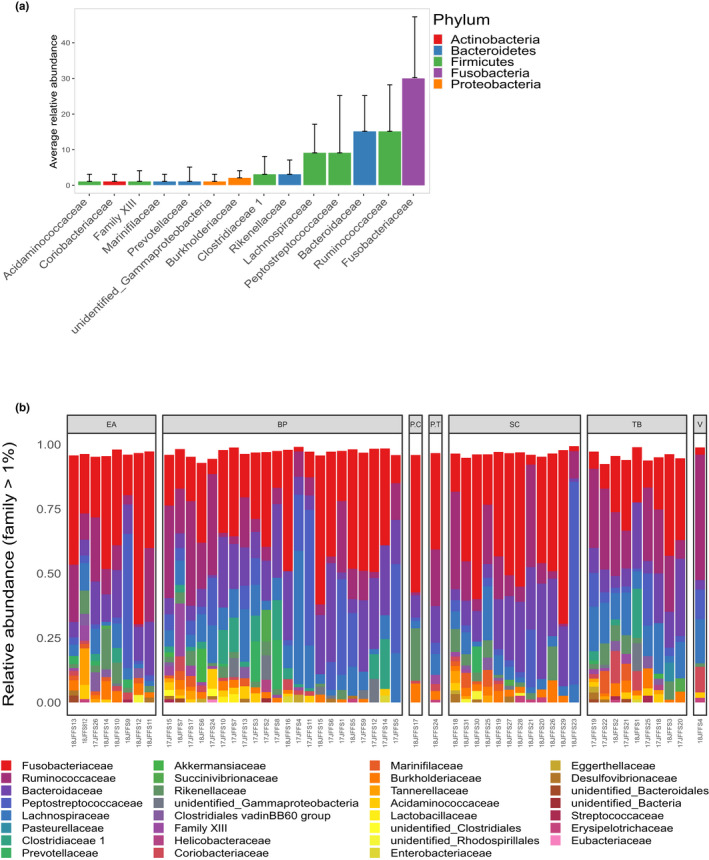
Composition of the Juan Fernandez fur seal fecal microbiome at the family level. Only families with >1% relative abundance are shown. (a) Average relative abundance across all samples with standard deviations. (b) Relative abundance per sample grouped by location: EA = El Arenal, BP = Bahia El Padre, PC = Piedra Carvajal, PT = Punta Truenos, SC = Santa Clara, TB = Tierras Blancas, V = Vaqueria
TABLE 1.
Amplicon sequence variants present in at least 90% of the samples. Only three were present in all the samples. Unrarefied data were used to build this table. Abundance (abun) was calculated based on the total ASVs counts
| ASV | Phylum | Family | Genus | Abun (%) |
|---|---|---|---|---|
| Present in all samples | ||||
| 57729b2b058d8d5253d3e56e4f6386ca | Fusobacteria | Fusobacteriaceae | Fusobacterium | 14.93 |
| e8b1922518029c50c69add839142db03 | Fusobacteria | Fusobacteriaceae | Fusobacterium | 6.52 |
| c0dc53aad260a1b951b7f99966251c7c | Fusobacteria | Fusobacteriaceae | Fusobacterium | 3.73 |
| Present in at least 90% of the samples | ||||
| f347c63fc5e4aeb97531e656e3765e2a | Firmicutes | Peptostreptococcaceae | Peptoclostridium | 8.29 |
| 57f9edc6542ce6b78ff352942d6774c6 | Bacteroidetes | Bacteroidaceae | Bacteroides | 4.28 |
| 31984a302fdfe46b5e852fa473e682a4 | Bacteroidetes | Bacteroidaceae | Bacteroides | 4.26 |
| 1153942c5cc40d6ba5609222ded586fe | Firmicutes | Lachnospiraceae | Coprococcus 3 | 2.98 |
| 65dd9f625700a97a1cce9f5eefe4e6cb | Firmicutes | Lachnospiraceae | Blautia | 2.18 |
| 435975b6d032d4b05233d8b94193b2ad | Firmicutes | Lachnospiraceae | [Ruminococcus] gauvreauii group | 1.93 |
| 03f74c0ea1f0654719b21d2701e9fa30 | Proteobacteria | Burkholderiaceae | Sutterella | 1.30 |
| 8e10797dedc288dbc0be61fe4b5a5dfb | Actinobacteria | Coriobacteriaceae | Collinsella | 1.16 |
3.2. Alpha diversity
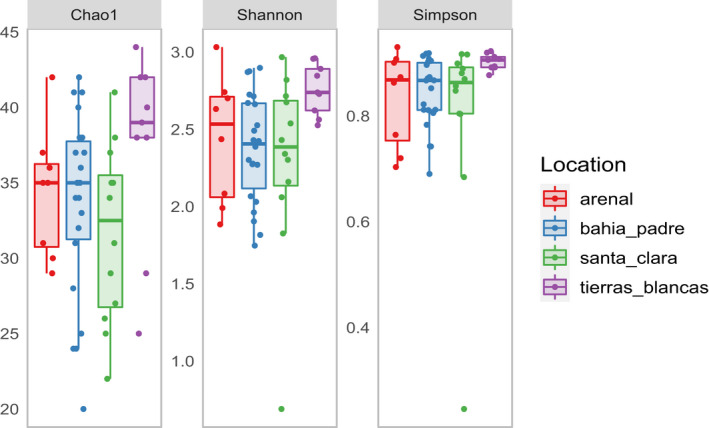
Three alpha diversity indices (Chao1, Shannon‐Weiner, and Simpson) were used to compare within‐sample diversity between locations (Table A5). Despite Tierras Blancas showing a trend towards higher diversity in all analyses, the one‐way ANOVA results showed no significant differences between locations according to Chao1 index (F (3/47) = 2.45, p = 0.07, ges = 0.08) and Shannon–Weiner index (F (3/46) = 2.65, p = 0.06, ges = 0.09). The Simpson index (chi‐squared = 8.26, p < 0.05, ges = not provided), on the other hand, showed a significant difference between locations. Post‐hoc Dunnet's and Dunn's tests consistently showed that samples from TB had higher mean and mean rank values (respectively) than the other locations, especially when compared to Tierras Blancas (Figure 4, Figure A2).
FIGURE 4.
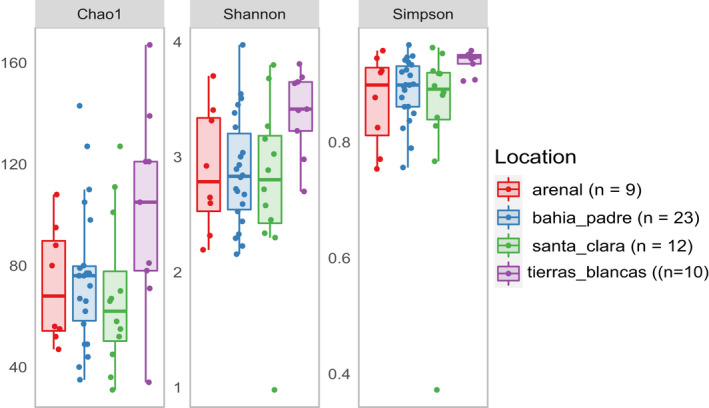
Comparison of three different alpha diversity indices between the four reproductive colonies in the Juan Fernandez archipelago. Samples collected from Tierras Blancas show a tendency to have higher levels of alpha diversity. Filtered rarefied data were used to calculate the diversity estimates
3.3. Beta diversity
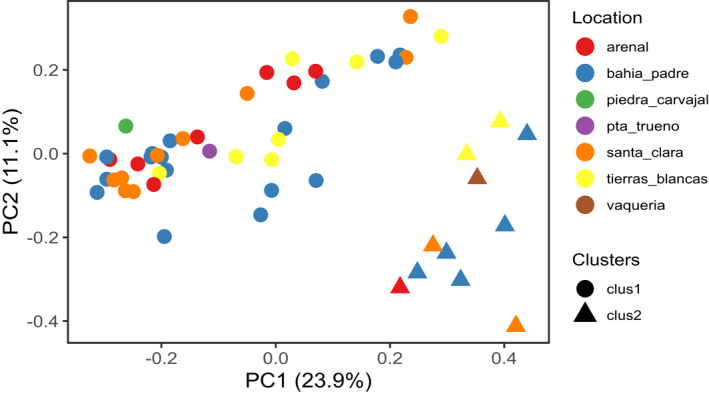
Based on weighted Unifrac dissimilarity distance, 51.0% (full dataset) and 53.8% (core dataset) of the total variation between samples could be explained by the first principal component (PC1). No clustering of individual samples by location or year of the collection was observed. Similarly, Bray‐Curtis dissimilarity, which quantifies the differences in ASV abundance, found that the first principal components in both the full and core datasets explained 23.9% and 29.8% of the total variation, respectively. In both data sets, a group of samples (cluster 2) was separated from the main cluster (cluster 1) along PC1 (Figure 5, Figure A3).
FIGURE 5.
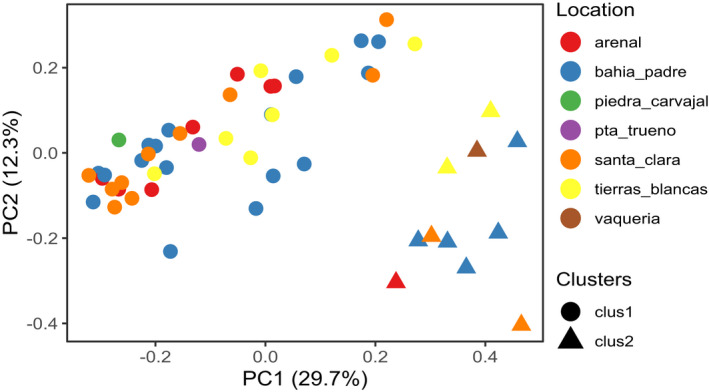
PCoA using Bray‐Curtis dissimilarity distance matrix using the filtered rarefied core dataset. Samples clustered in two groups. (circles = cluster 1, triangles = cluster 2). Location is not driving the clustering
Based on the relative average abundance of the dominant phyla, evident differences in the overall microbial composition were visualized between the two clusters (Figure 6). PERMANOVA evidenced a significant difference in the microbial composition between the two clusters. This was consistent in both full (F = 10.1, Pr (>F) = 0.001, R 2 = 16.3%) and core datasets (F = 13.6, Pr (>F) = 0.001, R 2 = 20.88%). SIMPER analysis identified five genera that together contributed 71% to the observed compositional difference between the clusters. As expected, both Fusobacterium and Peptoclostridium were the largest contributors (24% and 25%, respectively). Furthermore, the abundance of Fusobacterium and Peptoclostridium were significantly different between clusters. Full results of the SIMPER and Mann–Whitney U‐tests are summarised in Table 2.
FIGURE 6.
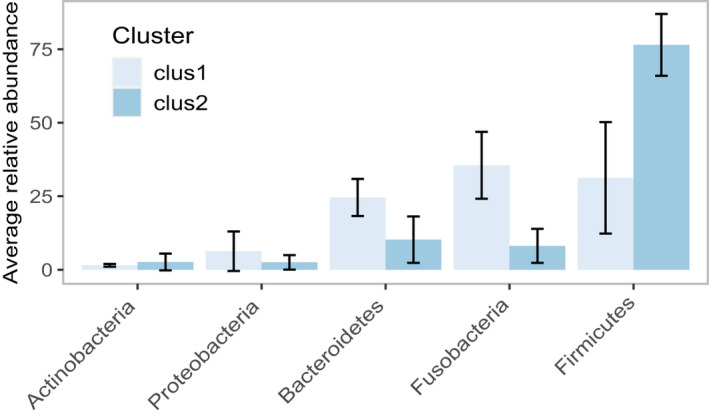
Relative average abundance of the dominant phyla according to the clusters identified with Bray Curtis dissimilarity. Showing only phyla with an average relative abundance ≥1%. The differences in microbial patterns can be identified from high taxonomic levels
TABLE 2.
SIMPER analysis comparing the fecal microbiota composition of Juan Fernadez fur seal at the genus level. The table showing up to a cumulative contribution of 70%. Cluster averages were calculated based on total counts. Kruskal‐Wallis results are only shown when reaching a significant difference
| Genus | Mean cluster 1 | Mean cluster 2 | Mean Diss | Contrib (%) | Cum (%) | w | P‐value |
|---|---|---|---|---|---|---|---|
| Peptoclostridium | 3% | 29% | 17% | 25 | 25 | 3 | <0.001 |
| Fusobacterium | 34% | 8% | 17% | 24 | 49 | 456 | <0.001 |
| Bacteroides | 14% | 6% | 7% | 10 | 59 | 365.5 | 0.006 |
| Ruminococcaceae UCG−005 | 4% | 7% | 4% | 6 | 65 | No sig | |
| [Ruminococcus] gauvreauii group | 1% | 6% | 4% | 5 | 70 | 124 | 0.06 |
3.4. Correlation analysis
Spearman correlation analysis revealed that the genera Bacteroides, Fusobacterium, and Peptoclostridium were strong drivers of PC1 in both Bray‐Curtis and Weighted Unifrac PCoA analyses. In addition, the genera Ruminoclostridium 9 and Ruminococcaceae NK4A214 were also found to be influential for PC1 in Bray‐Curtis analysis (Figure 7, Table A6). PCoA analyses showed strong negative correlations between PC1 and Bacteroides (Bray‐Curtis, ρ = −0.67, p ≤ 0.001) and between PC1 and Fusobacterium (Bray‐Curtis, ρ = −0.92, p ≤ 0.001 and weighted Unifrac, ρ = −0.94, p ≤ 0.001). Peptoclostridium, on the other hand, was positively correlated with PC1 (Bray‐Curtis, ρ = 0.81, p ≤ 0.001, and weighted Unifrac, ρ = −0.75, p ≤ 0.001).
FIGURE 7.
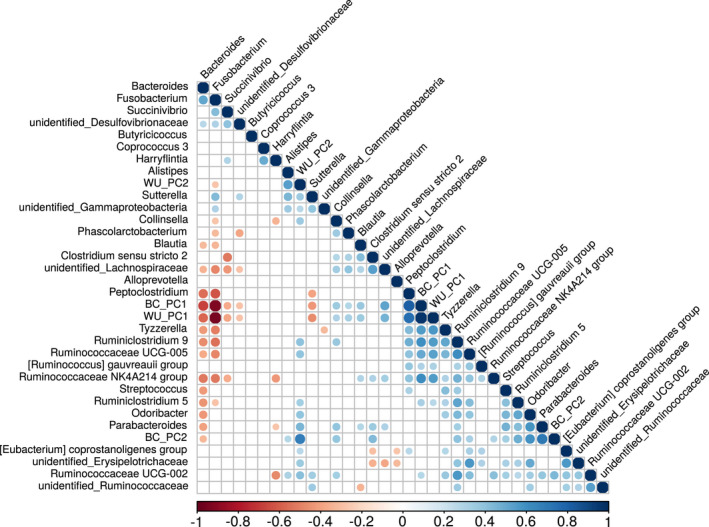
Spearman rank correlation correlogram between bacterial genera and the first two principal components generated from Unifraq WU_PC1 and WU_PC2 and Bray‐Curtis (BC_PC1 and BC_PC2) distances. The plot shows the direction (blue = positive, red = negative) and the strength (larger = stronger) of the correlation between each pair combination. Only significant correlations (p ≤ 0.05) are represented with circles
4. DISCUSSION
Marine mammal microbiome studies of free‐ranging wild populations are rare, with many of these studies being limited to a small number of individuals. Instead, most studies of marine mammals have relied on data from dead or captive animals. To our knowledge, this is one of the most extensive studies of the fecal microbiome in free‐ranging pinnipeds and the first of JFFS. Our approach focused on characterizing the core members of the JFFS fecal microbiome, identified at the genus level, providing a baseline for understanding host–microbial interactions in this species. However, interpreting unexpected phenomena in a dataset such as ours is made difficult by a lack of literature with results generated using similar methodologies, as well as the various uncontrollable factors influencing wild populations.
Consistent with previous reports in other pinniped species, five phyla dominated the JFFS fecal microbiome: Firmicutes, Fusobacteria, Bacteroidetes, Proteobacteria, and Actinobacteria (Bik et al., 2016; Kim et al., 2020; Nelson, Rogers, & Brown, 2013; Numberger et al., 2016; Pacheco‐Sandoval et al., 2019; Stoffel et al., 2020). When comparing our results to other southern pinnipeds, different microbial patterns were found in feces from other fur seal species (Medeiros et al., 2016; Smith et al., 2013). The fecal microbiome described for both the South American (Arctophoca australis australis) and the sub‐Antarctic fur seals (Arctophoca tropicalis) is almost entirely dominated by Firmicutes (88.56% and 85.02%). Fusobacteria, on the other hand, represents less than 1% of the bacterial community for both species (Medeiros et al., 2016). The study involving these two species collected samples from dead juvenile individuals. Thus, it is expected to find altered microbiomes. Smith et al. (2013) characterized the fecal microbiome of Australian fur seal (Arctocephalus pusillus doriferus) pups and adult females. The adult samples showed similar proportions of Firmicutes, Bacteriodetes, and Actinobacteria as those observed here for JFFS. Fusobacteria was not detected in any of the adults. However, the authors only relied on fluorescent in situ hybridization (FISH) to detect these bacteria for this age group.
Overall, pinniped gut microbiomes are very variable between and within species, possibly due to differences in their geographic range (e.g., polar versus subtropical), diet (benthic vs pelagic hunters, generalist versus specialist), or mating systems. One or more of Fusobacteria, Firmicutes, and Bacteroides (all three in the case of JFFS and harbor seals) have been found to consistently dominate the overall microbial composition of pinnipeds, followed by Proteobacteria and Actinobacteria (Nelson, Rogers, & Brown, 2013; Pacheco‐Sandoval et al., 2019). The latter two are usually at the lower abundance, and Actinobacteria, in particular, has not been described in every pinniped species studied. Another interesting observation, common to all the studies reviewed, including ours, is that, when Firmicutes dominates, the abundance of Fusobacteria and Bacteroidetes decreases, suggesting some degree of competition. The Firmicutes: Bacteroidetes ratio has been well documented in humans and mice. In these land mammals, the ratio increases in response to diets high in lipids and decreases in response to large amounts of protein (Hildebrandt et al., 2009; Pu et al., 2016; Turnbaugh et al., 2006). We also observed changes in the relative abundance of Fusobacteria were similar to those observed in Bacteroidetes. This suggests some functionally redundant roles.
The phylum Firmicutes is common in mammalian gut microbiomes (Ley et al., 2008a, 2008b). Members of this taxonomic group are well known for their role in obesity in humans and mice, which is associated with an increase in Firmicutes and a decrease in Bacteroidetes (Hildebrandt et al., 2009; Pu et al., 2016; Turnbaugh et al., 2006). The energy harvesting role of Firmicutes has also been identified in the zebrafish gut microbiome, where these bacteria are associated with an increase in lipid droplet numbers in epithelial cells (Semova et al., 2012). Fat is fundamental for marine mammal survival, as it is needed for energy storage and thermoregulation and may explain why Firmicutes is consistently among the most dominant phyla across all pinniped species (Guerrero & Rogers, 2019).
The phylum Fusobacteria consists of facultative or strict anaerobes that produce various organic acids from amino acids or carbohydrates fermentation (Olsen, 2014). This phylum is usually found at the high relative abundance in the gut microbiomes of strict carnivores adapted to diets rich in proteins, purines, and polyunsaturated fatty acids (Guo et al., 2020; Zhu et al., 2018). Similar to other marine carnivores, Fusobacteria was one of the most abundant phyla in JFFS (Pacheco‐Sandoval et al., 2019). Most of the knowledge generated around the specific role Fusobacteria may play in mammalian intestinal tracts is based on human‐centered research. Even though some genus members seem to play a beneficial role in the human gut microbiome, the presence of relatively high levels of the genus Fusobacterium is more often associated with health issues (Garrett & Onderdonk, 2014; Huh & Roh, 2020; Potrykus et al., 2008). Conversely, the high relative abundance of this bacterial genus in the gut of carnivores suggests a rather symbiotic relationship where Fusobacterium is likely to play a role in protein metabolism (Potrykus et al., 2008).
Similar to Fusobacteria, the phylum Bacteroidetes, especially members of the genus Bacteroides, are associated with diets high in animal proteins (Guo et al., 2020; Zhu et al., 2018). This genus, known for its capacity to degrade animal‐derived glycans (Eilam et al., 2014), was the most abundant Bacteroidetes. Similar to previous reports, JFFS samples high in Firmicutes contained lower relative abundances of Bacteriodetes and Fusobacteria. This phenomenon suggests differences in nutritional needs and will be discussed later in the text.
4.1. Within sample diversity
Initially, we hypothesized that the alpha diversity of samples collected from BP, a key access point to Robinson Crusoe Island, was going to be different from other colonies. BP is the most transited area in this study; it connects the airfield with the town and is a popular leisure location for the local community (Figure 1). We found instead that BP did not differ from other less‐visited locations such as EA and SC. Therefore, this finding is different from a previous report showing an association between exposure to anthropogenic stressors and reduced alpha diversity in harbor seals (Pacheco‐Sandoval et al., 2019). The colony at TB was the only location with higher alpha diversity, indicating that samples collected from TB had a richer and more evenly distributed microbial composition than other samples. Bacterial richness has been previously associated with population density due to the increase in microbial sharing (Li et al., 2016). Alternative studies have suggested that overcrowding might also negatively affect microbial diversity due to higher levels of stress (Li et al., 2016; Partrick et al., 2018). Lower diversity of the skin microbiome in denser populations was also observed in Arctocephalus gazella, a closely related species (Grosser et al., 2019). The population density of JFFS and its effects on the microbiome have not been studied. However, superficial observations from the field did not suggest differences in population density between the colonies. It may therefore be that other stressors were limiting alpha diversity in the other locations. For instance, the colony on TB was relatively sheltered compared to the other colonies, as it was situated on an open platform a few meters above sea level; in contrast, the other colonies were on narrow strips of land with greater exposure to sea storms, rockfalls, and landslides. Additionally, the colony on TB is rarely visited by humans due to the complicated access. However, the effects of location on alpha diversity were marginal. Nevertheless, the stress hypothesis could be tested in future studies by measuring markers of stress (e.g., cortisol) in the feces (Wasser et al., 2000).
Despite the trend showing how TB differed from the other locations, only one of the three alpha diversity estimates (Simpson) showed TB to be statistically significantly different from the other locations. The other two diversity estimates (Chao1 richness and Shannon‐Weiner) did not reach our significance cut‐off. Both these estimates are affected by the detection of rare taxa, and larger libraries and sample sizes are more likely to input rare taxa into the data set. ANOVA was also used to compare locations with these diversity estimates. ANOVA is sensitive to differences in sample size, and therefore small group sizes may have affected statistical power.
4.2. Variation between samples
The Bray‐Curtis dissimilarity PCoA revealed two distinct clusters. Seventy‐five percent of the samples clustered together in what we named cluster 1. The remaining samples were grouped as cluster 2. This variation between clusters was mostly explained by the differences in the relative abundance of the genera Fusobacterium and Peptoclostridium. Samples in cluster 1 had a high relative abundance of Fusobacterium and very low Peptoclostridium relative abundance, whilst samples in cluster 2 showed the opposite pattern: increased Peptoclostridium and a significant drop in Fusobacterium relative abundance. To our knowledge, this is the first time the genus Peptoclostridium (phylum Firmicutes, class Clostridia) has been reported in a pinniped gut microbiome. The family Peptostreptococcaceae, to which Peptoclostridium belongs, has been reported in previous studies, but representing no more than 8% of the total composition and more often less than 4% (Delport et al., 2016; Nelson, Rogers, & Brown, 2013; Pacheco‐Sandoval et al., 2019). On average, Peptoclostridium represented 29% of the microbial composition observed in Cluster 2 versus the average 3% observed in Cluster 1.
The genus Peptoclostridium was initially proposed in 2013 and validated in 2016 (Galperin et al., 2016). This poorly characterized taxonomic group is believed to metabolize amino acids and oligopeptides and has been isolated from both wastewater mud and marine sediments (Galperin et al., 2016). The SILVA 132 taxonomy reference database used in this study included 144 members in the Peptoclostridium clade from which only 11 were classified within the four known species of this genus (P. litorale, P. acidaminophilum, P. paradoxum, and P. thermoalcaliphilum). The remaining clade members were classified as uncultured bacteria. It should be noted that, depending on the taxonomic reference database used, the taxonomic classification regarding members of the genus Peptoclostridium may differ between studies. For instance, some studies may refer to species such as Clostridoides difficile (previously known as Clostridium) as Peptoclostridium difficile (Pereira et al., 2016). All four species included in the SILVA 132 database have been isolated from environments with little or no oxygen (Galperin et al., 2016). Despite these species being linked to environmental samples, Peptoclostridium was found in at least 90% of the samples. The particular condition required for this bacterial species to thrive makes it unlikely that the Peptoclostridium members found in JFFS feces originated from sample contamination by surrounding environmental bacteria. Such high prevalence may be a sign of a deeper relationship between these uncharacterized bacteria and the host.
The microbiome is constantly reshaping through an individual's lifetime. Most of the changes occur within symbiotic margins responding to factors such as diet, reproductive state, and age, but some changes may also result in dysbiosis and disease (Ley et al., 2008b; Nicholson et al., 2012). Despite the limited information available on free‐range pinnipeds, a few hypotheses may be suggested to explain the significant changes observed between the two clusters reported in our study.
There is evidence that the mammalian gut microbiota changes over time. This difference is particularly evident between suckling and post‐weaning stages, possibly due to dietary changes (milk vs solids). As discussed earlier, Firmicutes are known for their capacity to regulate lipid absorption (Semova et al., 2012). Juan Fernandez fur seal milk composition contains a higher proportion of lipids in comparison to many pinnipeds (~ 41%) (Ochoa‐Acuña et al., 1999). Thus, if the fecal samples from Cluster 2 were collected from pre‐weaning pups (7–10 months old), it may be expected that a higher relative abundance of members of the phylum Firmicutes would be found. Similar to the microbial pattern observed in Cluster 2, samples analyzed from Australian fur seals were dominated by the class Clostridia in six and nine months old pups (Smith et al., 2013). In the same study, the families Lachnospiraceae and Ruminococcaceae were the most dominant family within this Class, while the overall relative abundance of Peptostreptococcaceae was less than 4%. Despite age (preweaning diet) being a reasonable explanation for the difference observed in our dataset, this hypothesis arrives with a critical bias. Samples were collected between February and March, and, at this point, pups would be no older than four months. At this stage, pup feces are still distinguishable from older individuals in color and consistency. Individuals from the previous reproductive season would be older than a year and milk would no longer form a part of their diet. This suggests that a pre‐weaning diet is not the explanation for the abundance of Peptoclostridium.
Differences between sexes may also be an explanation of the difference in samples. Otarids and Phocids such as northern and southern elephant seals exhibit an important degree of sexual size dimorphism (Ralls & Mesnick, 2009). Sex differences in foraging behavior and prey selection have also been reported (Andersen et al., 2013; Lewis et al., 2006; Ochoa Acuña & Francis, 1995). Based on the differences in diets, it is not surprising to find studies in gut microbial composition also showing sex‐based differences. Samples collected from adult Southern elephant seals evidenced significant differences between adult males and females (Kim et al., 2020; Nelson, Rogers, & Brown, 2013). The same studies did not find differences in leopard or Weddel seals, less sexually dimorphic phocids. Adult southern elephant seal females showed a significantly higher relative abundance of Firmicutes and less Fusobacteria and Bacteriodetes than males (Kim et al., 2020; Nelson, Rogers, & Brown, 2013). The proportional changes are very similar to the one observed between clusters 1 and 2 here. Cluster 2 shows patterns similar to those observed in females. It seems that the microbial community diverges early in life based on sex as reported in northern elephant seal pups under a naturally controlled diet (Stoffel et al., 2020). Sexual dimorphism is a common mating strategy in otariids. Thus, otariids such as JFFS may show similar differences as the ones observed in elephant seals. This hypothesis could be confirmed using molecular methods for sex identification.
A commonality between the sex and age hypotheses is their relationship to the diet. Differences in diet have been identified as one of the main drivers of gut microbiome diversity (Ley et al., 2008a; Nelson, Rogers, & Brown, 2013; Nishida & Ochman, 2018). While pups rely on lipid‐rich milk, fish from the family Myctophidae are the most important prey of adult female JFFS (Francis et al., 1998). Myctophids are known to be rich in fatty acids (Baby et al., 2014; Lea et al., 2002). Pacheco‐Sandoval et al. (2019) showed that harbor seal fecal samples containing more lipid‐rich preys had a much higher abundance of Firmicutes and lower Fusobacteria and Bacteriodetes (Pacheco‐Sandoval et al., 2019). Molecular identification of prey species in fecal samples may therefore help to determine whether the diet is the driving factor behind the microbial differences observed here.
5. CONCLUSION
This study characterized the fecal microbiome of the Juan Fernandez fur seal for the first time, including colonies from two of the three islands of the Juan Fernandez archipelago to which the species is endemic. Our findings showed that the overall microbiome composition was similar to compositions described for other pinnipeds. However, some of the samples showed a very different microbial composition pattern. This difference was mostly explained by an inverse relationship between Peptoclostridium and Fusobacterium abundance. Sex and its relationship to foraging behavior seem to be the most likely explanation of this phenomenon. However, additional studies investigating the relationship between sex, age, and prey are required to test this hypothesis. Overall, the results of this study provide a good baseline from which future hypothesis‐based studies can be carried out, and it contributes to the understanding of host–microbial interaction in free‐ranging, wild populations of pinnipeds. We highlight the need to expand knowledge in this field, particularly on microbial functionality, to understand its different members’ roles and compare microbial patterns between and within species.
CONFLICT OF INTEREST
The authors confirm that they have no conflicts of interest related to the content of this article.
AUTHOR CONTRIBUTIONS
Constanza Toro‐Valdivieso: Conceptualization (equal); Formal analysis (lead); Funding acquisition (equal); Investigation (lead); Methodology (equal); Visualization (lead); Writing‐original draft (lead); Writing‐review & editing (equal). Frederick Toro: Formal analysis (supporting); Writing‐review & editing (supporting). Samuel Stubbs: Formal analysis (supporting); Writing‐original draft (supporting); Writing‐review & editing (equal). Eduardo Castro‐Nallar: Formal analysis (supporting); Funding acquisition (equal); Writing‐review & editing (equal). Barbara Blacklaws: Conceptualization (equal); Formal analysis (supporting); Funding acquisition (equal); Methodology (equal); Supervision (lead); Writing‐original draft (supporting); Writing‐review & editing (equal).
ETHICS STATEMENT
All fecal samples were collected from the environment in a non‐invasive manner. Disturbance of the colonies was kept to a minimum and no animal was handled or harmed in the process. Permits for the collection of samples were given by CONAF (Certificate 009217) and SERNAPESCA (R.E.X.N 43). Permission for the importation of samples into the United Kingdom was also obtained (ITIMP16.1158).
ACKNOWLEDGMENTS
We want to thank SERNAPESCA and the Chilean National Forestry Commission (CONAF) in particular Guillermo Araya (Director of the Juan Fernandez National Park) and the rangers Ángela García, Ramón Schiller, and Danilo Arredondo for the crucial support, teaching, and friendship provided during fieldwork. Many thanks to Aerocardal for sponsoring flights to the Juan Fernández Archipelago, and OIKONOS, especially Héctor Gutierrez and Pablo Marríquez Angulo, for always giving a hand when needed. Ph.D. scholarship was provided to Constanza Toro‐Valdivieso by The National Research and Development Agency of Chile (ANID) in partnership with Cambridge Trust. Financial support was provided by Newnham College and the Department of Veterinary Medicine, University of Cambridge. Further support was provided by more than 60 people who donated to our study via the Crowdfunding platform.
FIGURE A1.
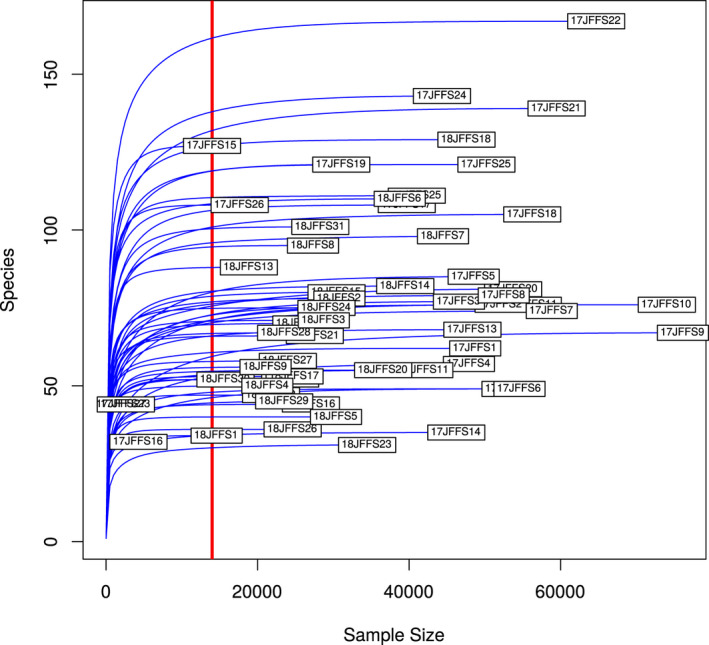
Rarefaction curve estimating the number of ASVs (y‐axis) for a given read count (x‐axis). The vertical line indicates the cutoff at which samples were retained and rarefied
FIGURE A2.
Comparison of three different alpha diversity indices between four reproductive colonies in the Juan Fernandez archipelago. Samples collected from Tierras Blancas show a tendency to have higher levels of alpha diversity. Core rarefied data were used to calculate the diversity estimates
FIGURE A3.
PCoA using Bray‐Curtis dissimilarity distance matrix using the filtered rarefied full dataset. Samples clustered in two groups (circles = cluster 1, triangles = cluster 2). Location is not driving the clustering
TABLE A1.
Inputs and outputs of each preprocessing step
| Preprocessing steps | Number of samples | Number of ASVs | Min. number of reads per sample | Max. number of read per sample | Filtered reads | Total |
|---|---|---|---|---|---|---|
| Raw | 57 | 595 | 2042 | 76,134 | 0 | 2,074,038 |
| Filter ASVs (non‐bacterial and ambiguous) | 57 | 577 | 2042 | 76,134 | 2081 | 2,071,957 |
| Filter samples | 54 | 577 | 13,981 | 76,134 | 8916 | 2,063,041 |
| Filter Contaminants | 54 | 558 | 13,981 | 76,134 | 278 | 2,062,763 |
| Rarefaction | 54 | 518 | 13,981 | 13,981 | 1,307,789 | 754,974 |
TABLE A2.
Bacterial phyla detected in Juan Fernandez fur seal feces
| Family counts | Total | Total counts rel. ab (%) | Mean rel.ab (%) | Rel. ab SD | Total ASV |
|---|---|---|---|---|---|
| Firmicutes | 863,365 | 41.85 | 40 | 24 | 296 |
| Fusobacteria | 582,406 | 28.23 | 30 | 17 | 46 |
| Bacteroidetes | 455,251 | 22.07 | 22 | 10 | 94 |
| Proteobacteria | 113,805 | 5.52 | 6 | 4 | 74 |
| Actinobacteria | 30,597 | 1.48 | 2 | 3 | 21 |
| Verrucomicrobia | 6653 | 0.32 | 0 | 2 | 3 |
| Epsilonbacteraeota | 6554 | 0.32 | 0 | 1 | 10 |
| Unidentified | 2204 | 0.11 | 0 | 0 | 2 |
| Tenericutes | 1005 | 0.05 | 0 | 0 | 8 |
| Lentisphaerae | 900 | 0.04 | 0 | 0 | 3 |
| Spirochaetes | 34 | 0.00 | 0 | 0 | 3 |
TABLE A3.
Summary of the bacterial family detected in feces of Juan Fernandez fur seal. Data are arranged in decreasing order based on counts mean
| Family | Total counts | Counts rel. ab (%) | Counts mean | Counts SD | Mean rel.ab (%) | Re. ab SD | Total ASV |
|---|---|---|---|---|---|---|---|
| Fusobacteriaceae | 582,404 | 28.23 | 10,785.26 | 6958.72 | 30 | 17 | 45 |
| Bacteroidaceae | 320,047 | 15.52 | 5926.8 | 5319.43 | 15 | 10 | 28 |
| Ruminococcaceae | 310,109 | 15.03 | 5742.76 | 5206.41 | 15 | 13 | 139 |
| Lachnospiraceae | 213,725 | 10.36 | 3957.87 | 4195.97 | 9 | 8 | 61 |
| Peptostreptococcaceae | 193,151 | 9.36 | 3576.87 | 6353.37 | 9 | 16 | 16 |
| Rikenellaceae | 65,548 | 3.18 | 1213.85 | 1543.63 | 3 | 4 | 20 |
| Clostridiaceae 1 | 60,276 | 2.92 | 1116.22 | 2385.25 | 3 | 5 | 16 |
| Burkholderiaceae | 47,544 | 2.3 | 880.44 | 849.59 | 2 | 2 | 8 |
| unidentified_Gammaproteobacteria | 27,169 | 1.32 | 503.13 | 1116.46 | 1 | 2 | 9 |
| Acidaminococcaceae | 27,237 | 1.32 | 504.39 | 734.56 | 1 | 2 | 3 |
| Marinifilaceae | 25,673 | 1.24 | 475.43 | 1022.21 | 1 | 2 | 13 |
| Prevotellaceae | 24,111 | 1.17 | 446.5 | 1666.42 | 1 | 4 | 4 |
| Coriobacteriaceae | 23,956 | 1.16 | 443.63 | 688.99 | 1 | 2 | 1 |
| Family XIII | 22,734 | 1.1 | 421 | 1294.81 | 1 | 3 | 11 |
| Clostridiales vadinBB60 group | 16,935 | 0.82 | 313.61 | 586.95 | 1 | 2 | 2 |
| Tannerellaceae | 15,153 | 0.73 | 280.61 | 647.81 | 1 | 2 | 8 |
| Succinivibrionaceae | 14,801 | 0.72 | 274.09 | 931.79 | 1 | 2 | 7 |
| Desulfovibrionaceae | 12,759 | 0.62 | 236.28 | 296.88 | 1 | 1 | 10 |
| Erysipelotrichaceae | 6926 | 0.34 | 128.26 | 161.05 | 0 | 1 | 7 |
| Akkermansiaceae | 6644 | 0.32 | 123.04 | 696.94 | 0 | 2 | 2 |
| Eggerthellaceae | 5951 | 0.29 | 110.2 | 241.78 | 0 | 1 | 4 |
| Helicobacteraceae | 5185 | 0.25 | 96.02 | 371.85 | 0 | 1 | 7 |
| Streptococcaceae | 4000 | 0.19 | 74.07 | 192.29 | 0 | 0 | 6 |
| unidentified_Rhodospirillales | 3691 | 0.18 | 68.35 | 153.19 | 0 | 1 | 4 |
| Lactobacillaceae | 3649 | 0.18 | 67.57 | 336.59 | 0 | 1 | 3 |
| unidentified_Bacteroidales | 3395 | 0.16 | 62.87 | 168.66 | 0 | 0 | 5 |
| Enterobacteriaceae | 3289 | 0.16 | 60.91 | 195.2 | 0 | 1 | 6 |
| unidentified_Clostridiales | 2650 | 0.13 | 49.07 | 186.76 | 0 | 1 | 10 |
| unidentified_Bacteria | 2204 | 0.11 | 40.81 | 129.68 | 0 | 0 | 2 |
| Pasteurellaceae | 2192 | 0.11 | 40.59 | 285.04 | 0 | 1 | 6 |
| Campylobacteraceae | 1369 | 0.07 | 25.35 | 92.27 | 0 | 0 | 3 |
| Spongiibacteraceae | 1064 | 0.05 | 19.7 | 79.73 | 0 | 0 | 1 |
| Nitrosomonadaceae | 888 | 0.04 | 16.44 | 53.79 | 0 | 0 | 1 |
| Mycoplasmataceae | 881 | 0.04 | 16.31 | 92.11 | 0 | 0 | 7 |
| Eubacteriaceae | 806 | 0.04 | 14.93 | 70.4 | 0 | 0 | 1 |
| Victivallaceae | 662 | 0.03 | 12.26 | 71.52 | 0 | 0 | 2 |
| Flavobacteriaceae | 568 | 0.03 | 10.52 | 38.49 | 0 | 0 | 4 |
| Barnesiellaceae | 639 | 0.03 | 11.83 | 41.46 | 0 | 0 | 2 |
| Peptococcaceae | 438 | 0.02 | 8.11 | 22.97 | 0 | 0 | 2 |
| Enterococcaceae | 325 | 0.02 | 6.02 | 26.47 | 0 | 0 | 4 |
| Vibrionaceae | 113 | 0.01 | 2.09 | 9.78 | 0 | 0 | 2 |
| vadinBE97 | 238 | 0.01 | 4.41 | 28.16 | 0 | 0 | 1 |
| unidentified_Mollicutes RF39 | 124 | 0.01 | 2.3 | 16.87 | 0 | 0 | 1 |
| Shewanellaceae | 108 | 0.01 | 2 | 14.7 | 0 | 0 | 1 |
| Corynebacteriaceae | 246 | 0.01 | 4.56 | 14.72 | 0 | 0 | 3 |
| Coriobacteriales Incertae Sedis | 200 | 0.01 | 3.7 | 16.27 | 0 | 0 | 1 |
| Christensenellaceae | 180 | 0.01 | 3.33 | 8.89 | 0 | 0 | 2 |
| Actinomycetaceae | 188 | 0.01 | 3.48 | 9.79 | 0 | 0 | 6 |
| Veillonellaceae | 96 | 0 | 1.78 | 10.53 | 0 | 0 | 2 |
| unidentified_Verrucomicrobiae | 9 | 0 | 0.17 | 0.86 | 0 | 0 | 1 |
| unidentified_Firmicutes | 8 | 0 | 0.15 | 1.09 | 0 | 0 | 1 |
| unidentified_Bacteroidia | 2 | 0 | 0.04 | 0.27 | 0 | 0 | 1 |
| unidentified_Actinobacteria | 13 | 0 | 0.24 | 1.18 | 0 | 0 | 1 |
| Thioalkalispiraceae | 2 | 0 | 0.04 | 0.27 | 0 | 0 | 1 |
| Staphylococcaceae | 35 | 0 | 0.65 | 2.84 | 0 | 0 | 1 |
| SC‐I−84 | 3 | 0 | 0.06 | 0.41 | 0 | 0 | 1 |
| Saprospiraceae | 2 | 0 | 0.04 | 0.27 | 0 | 0 | 1 |
| Rhodobacteraceae | 8 | 0 | 0.15 | 0.79 | 0 | 0 | 2 |
| Rhodanobacteraceae | 9 | 0 | 0.17 | 1.22 | 0 | 0 | 1 |
| Rhizobiales Incertae Sedis | 4 | 0 | 0.07 | 0.54 | 0 | 0 | 1 |
| Pseudomonadaceae | 16 | 0 | 0.3 | 1.24 | 0 | 0 | 3 |
| Porphyromonadaceae | 2 | 0 | 0.04 | 0.27 | 0 | 0 | 1 |
| OCS116 clade | 2 | 0 | 0.04 | 0.27 | 0 | 0 | 1 |
| Nocardioidaceae | 5 | 0 | 0.09 | 0.68 | 0 | 0 | 1 |
| Neisseriaceae | 80 | 0 | 1.48 | 9.69 | 0 | 0 | 2 |
| Muribaculaceae | 2 | 0 | 0.04 | 0.27 | 0 | 0 | 1 |
| Moraxellaceae | 12 | 0 | 0.22 | 1.16 | 0 | 0 | 3 |
| Micrococcaceae | 32 | 0 | 0.59 | 2.26 | 0 | 0 | 1 |
| Leptotrichiaceae | 2 | 0 | 0.04 | 0.27 | 0 | 0 | 1 |
| Halomonadaceae | 36 | 0 | 0.67 | 3.62 | 0 | 0 | 2 |
| Gracilibacteraceae | 29 | 0 | 0.54 | 2.96 | 0 | 0 | 2 |
| Family XI | 6 | 0 | 0.11 | 0.57 | 0 | 0 | 2 |
| Dietziaceae | 4 | 0 | 0.07 | 0.38 | 0 | 0 | 2 |
| Desulfobulbaceae | 3 | 0 | 0.06 | 0.41 | 0 | 0 | 1 |
| Crocinitomicaceae | 6 | 0 | 0.11 | 0.82 | 0 | 0 | 1 |
| Chitinophagaceae | 101 | 0 | 1.87 | 8.26 | 0 | 0 | 4 |
| Carnobacteriaceae | 10 | 0 | 0.19 | 0.97 | 0 | 0 | 2 |
| Cardiobacteriaceae | 12 | 0 | 0.22 | 1.21 | 0 | 0 | 1 |
| Brachyspiraceae | 34 | 0 | 0.63 | 3.02 | 0 | 0 | 3 |
| Bacillaceae | 40 | 0 | 0.74 | 3.6 | 0 | 0 | 3 |
TABLE A4.
Amplicon sequence variants present in at least 27 of the samples (50%). Relative abundance was calculated from the unrarefied data
| ASV | Phylum | Family | Genus | Abundance (%) |
|---|---|---|---|---|
| 57729b2b058d8d5253d3e56e4f6386ca | Fusobacteria | Fusobacteriaceae | Fusobacterium | 14.93 |
| f347c63fc5e4aeb97531e656e3765e2a | Firmicutes | Peptostreptococcaceae | Peptoclostridium | 8.29 |
| e8b1922518029c50c69add839142db03 | Fusobacteria | Fusobacteriaceae | Fusobacterium | 6.52 |
| 57f9edc6542ce6b78ff352942d6774c6 | Bacteroidetes | Bacteroidaceae | Bacteroides | 4.28 |
| 31984a302fdfe46b5e852fa473e682a4 | Bacteroidetes | Bacteroidaceae | Bacteroides | 4.26 |
| b8d6a5a80d025861f2afccb79e0a1aaf | Bacteroidetes | Bacteroidaceae | Bacteroides | 3.80 |
| c0dc53aad260a1b951b7f99966251c7c | Fusobacteria | Fusobacteriaceae | Fusobacterium | 3.73 |
| 1153942c5cc40d6ba5609222ded586fe | Firmicutes | Lachnospiraceae | Coprococcus 3 | 2.98 |
| 65dd9f625700a97a1cce9f5eefe4e6cb | Firmicutes | Lachnospiraceae | Blautia | 2.18 |
| e176cb3e4c2f33cee5d529c21ff5534e | Firmicutes | Clostridiaceae 1 | Clostridium sensu stricto 2 | 1.95 |
| 435975b6d032d4b05233d8b94193b2ad | Firmicutes | Lachnospiraceae | [Ruminococcus] gauvreauii group | 1.93 |
| 1a73c668a4bb92b74a18b79f9ae63460 | Firmicutes | Ruminococcaceae | Ruminococcaceae UCG−005 | 1.75 |
| 5b87f47a447ef9a905807d2abed5b638 | Bacteroidetes | Rikenellaceae | Alistipes | 1.68 |
| bf4112a100b11b4cbe9a25bdc591ea52 | Fusobacteria | Fusobacteriaceae | Fusobacterium | 1.38 |
| 03f74c0ea1f0654719b21d2701e9fa30 | Proteobacteria | Burkholderiaceae | Sutterella | 1.30 |
| 1188ef0238977f665e179642f287aead | Firmicutes | Ruminococcaceae | Ruminococcaceae UCG−005 | 1.29 |
| 25699f81befd34e0c9d81cfa84f4e751 | Firmicutes | Lachnospiraceae | unidentified_Lachnospiraceae | 1.27 |
| 8e10797dedc288dbc0be61fe4b5a5dfb | Actinobacteria | Coriobacteriaceae | Collinsella | 1.16 |
| 2553bcb6afcdea16b173909555484369 | Firmicutes | Ruminococcaceae | [Eubacterium] coprostanoligenes group | 1.15 |
| b15e41c7f20b8dcd4b0ed9f6c526885d | Bacteroidetes | Prevotellaceae | Alloprevotella | 1.14 |
| ca28c391514fb33d2d2df1c3c8e12317 | Firmicutes | Ruminococcaceae | Ruminococcaceae UCG−005 | 1.12 |
| 76ded93fadbc4155db4e9dcba2012c81 | Firmicutes | Ruminococcaceae | Ruminococcaceae UCG−002 | 1.07 |
| c45b2a8ebeca2fca503c6312e8611416 | Bacteroidetes | Marinifilaceae | Odoribacter | 1.07 |
| ce3476a906008973a3ab56de06817d56 | Proteobacteria | Burkholderiaceae | Sutterella | 0.87 |
| 975258836de3a001cb4d91cf6cf7de06 | Firmicutes | Acidaminococcaceae | Phascolarctobacterium | 0.72 |
| 1cde608d0a8b17d6fed116653581f050 | Proteobacteria | Succinivibrionaceae | Succinivibrio | 0.68 |
| 6c4c9e8ad2f56316cfffac9587c173ec | Firmicutes | Ruminococcaceae | Ruminococcaceae UCG−005 | 0.58 |
| 58514f20ebf4be2b13d619ba3bd2cf83 | Bacteroidetes | Bacteroidaceae | Bacteroides | 0.55 |
| a0eee6d353d432299b53c9663cf05597 | Bacteroidetes Bacteroidaceae | Bacteroides | 0.54 | |
| 0ac8214c377877609cd0f88567086b2e | Firmicutes | Lachnospiraceae | Tyzzerella | 0.44 |
| 420f3edebd00de18846a5941b55a6d5e | Bacteroidetes | Rikenellaceae | Alistipes | 0.44 |
| 0e7fdaa233c333cb8363b63a41bbfc32 | Firmicutes | Ruminococcaceae | Ruminococcaceae NK4A214 group | 0.43 |
| df408056297f20c5ce5cc68907e39cc8 | Firmicutes | Lachnospiraceae | Tyzzerella | 0.41 |
| c00129ca877cb31776ad4e4e03a9091d | Fusobacteria | Fusobacteriaceae | Fusobacterium | 0.41 |
| 93623ff4fe3615ce4aa4f0a9554fd4de | Proteobacteria | Desulfovibrionaceae | unidentified_Desulfovibrionaceae | 0.35 |
| a672e8b3efeb3a28e5beabe661606ad2 | Firmicutes | Ruminococcaceae | unidentified_Ruminococcaceae | 0.33 |
| 0c3d2038714f019f70fdc3b6f4b40419 | Firmicutes | Ruminococcaceae | Ruminiclostridium 9 | 0.30 |
| b6578d861d1c0e923087c8a5a81c8501 | Proteobacteria | unidentified_Gammaproteobacteria | unidentified_Gammaproteobacteria | 0.25 |
| 57f0c2ba2627cebfea197aa991777cb0 | Bacteroidetes | Tannerellaceae | Parabacteroides | 0.24 |
| 305caa259fb99e3e9aa1eb5dac615002 | Firmicutes | Erysipelotrichaceae | unidentified_Erysipelotrichaceae | 0.19 |
| cbeb8d4b3d3f4b0bfa328178582220a5 | Firmicutes | Streptococcaceae | Streptococcus | 0.17 |
| 66c7c850483807f63638f7e03975cf27 | Proteobacteria | unidentified_Gammaproteobacteria | unidentified_Gammaproteobacteria | 0.15 |
| 78990f6a6e53bd64b9371e316ad97362 | Firmicutes | Ruminococcaceae | Butyricicoccus | 0.14 |
| e8f48023e5081f948df1291acd8d356a | Firmicutes | Lachnospiraceae | unidentified_Lachnospiraceae | 0.09 |
| 1c4ff74a77a35261b972eb21737647e9 | Firmicutes | Ruminococcaceae | Ruminiclostridium 5 | 0.07 |
| 99ea1bdfe0e8b83616c6178b8fdbf1e4 | Firmicutes | Ruminococcaceae | Harryflintia | 0.05 |
TABLE A5.
Table reporting the mean values of Chao‐1, Shannon‐Weiner and Simpson (D) indexes and their standard deviation for each location. Tierras Blancas consistently show higher values than the other three locations. Simpson here is used as 1‐D. Thus, the higher the number, the more diverse. Non‐normalized data were used to build this table
| Location | Chao1 | Shannon‐Weiner | Simpson |
|---|---|---|---|
| Arenal | 72.6 ± 23.0 | 2.9 ± 0.5 | 0.87 ± 0.08 |
| Bahia El Padres | 75.7 ± 27.4 | 2.9 ± 0.5 | 0.89 ± 0.05 |
| Santa Clara | 68.3 ± 30.0 | 2.8 ± 0.7 | 0.85 ± 0.16 |
| Tierras Blancas | 101.9 ± 40.1 | 3.4 ± 0.4 | 0.94 ± 0.02 |
TABLE A6.
The selected value of the Spearman's rank correlation performed on the rarefied core data including PC1 and 2 for each dissimilarity distance. The table reporting only the correlation that showed to be strong (0.6 ≤ |ρ| ≤0.79) and very strong, (0.8 ≤ |ρ| ≤1)
| Correlation pair | ρ | Strength | p | |
|---|---|---|---|---|
| Bacteroides | Bray‐Curtis PC1 | −0.67 | Strong | <0.001 |
| Fusobacterium | Bray‐Curtis PC1 | −0.92 | Very strong | <0.001 |
| Peptoclostridium | Bray‐Curtis PC1 | 0.81 | Very strong | <0.001 |
| Ruminiclostridium 9 | Bray‐Curtis PC1 | 0.63 | Strong | <0.001 |
| Ruminococcaceae NK4A214 group | Bray‐Curtis PC1 | 0.61 | Strong | <0.001 |
| Odoribacter | Bray‐Curtis PC2 | 0.62 | Strong | <0.001 |
| Parabacteroides | Bray‐Curtis PC2 | 0.71 | Strong | <0.001 |
| Fusobacterium | Peptoclostridium | −0.63 | Strong | <0.001 |
| Ruminiclostridium 9 | Ruminococcaceae UCG−005 | 0.61 | Strong | <0.001 |
| Fusobacterium | Weighted Unifrac PC1 | −0.94 | Very strong | <0.001 |
| Peptoclostridium | Weighted Unifrac PC1 | 0.75 | Strong | <0.001 |
Toro‐Valdivieso, C. , Toro, F. , Stubbs, S. , Castro‐Nallar, E. , & Blacklaws, B. (2021). Patterns of the fecal microbiota in the Juan Fernández fur seal (Arctocephalus philippii). MicrobiologyOpen, 10, e1215. 10.1002/mbo3.1215
DATA AVAILABILITY STATEMENT
Raw reads data are publicly available in the European Nucleotide Archive (ENA) under the study accession PRJEB36555: https://www.ebi.ac.uk/ena/browser/view/PRJEB36555. All scripts used in this study can be accessed in GitHub at https://github.com/Cotissima/JFFS_microbiome_first_characterisation.
REFERENCES
- Aguayo, A. , Maturana, R. , & Torres, D. (1971). El lobo fino de Juan Fernández. Revista de Biología Marina Y Oceanografía, 14(3), 125–149. [Google Scholar]
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Andersen, J. M. , Skern‐Mauritzen, M. , Boehme, L. , Wiersma, Y. F. , Rosing‐Asvid, A. , Hammill, M. O. , & Stenson, G. B. (2013). Investigating annual diving behaviour by hooded seals (Cystophora cristata) within the northwest Atlantic Ocean. PLoS One, 8(11), e80438. 10.1371/journal.pone.0080438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2010). Babraham bioinformatics – FastQC a quality control tool for high throughput sequence data. Retrieved from https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- Baby, L. , Sankar, T. V. , & Anandan, R. (2014). Comparison of lipid profile in three species of myctophids from the south west coast of Kerala, India. National Academy Science Letters, 37(1), 33–37. 10.1007/s40009-013-0185-4 [DOI] [Google Scholar]
- Bik, E. M. , Costello, E. K. , Switzer, A. D. , Callahan, B. J. , Holmes, S. P. , Wells, R. S. , Carlin, K. P. , Jensen, E. D. , Venn‐Watson, S. , & Relman, D. A. (2016). Marine mammals harbor unique microbiotas shaped by and yet distinct from the sea. Nature Communications, 7, 10516. 10.1038/ncomms10516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekhman, R. , Tang, K. , Archie, E. A. , Barreiro, L. B. , Johnson, Z. P. , Wilson, M. E. , Kohn, J. , Yuan, M. L. , Gesquiere, L. , Grieneisen, L. E. , & Tung, J. (2016). Common methods for fecal sample storage in field studies yield consistent signatures of individual identity in microbiome sequencing data. Scientific Reports, 6, 31519. 10.1038/srep31519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen, E. , Rideout, J. R. , Dillon, M. R. , Bokulich, N. A. , Abnet, C. C. , Al‐Ghalith, G. A. , Alexander, H. , Alm, E. J. , Arumugam, M. , Asnicar, F. , Bai, Y. , Bisanz, J. E. , Bittinger, K. , Brejnrod, A. , Brislawn, C. J. , Brown, C. T. , Callahan, B. J. , Caraballo‐Rodríguez, A. M. , Chase, J. , … Caporaso, J. G. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37, 852–857. 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossart, G. D. (2011). Marine mammals as sentinel species for oceans and human health. Veterinary Pathology, 48(3), 676–690. 10.1177/0300985810388525 [DOI] [PubMed] [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , Rosen, M. J. , Han, A. W. , Johnson, A. J. A. , & Holmes, S. P. (2016). DADA2: High‐resolution sample inference from Illumina amplicon data. Nature Methods, 13(7), 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport, T. C. , Power, M. L. , Harcourt, R. G. , Webster, K. N. , & Tetu, S. G. (2016). Colony location and captivity influence the gut microbial community composition of the Australian sea lion (Neophoca cinerea). Applied and Environmental Microbiology, 82(12), 3440–3449. 10.1128/AEM.00192-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilam, O. , Zarecki, R. , Oberhardt, M. , Ursell, L. K. , Kupiec, M. , Knight, R. , Gophna, U. , & Ruppin, E. (2014). Glycan degradation (GlyDeR) analysis predicts mammalian gut microbiota abundance and host diet‐specific adaptations. mBio, 5(4), 2557–2568. 10.1128/mBio.01526-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, J. , Boness, D. , & Ochoa‐Acuña, H. (1998). A protracted foraging and attendance cycle in female Juan Fernandez fur seals. Marine Mammal Science, 14(3), 552–574. 10.1111/j.1748-7692.1998.tb00742.x. [DOI] [Google Scholar]
- Friedlander, A. M. , Ballesteros, E. , Caselle, J. E. , Gaymer, C. F. , Palma, A. T. , Petit, I. , Varas, E. , Muñoz Wilson, A. , & Sala, E. (2016). Marine biodiversity in Juan Fernández and Desventuradas Islands, Chile: Global endemism hotspots. PLoS One, 11(1), e0145059. 10.1371/journal.pone.0145059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin, M. Y. et al (2016). Phylogenomic analysis of the family peptostreptococcaceae (Clostridium cluster xi) and proposal for reclassification of Clostridium litorale (Fendrich et al 1991) and Eubacterium acidaminophilum (Zindel et al 1989) as peptoclostridium litorale gen. nov. International Journal of Systematic and Evolutionary Microbiology, 66(12), 5506–5513. 10.1099/ijsem.0.001548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett, W. S. , & Onderdonk, A. B. (2014). Bacteroides, Prevotella, Porphyromonas, and Fusobacterium species (and other medically important anaerobic gram‐negative bacilli). In Bennet J. E., Dolin R., & Blaser M. J. (Eds.), Mandell, Douglas, and Bennett’s principles and practice of infectious diseases (pp. 2773–2780). Elsevier Inc. [Google Scholar]
- Grosser, S. , Sauer, J. , Paijmans, A. J. , Caspers, B. A. , Forcada, J. , Wolf, J. B. W. , & Hoffman, J. I. (2019). Fur seal microbiota are shaped by the social and physical environment, show mother–offspring similarities and are associated with host genetic quality. Molecular Ecology, 28(9), 2406–2422. 10.1111/mec.15070 [DOI] [PubMed] [Google Scholar]
- Guerrero, A. I. , & Rogers, T. L. (2019). From low to high latitudes: Changes in fatty acid desaturation in mammalian fat tissue suggest a thermoregulatory role. BMC Evolutionary Biology, 19(1), 155. 10.1186/s12862-019-1473-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, X. , Lei, H. , Zhang, K. , Ke, F. , & Song, C. (2020). Diversification of animal gut microbes and NRPS gene clusters in some carnivores, herbivores and omnivores. Biotechnology and Biotechnological Equipment, 34(1), 1280–1287. 10.1080/13102818.2020.1835536 [DOI] [Google Scholar]
- Halpern, B. S. , Frazier, M. , Afflerbach, J. , Lowndes, J. S. , Micheli, F. , O’Hara, C. , Scarborough, C. , & Selkoe, K. A. (2019). Recent pace of change in human impact on the world’s ocean. Scientific Reports, 9(1), 1–8. 10.1038/s41598-019-47201-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen, E. L. , Abrahms, B. , Brodie, S. , Carroll, G. , Jacox, M. G. , Savoca, M. S. , Scales, K. L. , Sydeman, W. J. , & Bograd, S. J. (2019). Marine top predators as climate and ecosystem sentinels. Frontiers in Ecology and the Environment, 17(10), 565–574. 10.1002/fee.2125 [DOI] [Google Scholar]
- Hildebrandt, M. A. , Hoffmann, C. , Sherrill–Mix, S. A. , Keilbaugh, S. A. , Hamady, M. , Chen, Y. Y. , Knight, R. , Ahima, R. S. , Bushman, F. , & Wu, G. D. (2009). High‐fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology, 137(5), 1716–1724. 10.1053/j.gastro.2009.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh, J. W. , & Roh, T. Y. (2020). Opportunistic detection of Fusobacterium nucleatum as a marker for the early gut microbial dysbiosis. BMC Microbiology, 20(1), 1–17. 10.1186/s12866-020-01887-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT Multiple Sequence Alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M. , Cho, H. , & Lee, W. Y. (2020). Distinct gut microbiotas between southern elephant seals and Weddell seals of Antarctica. Journal of Microbiology, 58(12), 1018–1026. 10.1007/s12275-020-0524-3 [DOI] [PubMed] [Google Scholar]
- Kozich, J. J. , Westcott, S. L. , Baxter, N. T. , Highlander, S. K. , & Schloss, P. D. (2013). Development of a dual‐index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and Environmental Microbiology, 79(17), 5112–5120. 10.1128/AEM.01043-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea, M. A. , Nichols, P. D. , & Wilson, G. (2002). Fatty acid composition of lipid‐rich myctophids and mackerel icefish (Champsocephalus gunnari) – Southern Ocean food‐web implications. Polar Biology, 25(11), 843–854. 10.1007/s00300-002-0428-1 [DOI] [Google Scholar]
- Lewis, R. et al (2006). Sex‐specific foraging strategies and resource partitioning in the southern elephant seal (Mirounga leonina). Proceedings of the Royal Society B: Biological Sciences, 273(1603), 2901–2907. 10.1098/rspb.2006.3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R. , Hamady, M. et al (2008a). Evolution of mammals and their gut microbes. Science, 320(5883), 1647–1651. 10.1073/pnas.102164299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R. E. , Lozupone, C. A. , Hamady, M. , Knight, R. , & Gordon, J. I. (2008b). Worlds within worlds: Evolution of the vertebrate gut microbiota. Nature Reviews Microbiology, 6(10), 776–788. 10.1038/nrmicro1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Qu, J. , Li, T. , Li, J. , Lin, Q. , & Li, X. (2016). Pika Population density is associated with the composition and diversity of gut microbiota. Frontiers in Microbiology, 7, 758. 10.3389/fmicb.2016.00758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight, D. T. , Huerlimann, R. , Bower, D. S. , Schwarzkopf, L. , Alford, R. A. , & Zenger, K. R. (2019). Methods for normalizing microbiome data: An ecological perspective. Methods in Ecology and Evolution, 10(3), 389–400. 10.1111/2041-210X.13115 [DOI] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One, 8(4), e61217. 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros, A. W. et al (2016). Characterization of the faecal bacterial community of wild young South American (Arctocephalus australis) and Subantarctic fur seals (Arctocephalus tropicalis). FEMS Microbiology Ecology, 92(3). fiw029. 10.1093/femsec/fiw029 [DOI] [PubMed] [Google Scholar]
- Nelson, T. M. , Rogers, T. L. , & Brown, M. V. (2013). The gut bacterial community of mammals from marine and terrestrial habitats. PLoS One, 8(12), e83655. 10.1371/journal.pone.0083655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, T. M. , Rogers, T. L. , Carlini, A. R. , & Brown, M. V. (2013). Diet and phylogeny shape the gut microbiota of Antarctic seals: A comparison of wild and captive animals. Environmental Microbiology, 15(4), 1132–1145. 10.1111/1462-2920.12022 [DOI] [PubMed] [Google Scholar]
- Nicholson, J. K. , Holmes, E. , Kinross, J. , Burcelin, R. , Gibson, G. , Jia, W. , & Pettersson, S. (2012). Host‐gut microbiota metabolic interactions. Science, 336(6086), 1262–1267. 10.1126/science.1223813 [DOI] [PubMed] [Google Scholar]
- Nishida, A. H. , & Ochman, H. (2018). Rates of gut microbiome divergence in mammals. Molecular Ecology, 27(8), 1884–1897. 10.1111/mec.14473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numberger, D. et al (2016). Comparative analysis of the fecal bacterial community of five harbor seals (Phoca vitulina). MicrobiologyOpen, 5(5), 782–792. 10.1002/mbo3.369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara, A. M. , & Shanahan, F. (2006). The gut flora as a forgotten organ. EMBO Reports, 7(7), 688–693. 10.1038/sj.embor.7400731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa Acuña, H. , & Francis, J. M. (1995). Spring and summer prey of the Juan Fernandez fur seal, Arctocephalus philippii . Canadian Journal of Zoology, 73(8), 1444–1452. 10.1139/z95-170 [DOI] [Google Scholar]
- Ochoa‐Acuña, H. , Francis, J. M. , & Oftedal, O. T. (1999). Influence of long intersuckling interval on composition of milk in the Juan Fernández fur seal, Arctocephalus philippii . Journal of Mammalogy, 80(3), 758–767. 10.2307/1383245 [DOI] [Google Scholar]
- Olsen, I. et al (2014). The family fusobacteriaceae. In Rosenberg E. (Ed.), The prokaryotes (pp. 109–132). Springer. [Google Scholar]
- Pacheco‐Sandoval, A. , Schramm, Y. , Heckel, G. , Brassea‐Pérez, E. , Martínez‐Porchas, M. , & Lago‐Lestón, A. (2019). The Pacific harbor seal gut microbiota in Mexico: Its relationship with diet and functional inferences. PLoS One, 14(8), e0221770. 10.1371/journal.pone.0221770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partrick, K. A. , Chassaing, B. , Beach, L. Q. , McCann, K. E. , Gewirtz, A. T. , & Huhman, K. L. (2018). Acute and repeated exposure to social stress reduces gut microbiota diversity in Syrian hamsters. Behavioural Brain Research, 345, 39–48. 10.1016/j.bbr.2018.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, F. L. , Oliveira Júnior, C. A. , Silva, R. O. S. , Dorella, F. A. , Carvalho, A. F. , Almeida, G. M. F. , Leal, C. A. G. , Lobato, F. C. F. , & Figueiredo, H. C. P. (2016). Complete genome sequence of Peptoclostridium difficile strain Z31. Gut Pathogens, 8(1), 11. 10.1186/s13099-016-0095-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompa, S. , Ehrlich, P. R. , & Ceballos, G. (2011). Global distribution and conservation of marine mammals. Proceedings of the National Academy of Sciences of the United States of America, 108(33), 13600–13605. 10.1073/pnas.1101525108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potrykus, J. , White, R. L. , & Bearne, S. L. (2008). Proteomic investigation of amino acid catabolism in the indigenous gut anaerobe Fusobacterium varium . Proteomics, 8(13), 2691–2703. 10.1002/pmic.200700437 [DOI] [PubMed] [Google Scholar]
- Price, M. N. , Dehal, P. S. , & Arkin, A. P. (2010). FastTree 2 – Approximately maximum‐likelihood trees for large alignments. PLoS One, 5(3), e9490. 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu, S. , Khazanehei, H. , Jones, P. J. , & Khafipour, E. (2016). Interactions between obesity status and dietary intake of monounsaturated and polyunsaturated oils on human gut microbiome profiles in the Canola Oil Multicenter Intervention Trial (COMIT). Frontiers in Microbiology, 7, 1612. 10.3389/fmicb.2016.01612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , Peplies, J. , & Glöckner, F. O. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic, 41(D1), D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2019). R: The R project for statistical computing. R Foundation for Statistical Computing. Retrieved from https://www.r‐project.org/ [Google Scholar]
- Ralls, K. , & Mesnick, S. L. (2009). Sexual dimorphism. In Perrin W. F., Bernd W., & Thewissen J. G. M. (Eds.), Encyclopedia of marine mammals (2nd ed.). Elsevier Science & Technology. [Google Scholar]
- Redford, K. H. , Segre, J. A. , Salafsky, N. , del Rio, C. M. , & McAloose, D. (2012). Conservation and the microbiome. Conservation Biology, 26(2), 195–197. 10.1111/j.1523-1739.2012.01829.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter, S. J. , Cox, M. J. , Turek, E. M. , Calus, S. T. , Cookson, W. O. , Moffatt, M. F. , Turner, P. , Parkhill, J. , Loman, N. J. , & Walker, A. W. (2014). Reagent and laboratory contamination can critically impact sequence‐based microbiome analyses. BMC Biology, 12(1), 87. 10.1186/s12915-014-0087-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semova, I. , Carten, J. D. , Stombaugh, J. , Mackey, L. C. , Knight, R. , Farber, S. A. , & Rawls, J. F. (2012). Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host and Microbe, 12(3), 277–288. 10.1016/j.chom.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shade, A. , & Handelsman, J. (2012). Beyond the Venn diagram: The hunt for a core microbiome. Environmental Microbiology, 14(1), 4–12. 10.1111/j.1462-2920.2011.02585.x [DOI] [PubMed] [Google Scholar]
- Smith, S. C. et al (2013). Age‐related differences revealed in Australian fur seal arctocephalus pusillus doriferus gut microbiota. FEMS Microbiology Ecology, 86(2), 246–255. 10.1111/1574-6941.12157 [DOI] [PubMed] [Google Scholar]
- Stoffel, M. A. , Acevedo‐Whitehouse, K. , Morales‐Durán, N. , Grosser, S. , Chakarov, N. , Krüger, O. , Nichols, H. J. , Elorriaga‐Verplancken, F. R. , & Hoffman, J. I. (2020). Early sexual dimorphism in the developing gut microbiome of northern elephant seals. Molecular Ecology, 29(11), 2109–2122. 10.1111/mec.15385 [DOI] [PubMed] [Google Scholar]
- Trevelline, B. K. , Fontaine, S. S. , Hartup, B. K. , & Kohl, K. D. (2019). Conservation biology needs a microbial renaissance: a call for the consideration of host‐associated microbiota in wildlife management practices. Proceedings of the Royal Society B: Biological Sciences, 286(1895), 20182448. 10.1098/rspb.2018.2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trites, A. W. (2019). Marine mammal trophic levels and trophic interactions. In Cochran J. K., Bokuniewicz J. H., & Yager P. L. (Eds.), Encyclopedia of ocean sciences (3rd ed., pp. 589–594). Elsevier Ltd. [Google Scholar]
- Turnbaugh, P. J. , Ley, R. E. , Mahowald, M. A. , Magrini, V. , Mardis, E. R. , & Gordon, J. I. (2006). An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature, 444(7122), 1027–1031. 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- Vlčková, K. , Mrázek, J. , Kopečný, J. , & Petrželková, K. J. (2012). Evaluation of different storage methods to characterize the fecal bacterial communities of captive western lowland gorillas (Gorilla gorilla gorilla). Journal of Microbiological Methods, 91(1), 45–51. 10.1016/j.mimet.2012.07.015 [DOI] [PubMed] [Google Scholar]
- Wasser, S. K. , Hunt, K. E. , Brown, J. L. , Cooper, K. , Crockett, C. M. , Bechert, U. , Millspaugh, J. J. , Larson, S. , & Monfort, S. L. (2000). A generalized fecal glucocorticoid assay for use in a diverse array of nondomestic mammalian and avian species. General and Comparative Endocrinology, 120(3), 260–275. 10.1006/gcen.2000.7557 [DOI] [PubMed] [Google Scholar]
- Zhu, L. , Wu, Q. I. , Deng, C. , Zhang, M. , Zhang, C. , Chen, H. , Lu, G. , & Wei, F. (2018). Adaptive evolution to a high purine and fat diet of carnivorans revealed by gut microbiomes and host genomes. Environmental Microbiology, 20(5), 1711–1722. 10.1111/1462-2920.14096 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw reads data are publicly available in the European Nucleotide Archive (ENA) under the study accession PRJEB36555: https://www.ebi.ac.uk/ena/browser/view/PRJEB36555. All scripts used in this study can be accessed in GitHub at https://github.com/Cotissima/JFFS_microbiome_first_characterisation.