

A polyoxometalate cluster integrating a photothermal and catalytic center is used to synergistically enhance olefin oxidation.
Abstract
A strategy integrating near infrared (NIR) photothermal and catalytic effects within one active center beyond ultraviolet and visible light is proposed without the combination of separated photothermal transformation components. A giant polyoxomolybdate, which has high NIR photothermal conversion efficiency, is selected as the model catalyst, while a cationic β-cyclodextrin is used to cover its negatively charged surface electrostatically. Under NIR light radiation, the designed catalyst increases catalytic activity of cyclohexene oxidation under O2 atmosphere in water. The conversion reaches about pentaploid of the reaction without NIR radiation. By excluding heating effect from the external heater at the same temperature, about twice as much enhancement, which can be attributed to the sole photothermal action, is still observed. While the catalytic center is shielded by the organic porous layer, the surface cavity allows the integrated catalyst to conduct a selective catalysis by screening the molecules in size over the surface channel.
INTRODUCTION
Light-promoted chemical transformations have received a lot of interest over recent years because of their indispensable characteristics in developing clean fuels from solar energy conversion and degrading persistent organic pollutants through a convenient and energy saving approach. Various light-driven chemical reactions have been realized either in the form of higher efficiency or green and sustainable processes. Such a strategy has been applied in fields such as CO2 reduction, water splitting, and organic synthesis to yield faster or more efficient reactions than normal methods (1–6). During the process, the separation states of photo-induced electrons and holes in photocatalysts trigger and/or accelerate the redox reactions to follow more effective paths, and the wavelength of light is extended from ultraviolet-to-visible (UV-vis) region by precise modulation of electronic energy band configuration of substrates (7–12). In contrast to this, the potentials of near infrared (NIR) light at longer wavelength range, which follows a different approach in energy usage, remain less exploited yet has comparable importance and unique features. For example, the photothermal effect of NIR based on the nonradiation delay of excited state is considered as one of the efficient pathways for energetic conversion and is currently used in life science and medical materials, such as antitumor treatments, sterilization, controlled drug release, etc., due to its capability for deeper tissue penetration (13, 14). The photothermal effect of some metal nanoparticles, which originates from plasmonic resonance absorption, is blooming to be used to assist or enhance the CO2 conversion, hydrogen evolution reaction, and so forth (15–17), although its catalytic reaction path is proposed to be different from those driven by UV-vis light due to the lower energy and mismatched bandgap positions. In comparison to the typical thermal- or photo-promoted reaction, however, the combination of nonradiative photothermal conversion with catalysis allows the catalysts to collect a heating source to reduce the temperature gradient near catalytic center and get a higher reactivity at local sites. These properties can, in principle, increase the endurance of substrates while the reaction route does not change, especially for those that could not suffer from a long time at higher temperature within the whole catalytic process. In some cases, photothermally synergistic catalysis effectively reduce the deactivation of the catalyst and increase the selectivity for the desired products (18). So far, the reported catalysts that combined photothermal effect involve various plasmonic metals (19), transition metal oxides (20, 21), organic conjugated polymers (22), etc. Although some successful examples are achieved in noble metal nanoparticles, which are mostly trapped in metal-organic frameworks and enzymes (23, 24), critical bottlenecks remain to be solved. For an efficient catalytic reaction, a direct integration of the photothermal center and the catalytic center becomes necessary. At present, the high effective catalysts bearing photothermal property are limited, while those known photothermal materials normally do not perform applicable catalysts (25). Therefore, rational designs of multifunctional catalysts to realize the integration of photothermal conversion and catalytic activity for green chemistry and energy-saving process are highly desired.
Polyoxometalates (POMs), as a class of well-defined and structurally diverse inorganic nanoclusters that are composed of oxygen-bridged early transition metal ions at high valence state, are of extensive potentials in the areas ranging from materials sciences and energy conversion to bio-related systems, medicine, and so forth (26). Owing to their intrinsic feature of Brønsted acidity and high capacity to gain or lose electrons without structural change of architectures, POMs have also been widely used as effective acid catalysts and redox catalysts for esterification, hydrolysis, and oxidations of various compounds, such as alkenes, alcohols, sulfides, and even for splitting of water (27). Many POMs at reduced state, which are prepared by direct synthesis or postreduction of sacrifice agents, display characteristic deep colors. The yielded NIR absorption at wavelength range from 600 to about 1000 nm has been applied in the photothermal conversion of POMs under the NIR laser irradiation because of the nonradiative characteristic of general POMs (28). Comparing to other photothermal materials, the photothermal behaviors deriving from NIR absorption of reduced POMs also exhibit the characteristic property as tumor-specific, antiviral, and antibacterial agents for combined diagnosis and therapy in nano medicine (29–31). Despite the POMs having the undoubted catalytic ability and excellent photothermal performance, the research integrating both photothermal and catalytic features in one cluster to realize photothermally assisted catalysis under NIR light radiation has not yet been involved up to now as far as we know.
In this context, we here would like to report the integration of photothermal center and catalytic center within one molecular POM to achieve the synergistic enhancement of catalytic reactions. By selecting a giant mixed-valent wheel-shaped polymolybdate cluster, Na14[MoVI126MoV28O462H14(H2O)70]·ca.400H2O (the anion is abbreviated as {Mo154}), in a diameter of 3.6 nm as the dual-functional catalyst, the oxidation of olefins is realized in aqueous system under mild conditions. While exhibiting catalytic capability in the esterolysis and oxidation reactions, their 28 MoV ions that are inlaid on the outside surface around equator also endow {Mo154} cluster with excellent photothermal transduction performance (32–34). To avoid unwanted aggregation and instability because of the large number of negative charges and cluster size, β-cyclodextrin (CD) grafting a pyridinium cation (CDC) was chosen to enwrap the bare cluster through an ion exchange reaction in this work (Fig. 1). Besides, the introduction of CD can play a role of molecular sieve for screening olefins with specific sizes through host-guest interaction. The obtained supramolecular photothermal catalyst (C47H74O34N)12Na2[Mo154O462H14(H2O)70] (named as {Mo154}@CDC·20H2O) remains both preferred photothermal performance and satisfactory stability against the catalytic conditions. Considering that the oxidation of cyclohexene is a very typical reaction model, and the series products are very useful intermediates, together with the synthetic value for potential applications in multiple areas, such as organic synthesis, medicinal, and drug preparations, it is selected to evaluate the photothermal catalytic ability of the prepared cluster catalyst. Although many POMs have been intensively studied as catalysts for the same reaction, the results here represent a previously untapped route that integrates photothermal effect and catalytic ability for enhanced catalytic reactions, which will be applicable for precise preparation in green synthesis and local reaction in biosystem.
Fig. 1. Preparation and photothermal catalysis of {Mo154}@CDC.
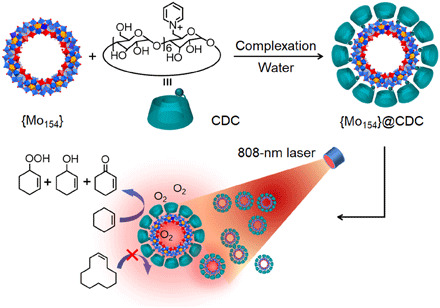
Schematic drawing of the molecular structure of the complex consisting of inorganic cluster and organic cation and photothermal catalysis for the oxidation of olefins.
RESULTS AND DISCUSSION
Structural characterizations
Because of several tens of reduced MoV ions that yield strong intervalence charge transfer (IVCT) transition for strong NIR absorption, the mixed-valent giant wheel {Mo154} cluster can, in principle, sustain a relatively long time under the gentle oxidation environment to carry out the photothermal transduction while the catalytic reaction in progress. However, suitable protection of the POM cluster is still necessary for both the initially reduced state and the complete chemical architecture bearing catalytic sites during the catalytic oxidation. On the basis of the previously published results (35, 36), organic cations attaching to the cluster surface electrostatically played a key role in matching the requirement. On the other hand, to ensure green catalytic conditions, the substituted organic counter cations that are used to protect the {Mo154} catalyst should be water-soluble and do not dissociate from the cluster surface. With the help of the additional lateral hydrogen bonding (37), CDC and its analogs are proved to cover the surface of cluster catalyst tightly and enhance the whole solubility of the formed ionic complexes in the aqueous system. In particular, the surface attachment of CD cations makes the supramolecular host layer performs a selective catalysis by promoting the dissolution of immiscible substrates. Through rapidly mixing the aqueous solutions of the two components together at room temperature, the ionic exchange reaction allows the CDC to bind onto {Mo154} polyanion electrostatically following a charge ratio in the case of enough space on the cluster surface. The formed ionic complex {Mo154}@CDC precipitated through the addition of methanol, and it was then purified by a simple filtration while their counterionic inorganic salt Na∙OTs (OTs denotes p-toluene sulfonate) from the individual components in filtrate was discarded. However, in comparison to other conventional photothermal catalysts, the {Mo154}@CDC can well dissolve in aqueous solution as a homogenous catalyst. The 1H nuclear magnetic resonance (NMR) spectra (Fig. 2A) show obvious changes of the proton peaks deriving from the CDC precursor. In detail, the proton peaks, Ha, Hb, and Hc, that are ascribed to the pyridinium group in CDC located in the cluster move downfield and become broadened, while the proton signals (Hd, He, and Hf), which belong to the OTs group referring to the counterion of CDC, disappear completely, confirming the full electrostatic combination of CDC with {Mo154}.
Fig. 2. Structural characterizations and comparison of the complex catalyst.

(A) 1H NMR spectra (500 MHz, D2O) of CDC∙OTs and catalyst {Mo154}@CDC at 25°C, in which the peak marked with asterisk (*) is derived from the residual solvent methanol and XPS spectra of Mo content in (B) catalyst {Mo154}@CDC and (C) isolated {Mo154}, which are summarized in table S2.
Fourier transform infrared (FTIR) spectrum of {Mo154}@CDC displays the characteristic vibration bands at around 969, 913, 820, 754, 630, and 557 cm−1 (fig. S1A), which can be attributed to the modes of terminal Mo═O, bridging Mo─O, and Mo─O─Mo stretching from {Mo154} cluster (33, 38), consistent with the cluster framework structure after the surface encapsulation. Meanwhile, the absorption bands of CDC at 3348, 2918, 1157, and 1038 cm−1, which correspond to the symmetric and asymmetric stretching vibrations of O─H, C─H, C─O, and C─O─C of the CDC part, and the bands at 1579 and 1490 cm−1 that are derived from pyridinium unit (39), support its ionic complexation. The elemental analysis result suggests a structural formula of (C47H74O34N)12Na2[Mo154O462H14(H2O)70]·20H2O, implying that 12 of the counterions in one {Mo154} cluster have been replaced by CDC, while two residue sodium ions remain average. The matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF) mass spectrum (fig. S1B) of freshly prepared complex via the negative mode presents the molecular weight 1805.08 of [Na{Mo154}]13−, after losing 12 CDCs and a sodium ion as well as 20 water molecules based on the chemical formula from elemental analysis. At the same time, the mass/charge ratio (m/z) peaks at 1196, 1197, 1198, and 1199 attributed to the CDC were observed in the positive mode, proving that the CDC binds to the {Mo154} cluster (fig. S1C). The thermogravimetric analysis (TGA) curve of {Mo154}@CDC presents a weight loss of 4.01% in the temperature range from 29° to 200°C, which fits the elimination of ca. 90 of water molecules (4.2%), in agreement with the sum of coordination water and hydrate (fig. S1D). A large weight loss of about 37.23%, with further increasing the temperature up to 620°C, corresponds well to the calculated weight of the total CDC cations (37.59%) based on the molecular formula given by elemental analysis. Considering the sublimation of MoO3 at the temperature over 600°C, the residual weight could not be used to analyze the precise content of cluster component in the prepared complex, but the total elemental analysis calculated (%) for C564H1082N12O960Na2Mo154 matches well to the experiment results referring to all individual elements, supporting that 12 CDCs bind to one {Mo154} cluster in average. In addition, isothermal titration calorimetric (ITC) results also proved a 12:1 ratio of CDC to {Mo154} with an electrostatic binding constant of 5.54 × 10−4 M−1, although the charged components are initially mixed in an equivalent charge ratio (fig. S1E). On the basis of the data of the molecules geometry in published results, in which the equatorial surface area of {Mo154} is S{Mo154} = 21.01 nm2, and the area at primary face of CDC is SCDC = 1.74 nm2, the estimated largest possible number (n) of CDC occupying on {Mo154} should be n = S{Mo154}/SCDC = 12.07, in good agreement with the values from above series measurements.
The x-ray photoelectron spectroscopy (XPS) measurement is carried out to further identify the chemical composition and the valence states of the obtained catalyst {Mo154}@CDC. As presented in Fig. 2B, the binding energy peaks of Mo 3d3/2 and Mo 3d5/2 in doublets at 236.07 and 232.97 eV can be rationally ascribed to the highest oxidation states of MoVI, while those peaks that appeared at 234.8 and 231.69 eV match up to the lower oxidation states of MoV doublets, and the energy gap between them is calculated to be 3.2 eV, in good agreement with the freshly prepared {Mo154} in Fig. 2C and the data in literature (40). Moreover, it is seen that the quantitative calculation for peak area ratio of MoV to MoVI remains at about 28:126, identically the same as that of the maintained cluster structure. The cyclic voltammetry (CV) measurement was carefully performed to confirm the presence of mixed valence. The redox couple MoV/MoVI is observed at E1/2 = −320 mV versus Ag/AgCl, and the separation between the oxidation and the reduction peaks is ca. 360 mV. The peak current of MoV/MoVI versus the square root of scan speed indicates a diffusion controlled dynamic process during the electrochemical redox process (fig. S2). Moreover, detailed electrochemical measurements give the molar ratio of MoV/MoVI (6.53/25.98 = 0.25), which closes to the ideal ratio of mixed-valence MoV/MoVI (28/126 = 0.22) in crystal structure.
Although the {Mo154} cluster was pointed out to have unusual potentials (41–43), the detailed applications are still under development due to the unsatisfied stability. A rapid elimination of coordination of water molecules made the {Mo154} cluster unstable. In contrast to that, a rapid weight loss (25.8%) of isolate {Mo154} cluster is observed with the increase of temperature to 100°C, and the catalyst {Mo154}@CDC performs higher thermal stability (fig. S1D) as the temperature is raised to about 200°C since only water loss was observed. The result indicates that the surface covering of CDC contributes to the improvement of cluster thermal stability, which provides a favorable condition for the following catalytic performance. Also, the stability of {Mo154}@CDC in aqueous solution against oxidation (44) was compared with that of isolate {Mo154} cluster with respect to time-dependent UV-vis-NIR spectra. Excess oxygen was bubbled in a 10-ml glass tube containing a sample solution, and the absorption changes were recorded with the time (fig. S3). The characteristic absorptions of both isolate {Mo154} and the catalyst {Mo154}@CDC at λmax = 745 nm show a decline after 180 min of bubbling. When the two sample aqueous solutions are exposed in air instead, the spectral change of {Mo154}@CDC becomes much slower than that of isolate {Mo154} cluster. The absorption of {Mo154}@CDC maintains over 60% of intensity after 7 days, while in contrast, the absorption intensity of isolate {Mo154} decreases to about 20% after the same number of days in air, suggesting the strong protection of CDC shielding from the oxidation of {Mo154} cluster.
Existing state of {Mo154}@CDC in solution
The bare {Mo154} cluster is confirmed to experience a dynamic evolution from a monodispersed state to a hollow sphere-like blackberry assembly in a several days’ process or even longer (45). To confirm the existing state of the prepared catalyst in aqueous solutions, dynamic light scattering (DLS) and transmission electronic microscope (TEM) characterization are carried out at different temperatures. The TEM image suggests an irregularly shaped aggregation of {Mo154}@CDC in size within 150 to 200 nm at room temperature (25°C) (Fig. 3A), and the DLS result shows the average hydrodynamic diameter ca. 168 nm (Fig. 3B), implying the aggregation of the prepared catalyst because the individual clusters can still be seen on the assembly clearly. In contrast, when heating to about 65°C, such an aggregation in solution disappears, and instead, a large number of well-dispersed dark spots with uniformed size distribution are observed close to the dimension of monodispersed {Mo154} (Fig. 3C), indicating the dissociation of the aggregate formed at room temperature. High-resolution images point out that these spot objects in ca. 3.5 to 4.0 nm can be ascribed to the isolated {Mo154} cluster bearing a size of 3.6 nm, which is reported in crystal structure owing to its strong contrast with CDC. At the same time, the corresponding DLS result of the sample at the temperature shows the average hydrodynamic diameter of {Mo154}@CDC in aqueous solution reaching ca. 5.8 nm (Fig. 3D), just matching the ideal complex size (5.4 nm) after considering the cluster surrounded by CDC with a cationic height (ca. 0.9 nm), very near to the ideal monodispersed catalyst. As a comparison, the hydrodynamic behaviors of isolate {Mo154} and CDC are also evaluated at room and reaction temperatures (fig. S4, A to D). The isolated {Mo154} is in size ca. 43 nm at 25°C, approaching to the size of blackberry reported by Liu et al. (46), but the hydrodynamic diameter decreases to ca. 3.8 nm at reaction temperatures, close to the size of single {Mo154} cluster at monodispersed state. Similarly, the isolated CDC in aqueous solution is in an aggregate of ca. 50 nm at room temperatures. However, the hydrodynamic diameter becomes ca. 1 nm at reaction temperatures, approaching to the single CDC molecule. This means that the observed particle size in the complex solution at 65°C does not originate from either {Mo154} or CDC colloids because any of its aggregation has already dissociated into monodispersed state at the reaction conditions. The behaviors of the complex catalyst in solution provide obvious advantages for both convenient separation of aggregates at room temperature and the largest increase of specific surface at the reaction temperature raised by NIR light radiation and external heating. Meanwhile, the hydrodynamic diameters of complex catalyst at room and reaction temperatures remain unchanged versus the time within 3 hours (fig. S4E), demonstrating the stability of catalyst complex in the reaction solutions.
Fig. 3. Assembled state characterizations of the synthetic complex catalyst.

(A) TEM image and (B) DLS diagram of {Mo154}@CDC solution at room temperature. (C) TEM image and (D) DLS diagram of the same sample at 65°C.
NIR photothermal transformation
With the benefit of maintained cluster structure and ionic complexation, the intense and broad absorption band centered at 745 nm sourcing from {Mo154} in the complex extends up to over 1000 nm in the NIR region (Fig. 4A). Although the absorptions at this region with the bandgap much smaller than that of ligand-to-metal charge transfer transition are assigned to d-d transition of the reduced electrons in some POMs (47, 48), the absorption bands were definitely ascribed to the IVCT (33). As no fluorescence is detected for most POMs, the excited electrons should go back to ground state following the nonradiative inner and exter conversions. Both paths transform excitation energy to heat quickly under laser radiation, and this makes {Mo154} a photothermal center. On the basis of the crystal structure analysis of this cluster that 4d electrons from 28 MoV centers locate within the 14 incomplete double-cubane–type compartments surrounding the quatorial plane (43), the NIR absorption and photothermal transformation should also occur at the same place. Considering the influence of the absorption coefficient on the photothermal conversion efficiency, the laser with a wavelength of 808 nm is selected as the NIR light source to evaluate the photothermal performance of catalyst {Mo154}@CDC. The time-dependent temperature profiles of the aqueous samples with and without catalyst {Mo154}@CDC (0.204 mM) in air are conducted under the laser light with an intensity of 1000 mW cm−2 (Fig. 4B). In comparison to the blank solution showing only ca. 5°C increasing within 180 min, the temperature of catalyst {Mo154}@CDC solution rises fast and reaches to ca. 65°C within 5 min upon the NIR laser radiation. In a longer term up to 180 min, the temperature does not show a linear increase and maintains at 67° ± 2°C constantly under the experimental conditions. The photothermal conversion efficiency (η) at the given conditions was calculated by exposing the aqueous solution on the laser light to a steady-state temperature increase (∆T) (fig. S5) (37). From the typical temperature change profile versus the time (Fig. 4C), the η is calculated to be 48.1% with a ∆Tmax of 42°C, indicating a comparable heating effect of the present catalyst to metal and metal oxide nanoparticles. The evaluation on the cycles of temperature change illustrates that the photothermal conversion could be repeated without obvious photobleaching for several times (Fig. 4D), demonstrating the sustainable photothermal stability in air.
Fig. 4. Photothermal performances of the synthetic complex catalyst.

(A) UV-vis-NIR spectra of isolate {Mo154} cluster (black) and catalyst {Mo154}@CDC (red) in aqueous solution, (B) time-dependent temperature profiles of the aqueous solutions with and without catalyst {Mo154}@CDC, (C) temperature-dependent plots of the aqueous solutions with and without catalyst {Mo154}@CDC versus the time under the irradiation of laser light under the irradiation of an 808-nm laser light with intensity of 1000 mW cm−2, and (D) temperature plots versus the irradiation cycles with the same light intensity used in (B) and (C).
NIR photothermally enhanced catalysis
While the electron transitions between MoV and MoVI through μ2-O atom in {Mo154} cluster contribute to photothermal conversion property (20), the rich bridging structure sites can also provide catalytically active centers for accelerating chemical reactions in the meantime. Thus, the catalytic oxidation of cyclic olefins, using gentle O2 agent other than those stronger oxidants, such as hydrogen peroxide or organic peroxides as a safe and green oxidant, is performed in aqueous solution. In the present study, cyclohexene is taken as the model substrate, first to demonstrate the photothermal transformation-promoted catalytic oxidation activity of {Mo154}@CDC. All detected products are shown in Fig. 5, and the main results are summarized in Table 1, while some related individual components are carried out parallelly for comparison. In the case of without a catalyst, both external heating and NIR radiation cannot accelerate the oxidation reaction of the substrate, and very little oxidized products are formed (Table 1, entries 1 to 3). In contrast, the {Mo154}@CDC catalyst exhibits an evident enhancement of the catalytic performance upon NIR irradiation in comparison to the same catalytic reaction without laser light radiation. The conversion of the photothermally supported catalytic reaction reaches ca. 38.5% within 3 hours (Table 1, entry 4), while the conversion of the same catalytic reaction under conventional external heating at 65°C and room temperature only comes to 18.34 and 6.70% (Table 1, entries 5 to 6). Apparently, the photothermal effect plays a crucial role in raising catalytic conversion, and we speculate that the main reason for the conversion efficiency increases sources from the local heating effect induced by the photothermal transduction at the catalytic center. The kinetic behavior of cyclohexene oxidation under different reaction conditions in the presence of catalyst {Mo154}@CDC was further characterized (Fig. 6A). The conversion upon NIR irradiation is surely much faster than those at room temperature and upon external heating, indicating that the temperature increase induced by photothermal transduction is not limited to the conventional heating pathway. According to the kinetic equation, ln (C/C0) = −kt, where C means the concentration of substrate at time t, while C0 is the concentration at the beginning, a proximate straight line from the ln (C/C0) plot versus the time (Fig. 6B) suggests a pseudo first-order kinetics of cyclohexene in the presence of catalyst {Mo154}@CDC under the photothermal condition with the kinetic constant of k = 3.18 × 10−3 min−1. As a comparison, although the reaction follows pseudo first-order kinetics as well under external heating, the dynamic constant decreases to k = 5.37 × 10−4 min−1. By comparing the conversions of the photothermal route with those at room temperature and encountering external heating up to 65°C, respectively, the enhancement factor of photothermal catalysis, in terms of the conversion ratio under the photothermal condition versus that under room temperature, is calculated to be in the range of 4.8 to 5.1 with less fluctuation throughout the given time limit with respect to the reaction at room temperature (Fig. 6C). By comparing to the conversion of this catalytic reaction with the one undergoing an external heating to the same temperature, the enhancement factor still reaches about 1.9 to 2.1, which can be attributed to the local photothermal effect that excludes the influence from environment temperature. These results demonstrate that the photothermal conversion of catalyst {Mo154}@CDC not only contributes to the suitable increase of temperature in the reaction system but also accelerates the catalytic reaction rate at catalytic sites. To further identify the influence of photothermal effects on the catalytic reaction, the catalytic performance of {Mo154}@CDC under different light intensities were conducted (Fig. 6D). The reaction conversion of cyclohexene under 1 atm of O2 atmosphere is almost linearly growth with regard to the laser light intensity. Although similar tendency of conversion versus the temperature increase upon external heating is observed, the conversion values of catalytic oxidation at different temperatures are always lower than the corresponding values under photothermal conditions at the same temperature. Typically, the conversion of catalytic oxidation at 95°C under external heating is just comparable to the value under NIR irradiation with light intensity of 1000 mW cm−2, revealing that the higher photothermal transduction has much better effect to enhance the catalytic reaction.
Fig. 5. Catalytic reaction of cyclohexene.

The oxidation of cyclohexene by O2 in the presence of the prepared catalyst in aqueous solution.
Table 1. The catalytic performance of the complex catalyst and its isolated components.
The summary of oxidation of cyclohexene catalyzed by the prepared catalyst and references with O2 as the oxidant.* n.d., not detected.
| Entry | Catalyst | Condition† | Conv. %‡ | Product selectivity % | |||
| D | E | F | G | ||||
| 1 | Blank | PT condition | 2.50 | 51.77 | 24.06 | 22.37 | 1.08 |
| 2 | Ex. heating 65°C | 1.20 | 53.84 | 22.31 | 22.72 | 1.13 | |
| 3 | R.T. | n.d. | n.d. | n.d. | n.d. | n.d. | |
| 4 | {Mo154}@CDC | PT condition | 38.53 | 25.28 | 20.68 | 52.94 | 1.10 |
| 5 | Ex. heating 65°C | 18.34 | 25.91 | 23.21 | 49.87 | 1.01 | |
| 6 | R.T. | 6.70 | 26.38 | 24.11 | 48.43 | 1.08 | |
| 7 | CDC∙OTs | Ex. heating 95°C | 6.84 | 21.15 | 25.42 | 52.23 | 1.20 |
| 8 | Ex. heating 65°C | 5.53 | 20.83 | 27.16 | 51.03 | 0.98 | |
| 9 | R.T. | n.d. | n.d. | n.d. | n.d. | n.d. | |
| 10 | Na14{Mo154} | PT condition | 12.25 | 21.67 | 28.34 | 48.89 | 1.10 |
| 11 | Ex. heating 65°C | 7.01 | 22.34 | 26.51 | 49.89 | 1.26 | |
| 12 | R.T. | 2.30 | 23.38 | 26.98 | 48.36 | 1.28 | |
| 13 | CD/{Mo154} | PT condition | 18.61 | 24.31 | 25.70 | 48.79 | 1.20 |
| 14 | Ex. heating 65°C | 10.18 | 23.84 | 25.14 | 49.67 | 1.35 | |
| 15 | R.T. | 4.50 | 24.67 | 25.87 | 48.33 | 1.13 | |
*Reaction conditions: cyclohexene (2.45 mM), catalyst (0.204 mM), 1 atm O2 (balloon); reaction time: 180 min.
†“Room temperature (R.T.)” and “External (Ex.) heating 65°C″ represent the reaction and was carried out at room temperature and 65°C in an oil bath, and “Photothermal (PT) condition” means that the reaction was conducted under NIR irradiation. All the control reactions were performed in dark to ensure that the NIR light used in the experimental group is the only light source.
‡Conversion and selectivity were determined by GC using naphthalene as the internal standard.
Fig. 6. The catalytic performance of the complex catalyst versus the reaction time.

(A) The total conversion plots and (B) catalytic kinetic curves of the substrate cyclohexene catalyzed by {Mo154}@CDC versus the reaction time under different temperature conditions, (C) the calculated enhanced factors of Con.(P.T.)/Con.(R.T.) in red square and Con.(P.T.)/Con.(Ex.H.) in black square versus the reaction time, and (D) the conversion plots of the same catalytic reaction versus the temperature increase under different light intensity of NIR radiation and external heating to 65°C.
Synergistic effect of CDC
The control experiments for the oxidation reaction in the absence and presence of independent components, CDC, and bare {Mo154} are carried out under the conditions without and with NIR irradiation. Because of the nonphotothermal center, the CDC-joined reaction is performed at room temperature and under external heating up to 65° and 95°C (Table 1, entries 7 to 9). Very little conversions are checked out under all these conditions. The bare {Mo154} exhibits a little bit increased catalytic activity, and the conversion after NIR radiation becomes double of the value undergoing external heating treatment (Table 1, entries 10 to 12), confirming that the cluster serves as the catalytic center. For the mixture of free {Mo154} and CD without specific interaction, a further raised catalytic effect emerges at a scale of simple sum of the conversions catalyzed by each of the two isolated additives (Table 1, entries 13 to 15). Therefore, in any case of the isolate components or their mixture, we do not observe the excess increase of catalytic efficiency (Fig. 7A). These results confirm that only the tight covering of CDC on the surface of {Mo154} directs the highest catalytic conversion, which means there is a synergistic effect between the two components for the catalytic oxidation of cyclohexene. The process can be explained as that, in aqueous environment, the immiscible substrates is trapped by CDC onto the cluster surface to accomplish the catalytic process. The fixed CDC on the {Mo154} conducts the roles of both transportation agent of substrate into aqueous phase and anchoring it at the active sites in the present catalyst.
Fig. 7. Conversion plots of series substrates under different catalytic conditions.

(A) Conversion plots of cyclohexene oxidation in the presence of {Mo154}@CDC and bare {Mo154} under the NIR irradiation, while in the case of isolated CDC∙OTs as catalyst, the reaction was conducted upon external heating to 95°C, (B) conversions of the oxidation of series alkenes (C-hex, cyclohexene; C-oct, cyclooctene; C-dod, cyclododecene) over catalyst {Mo154}@CDC and isolate Na14{Mo154} cluster, (C) catalytic oxidation of {Mo154}@CDC for cyclohexene quenched by radical trapping agents, and (D) conversion curves of cyclohexene oxidation over {Mo154}@CDC without (square line) and with Ada-COOH (circle line).
The CD bearing hydrophobic cavity is known to include organic guest molecules by noncovalent interaction in aqueous solution, and this feature makes it a good carrier to build a close relation of substrates to the cluster for the subsequent catalytic oxidation in a more favorable state, as verified by NMR spectra (fig. S6). By referring to free CD, the changed chemical shifts of H3 [∆δ = 0.01 parts per million (ppm)] and H5 (∆δ = 0.01 ppm) in catalyst {Mo154}@CDC ascribing to the protons inside the cavity move upfield accompanied by the addition of cyclohexene, revealing the inclusion interaction of CDC with the substrate molecule. The strong correlation signals of H3 and H5 of CD with cyclohexene in two-dimensional (2D) NOESY (nuclear Overhauser effect spectroscopy) spectrum further confirm the host-guest inclusion and the trapping property of catalyst {Mo154}@CDC for substrate molecules. Therefore, it can be concluded that only the tight connection of CDC and {Mo154} helps the solvation of cyclohexene in water and delivers the substrate to the catalytic center, because the hydrophobic cyclohexene itself hardly accesses the hydrophilic surface of {Mo154} cluster through the bulk water. The reason that the simple mixture could not raise the catalytic conversion is that although free CD phase transfers the cyclohexene into aqueous solution via host-guest inclusion, the effective diffusion to the active surface of the catalyst is still a necessary procedure. Apparently, the attaching of CDC on {Mo154} becomes beneficial to the mass transfer of substrate. As a result, the catalytic conversion for the same oxidation reaction of the used substrate in the CD/{Mo154} mixture is far less than that of complex catalyst.
To disclose another role of CDC covering layer for selective catalysis, a series of olefin homologs with different molecular sizes from cyclohexene are used to conduct the catalytic oxidation of catalyst {Mo154}@CDC under the same conditions. For cis-cyclooctene with a flexible structure state, its size (5.2 Å) is only slightly smaller than the CD cavity diameter (ca. 6.0 Å for primary face) (18, 49). Although the guest molecule can be recognized at a favorable orientation (50), the small association constant and the higher modulation energy inside the cavity to get close to the catalytic surface of {Mo154} due to the unfavorable size causes the conversion to decrease greatly to 13.3% within 180 min (Fig. 7B and Table 2, entry 1). In the case of cyclodecene with a larger 3D size (6.5 Å × 4.9 Å × 3.1 Å, calculated from ChemBioOffice), the twisting conformation leads to the embedding of the neighboring groups of unsaturated double bond inside the cavity, which results in a blocking effect to the oxidation of this substrate. Consequently, the unfavorable inclusion yields a much lower conversion of 8.6% (Table 2, entry 2). As for the cyclododecene, the much larger molecular size and lower solubilization in water cause unfavorable inclusion, and thus, negligible conversion (~3.1%) is checked out (Fig. 7B and Table 2, entry 3). These results reveal that the CDC on the shielding surface dominates the catalytic selectivity of the catalyst {Mo154}@CDC by screening those molecules with molecular sizes out of the range that the cavity can accommodate. In general, when the substrates are larger than the cavity diameter of CDC protective layer, the transportation to the catalytic surface will be selectively restrained, and the observed catalytic conversion becomes lower than that of bare {Mo154} (Table 2, entries 1 to 3 in brackets). However, when the substrates are smaller than the cavity, the transportation to the reaction center will no longer be blocked even if the host-guest interactions are not as strong as that of cyclohexene, because the consecutive phase transfer behavior can offset the weak binding (Table 2, entries 4 to 6). Because the molecular size of the corresponding substrates in entries 4 to 6 matches the cavity of the host CD better than the cyclooctene, their catalytic oxidation presents the allylic products with even increased conversions to those in the case of bare {Mo154}.
Table 2. The summary of conversion and selectivity.
Oxidation of various substrates with O2 catalyzed by {Mo154}@CDC.*
| Entry | Substrate | Conv. %† | Product selectivity %† | ||
| ortho ─OH | ortho ═O | Others‡ | |||
| 1 |  |
13.3 (17.3)§ | 27.98 | 45.69 | 26.33 |
| 2 |  |
8.6 (16.5) | 33.92 | 66.08 | – |
| 3 |  |
3.1 (15.1) | 36.31 | 63.69 | – |
| 4 |  |
20.4 (12.8) | 35.01 | 64.99 | – |
| 5 |  |
18.5 (11.3) | 22.89 | 77.11 | – |
| 6 |  |
13.2 (10.1) | 30.23 | 69.77 | – |
*Reaction conditions: substrate (2.45 mM), catalyst (0.204 mM), 1 atm O2 (balloon), 180 min under NIR irradiation.
†Conversion and selectivity were examined by GC using naphthalene as the internal standard at 180 min.
‡The other substituted products including peroxide and epoxide were only checked in entry 1.
§Calculated conversion values of oxidation of series alkenes over catalyst {Mo154}@CDC and isolate Na14{Mo154} cluster in parenthesis.
Possible catalytic mechanism
The main products of cyclohexene oxidation catalyzed by catalyst {Mo154}@CDC under the photothermal condition are cyclohexenyl hydroperoxide, 2-cyclohexen-1-ol and 2-cyclohexen-1-one, and a very small portion of epoxidation in different yields (Table 1). On account of having little epoxidized products, a radical mechanism can be speculated by referring to the published results on the reaction (51–53). Note that even under the covering condition of CDC cations, the substrate molecules can get close to the catalytic sites on the surface of {Mo154} cluster through the primary face of CD cavity, accomplish the catalytic oxidation, and then leave the catalytic position due to the increased polarity of the formed product molecules. For those POMs at fully oxidized state, the surface MVI = O (M: transition metals, such as Mo, W, V, etc.) groups accept an oxygen atom from peroxides or oxygen to form a transition state MVI─O─O∙ and then transfer the oxygen to the adsorbed substrate to accomplish the oxidation (54, 55). Such a catalytic route usually causes epoxy products of olefin substrates in the presence of peroxide oxidants. In the case of catalytic center at a locally reduced state, the bridging structure MoV─O─MoVI acting as the catalytic sites often offers a radical path in the catalytic oxidation reaction, especially when O2 is used as the oxidant (56, 57). On the other hand, the photothermal conversion in molecular system including POMs can be well explained to derive from the thermal vibration delay in the present system during the nonradiation transition of the excited electrons from the excited state back to the ground state (58, 59) and can be taken as the parallel effect with nonrelevant influence to the cluster’s catalysis. In a further step to check the locations of proposed free radicals, the trapping experiments in the presence of N-tert-butyl-α-phenylnitrone (PBN) and para-benzoquinone (PBQ) as carbon and reactive oxygen radical quencher are carried out, respectively. To exclude the influence of CD on the signal of free radical adducts, isolate {Mo154} was used as the control catalyst to perform the catalytic reaction under the conditions with and without the scavengers (60). The results presented in Fig. 7C indicate that the total conversion was dramatically hindered from 12.25 to 1.3% before and after the presence of PBN, and the detailed products are too little to be detected.On the other hand, when PBQ is added into the catalytic reaction of cyclohexene oxidation, it also retards the conversion greatly, suggesting that oxygen radicals emerge during the reaction. The strong depressing effect of the two quenchers for the production of final products illustrates two important facts: The reaction involves the radical production, and the reaction process deals with the formation of both carbon and oxygen radicals. According to the reported results, the carbon radical C6H9∙ and oxygen radicals, C6H9O2∙ and C6H9O∙, can be inferred to emerge as the intermediates during the reaction. The electron paramagnetic resonance (EPR) spin-trap of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and PBN is further used to check the reactive species. Definitely, on the basis of the appearing signals of radical adducts, DMPO─C6H9O with aN = 13.15 G and aH = 6.4 G, PBN─C6H9O2 with aN = 14.05 G and aH = 11.7 G, and PBN─C6H9 with aN = 13.8 G, the coincident EPR spin-trap spectra confirm the radical species (fig. S7) (61). With these results, a possible catalytic mechanism for the oxidation reaction is proposed to start from the generation of α carbon radical, addition of oxygen, and radical transfer to final multiple products (Fig. 8). In detail, the substrate is first captured to the surface of {Mo154}, and then the vicinal H from allylic C─H is captured by the bridging MoV─O─MoVI moiety at equatorial face to form additive μ2-O─H bond, while MoVI is then reduced to MoV (62, 63). Meanwhile, the cyclohexene (S) is split into a cyclohexene radical (A) at α carbon. The subsequent insertion of the activated O2 allows the formed primary C6H9∙ radical (A) to yield a C6H9O2∙ peroxy radical (B), which can easily capture H from catalyst to form cyclohexenyl hydrogen peroxide C6H9O2H (D). Meanwhile, the peroxy radical (B) can recombine the primary radical (A) to generate another additive oxygen radical (C). Note that besides binding a proton radical from catalyst, the product (C) can disproportionate into the 2-cyclohexen-1-ol (E) and 2-cyclohexen-1-one (F). Because the product (D) can form additional stable ketone (F) by losing one water molecule, the proposed mechanism explains the reason that this product is approximately doubled of the other products, (D) and (E). Considering that the substrate and reactive intermediates mainly existed in the included state, the free contacts between them should be highly restrained. Thus, the incomplete consumption of the {Mo154} at reduced state formed in the catalytic process becomes an advantage to block the oxidation from oxygen, which may cause the weakening of photothermal effect. Besides these products, a very small amount of cyclohexene epoxide (G) are found as well at the end of the reaction, implying that there is a different oxidation process due to the existence of MVI═O catalytic center.
Fig. 8. Proposed catalytic mechanism.

Oxidation path of the cyclohexene catalyzed by complex catalyst {Mo154}@CDC.
To ascertain whether there is a possibility of radical iniation, we add the competitive guest adamantane formic acid (Ada-COOH) to the ongoing catalytic reaction. Because the host-guest interaction of Ada-COOH with binding constant 2 × 104 M−1 (64) and β-CD is much stronger than cyclohexene with binding constant 3.4 × 102 M−1 (65), the addition of Ada-COOH will block the catalytic oxidation of cyclohexene if it follows a general catalytic mechanism. Otherwise, radical iniation will be more possible. 1H NMR spectra provide the evidence for the competitive substitution of Ada-COOH to cyclohexene (fig. S6, A and B). The proton peaks of H3 and H5 belonging to CDC and the peaks attributed to Ada-COOH both show an obvious shifting at included state, while the proton shifts of cyclohexene move toward nonincluded state. On the basis of this result, the catalytic reaction under NIR irradiation (Fig. 7D) is conducted. When the reaction comes to 30 min, and Ada-COOH (0.017 mM, equivalent molar ratio to CDC) is added to the reaction, it is found that the reaction conversion rate gradually decreased, compared with the virgin catalytic reaction without addition of Ada-COOH. Within 80 min, the reaction conversion rate no longer rises, indicating the reaction suspended. This result strongly removes the possibility of {Mo154}@CDC acting as a radical initiator.
Stability of the catalyst complex
Although the catalyst dissolves well in aqueous solution, it is facilely separated via a hot filtration through the precipitation from insoluble solvents. After the separation, the conversion of substrate no longer proceeds upon NIR radiation (Fig. 9A). Therefore, there are not any soluble components that can catalyze the reaction. To evaluate the stability and sustainability, the used catalyst {Mo154}@CDC is collected by precipitation and then a centrifugation treatment, washing with acetone for the next catalytic cycle. The catalytic activity is performed under the same photothermal and conventional heating conditions. The conversions over 30% are detected in three consecutive runs without any further activation treatment (Fig. 9B). The observed conversion decrease can be attributed to the reduced weight of catalyst during the extraction from the reaction solution. The very similar characteristic vibrations in IR spectra of the used catalyst with the fresh one reveal that the {Mo154} cluster structure maintains well after the NIR radiation–related catalytic oxidation (fig. S8A). The broad absorption band at 600 to 1000 nm in the UV-vis-NIR spectra shows little change in both intensity and shape, further implying the stabilized cluster structure and valent state of {Mo154} in the catalyst (fig. S8B). The MALDI-TOF mass spectrum of the used complex presents the molecular weight 1809.39 via the negative mode (fig. S8C), implying the cluster structure completeness. The slight weight difference is from the change of coordination water in the posttreatment of the catalyst. The observed m/z peaks at 1196, 1197, 1198, and 1199 under positive mode well fit to the molecular weight of CDC+, supporting its role as the counterion accompanying the catalyst after the separation from reaction system (fig. S8D). The structural formula of used catalyst {Mo154}@CDC is examined with organic analysis (table S1) and TGA spectra (fig. S8E). The results prove that there are still 12 CDCs bearing in the used catalyst {Mo154}@CDC. The diffusion coefficient values of CDC, fresh {Mo154}@CDC, and used {Mo154}@CDC at reaction temperatures are evaluated with 2D diffusion-ordered spectroscopy (DOSY) spectra (fig. S8, F to H). The estimated diffusion coefficient of isolated CDC is about 6 × 10−10 m2/s, larger than the fresh {Mo154}@CDC at about 4 × 10−10 m2/s, consistent to the molecular weight and size difference. The used {Mo154}@CDC displays almost identical diffusion coefficient to the fresh one, and no signals of isolated CDC appear at the same time, confirming the stability of ionic complexation. To evaluate the structural integrity of {Mo154} in the complex after the catalysis and reduce the damage of electron beam in focusing, the ex situ atomic force microscopy (AFM) is conducted by dropping the {Mo154}@CDC sample solution at reaction temperature onto a silicon wafer or a mica, which is preheated to the same temperature. A huge amount of particles are observed to disperse with even size (fig. S8I). It is difficult to discern the particle morphology and size precisely, but the observed spots are consistent with the assigned assemblies after considering the influence of the AFM tip. The local magnification to one of the spots represents the ring-like morphology as that of {Mo154} (fig. S8J). These characterizations suggest that the CDC still attach on the cluster surface, and its surface covering protects the cluster structure from degradation during the catalytic process. The Mo content of the used catalyst that suffers three cycles of catalysis is examined, and the inductively coupled plasma (ICP) data show that almost no Mo in total (~0.02%) leaks out, demonstrating the whole structure stability of the catalyst during the catalytic reaction (table S1). However, it is seen that the content of MoV increases after catalytic cycles, which means that the cluster has been reduced partially. The XPS analysis is conducted to characterize the valent state change of Mo after the catalyst is recovered. The coexistence of MoV and MoVI is seen by deconvolution treatment of Mo spectrum, but the ratio of MoV to MoVI becomes ca. 30:70 (Fig. 9C). By comparing to the Mo valence ratio of 21.2:78.8 calculated from fresh catalyst (table S2), the present simulated result of the used catalyst implies that approximate 9% MoVI was reduced to MoV after three catalytic cycles, which is consistent with that checking out from ICP calculation.
Fig. 9. Sustainability characterizations of the complex catalyst.

(A) Conversion curves of cyclohexene oxidation catalyzed with {Mo154}@CDC (black line) and after taking out the catalyst via the hot filtration during the reaction (red line), (B) conversion column plots versus catalytic recycles of the same catalytic reaction under NIR irradiation (red) and conventional external heating (green), and (C) XPS spectrum of Mo in the used {Mo154}@CDC after three catalytic cycles.
In conclusion, we demonstrated a photothermal catalytic principle for multifunctional POM catalysts, which can integrate catalytic center and photothermal center together based on the surface metal oxide bonds and NIR absorption performance sourcing from IVCT transition. By taking the giant wheel-like {Mo154} cluster as the catalyst, an environment friendly catalytic oxidation of cyclohexene with oxygen at 1 atm in aqueous system was conducted in the presence of NIR radiation. The observed conversion became much higher than that of the same reaction under generally external heating, clearly proving the enhancement of direct photothermal effect yielded by the same cluster catalyst. Although the final conversion still did not reach a satisfactory level, the present results provided the first example, which is the outstanding catalytic property of POMs combined its NIR absorption feature at the catalytic center MoV─O─MoVI. In general, besides those bearing lower-valence metal substituted clusters, many POMs that perform excellent catalytic property are applicable for such kind of photothermal catalysts only if these clusters can be reduced to the sates holding photothermal effect at the visible and NIR region. The recovery of catalyst is also applicable through a simple filtration procedure.The organic modification via ionic interaction plays an important role for stabilizing the cluster architecture and MoV at reduced state for sustainable photothermal transduction against the catalytic conditions. When a supramolecular host like CD was used for such a purpose, synergistic functions in green catalysis and substrate selectivity via the cavity screening can be collected in the cluster catalyst. It can be envisioned that such an integrated catalyst system is also expected to apply for the catalytic hydrolysis and other nonoxidation reactions such as addition reactions.
MATERIALS AND METHODS
Materials
The CD used in the present study was purchased from Sinopharm Chemical Reagent Co. Ltd. and recrystallized three times before use. p-Toluene sulfonyl chloride and pyridine were products of Aladdin Co. Ltd. Dichloromethane (high-performance liquid chromatography grade) was from Sigma-Aldrich. Cyclohexene (99%), cyclohexene oxide (98%), 2-cyclohexene-1-ol (≥95%), cis-cyclooctene (95%), cyclododecene (95%), and naphthalene (99%) were received from J&K Chemical Co. Ltd. 2-Cyclohexen-1-one (97%), PBN, and PBQ were purchased from Energy Chemical Co. Ltd. Cyclooctene oxide (97%) was obtained from 9-Ding Chemistry Co. Ltd. Unless otherwise stated, these reagents were all used without further treatment. Acetone was distilled over P2O5 before use. Deionized water with a specific resistance of 18.25 megohm·cm was used throughout the experiments.
Measurements
1H NMR, 2D NOSEY NMR, and 2D DOSY NMR were conducted on a Bruker Avance 500 or 600-MHz NMR spectrometer (Germany) using tetramethylsilane as an internal reference. Chemical shifts were referenced to the solvent values (like δ = 4.79 ppm for D2O). FTIR spectra were obtained on a Bruker Vertex 80 V spectrometer equipped with a deuterated triglycine sulfate detector (32 scans) with a resolution of 4 cm−1 on a potassium bromide pellet. Organic elemental analysis (C, H, and N) was performed on a Vario microcube from Elementar Germany. ICP analysis was carried out on an OPTIMA 3300DV (PerkinElmer). TGA was recorded on a PerkinElmer Diamond TG/differential thermal analysis instrument with a heating rate of 10°C min−1. MALDI-TOF mass spectra were recorded on a Bruker Autoflex speed TOF/TOF mass spectrometer (Germany), equipped with a nitrogen laser (337 nm, 3-ns pulse). The m/z range during datum acquisition is from 700 to 3500 Da for linear negative mode. CV measurements were carried out on CHI 660C electrochemical workstation at room temperature under nitrogen atmosphere. A three-electrode system containing indium tin oxide–coated glass slide as the working electrode, a platinum wire as the counter electrode, and an Ag/AgCl as the reference electrode were used in the measurement. UV-vis absorption spectra were carried out on a Varian CARY 50 probe spectrometer (USA) at room temperature. ITC data were obtained from a MicroCal VP-isothermal titration calorimeter (Malvern, UK). DLS measurements were performed on a Zetasizer NanoZS instrument (Malvern, UK). XPS spectra were acquired on an ESCALAB 250 spectrometer (Thermo Fisher Scientific, USA) with a monochromic x-ray source (Al Kα line, 1486.6 eV). NIR laser is generated from an 808-nm monochromatic source with an optical fiber coupler laser and adjustable output power from Xi’an Hxrld Laser Tech. Co. Ltd. The output power was calibrated by an optical power meter (CEL-NP2000-2) from Beijing Au-Light Tech Co. Ltd. EPR (JESFA200, Japan) was used to detect the carbon radical and reactive oxygen species generated in the catalytic oxidation system with DMPO and PBN as the radical spin-trapped reagents. TEM images were obtained on a field emission electron microscope (JEOL JEM-2100F, Japan) with an accelerating voltage of 200 kV. AFM images were taken with a Dimension FastScan TM AFM (Bruker, USA) and a Cypher ES (Asylum Research, an Oxford Instruments company, Santa Barbara, CA, USA). All the reagents and products were analyzed by gas chromatography (GC) (Shimadzu GC-14C, Japan) contained HP-FFAP capillary column.
Sample preparation for TEM measurement
The samples for TEM characterization are prepared by putting a drop of a highly diluted aqueous solution of the catalyst {Mo154}@CDC (0.001 mg/ml) onto an ultrathin micro grid and allowed the water to evaporate at room temperature. When characterizing the morphology of the catalyst at the reaction temperature of 65°C, the preparations of all samples are carried out in an incubator maintaining the target conditions to ensure the existing state in the solution closing to the reaction environment.
Synthesis of photothermal catalyst complex
The Na14{Mo154} was freshly prepared according to the published procedures (33, 38). The precursor, mono-6-deoxy p-toluene sulfonate (2.58 g, 2 mmol), which was synthesized first following a similar procedure in literatures (66, 67), was dissolved in anhydrous pyridine (170 ml) and heated to 90°C with stirring for 72 hours. The reaction mixture was cooled to room temperature and added dropwise to the dry acetone (500 ml). The formed precipitate was filtered off and washed three times with excess dry acetone, giving the product mono-6-deoxy-6-pyridinium-β-cyclodextrin p-toluene sulfonate (CDC∙OTs) a yield of 80% (2.19 g). 1H NMR (500 MHz, D2O, 25°C): δ (ppm) = 8.94 (d, 2H, Ar H), 8.68 (t, 1H, Ar H), 8.17 (t, 2H, Ar H), 7.71 (d, 2H, Ar H), 7.37 (d, 2H, Ar H), 5.27 to 4.97, 4.28 to 2.57 (m, 42H, CH), and 2.41 (s, 3H, CH3). MALDI-TOF MS (m/z) [M+]: calculated for C47H74O34N: 1197.08, found: 1197.6.
The catalyst {Mo154}@CDC was prepared according to an ionic exchange method described in literature (14). {Mo154} (0.3 g, 9.7 × 10−3 mmol) dissolved in deionized water (10 ml) was added dropwise into the vigorously stirred aqueous solution (10 ml) of CDC∙OTs (0.186 g, 0.136 mmol) at the initial molar ratio of Mo154 to CDC at 1:14. After 12 hours of stirring, the reaction mixture was added dropwise to anhydrous methanol (500 ml), and the formed precipitate was filtered off and washed with a large amount of anhydrous methanol several times. Drying the precipitate in vacuum until the weight remained constant gave the product (0.185 g, 50%). The 1H NMR spectrum confirmed the counter ion p-toluene sulfonate having been eliminated. Elemental analysis for C564H1082N12O960Na2Mo154 (38212.87 g mol−1) Calcd. C 17.73, H 2.85, N 0.44, Na 0.12, Mo 38.66 (where MoV 7.03, cerimetric titration); found: C 17.59, H 2.98, N 0.44, Na 0.14, Mo 38.24 (where MoV 6.95).
Photothermal property measurement
During the measurement of the photothermal conversion efficiency η, the aqueous solution (5 ml) of catalyst {Mo154}@CDC (39 mg) was irradiated by a NIR light with a power density of 1000 mW cm−2 for 120 min starting from room temperature. The temperature changes were recorded at an interval of 30 s. An equivalent amount of water under the same condition was set as a control. The temperature of the sample solutions was monitored by a K/J type thermometer. Each of the experiments was parallelly repeated three times.
Catalytic activity measurement
The photothermal catalytic activity of {Mo154}@CDC for the oxidation of olefins in aqueous solution was assessed. Typically, the substrate (2.45 mM) was added to a 25-ml two-necked rubber-sealed quartz flask containing an aqueous solution (5 ml) of {Mo154}@CDC (39 mg), including cyclohexene, cyclooctene, and cyclododecene, respectively. The reactor was initially flushed with N2 for several times to avoid the disturbance of residue air and maintained at the atmosphere for 2 hours to ensure the host-guest interaction between the catalyst and substrates. Subsequently, pure O2 was fed into the reactor for 2 min at a rate of 100 ml min−1 to replace N2 and then sustained the atmosphere with an O2 balloon (1 atm). The moment was defined at reaction time t = 0. The catalytic oxidation was carried out under NIR laser irradiation (808 nm) with power densities of 800, 1000, and 1200 mW cm−2 with vigorous stirring (1200 rpm), respectively. The reaction progress was monitored through the thin layer chromatography method. Once the reaction finished, the prepared catalyst was separated from the reaction mixture by precipitation from the addition of acetone (10 ml). After removing the excess solvent in the filtrate under the reduced pressure, the product mixture was obtained and encountered a quantitative analysis by GC using naphthalene as the internal standard. Under conditions of GC measurement, an HP-FFAP capillary column, a flame ionization detector, was used. The temperature program was set as follows: initial temperature, 50°C kept for 5 min; the second temperature setting at 200°C with heating rate of 25°C/min; the next temperature setting at 210°C upon heating rate of 2°C/min, kept for 0.2 min; the final temperature maintaining at 220°C with heating rate of 1°C/min. The temperature of injector is set at 300°C, and the temperature of detector is 250°C. The D cannot be measured directly by using GC because of its thermal instability. Instead, we quantified it by a control reaction in which triphenylphosphine (PPh3; 26.2 mg, 1 mole percent) was added to exhaust it. After the sample was shaken for 5 min, the PPh3 reacted with D to form PPh3O and E, which was then analyzed by GC and compared with the untreated counterpart. The subtraction of the initial amount of the E formed in the control reaction from the content of the alcohol after the addition of PPh3 affords the quantity of D in original sample. The catalyst as a precipitate was washed three times with acetone and reused for the next catalytic cycle after drying in a vacuum oven at 50°C for 12 hours. As a control, the thermal catalytic activity of {Mo154}@CDC was assessed following the same oxidation process of olefins, except that NIR light irradiation was substituted by external heating under an oil bath.
Acknowledgments
We thank J. M. Poblet for useful discussion on electron transfer transitions of the catalyst at the NIR region. We also thank W. K. Zhang and Z. W. Ma for helping with AFM measurement. Funding: This work was supported by the National Natural Science Foundation of China (91961117) and the Program for JLU Science and Technology Innovative Research Team (2017TD-10). Author contributions: L.W. designed and guided the project. X.C. performed most experiments, data analysis, and preparation of a draft manuscript. G.Z. carried out TEM measurement. L.W., B.L., and X.C. finished the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/30/eabf8413/DC1
REFERENCES AND NOTES
- 1.Ravetz B. D., Pun A. B., Churchill E. M., Congreve D. N., Rovis T., Campos L. M., Photoredox catalysis using infrared light via triplet fusion upconversion. Nature 565, 343–346 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng X., Pi Y., Song Y., Brzezinski C., Xu Z., Li Z., Lin W., Metal-organic frameworks significantly enhance photocatalytic hydrogen evolution and CO2 reduction with earth-abundant copper photosensitizers. J. Am. Chem. Soc. 142, 690–695 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Pagire S. K., Foll T., Reiser O., Shining visible light on vinyl halides: Expanding the horizons of photocatalysis. Acc. Chem. Res. 53, 782–791 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Jin J., Chen S., Wang J., Chen C., Peng T., One-pot hydrothermal preparation of PbO-decorated brookite/anatase TiO2 composites with remarkably enhanced CO2 photoreduction activity. Appl. Catal. B Environ. 263, 118353 (2020). [Google Scholar]
- 5.Wang Z., Li C., Domen K., Recent developments in heterogeneous photocatalysts for solar-driven overall water splitting. Chem. Soc. Rev. 48, 2109–2125 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Cheung P. L., Kapper S. C., Zeng T., Thompson M. E., Kubiak C. P., Improving photocatalysis for the reduction of CO2 through non-covalent supramolecular assembly. J. Am. Chem. Soc. 141, 14961–14965 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Ha S., Lee Y., Kwak Y., Mishra A., Yu E., Ryou B., Park C.-M., Alkyne-alkene [2 + 2] cycloaddition based on visible light photocatalysis. Nat. Commun. 11, 2509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uchikura T., Oshima M., Kawasaki M., Takahashi K., Iwasawa N., Supramolecular photocatalysis by utilizing the host-guest charge-transfer interaction: Visible-light-induced generation of triplet anthracenes for [4+2] cycloaddition reactions. Angew. Chem. Int. Ed. 59, 7403–7408 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Crake A., Christoforidis K. C., Godin R., Moss B., Kafizas A., Zafeiratos S., Durrant J. R., Petit C., Titanium dioxide/carbon nitride nanosheet nanocomposites for gas phase CO2 photoreduction under UV-visible irradiation. Appl. Catal. B Environ. 242, 369–378 (2019). [Google Scholar]
- 10.Huang W., Huber N., Jiang S., Landfester K., Zhang K. A. I., Covalent triazine framework nanoparticles via size-controllable confinement synthesis for enhanced visible-light photoredox catalysis. Angew. Chem. Int. Ed. 59, 18368–18373 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu X. Y., Li M. M., Li J., Zhang G. K., Yin S., A sillenite-type Bi12MnO20 photocatalyst : UV, visible, and infrared lights responsive photocatalytic properties induced by the hybridization of Mn 3d and O 2p orbitals. Appl. Catal. B Environ. 219, 132–141 (2017). [Google Scholar]
- 12.Ma J. Z., Wang C. X., He H., Enhanced photocatalytic oxidation of NO over g-C3N4-TiO2 under UV and visible light. Appl. Catal. B Environ. 184, 28–34 (2016). [Google Scholar]
- 13.Ma M., Gao N., Sun Y., Du X., Ren J., Qu X., Redox-activated near-infrared-responsive polyoxometalates used for photothermal treatment of Alzheimer’s disease. Adv. Healthc. Mater. 7, 1800320 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Zhang S., Chen H., Zhang G., Kong X., Yin S., Li B., Wu L., An ultra-small thermosensitive nanocomposite with a Mo154-core as a comprehensive platform for NIR-triggered photothermal-chemotherapy. J. Mater. Chem. B 6, 241–248 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Meng X., Wang T., Liu L., Ouyang S., Li P., Hu H., Kako T., Iwai H., Tanaka A., Ye J., Photothermal conversion of CO2 into CH4 with H2 over group VIII nanocatalysts: An alternative approach for solar fuel production. Angew. Chem. Int. Ed. 53, 11478–11482 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Jin B., Li Y., Wang J., Meng F., Cao S., He B., Jia S., Wang Y., Li Z., Liu X., Promoting oxygen evolution reaction of Co-based catalysts (Co3O4, CoS, CoP, and CoN) through photothermal effect. Small 15, 1903847 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Li X., Zhao S., Duan X., Zhang H., Yang S.-Z., Zhang P., Jiang S. P., Liu S., Sun H., Wang S., Coupling hydrothermal and photothermal single-atom catalysis toward excellent water splitting to hydrogen. Appl. Catal. B Environ. 283, 119660 (2021). [Google Scholar]
- 18.Yang Q., Xu Q., Yu S.-H., Jiang H.-L., Pd nanocubes@ZIF-8: Integration of plasmon-driven photothermal conversion with a metal-organic framework for efficient and selective catalysis. Angew. Chem. Int. Ed. 55, 3685–3689 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Chen Y. Z., Wang Z. U., Wang H., Lu J., Yu S. H., Jiang H. L., Singlet oxygen–engaged selective photo-oxidation over Pt nanocrystals/porphyrinic MOF: The roles of photothermal effect and Pt electronic state. J. Am. Chem. Soc. 139, 2035–2044 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Chou S. S., Kaehr B., Kim J., Foley B. M., De M., Hopkins P. E., Huang J., Brinker C. J., Dravid V. P., Chemically exfoliated MoS2 as near-infrared photothermal agents. Angew. Chem. Int. Ed. 125, 4254–4258 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z., Wang Q., Wang H., Zhang L., Song G., Song L., Hu J., Wang H., Liu J., Zhu M., Zhao D., Ultrathin PEGylated W18O49 Nanowires as a new 980 nm-Laser-Driven photothermal agent for efficient ablation of cancer cells in vivo. Adv. Mater. 25, 2095–2100 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Lovell J. F., Jin C. S., Huynh E., Jin H., Kim C., Rubinstein J. L., Chan W. C. W., Cao W., Wang L. V., Zheng G., Porphysome nanovesicles generated by porphyrin bilayers for use as multimodal biophotonic contrast agents. Nat. Mater. 10, 324–332 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Xiao J. D., Jiang H. L., Metal-organic frameworks for photocatalysis and photothermal catalysis. Acc. Chem. Res. 52, 356–366 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Wang C. P., Zhang Q., Wang X. Y., Chang H., Zhang S. J., Tang Y. K., Xu J. H., Qi R. J., Cheng Y. Y., Dynamic modulation of enzyme activity by near-infrared light. Angew. Chem. Int. Ed. 56, 6767–6772 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Li J., Cushing S. K., Zheng P., Meng F., Chu D., Wu N., Plasmon-induced photonic and energy-transfer enhancement of solar water splitting by a hematite nanorod array. Nat. Commun. 4, 2651 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Pope M. T., Müller A., Polyoxometalate chemistry: An old field with new dimensions in several disciplines. Angew. Chem. Int. Ed. 30, 34–48 (1991). [Google Scholar]
- 27.Wang S. S., Yang G. Y., Recent advances in polyoxometalate-catalyzed reactions. Chem. Rev. 115, 4893–4962 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Buckley R. I., Clark R. J. H., Structural and electronic properties of some polymolybdates reducible to molybdenum blues. Coord. Chem. Rev. 65, 167–218 (1985). [Google Scholar]
- 29.Li J., Chen Z., Zhou M., Jing J., Li W., Wang Y., Wu L., Wang L., Wang Y., Lee M., Polyoxometalate-driven self-assembly of short peptides into multivalent nanofibers with enhanced antibacterial activity. Angew. Chem. Int. Ed. 55, 2592–2595 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Shi Y. H., Zhang J. J., Huang H., Cao C. Y., Yin J. J., Xu W. J., Wang W. J., Song X. J., Zhang Y. W., Dong X. C., Fe-doped polyoxometalate as acid-aggregated nanoplatform for NIR-II photothermal-enhanced chemodynamic therapy. Adv. Healthc. Mater. 9, 2000005 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Zhou J. H., Zhao W. C., Miao Z. H., Wang J. G., Ma Y., Wu H. T., Sun T. D., Qian H. S., Zha Z. B., Folin-Ciocalteu assay inspired polyoxometalate nanoclusters as a renal clearable agent for noninflammatory photothermal cancer therapy. ACS Nano 14, 2126–2136 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Noro S. I., Tsunashima R., Kamiya Y., Uemura K., Kita H., Cronin L., Akutagawa T., Nakamura T., Adsorption and catalytic properties of the inner nanospace of a gigantic ring-shaped polyoxometalate cluster. Angew. Chem. Int. Ed. 48, 8703–8706 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Müller A., Das S. K., Fedin V. P., Krickemeyer E., Beugholt C., Bögge H., Schmidtmann M., Hauptfleish B., Rapid and simple isolation of the crystalline molybdenum-blue compounds with discrete and linked nanosized ring-shaped anions: Na15[MoVI126MoV28O462H14(H2O)70]0.5[MoVI124MoV28O457H14(H2O)68]0.5·ca.400H2O. Z. Anorg. Allg. Chem. 625, 1187–1192 (1999). [Google Scholar]
- 34.Müller A., Krickemeyer E., Meyer J., Bögge H., Peters F., Plass W., Diemann E., Dillinger S., Nonnenbruch F., Randerath M., Menke C., [Mo154(NO)14O420(OH)28(H2O)70](25± 5)−: A water-soluble big wheel with more than 700 atoms and a relative molecular mass of about 24000. Angew. Chem. Int. Ed. 34, 2122–2124 (1995). [Google Scholar]
- 35.Li B., Li W., Li H. L., Wu L. X., Ionic complexes of metal oxide clusters for versatile self-assemblies. Acc. Chem. Res. 50, 1391–1399 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Zhang G., Li B., Zhou Y., Chen X., Li B., Lu Z.-Y., Wu L., Processing supramolecular framework for free interconvertible liquid separation. Nat. Commun. 11, 425 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yue L., Wang S., Zhou D., Zhang H., Li B., Wu L. X., Flexible single-layer ionic organic-inorganic frameworks towards precise nano-size separation. Nat. Commun. 7, 10742 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.G. Becker, H. Schmidt, G. Uhl, W. Uhl, M. Regitz, W. Rösch, U. J. Vogelbacher, Inorganic Syntheses (Wiley, 2007), pp. 249. [Google Scholar]
- 39.Ni L., Li H., Xu H., Shen C., Liu R., Xie J., Zhang F., Chen C., Zhao H., Zuo T., Diao G., Self-assembled supramolecular polyoxometalate hybrid architecture as a multifunctional oxidation catalyst. ACS Appl. Mater. Interfaces 11, 38708–38718 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Kibsgaard J., Chen Z. B., Reinecke B. N., Jaramillo T. F., Engineering the surface structure of MoS2 to preferentially expose active edge sites for electrocatalysis. Nat. Mater. 11, 963–969 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Passadis S., Kabanos T., Song Y. F., Miras H., Self-assembly in polyoxometalate and metal coordination-based systems: Synthetic approaches and developments. Inorganics 6, 71–95 (2018). [Google Scholar]
- 42.Zhong D. Y., Sousa F. L., Muller A., Chi L. F., Fuchs H. A., A nanosized molybdenum oxide wheel with a unique electronic-necklace structure: STM study with submolecular resolution. Angew. Chem. Int. Ed. 50, 7018–7021 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Müller A., Krickemeyer E., Bögge H., Schmidtmann M., Beugholt C., Das S. K., Peters F., Giant ring-shaped building blocks linked to form a layered cluster network with nanosized channels: [MoVI124MoV28O429 (μ3-O)28H14(H2O)66.5]16−. Chem. Eur. J. 5, 1496–1502 (1999). [Google Scholar]
- 44.Callahan C. M., Foti S. C., Lai J. R., Cerimetric Determination of molybdenum in high chloride media using molybdenum blue reaction. Anal. Chem. 32, 635–637 (1960). [Google Scholar]
- 45.Liu T. B., An unusually slow self-assembly of inorganic ions in dilute aqueous solution. J. Am. Chem. Soc. 125, 312–313 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Liu T., Diemann E., Li H., Dress A. W. M., Müller A., Self-assembly in aqueous solution of wheel-shaped Mo154 oxide clusters into vesicles. Nature 426, 59–62 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Gumerova N. I., Rompel A., Synthesis, structures and applications of electron-rich polyoxometalates. Nat. Rev. Chem. 2, 0112 (2018). [Google Scholar]
- 48.Ci C. G., Carbó J. J., Neumann R., Graaf C. D., Poblet J. M., Photoreduction mechanism of CO2 to CO catalyzed by a Rhenium(I)–Polyoxometalate hybrid compound. ACS Catal. 6, 6422–6428 (2016). [Google Scholar]
- 49.Szejtli J., Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 98, 1743–1754 (1998). [DOI] [PubMed] [Google Scholar]
- 50.Yujuan C., Runhua L., 1H NMR Titration and quantum calculation for the inclusion complexes of cis-cyclooctene, cis, cis-1, 3-cyclooctadiene, and cis, cis-1, 5-cyclooctadiene with β-cyclodextrin. Spectrochim. Acta. Part A 73, 713–718 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Denekamp I. M., Antens M., Slot T. K., Rothenberg G., Selective catalytic oxidation of cyclohexene with molecular oxygen: Radical versus nonradical pathways. ChemCatChem 10, 1035–1041 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao Y. H., Yu H., Peng F., Wang H. J., Selective allylic oxidation of cyclohexene catalyzed by nitrogen-doped carbon nanotubes. ACS Catal. 4, 1617–1625 (2014). [Google Scholar]
- 53.Wang S., Liu Y., Zhang Z., Li X., Tian H., Yan T., Zhang X., Liu S., Sun X., Xu L., Luo F., Liu S., One-step template-free fabrication of ultrathin mixed-valence polyoxovanadate-incorporated metal-organic framework nanosheets for highly efficient selective oxidation catalysis in air. ACS Appl. Mater. Interfaces 11, 12786–12796 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Mimoun H., Roch I. S., Sajus L., Epoxydation des olefines par les complexes peroxydiques covalents Du Molybdene-VI. Tetrahedron 26, 37–50 (1970). [Google Scholar]
- 55.Sharpless K. B., Townsend J. M., Williams D. R., On the mechanism of epoxidation of olefins by covalent peroxides of molybdenum(VI). J. Am. Chem. Soc. 94, 295–296 (1972). [Google Scholar]
- 56.Liu K. T., Huang X. M., Pidko E. A., Hensen E. J. M., MoO3–TiO2 synergy in oxidative dehydrogenation of lactic acid to pyruvic acid. Green Chem. 19, 3014–3022 (2017). [Google Scholar]
- 57.Wang G. M., Yang Y., Han D. D., Li Y., Oxygen defective metal oxides for energy conversion and storage. Nano Today 13, 23–39 (2017). [Google Scholar]
- 58.Alcaraz M. R., Monago-Marana O., Goicoechea H. C., De la Pena A. M., Four- and five-way excitation-emission luminescence-based data acquisition and modeling for analytical applications: A review. Anal. Chim. Acta 1083, 41–57 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Brown A. M., Sundararaman R., Narang P., Goddard W. A. III, Atwater H. A., Nonradiative plasmon decay and hot carrier dynamics: Effects of phonons, surfaces, and geometry. ACS Nano 10, 957–966 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Spulber M., Schlick S., Using cyclodextrins to encapsulate oxygen-centered and carbon-centered radical adducts: The case of DMPO, PBN, and MNP spin traps. J. Phys. Chem. A 114, 6217–6225 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Sankar M., Nowicka E., Carter E., Murphy D. M., Knight D. W., Bethell D., Hutchings G. J., The benzaldehyde oxidation paradox explained by the interception of peroxy radical by benzyl alcohol. Nat. Commun. 5, 3332 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Deshlahra P., Iglesia E., Reactivity and selectivity descriptors for the activation of C−H bonds in hydrocarbons and oxygenates on metal oxides. J. Phys. Chem. C 120, 16741–16760 (2016). [Google Scholar]
- 63.Deshlahra P., Iglesia E., Methanol oxidative dehydrogenation on oxide catalysts: Molecular and dissociative routes and hydrogen addition energies as descriptors of reactivity. J. Phys. Chem. C 118, 26115–26129 (2014). [Google Scholar]
- 64.Cromwell W. C., Byström K., Eftink M. R., Cyclodextrin-adamantane carboxylate inclusion complexes: Studies of the variation in cavity size. J. Phys. Chem. 89, 326–332 (1984). [Google Scholar]
- 65.Fourmentin S., Ciobanu A., Landy D., Wenz G., Space filling of β-cyclodextrin and β-cyclodextrin derivatives by volatile hydrophobic guests. Beilstein J. Org. Chem. 9, 1185–1191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fetter R. C., Salek J. S., Sikorski C. T., Kumaravel G., Lin F. T., Cooperative binding by aggregated mono-6-(alkylamino)−/3-cyclodextrins. J. Am. Chem. Soc. 112, 3860–3868 (1990). [Google Scholar]
- 67.Yue L., Ai H., Yang Y., Lu W. J., Wu L. X., Chiral self-assembly and reversible light modulation of a polyoxometalate complex via host-guest recognition. Chem. Commun. 49, 9770–9772 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Roper D. K., Ahn W., Hoepfner M., Microscale heat transfer transduced by surface plasmon resonant gold nanoparticles. J. Phys. Chem. C 111, 3636–3641 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/30/eabf8413/DC1