

Paracrine TGF-β signaling is crucial to maintain robust and coherent circadian rhythms of peripheral oscillator networks.
Abstract
Coupling between cell-autonomous circadian oscillators is crucial to prevent desynchronization of cellular networks and disruption of circadian tissue functions. While neuronal oscillators within the mammalian central clock, the suprachiasmatic nucleus, couple intercellularly, coupling among peripheral oscillators is controversial and the molecular mechanisms are unknown. Using two- and three-dimensional mammalian culture models in vitro (mainly human U-2 OS cells) and ex vivo, we show that peripheral oscillators couple via paracrine pathways. We identify transforming growth factor–β (TGF-β) as peripheral coupling factor that mediates paracrine phase adjustment of molecular clocks through transcriptional regulation of core-clock genes. Disruption of TGF-β signaling causes desynchronization of oscillator networks resulting in reduced amplitude and increased sensitivity toward external zeitgebers. Our findings reveal an unknown mechanism for peripheral clock synchrony with implications for rhythmic organ functions and circadian health.
INTRODUCTION
Circadian clocks regulate rhythmic physiological processes in accordance with periodically reoccurring environmental zeitgebers, providing an adaptive advantage (1, 2). In mammals, the suprachiasmatic nucleus (SCN), as central clock, senses external time, drives behavior, and aligns tissue clocks with the daily light-dark cycle. Nevertheless, not only the SCN but also virtually all mammalian cell types constitute autonomous circadian oscillators that generate circadian rhythms through transcriptional-translational feedback loops (TTFLs) among molecular clock components (3). The transcriptional circuit underlying mammalian circadian rhythms is driven by cis-regulatory elements regulating cyclic activation and repression of clock and clock-controlled genes (4). In addition, endogenous and exogenous zeitgeber signals can input to the molecular clock machinery and alter clock gene expression (5, 6). Since circadian clocks are cell autonomous, single cells cycle with self-sustained, intrinsic circadian periods (7–9) and must be synchronized to maintain coherent tissue rhythms. While the SCN displays synchronized circadian rhythms for long durations ex vivo, oscillations of peripheral tissue explants and cellular ensembles have been described to dampen quickly. Traditionally, this difference has been attributed to a lack of intercellular coupling within peripheral tissue clocks, resulting in desynchronization of single cells and damping of network rhythms. However, methodological advances have yielded evidence indicating that peripheral oscillators also maintain intercellular synchrony: (i) Peripheral clock rhythms in vivo and ex vivo persist independently, although with reduced amplitudes, of SCN-derived or external entrainment signals, suggesting that cell-autonomous oscillators couple (10–13); (ii) embryonic tissue explants cultured in toto and organoid models of peripheral clocks display robust circadian rhythmicity (14, 15), suggesting that damped rhythms of adult tissues result from disturbed tissue integrity upon slice preparation; and (iii) rhythms of cultured peripheral cells show density dependence (16, 17), local coupling (18), and phase cross-talk (19), suggesting that peripheral oscillators partially synchronize in vitro. Nevertheless, contradictory results from in vivo studies relying on population sampling of multiple animals (20, 21) and from coculture studies using conventional in vitro models (18, 22, 23) have rendered peripheral coupling, and autonomy of non-SCN tissue clocks a highly debated topic in chronobiology with far-reaching consequences for circadian health: Intercellular coupling governs features of oscillator networks, i.e., ensemble amplitude, response to zeitgebers and entrainment, and transients upon phase shifting (24, 25) that control rhythmic physiology and behavior, as well as the alignment of body clocks.
While neuronal coupling within the SCN is achieved via secreted neurotransmitters (26, 27) or gap junction signaling (28), mechanisms of coupling among peripheral clock cells and corresponding intercellular communication pathways are currently unknown. Thus, in this study, we addressed two questions: First, do peripheral circadian oscillators couple intercellularly to maintain robust and coherent ensemble rhythms? Second, how is such coupling achieved molecularly? Using primarily human osteosarcoma (U-2 OS) cells, an established in vitro model of human peripheral clocks (29, 30) and other immortalized and primary mammalian cell types and tissue explants, we demonstrate that peripheral oscillators indeed couple intercellularly. Moreover, we find that perturbation of the secretory pathway results in disrupted ensemble rhythms and that secreted signaling factors input to the molecular clock machinery, promoting high-amplitude, robust, and coherent ensemble rhythms. Therefore, we reasoned that intercellular coupling among cell-autonomous oscillators in the periphery is mediated by paracrine communication pathways and identify transforming growth factor–β (TGF-β) as peripheral coupling factor that transmits paracrine synchronization signals to the molecular clock machinery through adenosine 3′,5′-monophosphate (cAMP) response element (CRE)–driven, immediate-early expression of the core-clock gene PER2.
RESULTS
Peripheral oscillator cells couple intercellularly
To study whether peripheral circadian oscillator cells couple intercellularly, we performed coculture experiments with U-2 OS populations that differed in circadian phase or period. If peripheral oscillators couple, then time information should be exchanged between cells leading to the adjustment of individual phases or periods toward a common phase or period of the ensemble (Fig. 1, A to C).
Fig. 1. Peripheral circadian oscillators couple intercellularly.
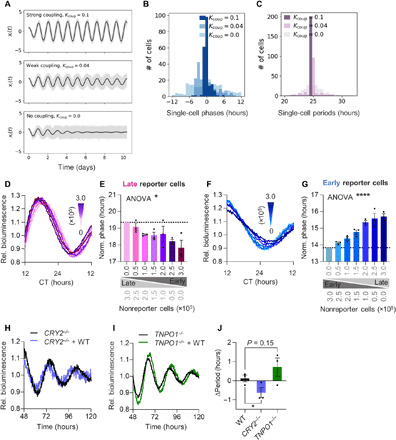
(A to C) Circadian oscillations of 200 stochastic, heterogeneous, amplitude-phase oscillators were modeled for three different intercellular coupling strengths Kcoup. (A) Single-cell traces (gray lines) with ensemble averages (black line). (B) Single-cell phase and (C) period distributions. (D to G) Low-density (0.25 × 105 cells per 35-mm dish) U-2 OS Per2:Luc reporter cells were cocultured with increasing numbers of 6-hour phase-delayed or -advanced nonreporter cells, as well as phase-equal nonreporter cells to keep total cell numbers constant (fig. S1A). Shown are detrended representative time series (D and F) and phases (E and G) of Per2:Luc reporter cells cocultured with phase-different nonreporter cells. Phases were normalized to data for 0.0 × 105 cocultured nonreporter cells (n = 3 with three technical replicates, means ± SEM, one-way analysis of variance (ANOVA), *P < 0.05 and ****P < 0.0001). CT; circadian time. (H to J) 3D spheroids were prepared from long-period CRY2−/−, short-period TNPO1−/−, or WT U-2 OS Bmal1:Luc reporter cells either as pure or mixed cultures with WT nonreporter cells (fig. S1J). Shown are representative detrended time series of pure and mixed CRY2−/− (H) or TNPO1−/− (I) spheroids and the quantification (J) of period changes upon coculture of knockout with WT cells (relative to periods of pure spheroids, n = 3 with two technical replicates, means ± SEM, unpaired one-tailed t test, *P < 0.05).
To test for phase synchronization between peripheral oscillators, we monitored circadian bioluminescence rhythms of U-2 OS cells harboring a Per2:luciferase (Per2:Luc) reporter that were cocultured with increasing amounts of phase-different nonreporter cells. To control for effects of cell density, we kept total cell numbers identical in the cocultures (fig. S1A). To avoid resynchronization by experimental handling, we synchronized cells in solution and added them carefully to the existing phase-different cultures in prewarmed medium (fig. S1, B and C). Phases of late reporter cells cocultured with 6-hour advanced (early) nonreporter cells became increasingly earlier (Fig. 1, D and E), while the phases of early reporter cells cocultured with 6-hour delayed (late) nonreporter cells became increasingly later (Fig. 1, F and G). In contrast, these coculture experiments with essentially arrhythmic nonreporter cells (due to BMAL1 knockdown) (fig. S1, D and E) strongly attenuated phase-pulling effects (fig. S1, F to I). Together, this indicates that rhythmic nonreporter cells convey time information to reporter cells. Nevertheless, the extent of phase adjustment was at most 2 hours (despite a 6-hour phase difference) indicating incomplete phase synchronization (weak coupling) in our two-dimensional (2D) in vitro model.
To test for period adjustment, we cocultured reporter and nonreporter U-2 OS cells with different periods. In agreement with previous reports (18, 22, 23), we did not observe period changes of peripheral clock cells under conventional 2D culture. Since monolayer cultures cannot completely reproduce characteristics of tissue networks, we performed coculture experiments using 3D spheroids (fig. S1J) to increase the complexity of cell-cell interactions and local concentrations of hypothetical coupling factors. We cocultured Bmal1:Luc long-period CRY2−/− (31), short-period U-2 OS TNPO1−/− (32), or wild-type (WT)–period U-2 OS cells with a fivefold excess of U-2 OS WT nonreporter cells and monitored circadian bioluminescence rhythms. Long periods of CRY2−/− reporter cells were shortened, and short periods of TNPO1−/− reporter cells lengthened, while intermediate periods of WT reporter cells were unaffected (compared to noncocultured spheroids) (Fig. 1, H to J), again suggesting that nonreporter cells convey time information to the reporter cell population.
Overall, bidirectional phase- and period-pulling effects suggest that peripheral oscillators at least partially synchronize via intercellular coupling. In addition, coupling strength appears to be enhanced by the formation of 3D tissue networks that are similar to tissues in vivo.
Coupling among peripheral oscillators is achieved by secreted factors
Published studies show that coherent circadian rhythmicity of cellular networks depends on culture density (16, 33). Concordantly, mathematical modeling predicts that strong intercellular coupling promotes high-amplitude population rhythms with low damping rates (Fig. 2, A and B). Thus, to study whether cell-cell communication among adjacent oscillators is required for peripheral coupling, we cultured U-2 OS reporter cells at decreasing culture densities in serum-free medium and monitored circadian bioluminescence rhythms. Reducing culture density led to amplitude reduction, increased damping, and period lengthening (Fig. 2, C to F).
Fig. 2. Peripheral oscillators communicate via secreted factors.
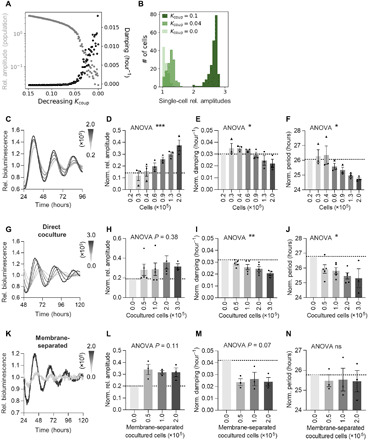
(A and B) Circadian oscillations of 200 stochastic, heterogeneous, amplitude-phase oscillators were modeled. (A) Shown is the dependence of ensemble amplitude and damping, as well as (B) the distribution of single-cell amplitudes. (C to F) U-2 OS Bmal1:Luc (circles) or Per2:Luc (triangles) reporter cells were seeded at decreasing culture density in 35-mm dishes and were synchronized before imaging in serum-free medium. (C) Detrended representative time series of Bmal1:Luc reporter cells plated at different densities. (D) Relative amplitudes, (E) damping, and (F) circadian periods of reporter cells [normalized to the 0.2 × 105 cells group (n = 2); all others n = 3 with two to three technical replicates, means ± SEM, one-way ANOVA, *P < 0.05 and ***P < 0.001]. (G to J) Low-density (0.3 × 105 cells) U-2 OS Bmal1:Luc reporter cells were seeded into 35-mm dishes or (K to N) on 30-mm membrane inserts. Increasing numbers of nonreporter cells were coseeded (fig. S2, G and H), and cells were synchronized before imaging in serum-free medium. (G and K) Detrended representative time series of Bmal1:Luc reporter cells cocultured with different numbers of nonreporter cells, (H and L) relative amplitudes, (I and M) damping, and (J and N) circadian periods of reporter cells upon coculture (normalized to the respective 0.0 ×105 cocultured cells groups, n = 3 to 5 (F to I) or 0.0 × 105 cells group (n = 2); all others n = 3 (J to M) with two technical replicates, means ± SEM, one-way ANOVA, *P < 0.05 and **P < 0.01).
To investigate whether cell division–dependent phase noise contributes to damping of circadian rhythms on the population level, we studied the effect of cell division on oscillator coherence at the single-cell level. To this end, we plated U-2 OS cells harboring an NR1D1::VNP fluorescent reporter [Venus-NLS-PEST driven by Nuclear Receptor Subfamily 1 Group D Member 1 regulatory sequences (23)] at different densities and monitored circadian fluorescence rhythms over 5 days. As expected, higher cell division rates resulted in decreased amplitudes and reduced number of rhythmic cells (fig. S2, A and B). Yet, in agreement with the population experiments (Fig. 2, C to F), we observed lower phase coherence with reduced culture density (fig. S2, C and D), even though cell division rate was also reduced in low density cultures (fig. S2, E and F). This suggests that impaired intercellular communication, rather than increased cell division rate, led to the density-dependent effects on amplitude and damping.
In addition, we could gradually rescue these low-density phenotypes on the population level by coculture with increasing amounts of nonreporter cells (Fig. 2, G to J, and fig. S2G), further indicating that overall culture density rather than the absolute number of reporter cells governs circadian dynamics of the population. Poor rhythmicity (low amplitude and high damping) of the sparse reporter cell populations could also be rescued by coculture with nonreporter cells that were physically separated from the reporter cell population allowing communication only via diffusible factors (Fig. 2, K to N, and fig. S2H). This further suggests that intercellular coupling is achieved by paracrine communication.
Thus, to investigate whether a functional secretory pathway is important for normal circadian dynamics of cellular ensembles, we perturbed early secretory pathway components, i.e., the endoplasmic reticulum (ER), coat protein complex I (COPI) and COPII transport vesicles, and the Golgi apparatus, either by treatment with pharmacological inhibitors or by targeted RNA interference (RNAi) mediated knockdown of essential genes (Fig. 3A). Treatment of U-2 OS cells or mouse lung explants with inhibitors of ER translocation and exit, glycoprotein synthesis, and COP vesicle formation and trafficking led to disruption of circadian dynamics, i.e., amplitude reduction, increased damping, and period lengthening (Fig. 3, B to D, and fig. S3, A to C). In addition, RNAi knockdown of genes essential for the early secretory pathway (GBF1, SEC13, and TMED2/10) also severely disrupted U-2 OS population rhythms, resulting in altered clock gene expression, amplitude reduction, increased damping, and period lengthening (Fig. 3, E to H). While these knockdown phenotypes were associated with altered Golgi morphology (fig. S3, D and E), neither cell viability (fig. S3, F and G) nor unfolded protein response (fig. S3, H and I) was affected in RNAi knockdown cells, indicating that functional ER-Golgi transport of secreted molecules is crucial for coherent circadian dynamics of peripheral oscillators networks. Overall, these data indicate that intercellular coupling among peripheral circadian oscillators is important to maintain coherent, high-amplitude ensemble rhythms and is achieved by cell-cell communication via secreted factors.
Fig. 3. The secretory pathway is crucial for coherent dynamics of oscillator ensembles.
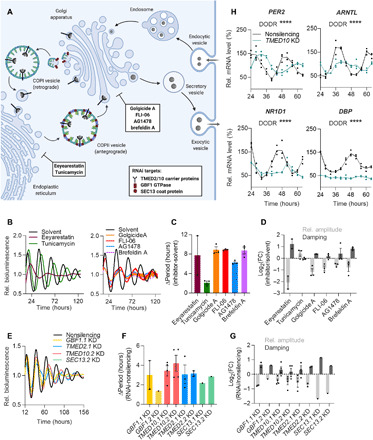
(A) Schematic representation of the secretory pathway and essential components targeted by RNAi knockdown or pharmacological inhibition [transmembrane p24 trafficking protein 2/10 (TMED2/10), Golgi brefeldin A–resistant guanine nucleotide exchange factor 1 (GBF1), SEC13 homolog (SEC13), nuclear pore, and COPII coat complex component]. GTPase, guanosine triphosphatase. (B to D) U-2 OS Bmal1:Luc reporter cells were synchronized and treated with 5 μM inhibitors (or respective solvent control) before imaging. (B) Detrended representative time series of inhibitor-treated reporter cells. (C) Quantification of changes in circadian period, as well as (D) amplitude and damping upon inhibitor treatment (relative to solvent control, n = 2 to 3 with four technical replicates, means ± SEM). FC, fold change. (E to G) U-2 OS Bmal1:Luc reporter cells were lentivirally transduced with short hairpin RNA (shRNA) targeting the indicated genes (two distinct RNAi constructs per gene) or with nonsilencing control. Cells were synchronized before imaging. (E) Detrended representative time series of Bmal1:Luc knockdown (KD) cells. (F) Quantification of changes in circadian period, as well as (G) amplitude and damping upon RNAi-mediated knockdown (relative to nonsilencing control, n = 1 to 4 with three technical replicates, means ± SEM). (H) U-2 OS cells were lentivirally transduced with shRNA targeting TMED10 or with nonsilencing control. Cells were synchronized, and RNA was isolated at indicated time points. Given are mRNA expression levels of clock genes analyzed by reverse transcription quantitative polymerase chain reaction (RT-qPCR) (relative to GAPDH and normalized to the mean of nonsilencing control, n = 1 with three technical replicates per time point, Detection of Differential Rhythmicity (DODR) ANOVA applied on harmonic regression (HANOVA) test for differential rhythmicity, ****P < 0.0001).
Secreted factors modulate circadian dynamics via CRE-mediated immediate-early induction of PER2
Coupling of circadian oscillator cells requires an appropriate input into the molecular clock machinery, i.e., coupling factors should act as zeitgebers (34, 35). To test whether factors secreted by peripheral oscillators influence the canonical TTFL network, we used conditioned medium (CM) derived from human and murine cells, including primary hepatocytes, to stimulate U-2 OS reporter cells, organotypic slices from period circadian regulator 2::luciferase (PER2::LUC) reporter mice, as well as PER2::LUC liver-derived organoids. Independent of the source of the CM, U-2 OS reporter cells responded with robust phase delays of 4 to 8 hours upon CM treatment (relative to control; Fig. 4A and fig. S4A) with maximal phase delays occurring around the trough of PER2 expression (fig. S4B). In addition, mouse tissue explants and mouse liver organoid rhythms displayed phase delays upon treatment with CM derived from human U-2 OS cells (fig. S4, C and D), suggesting (i) that secreted factors input to the molecular clock machinery to modulate circadian dynamics and (ii) that these factors are conserved across tissues and species.
Fig. 4. Secreted factors modulate circadian dynamics via CRE-mediated immediate-early induction of PER2.
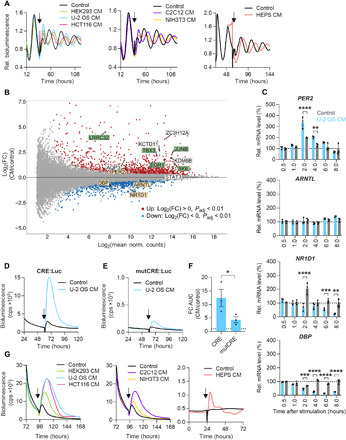
(A) Synchronized U-2 OS Per2:Luc reporter cells were stimulated with control medium or CM from indicated cell lines at the trough of PER2 expression (arrow). HEPS, primary hepatocytes). Given are detrended representative time series. (B and C) U-2 OS cells were synchronized and stimulated with U-2 OS CM or control medium at the trough of PER2 expression. (B) RNA was isolated 2 hours after stimulation and used for RNA-seq. MA plot of stimulation-induced log2 expression changes of the U-2 OS transcriptome (yellow boxes, clock genes; green and white boxes, top 10 differentially regulated genes; green boxes, genes associated with TGF-β signaling; n = 2 to 3 with one technical replicate). (C) RNA was isolated after indicated times and reversely transcribed. Given are mRNA expression levels of clock genes analyzed by RT-qPCR (relative to GAPDH and normalized to the unstimulated control, n = 3 with three technical replicates, unpaired t test, **P < 0.01, ***P < 0.001, and ****P < 0.0001). (D to F) U-2 OS CRE:Luc and mutCRE:Luc reporter cells were stimulated with U-2 OS CM or control medium (arrow). (D) Representative time series of CRE:Luc and (E) mutCRE:Luc reporter cells upon stimulation. (F) Quantification of reporter gene induction following CM stimulation (relative to control medium, n = 3 with three to four technical replicates, means ± SEM, unpaired one-tailed t test, *P < 0.05). cps, counts per second; AUC, area under the curve. (G) U-2 OS CRE:Luc reporter cells were stimulated with control medium or CM from indicated cell lines (arrow). Shown are representative time series.
To identify signaling pathways induced by potential secreted coupling factors, we performed RNA sequencing (RNA-seq) of U-2 OS cells stimulated with U-2 OS CM or control medium at the time when phase responses were strongest. Genes differentially expressed in response to CM were associated with protein, nucleic acid, and heterocyclic compound binding (fig. S4E and table S1). Most of the top 10 differentially regulated transcripts identified are known to be involved in growth factor signaling. Five of those transcripts were associated with TGF-β/SMAD signaling [LRRC32 (36), SKIL (37), TBX3 (38), JUNB (39), and EGR1 (40)] (Fig. 4B, green boxes), suggesting that this pathway plays a role in peripheral oscillator coupling. In addition, EGR1 and JUNB encode immediate-early transcription factors that have been reported to regulate circadian rhythms (41, 42). As expected from our phase-shift experiments, the expression of clock genes was also affected by CM: PER2 was significantly up-regulated, while DBP and NR1D1 were significantly down-regulated (Fig. 4B, yellow boxes). Thus, RNA-seq results led us to hypothesize that potential paracrine coupling factors activate immediate-early transcription factors to control the molecular clock machinery. Exposure of U-2 OS cells to increasing durations of CM and control medium resulted in a significant up-regulation of PER2 mRNA levels after 2 to 4 hours, while DBP and NR1D1 expression was suppressed starting at 2 to 4 hours with maximal suppression at 8 hours (Fig. 4C). This suggests that paracrine signals induce the immediate-early expression of PER2, resulting in the prolonged suppression of E-box–driven clock genes.
To further characterize transcriptional responses of CM-mediated immediate-early expression of PER2, we analyzed cis-regulatory elements that have been associated with clock gene transcriptional regulation. To this end, we equipped U-2 OS cells with luciferase reporter genes driven by synthetic promoters covering six to seven tandem repeats of canonical enhancer sites [E-box, D-box, RAR Related Orphan Receptor response elements (RRE), CRE, and serum response element (SRE); fig. S5A] and stimulated cells with CM or control medium. While E-box, D-box, RRE, and SRE reporter genes were not or only moderately induced by CM (fig. S5B,C), CREs provoked a very strong and specific reporter gene induction, i.e., mutation of its sequence significantly attenuated the response to CM (Fig. 4, D to F). In addition, CRE activation by secreted signaling molecules was conserved across species and cell types, since CM from various human and mouse cells induced CRE reporter gene transcription (Fig. 4G and fig. S5D).
Overall, these data indicate that peripheral oscillators secrete paracrine signaling molecules that alter circadian dynamics in adjacent cells by activating the CRE-driven immediate-early expression of PER2. Potentially, these molecules may belong to the family of growth factors, specifically the transforming growth factor (TGF-β) family.
TGF-β is a candidate coupling factor activating PER2 expression
To elucidate the identity of factor(s) mediating CM activity, we performed preliminary characterization of the chemical nature of the secreted molecules using the CRE:Luc induction assay as approximation for CM effects on circadian dynamics. Size fractionation and heat treatment (95°C for 10 min) suggested that active U-2 OS CM components are polypeptides of intermediate size that can be inactivated by heat (Fig. 5, A to C). This was further supported by ammonium sulfate precipitation, showing that about 60% of U-2 OS CM activity precipitated in 40 to 60% saturated (NH4)SO4 solutions (Fig. 5, D and E). In addition, tryptic digestion reduced CM activity to about 40% compared to nondigested control (fig. S6, A and B).
Fig. 5. Active CM factors are secreted proteins.
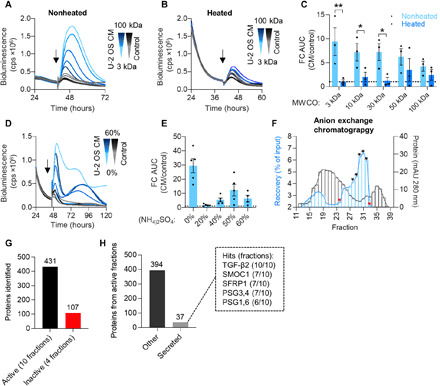
(A to C) U-2 OS CM and control medium were fractionated by ultrafiltration using centrifugal filters with different molecular weight cutoffs (MWCOs). Concentrates were used directly or were heated (95°C for 10 min) before stimulation of U-2 OS CRE:Luc reporter cells (arrow). (A) Representative time series of reporter cells stimulated with nonheated or (B) heated CM and control medium concentrates. (C) Quantification of reporter gene induction after CM stimulation (relative to control medium, n = 3 with two to three technical replicates, means ± SEM, unpaired t test, *P < 0.05 and **P < 0.01). (D and E) U-2 OS CM and control medium were saturated with increasing concentrations of (NH4)2SO4. Precipitates were resolubilized and used to stimulate U-2 OS CRE:Luc reporter cells (arrow; 0%, nonprecipitated control). (D) Representative time series and quantification (E) of reporter gene induction upon stimulation with CM and control medium precipitates or nonprecipitated controls (relative to control medium; n = 4 to 5 with three technical replicates, means ± SEM). (F) Two pools from initial GFX (see fig. S6C) were fractionated by AEX (see fig. S6D for pool 2). Fractions were used to stimulate U-2 OS CRE:Luc reporter cells and to quantify reporter gene induction relative to the nonfractionated chromatography input. Protein content of the fractions was approximated by measuring their absorbance at 280 nm. From each AEX pool, five active (black asterisks) and two inactive (red asterisks) fractions were selected for subsequent (G and H) mass spectrometry analysis. mAU, milli-absorbance units. (G) Total number of proteins uniquely identified in active and inactive chromatography fractions. (H) Number of active fraction proteins classified as MDSEC-predicted human secreted proteins (The Human Protein Atlas; table S3) or other. Proteins present in >50% of active fractions are given (dashed box).
To identify potential paracrine coupling factors, we fractionated U-2 OS CM using a two-step chromatography approach and analyzed active fractions by mass spectrometry (table S2). First-line gel filtration chromatography (GFX) yielded two active pools with enriched specific activity (fig. S6C, gray and black asterisks) that were further fractionated by anion exchange chromatography (AEX). From each AEX-fractionated pool, we selected five active fractions with the highest specific activity and two inactive fractions as negative controls (Fig. 5F and fig. S6D, black and red asterisks, respectively). Using mass spectrometry, we identified 431 proteins specifically present in active fractions with 37 being secreted proteins (Fig. 5, G and H). Having found indications that intercellular coupling among peripheral circadian oscillators is achieved by cell-cell communication via secreted factors (Fig. 2, J to M), we considered secreted proteins present in more than 50% of active fractions analyzed as candidate coupling factors (Fig. 5H, dashed box). While all seven candidates [TGF-β2, Pregnancy Specific Beta-1-Glycoprotein (PSG1), PSG3, PSG4, PSG6, SPARC Related Modular Calcium Binding 1 (SMOC1), and secreted frizzled related protein 1 (SFRP1)] are reported to modulate TGF-β signaling (43–47), TGF-β appeared to be the most likely candidate coupling factor, since (i) it was present in all active fractions analyzed, (ii) it is the only candidate acting as bona fide growth factor, and (iii) RNA-seq results indicated an involvement of the TGF-β pathway in CM responses (Fig. 4B).
To test whether TGF-β was the CM constituent responsible for mediating changes to the molecular clock machinery, i.e., CRE-mediated induction of PER2 transcription, we performed a series of loss-and-gain-of-function experiments. First, we used αTGF-β antibodies to immunodeplete or neutralize TGF-β in U-2 OS CM, which indeed abolished CRE:Luc reporter gene induction (Fig. 6, A and B, and fig. S6, E and F). In contrast, recombinant TGF-β dose-dependently induced CRE-mediated reporter gene transcription (Fig. 6, C and D) and activated the immediate-early expression of PER2 (fig. S6G) when used in physiological concentrations (48–50). Furthermore, pharmacological inhibition of TGF-β signaling by the selective TGF-β receptor blocker LY2109761 abolished transcriptional responses of CRE enhancer elements to CM (Fig. 6, E and F), as well as phase shifts of circadian rhythms upon CM stimulation (Fig. 6, G to I). Last, an RNAi knockdown screen in U-2 OS CRE:Luc reporter cells revealed extra- and intracellular components of the canonical TGF-β pathway as crucial for CM-mediated activity (fig. S6H). For example, knockdown of key intracellular transducers of TGF-β signaling, i.e., TGFBR1, SMAD4, and SKI, resulted in reduced responsiveness of U-2 OS CRE:Luc reporter cells to CM stimulation (Fig. 6, J and K). In contrast, down-regulation of LTBP1 and ITGAV, latent TGF-β binding protein and integrin subunit required for release of active TGF-β from its extracellular latent complex, led to increased induction of CRE:Luc expression upon CM stimulation (Fig. 6, J and K). This suggests (i) that perturbation of TGF-β signaling components downstream of the TGF-β receptor attenuates CM-induced transcriptional responses and (ii) that disturbance of extracellular binding partners of latent TGF-β alters the availability of active TGF-β forms, resulting in enhanced responsiveness to externally applied TGF-β.
Fig. 6. TGF-β is a candidate coupling factor activating CRE-driven expression of PER2.
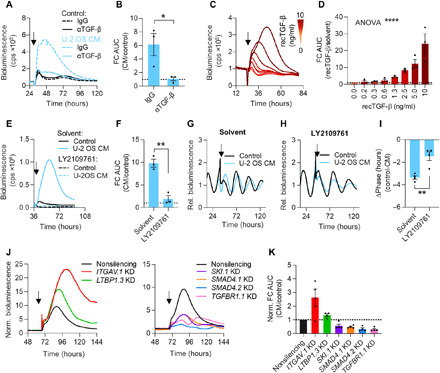
(A and B) U-2 OS CM and control medium were treated with αTGF-β or isotype control. Antibody complexes were removed by pulldown, and supernatants were used to stimulate U-2 OS CRE:Luc reporter cells (arrow). (A) Representative time series and (B) quantification of reporter gene induction by stimulation with αTGF-β– or immunoglobulin G (IgG)–treated CM (relative to control medium; n = 3 with three technical replicates, means ± SEM, unpaired one-tailed t test, *P < 0.05). (C and D) U-2 OS CRE:Luc reporter cells were stimulated (arrow) with increasing doses of recombinant TGF-β or solvent control. (C) Representative time series and (D) quantification of reporter gene induction upon stimulation (relative to solvent; n = 3 with two technical replicates, means ± SEM, one-way ANOVA, ****P < 0.0001). (E and F) U-2 OS CRE:Luc or (G to I) Bmal1:Luc reporter cells were treated with 5 μM TGF-β receptor inhibitor (LY2109761) or solvent control during seeding. Bmal1:Luc cells were synchronized before imaging. Cells were stimulated with CM or control medium containing 5 μM inhibitor or solvent (arrow). (E) Representative time series of CRE:Luc cells and (F) quantification of reporter gene induction following stimulation with CM containing inhibitor or solvent (relative to control medium; n = 3 with two technical replicates, means ± SEM, unpaired one-tailed t test, **P < 0.01). (G) Detrended representative time series of Bmal1:Luc cells upon stimulation with solvent or (H) inhibitor. (I) Phase shifts induced by CM containing inhibitor or solvent (relative to control medium, n = 4 with six to eight technical replicates, means ± SEM, unpaired one-tailed t test, **P < 0.01). (J and K) U-2 OS CRE:Luc reporter cells were lentivirally transduced with shRNA targeting the indicated genes or with nonsilencing control and were stimulated with U-2 OS CM or control medium (arrow). (J) Representative time series of knockdown cells (normalized to control medium) and (K) quantification of reporter gene induction by CM stimulation (relative to control medium; n = 4 with two technical replicates, means ± SEM).
To test whether TGF-β signaling is controlled by the molecular circadian oscillator, we generated luciferase reporters to monitor the dynamics of TGF-β promoter and SMAD enhancer site activity (fig. S7A). All reporters displayed circadian oscillations in synchronized U-2 OS cells (fig. S7A), suggesting that paracrine TGF-β signaling is controlled by the circadian clock. Overall, these data identify TGF-β as a signaling factor secreted by peripheral oscillator cells that can act as a rhythmic zeitgeber by inducing PER2, likely via CRE transcriptional enhancer sites.
TGF-β signaling promotes intercellular coupling among peripheral oscillators
If TGF-β acts as a coupling factor between peripheral circadian oscillators, then the following predictions arise (24, 25): (i) Perturbation of TGF-β signaling should promote intercellular desynchronization by broadening phase and period distributions of single-cell oscillators (Fig. 1, A to C). (ii) Thus, the damping rate of ensemble rhythms should increase in inhibition of TGF-β signaling leading to overall lower amplitudes (Fig. 2A). (iii) Coupled oscillator networks are more robust against perturbations; thus, inhibition of TGF-β signaling should render cellular ensembles more susceptible toward zeitgeber perturbation (fig. S8A). These predictions turned out to be true.
Ad (i): We performed real-time fluorescence imaging of U-2 OS NR1D1::VNP reporter cells with or without pharmacological perturbation of TGF-β signaling to quantify rhythms and network behavior of single-cell oscillators. Not only did TGF-β receptor inhibition result in faster decay of single-cell phase coherence (Fig. 7, A and B, and fig. S8B) but it also attenuated single-cell rhythmicity (fig. S8C) and induced a shift of period and amplitude distributions toward longer periods and reduced amplitudes (fig. S8, D and E), respectively. Ad (ii): An RNAi screen in U-2 OS Bmal1:Luc identified extra- and intracellular components of the TGF-β growth factor signaling as crucial for robust circadian rhythms on the population level (fig. S8F). For example, knockdown of TGFBR1/2 and BMPR1A, receptors for the TGF-β family of ligands; of SMAD4, transcriptional regulator of TGF-β signaling; of SMURF1 and RNF111, E3 ligases that regulate the degradation of activated SMADs, SMAD regulators, and TGF-β receptors; and of ITGAV, extracellular regulator of TGF-β activation, resulted in amplitude reduction, increased damping, and period alterations of U-2 OS ensemble rhythms (Fig. 7, C to E). In addition, pharmacological inhibition of TGF-β receptors led to amplitude reduction, increased damping, and period lengthening of U-2 OS ensemble rhythms, as well as of PER2::LUC organotypic tissue explants (Fig. 7, F to H). Ad (iii): To test whether robustness of the circadian oscillator network depends on intact TGF-β signaling, we applied cold temperature as a zeitgeber (20°C for 8 hours) to cellular ensembles with or without pharmacological TGF-β receptor blockage. While this zeitgeber did not elicit significant phase shifts in solvent treated cell cultures, it shifted the phase of circadian rhythms in TGF-β receptor inhibitor treated cells by about 8 hours (Fig. 7, I to K). Together, these data indicate that intact TGF-β signaling is essential for coherent, high-amplitude, and robust circadian rhythms of cellular ensembles via TGF-β acting as a coupling factor between single-cell oscillators in peripheral tissue clocks.
Fig. 7. TGF-β promotes coherent and robust circadian rhythms of peripheral oscillator ensembles.
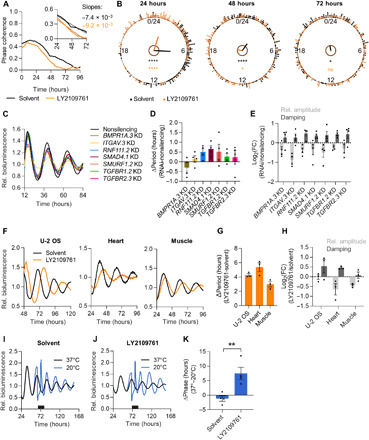
(A and B) Synchronized U-2 OS NR1D1::VNP cells were treated with TGF-β receptor inhibitor (LY2109761) or solvent control. Fluorescence was continuously monitored (movies S1 and S2). Rhythmic time series [meta2d false discovery rate (FDR), <0.05 (51); solvent, 215 cells; inhibitor, 180 cells] were used for phase prediction in pyBOAT (52). (A) Phase coherence (resultant vector) of single-cell instantaneous phases. Inset: Linear regression for days 1 to 3 of imaging reflects speed of desynchronization following initial synchronization (solvent, R2 = 0.95; inhibitor, R2 = 0.96). (B) Rayleigh phase plots. Dots indicate instantaneous phases of single cells, lines indicate that direction/length denote mean phase and phase coherence, respectively, and inner circles indicate Rayleigh significance threshold P < 0.01 (black, solvent; orange, inhibitor; Rayleigh test of uniformity, *P < 0.05 and ****P < 0.0001). (C to E) U-2 OS Bmal1:Luc cells were lentivirally transduced with shRNA targeting the indicated genes or with nonsilencing control. Cells were synchronized before imaging. (C) Detrended representative time series of knockdown cells. (D) Quantification of changes in circadian period and (E) amplitude and damping upon RNAi knockdown (relative to nonsilencing control, n = 4 with two technical replicates, means ± SEM). (F to H) U-2 OS Bmal1:Luc reporter cells and PER2::LUC organotypic slices were treated with LY2109761 or solvent control during seeding and imaging. Synchronization was performed before imaging. (F) Detrended representative time series and (G) quantification of changes in circadian period and (H) in amplitude and damping upon inhibitor treatment (relative to solvent; n = 3 with two to three technical replicates, means ± SEM). (I to K) U-2 OS Bmal1:Luc cells were treated with LY2109761 or solvent control during seeding and imaging. Cells were synchronized and exposed to a cold zeitgeber (20°C for 8 hours) at the peak of Bmal1:Luc expression (black bar). (I) Detrended representative time series of solvent and (J) inhibitor-treated cells upon temperature pulse. (K) Cold-induced phase shifts (relative to 37°C control; n = 4 with two technical replicates, means ± SEM, unpaired one-tailed t test, **P < 0.01).
DISCUSSION
In this study, we report two major findings. First, we show that peripheral circadian oscillators couple intercellularly and via paracrine signaling factors to maintain (partially) synchronized and robust ensemble rhythms. Second, we identify TGF-β as peripheral coupling factor that promotes coherent, high-amplitude rhythms of peripheral oscillators and renders them more robust against perturbation by external zeitgebers. Our work uncovers a molecular mechanism of peripheral coupling, which is in agreement with canonical TGF-β signaling (Fig. 8): Secreted TGF-β is activated extracellularly by the interaction with integrins, binds its transmembrane receptors, and induces complex formation and nuclear translocation of SMAD2 to SMAD4 proteins. In turn, SMAD complexes, together with important coregulators (51, 52), control the expression of TGF-β target genes by binding to defined enhancer sites (52). Concordantly, peripheral oscillators secrete TGF-β and RNAi knockdown of important extracellular and intracellular mediators of TGF-β signaling, and pharmacological block of TGF-β receptors perturb ensemble rhythms and phase coherence of peripheral cells and tissues. Likely, SMAD complexes recruit 5′-TGACGTCA-3′ (CRE)–binding transcription factors (53, 54) to induce the observed CRE-dependent, immediate-early expression of PER2 and subsequent suppression of E-box–driven clock genes, e.g., NR1D1 and DBP, thereby feeding into and phase-adjusting the molecular circadian clock machinery of neighboring cells. Given the mechanistic insight that our work has generated, it should enable further studies of how the targeted disruption of intercellular coupling affects peripheral clock function and circadian health in vivo.
Fig. 8. TGF-β acts as intercellular coupling factor in the periphery.
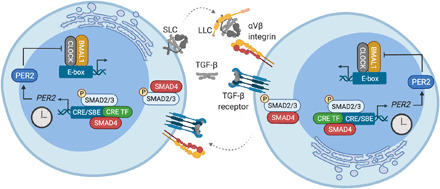
Schematic representation of TGF-β–dependent intercellular coupling between peripheral oscillator cells. SBE, SMAD-binding element; CRE TF, CRE-binding transcription factor; SLC, small latent TGF- β complex; LLC, large latent TGF- β complex.
Previously, the nature of coupling signals in peripheral oscillator networks has been unknown as peripheral cells secrete many diffusible factors. The TGF-β pathway controls numerous key cellular processes, including development, growth, differentiation, apoptosis, and cellular homeostasis. TGF-β has been shown to act as zeitgeber for peripheral clocks in organisms, ranging from rodents to zebrafish (55, 56). Concordantly, we show that CM, containing TGF-β as active factor, is able to phase shift circadian rhythms of various cell and tissue models, an effect that is attenuated upon TGF-β receptor blockage. Moreover, perturbation of TGF-β signaling results in weakened network rhythms (reduced amplitudes, increased damping, and loss of phase coherence) of human cells and murine tissue explants. Since coupling factors should act as zeitgebers, i.e., they should elicit time-dependent phase responses to induce phase alignment among oscillators, this further supports the role of TGF-β as peripheral coupling factor.
Theoretical models predict that rhythmic coupling factors drive oscillations of cellular ensembles (57). Whether signaling of active TGF-β is circadian rhythmic is currently unknown. Our work shows that period and phase pulling among U-2 OS populations is bidirectional, suggesting that coupling signals carry time information. This is consistent with the rhythmicity of the TGF-β promoter and SMAD-binding element reporters in U-2 OS cells. Moreover, many components of the canonical TGF-β pathway have been found to be rhythmically expressed in various organisms and tissues (fig. S9) (58, 59). Overall, this suggests that TGF-β acts as a rhythmic coupling signal to facilitate synchronization of cell-autonomous oscillators. Rhythmic control of TGF-β as peripheral coupling factor may also be important for the temporal regulation of TGF-β activity, which is highly context dependent. Therefore, our work also creates scope for further investigation of the mutual interactions between TGF-β and the circadian system in regulating physiological tissue functions.
Intercellular coupling promotes synchronization and amplitude resonance between single-cell oscillators (24), as well as governs the response of oscillator ensembles to zeitgeber/entrainment signals (25)—features that are crucial for the coherence and robustness of mammalian circadian rhythms. Not only SCN coupling is important for rhythmic behavior and entrainment to the environmental light-dark cycle but also non-SCN coupling is likely to play an important role in the mammalian circadian system. It has been reported that non-neuronal cellular networks in the brain, i.e., epithelial and ependymal cells of the choroid plexus and astrocytes of the SCN, display characteristics of locally coupled networks that influence SCN oscillations and rhythmic behavior in mice (60, 61), suggesting that this coupling is crucial for rhythm generation on the organismal level and thus circadian health and well-being. The peripheral coupling mechanism presented here may account for previous observations, showing that, in vivo and ex vivo, peripheral oscillators are able to maintain (partially) synchronized tissue rhythms independently of the SCN or external entrainment signals (10–15). Thus, TGF-β signaling may be crucial for controlling circadian tissue functions in the periphery. While further studies are needed to characterize the role of intercellular coupling for peripheral physiology, it is likely that peripheral coupling is important for the robustness of peripheral tissue rhythms and the response to zeitgeber/entrainment signals either from the external environment or from other body clocks, e.g., insulin-like growth factor 1, insulin, glucocorticoids, or temperature (62–65). Moreover, deregulation of TGF-β signaling is observed in many human diseases, including cancer (66, 67). Similarly, circadian rhythms are known to be disrupted in cancerous tissues, and loss of circadian control has been associated with poor treatment outcomes (68, 69). Thus, elucidating the role of peripheral coupling does have important implications not only for circadian tissue physiology but also for understanding pathologies associated with circadian disruption and will be an exciting future endeavor.
MATERIALS AND METHODS
Cloning of luciferase reporter constructs
The lentiviral reporter plasmids CRE:Luc, mutCRE:Luc, and Per2:Luc were cloned for real-time bioluminescence imaging as follows: (i) A sequence fragment containing seven tandem repeats of the cAMP enhancer element upstream of a minimal TATA promoter and luciferase was amplified from the pGL4.23_luc2/minP_CRE:Luc plasmid (gift from U. Schibler, University of Geneva) using polymerase chain reaction (PCR) with self-designed primers [GGCCATATCGATAGGTGCCAGAACATTTCTC (forward) and GGCATACGCGTTACCACATTTGTAGAGGTTTT (reverse)]. The PCR product was cleaned up by gel electrophoresis. Linearized inserts and pLenti6 backbone were generated by restriction enzyme digest of the PCR products and the pLenti6/V5-DEST Gateway (Thermo Fisher Scientific, #V49610) plasmid Cla I and Mlu I [New England Biolabs (NEB)] enzymes according to the manufacturer’s instructions. Fragments were cleaned up by gel electrophoresis, and ligation was performed with a T4 DNA ligase kit (Thermo Fisher Scientific, #K1422) according to the manufacturer’s instructions. (ii) A sequence containing seven mutated CRE elements (5′-TTAAACCA-3′) upstream of a minimal promoter (TATA) and 3′-flanking sequence was designed analogous to the sequence of the CRE:Luc expression plasmid and generated by gene synthesis (Bio Basic Inc.). Linearized mutCRE inserts and pLenti6_Luc backbone were generated by restriction enzyme digest of the pUC57_mutCRE and the pLenti6_CRE:Luc plasmids with Apa I and Nhe I– high fidelity (HF) (NEB) enzymes according to the manufacturer’s instructions. Fragments were cleaned up by gel electrophoresis, and ligation was performed with a T4 DNA ligase kit (Thermo Fisher Scientific, #K1422) according to the manufacturer’s instructions. (iii) A plasmid containing a ~3.4-kb fragment of the mouse Per2 promoter upstream of luciferase was a gift from O. Shigehiro (Kyushu University). Linearized Per2:Luc inserts were generated by restriction enzyme digest of the pGL3basic_Per2:Luc with Hpa I and Cla I (NEB) enzymes according to the manufacturer’s instructions. Linearized pLenti6 backbone was generated by restriction enzyme digest of the pLenti6/V5-DEST Gateway (Thermo Fisher Scientific, #V49610) plasmid with Mlu I (NEB) enzyme according to the manufacturer’s instructions. The Mlu I cut site was blunted using Klenow polymerase (NEB, #M0212S), and the linearized backbone was cut with Cla I (NEB) restriction enzyme according to the manufacturer’s instructions. Insert and backbone fragments were cleaned up by gel electrophoresis and ligation was performed with a T4 DNA ligase kit (Thermo Fisher Scientific, #K1422) according to the manufacturer’s instructions. Cloning procedures for other luciferase reports, not used in the main figures, are described in the Supplementary Materials.
Lentivirus production
Human embryonic kidney (HEK) 293T cells were plated to reach 70% confluence. The next day, cells were transfected with psPAX (Addgene, #12260; T-75-cm2 flask, 6 μg per flask; 96-well plate, 0.1 μg per well) and pMD2G (Addgene, #12259; T-75-cm2 flask, 3.6 μg per flask; 96-well plate, 0.06 μg per well) lentiviral packaging plasmids and lentiviral expression plasmids (T-75-cm2 flask, 8.4 μg per flask; 96-well plate, 0.14 μg per well) using a CalPhos transfection kit (Takara Bio, #631312; T-75-cm2 flask) or Lipofectamine 2000 (Thermo Fisher Scientific; 96-well plates) according to the manufacturer’s instructions. The next day, transfection medium was replaced by complete culture medium, and cells were incubated overnight. Subsequently, lentivirus containing supernatant was harvested, spun down (3000g for 10 min at 4°C), filtered through 0.45-μm sterile filters, and either used directly or stored at −80°C.
Lentivirus transduction
Cells were plated at 30 to 50% confluence and were transduced with lentiviral supernatant (T-75-cm2 flask, 4.5 ml per flask; 96-well plate, 100 μl per well) and protamine sulfate (8 μg/ml). Cells were incubated under standard tissue culture conditions overnight. The next day, viral transduction medium was replaced by complete culture medium with the appropriate selection antibiotic (10 μg/ml). Selection pressure was maintained for ~5 to 7 days. RNAi knockdown was performed with lentivirus-expressing GIPZ lentiviral short hairpin RNA (shRNA) or nonsilencing constructs (Horizon, #RHS6037).
RNA purification and reverse transcription quantitative PCR
RNA purification was performed using the Pure Link RNA Mini Kit (Thermo Fisher Scientific, #12183025) according to the manufacturer’s instructions with an additional on-column deoxyribonuclease I digestion. For reverse transcription quantitative PCR (RT-qPCR), RNA was reversely transcribed using the Moloney Murine Leukemia Virus Reverse Transcriptase Kit (Thermo Fisher Scientific, #28025013) with random hexamers (Thermo Fisher Scientific, #N8080127) according to the manufacturer’s instructions. RT-qPCR was performed using the SYBR Green Master Mix (Thermo Fisher Scientific, #A46112) and a CFX96 PCR system equipped with the CFX Manager software (Bio-Rad). The following primers were used for target amplifications: GAPDH [TGCACCACCAACTGCTTAGC (forward) and ACAGTCTTCTGGGTGGCAGTG (reverse)], PER2 [CACCAAATTGTTTGTTCCAGG (forward) and AACCGAATGGGAGAATAGTCG (reverse)], ARNTL (QuantiTect primer QT00011844), NR1D1 (QuantiTect primer QT00000413), DBP (QuantiTect primer QT00055755), XBP1 [TTACGAGAGAAAACTCATGGCC (forward) and GGGTCCAAGTTGTCCAGAATGC (reverse)], DNAJC3 (QuantiTect primer QT00011284), and HERPUD1 (QuantiTect primer QT00026418). Samples were measured in triplicates and cycle quantification values (Cq) were normalized to GAPDH. Relative expression was calculated using the 2−ΔCq method. Data were normalized to the mean expression of nonsilencing control or unstimulated control as indicated in the legends.
RNA sequencing
Cells were seeded at high (4 × 105 cells per well) or low (0.4 × 105 cells per well) culture density into six-well plates and maintained under experimental culture conditions described above. Cells were synchronized and, at the trough of PER2 expression (16 to 18 hours after sync), stimulated with CM and control medium for 2 hours. Total RNA was isolated as described. To provide sufficient RNA amounts for RNA-seq, RNA isolated from low density cells was pooled from three wells. Sequencing was performed by the Genomics Core Facility of the Berlin Institute of Health. Briefly, NEBNext polyadenylate mRNA magnetic isolation modules (NEB, #E7490S) were used to enrich polyadenylated mRNA, which was fragmented to approximately 200-nucleotide fragments. The NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, #E7530S) was used for complementary DNA synthesis and sequencing library preparation. Single-read RNA-seq was performed using the NextSeq 500/550 High-Output Kit v2 (Illumina, #FC-404-2005; 75 cycles). Quality control of raw reads was conducted using FastQC in Python. Mapping/alignment and counting of reads were conducted using STARaligner and subread featureCounts in Python. GRCh38.all.fa was used as reference genome with its respective annotation library Homo_sampiens_GRCh38.93.gft. During read counting, hits were called if overlaps of ≥1 base pair were found between raw reads and a single genomic feature (no multiple overlaps allowed). Quality control of aligned reads was conducted using MultiQC and RSeQC in Python. Read normalization and differential gene expression analysis was performed using DESeq2 in R [~x + y design matrix to test for the effect of medium (y) stimulation while controlling for culture density (x)]. Genes with <10 counts across samples were excluded from the differential gene expression analysis. Gene ontology (GO) analysis of significantly differentially expressed genes (Padj ≤ 0.01) was conducted with the web-based application GOrilla (70). All expressed genes in U-2 OS cells were used as background for enrichment analysis (table S1).
Cell lines
Human U-2 OS cells were a gift from C. Hagemeier (Charité Berlin), murine C2C12 were a gift from J. Bass (Northwestern University), and murine NIH3T3 cells were a gift from U. Schibler (University of Geneva). Human HEK293 [American Type Culture Collection (ATCC), CRL-1573], HEK293T (ATCC, CRL-3216), and HCT116 (ATCC, CCL-247) were purchased from ATCC. Cells were confirmed to be free of mycoplasma using the MycoAlert Detection Kit (Lonza, #LT07-418).
Fluorescent reporter cells
U-2 OS cells stably expressing an NR1D1::VNP [nuclear localization signal and PEST element fused to the open reading frame of Venus and with the 116 N-terminal amino acids of NR1D1, described in (23)] fusion protein were provided by M. Brunner (Heidelberg University). A single subclone was used for experiments.
Luciferase reporter cells
U-2 OS cells expressing firefly luciferase from a 0.9-kb mouse Bmal1 promoter fragment were generated by lentiviral transduction of the respective expression plasmid (gift from S. Brown, University of Zurich). A single subclone was used for experiments. U-2 OS cells expressing firefly luciferase from a ~3.4-kb mouse Per2 promoter fragment were generated by lentiviral transduction of the respective expression plasmid. U-2 OS cells expressing firefly luciferase from artificial promoters containing seven tandem repeats of the cAMP enhancer element CRE (5′-TGACGTCA-3′) or mutated CRE (5′-TTAAACCA-3′) were generated by lentiviral transduction of the respective expression plasmid. U-2 OS cells expressing Per2:Luc, CRE:Luc, and mutCRE:Luc were subjected to antibiotic selection with blasticidin (10 μg/ml) but not to clonal selection. U-2 OS Bmal1:Luc CRY2−/− and U-2 OS Bmal1:Luc TNPO1−/− cell lines were generated by CRISPR-Cas9–mediated gene knockout and clonal selection as previously described in (31, 32).
3D spheroids
3D spheroids were generated from U-2 OS Bmal1:Luc CRY2−/−, TNPO1−/−, (31, 32) or WT cells as described below.
Animal-derived models
Primary hepatocytes were collected from C57BL/6 J mice. Peripheral tissue explants for organotypic slice cultures were harvested from PER2::LUC knockin animals (10). Animal procedures were in accordance with the guidelines of the Federation for Laboratory Animal Science Associations and the Landesamt für Gesundheit und Soziales at the Charité Universitätsmedizin Berlin.
Maintenance culture
Cell lines and organotypic explant slices were maintained in standard tissue culture flasks or dishes. Primary hepatocytes were plated into collagen I–coated 175-cm2 flasks (Corning, #354487). Cells (except U-2 OS NR1D1::VNP cells) and organotypic slices were maintained in complete culture medium: Dulbecco’s modified Eagle’s medium (DMEM) [glucose (4500 mg/liter) and l-glutamine; Thermo Fisher Scientific #41965039], supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml)/streptomycin (100 μg/ml) (P/S), and 25 mM Hepes buffer (in-house). Fluorescent reporter cells were maintained in RPMI 1640 (l-glutamine; Capricorn, #RPMI-A) with 10% FBS and 1% P/S. Organoids were maintained in specialized media formulations as indicated in the Supplementary Materials. Spheroids were embedded in Cultrex growth factor–reduced basement membrane matrix (Trevigen, #3433-001-01) and maintained in complete culture medium. All model systems were maintained under standard tissue culture conditions in humidified 5% CO2 at 37°C tissue culture incubators.
Synchronization
Synchronization was performed by incubation with 1 μM dexamethasone (Sigma-Aldrich, #D4902) for 20 to 30 min under standard tissue culture conditions. Following incubation, cells or tissues were washed with 1× phosphate-buffered saline (PBS) and prepared for downstream applications.
Hepatocyte isolation
Livers of anesthetized [ketamine (80 mg/kg), Pfizer; and xylazine (12 mg/kg), Bayer] male or female C57BL/6 J mice were perfused with perfusion buffer (1× Earle’s balanced salt solution without CaCl2/MgCl2 containing 1% 50 mM EGTA), followed by digestion buffer (1× Hanks’ balanced salt solution containing 5000 U of collagenase type I, Worthington, #LS004197). After filtration and separation by Percoll gradients (Easycoll separating solution, Biochrom, #L6135), hepatocytes were seeded into collagen I–coated T-175-cm2 flasks (Corning, #354487) in complete culture medium and were maintained under standard tissue culture conditions.
Organotypic explant slices
PER2::LUC male or female mice were anesthetized by isoflurane (Abbott) inhalation and euthanized by cervical dislocation. Explanted tissues were transferred to cold 1× PBS and stored on ice until chopped into 300- to 500-μm-slice sections using a McIlwain tissue chopper (Campden Instruments). Slice sections were placed in Nunc Delta 35-mm dishes and maintained in complete culture medium under standard tissue culture conditions.
3D spheroid generation
Standard 100-mm tissue culture dishes were supplemented with 5 to 10 ml of 1× PBS. Cellular suspensions (3.0 ×106 cells/ml) were prepared in complete culture medium. Ten microliters of droplets were seeded onto the inverted lids of the 100-mm dishes. Lids were inverted again and placed onto the PBS-filled bottom without disrupting integrity of the droplets. Hanging drop cultures were maintained for ~1 week under standard tissue culture conditions until spheroids had formed. Spheroids were harvested by pipetting and transferred to 1 ml per well of 1× PBS in standard tissue culture six-well plates. PBS was carefully aspirated to remove as much volume as possible. Subsequently, spheroids were resuspended and seeded in Cultrex basement membrane extracts (Trevigen, #3433-001-01) in 35-mm Nunc Delta dishes. Basement matrix was solidified at 37°C for 1 hour before, and spheroids were supplemented with complete culture medium.
CM generation
Cell lines were seeded into standard T-175-cm2 flasks, hepatocytes into collagen I–coated T-175-cm2 flasks (Corning, #354487), at high density. Cells were incubated in complete culture medium to reach confluence. Then, cells were washed twice with 1× PBS and were incubated in 15 ml per flask of serum- and phenol red–free DMEM with 1% P/S for 18 to 24 hours and under standard tissue culture conditions. Following incubation, supernatants were harvested, spun down (3000g for 10 min), filtered through 0.2-μm sterile filters, and concentrated 60- to 100-fold using Amicon Ultra-15 centrifugal filters [Merck, #UFC90; 10-kDa molecular weight cutoff (MWCO)]. Control medium was produced by the same procedure using empty culture flasks. Conditioned and control medium from different preparations was pooled and stored at −80°C.
Density dependence
U-2 OS nonreporter cells were seeded at indicated densities into Nunc Delta 35-mm dishes. At the same time, U-2 OS reporter cells were coseeded at low density (0.3 × 105 cells per dish) in the same dishes or onto 30-mm organotypic membrane inserts (Merck, catalog no. PICMORG50) that were placed into dishes containing nonreporter cells. Cells were synchronized before luciferase imaging.
Phase pulling experiments
U-2 OS Per2:Luc reporter and U-2 OS nonreporter cells were split into two groups (“early” and “late”) that were synchronized 6 hours apart (fig. S1A). At the early time point, early Per2:Luc and early nonreporter cells were synchronized in suspension and, at the indicated densities, plated into Nunc Delta 35-mm dishes in 2 ml per dish of imaging medium. Dishes were incubated for 6 hours in a standard tissues culture incubator. At the late time point, Per2:Luc and nonreporter cells were synchronized in suspension and resuspended in prewarmed imaging medium. A total of 100 μl of cellular suspensions (either late Per2:Luc+ late nonreporter cells or late nonreporter cells only, as indicated in fig. S1A) were carefully seeded into dishes containing the early cells. Dishes were sealed and long-term imaging was started.
Period pulling experiments
“Pure” spheroids were generated from U-2 OS Bmal1:Luc WT, CRY2−/−, or TNPO1−/− cells as described. “Mixed” spheroids were generated from cellular suspensions that contained a 1:5 mixture of U-2 OS Bmal1:Luc WT, CRY2−/−, or TNPO1−/− cells and U-2 OS WT nonreporter cells. Spheroids were synchronized before imaging.
Real-time monitoring of luciferase reporters
Luciferase reporter models, i.e., cells, organotypic slices, organoids, and spheroids, were imaged in reporter medium: phenol red–free DMEM [glucose (4500 mg/liter), Hepes, and l-glutamine; Thermo Fisher Scientific, #21063029] supplemented with 10% FBS (if not otherwise indicated), 1% P/S, and 250 μM d-luciferin (PJK GmbH, #102112). Cells were plated at high culture density if not stated otherwise (35-mm dishes, 3 × 105 cells per dish; 96-well plates, 0.2 × 105 cells per well). Dishes or plates were placed in real-time luminometers, and luminescence intensity (counts per second) was recorded in regular intervals for multiple days. Four different real-time luminometer systems were used: (i) LumiCycle rotating luminometer with 32 positions and built-in visualization software (Actimetrics). (ii) Temperature-adjustable light-tight boxes containing single photomultiplier tubes (Hamamatsu Photonics) with PhotoNGraph visualization software (in-house) and JUMO IMAGO 500 temperature controllers (JUMO). (iii) TopCount NXT 96-well plate luminometer with a 20-plate stacker unit (PerkinElmer) and TopCount NXT software (PerkinElmer). (iv) Orion II 96-well plate luminometer with a single-plate unit and Simplicity visualization software (Berthold Ditection Systems). Measurements in the LumiCycle and the light-tight boxes were performed in Nunc Delta 35-mm dishes (Thermo Fisher Scientific, #153066) and nonhumidified 5% CO2 at 37°C standard tissue culture incubators. Measurements in the TopCount and Orion II were performed in in Nunc F96 MicroWell 96-well plates (Thermo Fisher Scientific, #136102) under ambient CO2 (no CO2 control unit) at 37°C incubation. Nunc dishes and plates were sealed with silicone grease and Parafilm or clear seal heat seals (Thermo Fisher Scientific, #AB-0812), respectively.
Bioluminescence data analysis
Raw data were exported, and ChronoStar software (71) was used for data analysis and estimation of rhythm parameters including period, phase, relative amplitude, damping, and average bioluminescence. Briefly, time series data were trend-eliminated by dividing by a 24-hour running average, and rhythm parameters were estimated by fitting a cosine wave function including an exponential decay term. For comparison of phases, phases were divided by the periods and multiplied by 24. For noncircadian reporter cells, raw times series data were used for downstream analysis in OriginPro (OriginLab) to determine the peak area under the curve following reporter induction and using the built-in peak integration script (baseline signal was set according to luminescence intensity before reporter gene induction and was subtracted).
Real-time monitoring of fluorescent reporters
U-2 OS NR1D1::VNP reporter cells were maintained in FluoroBrite medium: FluoroBrite DMEM (high glucose and no Hepes; Thermo Fisher Scientific #A1896701) supplemented with 2% FBS, 1% P/S, and l-glutamine (300 mg/liter; Thermo Fisher Scientific, #25030149). If not indicated otherwise, then cells were plated at high culture density (0.65 × 105 cells per well) into 24-well black plates (Ibidi, #82406). For inhibitor treatments, Fluobrite imaging medium was supplemented with indicated concentrations of LY2109761 or solvent control. Time-lapse imaging was accomplished by a high-resolution multichannel wide-field epi-fluorescence Nikon Ti2 microscope equipped with an environmental chamber for cell culture (Okolab) and using NIS-Elements visualization software (Nikon). Images were captured each 30 min using a 20× Plan Apo Ph2 DM λ magnification objective (Nikon), light-emitting diode illumination (Lumencor, SPECTRA X), and a scientific complementary metal-oxide semiconductor active-pixel sensor (sCMOS), PCO.edge camera (PCO) for multiple days.
Fluorescence image and data analysis
Tracking and analysis of images used the p53Cinema MATLAB code as described in (72). Single cells were tracked from the beginning of the experiment (t = 0) until the last time point or the time of death. Cell division events were recorded, and upon cell division, only one of the daughter cells was further tracked. Cells that left the field of view were discarded from the analysis. Intensity of fluorescent reporters was quantified using the tracking information from background-subtracted images by averaging 10 pixels around the cell nucleus. Fluorescent intensity recorded for single-cell traces was exported and analyzed for the full-time series. Missing data were filled via interpolation before rhythm analysis.
Rhythmicity analysis
Rhythmicity of single-cell time series was evaluated using the R package MetaCycle with the meta2d method as described in (73) [minper = 20, maxper = 35, cycMethod = c (“ARS”, “JTK”, “LS”)]. An upper period limit of 35 hours was chosen, because of the period lengthening effect of TGF-β receptor inhibitor. Time series that did not pass the rhythmicity test [meta2d Benjamini-Hochberg false discovery rate (FDR), ≥0.05] were excluded from further analysis. MetaCycle (meta2d method) was used to estimate period and amplitude parameters displayed in figs. S2A and S8 (D and E). Instantaneous phases and phase coherence over time (Fig. 7, A and B; and figs. S2, C and D, and S8B) were estimated using the Python-based application pyBOAT as described in (74) (minper = 20, maxper = 35, ridge threshold = 0, sinc detrending filter = 72 hours). Rayleigh plots were generated, and underlying parameters were computed, using the R package circular.
Pharmacological perturbations
Pharmacological inhibitors were prepared in recommended solvents according to the manufacturer’s instructions and stored at −20°C. Inhibitor treatments were performed with the indicated concentrations either during seeding and imaging or only during imaging (as stated in the legends). Inhibitors remained in the medium from their addition for the duration of the experiment. When performing stimulation experiments, TGF-β receptor inhibitor LY2109761 and solvent control were added to CM or control medium before stimulation. Inhibitors were purchased from the following manufacturers: eeyarestatin, tunicamycin (Cayman Chemical, #10012609 and #11445), FLI-06, golgicide A, AG1478, brefeldin A (Sigma-Aldrich, #SML0975, #G0923, #T4182, and #B7651), and LY2109761 (Cayman Chemical, #15409).
CM assays
For size fractionation, CM and control medium were generated as described, but Amicon Ultra-15 centrifugal filters with 3 to 100-kDa MWCO were used for concentration. Subsequently, medium was split into two groups (“heated” and “nonheated”). The heated group was boiled at 95°C for 10 min. Both groups were centrifuged (3000g for 5 min), and supernatants were either used directly for stimulations or stored at −20°C. Ammonium sulfate precipitation was performed by saturating CM and control medium with 10 to 60% (NH4)2SO4 at room temperature using a magnetic stirrer. Media were incubated at 4°C overnight. The next day, media were centrifuged (3000g for 45 min at 4°C) to pellet precipitates. Pellets were washed once with 1× PBS and resolubilized in serum- and phenol red–free DMEM with 1% P/S. Precipitates were concentrated 75-fold using Amicon Ultra-15 centrifugal filters (10-kDa MWCO) and were either used directly for stimulation or stored at −20°C. For 0% (NH4)2SO4 controls, CM and control medium were treated the same way, but supernatants not pellets were used following the centrifugation step. For tryptic digest, CM and control medium were treated with immobilized TPCK trypsin (Thermo Fisher Scientific, #20230). Before addition, immobilized tosyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin was washed three times with and resuspended in serum- and phenol red–free DMEM with 1% P/S and 20 mM EGTA. Immobilized TPCK trypsin was added to CM and control medium to prepare a solution containing 50 TAME units. Samples were incubated in a shaking incubator (300 rpm at 37°C) overnight. The next day, trypsin gel was separated from the digestion mixture by centrifugation (12,000g for 2 min), and supernatants were either used directly for stimulation or stored at −20°C. Nondigested control samples were treated the same way except that the serum- and phenol red–free DMEM with 1% P/S and 20 mM EGTA was added to the conditioned and control medium instead of immobilized TPCK trypsin.
Stimulation experiments
Experimental models were maintained in serum-free medium before stimulation (serum-free imaging medium for real-time monitoring experiments and serum-free culture medium for RT-qPCR and RNA-seq experiments). Conditioned and control medium were prepared as described. Recombinant TGF-β was prepared as 1:1:1 isotype mix of recombinant human TGF-β1 to TGF-β3 (Abcam, #ab50036, #ab84070, and #ab217402) according to the manufacturer’s instructions. Recombinant TGF-β stock solution and the respective solvent control were further diluted in serum- and phenol red–free DMEM with 1% P/S to reach the indicated concentrations. For bioluminescence imaging CM, control medium, recombinant TGF-β, and solvent were further supplemented with 250 μM d-luciferin (P.J.K, #102112). To perform stimulations during imaging, recording was paused, and plates/dishes were transferred to the tissue culture hood. Medium was aspirated and replaced with prewarmed stimulant (96-well plates, 20 to 50 μl per well; 35-mm dishes, 1 ml per dish; six-well plates, 500 μl per well). Subsequently, dishes/plates were resealed, and real-time monitoring was continued, or plates/dishes were placed back in the tissue culture incubator for indicated durations before RNA was harvested.
Cold zeitgeber
U-2 OS Bmal1:Luc reporter cells were treated with TGF-β receptor inhibitor or solvent control during seeding and imaging. Real-time monitoring was performed in temperature-adjustable light-tight boxes as described. JUMO IMAGO 500 temperature controllers were programmed to apply an 8-hour 20°C temperature pulse at the peak of Bmal1:Luc expression (i.e., 64 hours after synchronization for solvent and 70 hours after synchronization for inhibitor) using JUMO IMAGO 500 software (note that inhibitor treated cells display a 4- to 6-hour period lengthening). Subsequently, the temperature was reset to 37°C.
Immunodepletion and neutralization
αTGF-β antibody (10 μg/ml; Thermo Fisher Scientific, #15583482) or immunoglobulin G (IgG) isotype control (10 μg/ml; R&D Systems, #MAB002) was added to CM and control medium. Specificity of the αTGF-β antibody was determined by pulldown with subsequent mass spectrometry analysis (see table S2). Samples were incubated on a spinning wheel at 4°C overnight. The next day, samples were spun down (12,000g for 5 min). For neutralization, supernatants were used directly to stimulate reporter cells (or stored at −20°C). For immunodepletion, antibodies were pulled down using protein G plus agarose beads (Santa Cruz Biotechnology, #sc-2002). Before pulldown, beads were washed three times with serum- and phenol red–free DMEM with 1% P/S. A total of 20 to 30 μl of beads/1 μg of antibody was added to CM and control medium. Samples were incubated on a spinning wheel at 4°C overnight and then spun down (2500g for 3 min). Supernatants were either used directly for stimulation or stored at −20°C.
Chromatography and mass spectrometry
Chromatography and mass spectrometry were performed by the Protein Purification facility of the Max Planck Institute for Infectious Biology (Berlin, Germany). Briefly, GFX of 1000× CM (500 μl of input) was performed with Superdex HR-200 GL columns (Sigma-Aldrich; elution with 1× PBS). AEX of selected GFX pools (2.5 ml of input each) was performed with Poros HQ-20 columns [Thermo Fisher Scientific; elution with 20 mM tris (pH 8.0) and 1 M NaCl gradient]. AEX fractions were desalted and concentrated fivefold using Amicon Ultra-0.5 centrifugal filters (3-kDa MWCO; Merck, #UFC500396). Reporter cells were stimulated with chromatography fractions or input as described, except that medium was not aspirated, but samples were added directly to imaging medium in a 1:5 ratio. Recovery of activity was determined relative to chromatography input. Protein content was determined by standard Bradford protein assay (GFX) or by using the fractions’ absorbance maxima at 280 nm. Active fractions were defined as fractions with percent recovery > means ± SD of the percent recovery of all fractions, as well as with protein content < mean protein content of all fractions (excluding negative absorbance values at 280 nm). Selected AEX fractions were analyzed by mass spectrometry as follows: Samples were prepared by reduction, alkylation, and tryptic digest. Desalting was performed with C18 tips (Thermo Fisher Scientific, #8772), and samples were solubilized in 2% acetonitrile with 0.1% trifluoroacetic acid (v/v). Peptide separation was performed using an UltiMate 3000 high-performance liquid chromatography system (Thermo Fisher Scientific) with Acclaim PepMap 100 C18 nano-trap (Thermo Fisher Scientific, #164197; 2% acetonitrile/0.1% trifluoroacetic acid solvent) and Acclaim PepMap rapid separation liquid chromatography (Thermo Fisher Scientific, #164534; 0.1% formic acid, solvent A; and 80% acetonitrile/0.1% formic acid, solvent B) columns. Electrospray ionization mass spectrometry was performed using a Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific). Observed mass spectrometry spectra were compared to a contaminant, as well as the SwissProt (release 2018_11, taxonomy: Homo sapiens) primary sequence databases using Mascot software (Matrix Science). Resulting Mascot hits (FDR, 1%) (table S2) were filtered for human secreted proteins predicted by majority decision–based method for secreted proteins (MDSEC; Human Protein Atlas, accessed February 2019; table S3). Identified secreted proteins were considered candidate coupling factors if present in >50% of active but not inactive fractions.
Computational modeling
Mathematical simulations of single-cell oscillators were designed to explore how oscillation parameters (amplitude, damping rate, period, and phase), both at the single cell and bulk level, change with varying coupling strengths. The results presented in Figs. 1 (A to C) and 2 (A and B) were numerically calculated using an ensemble of n = 200 mutually coupled stochastic amplitude-phase oscillators in Cartesian coordinates, modeled with explicit twist (ε) describing amplitude-dependent periods (Eqs. 1 and 2)
| (1) |
| (2) |
The simulated phase response curve shown in fig. S8A was calculated in a similar way but using an ensemble of n = 100 mutually coupled deterministic (i.e., not stochastic) amplitude-phase oscillators in Cartesian coordinates, modeled with explicit twist (ε) describing amplitude-dependent periods (Eqs. 3 and 4)
| (3) |
| (4) |
Parameters represent single-cell oscillator period (τi = 24.9 ± 1.5 hours), amplitude (Ai = 1), amplitude relaxation rate (λ = 0.03 hour−1), and explicit twist (ε = −0.005). Noise was introduced by adding a Wiener process (dWt with σx = 0.1 and σy = 0.1). We assumed that the mean field (Eq. 5) additively couples solely to the x coordinate. The parameter Kcoup denotes the strength of the coupling between the mean field and the single oscillatory unit. In the case of fig. S8A, a perturbation of F = 0.5 was applied to the x coordinate for a duration of 2 hours. Single-cell amplitudes, phases, and periods from Figs. 1 (B and C) and 2B were estimated by fitting a cosine curve (Eq. 6) to the single-cell traces from the numerical simulations, after removing transients. In Fig. 2A, the mean signal of the ensemble was determined, and the mean amplitude was calculated by fitting Eq. 6 to the time series, after removing transients. Similarly, the damping rate of the ensemble was calculated by fitting an exponentially decaying cosine curve (Eq. 7)
| (5) |
| (6) |
| (7) |
where A is an amplitude, d is a damping constant, τ is a period, and φ is a phase.
Numerical simulations were obtained for a total integration time of 24.9 ×100 hours at Δt = 0.1-hour time steps using the odeint function of the scipy module in Python. Fits were performed using the curve_fit function of the scipy module in Python.
Quantification and statistical analysis
Circadian rhythmicity and bioinformatic analysis are described above in the corresponding sections. Statistical parameters, including statistical analysis, statistical significance, and n values, are reported in the figure legends. Data are presented as individual data points from biological repeats (independent experiments) and with means ± SEM calculated by GraphPad Prism. In rare instances, data are represented as individual data points with means ± SEM from technical replicates. Statistical analysis was performed using GraphPad Prism or R in RStudio for single-cell imaging data. All statistical details of experiments, including the number of biological repeats and technical replicates, are as described in the figure legends. For all statistical tests, P < 0.05 was considered to be statistically significant. P values are reported using the following symbolic representations: ns = P ≥ 0.1, #P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. When tests that correct for multiple comparisons were used, adjusted P values are presented. For real-time bioluminescence imaging, immunostainings, and DNA gels, representative time series and images from biological replicate experiments are depicted unless otherwise stated.
Acknowledgments
We thank C. Hagemeier (Charité Universitätsmedizin Berlin), J. Bass (Northwestern University), U. Schibler (University of Geneva), S. Brown (University of Zurich), M. Brunner (Heidelberg University), and J. Takahashi (Northwestern University) for the provision of cell lines, PER2::LUC animals, and reporter constructs. We thank M. Schupp and P. Weber for help in isolating primary hepatocytes and F. Heyd for support in Golgi apparatus microscopy. We thank the Protein Purification Core Facility of the Max Planck Institute for Infection Biology Berlin, the Genomics Core Facility of the Berlin Institute of Health, and the AMBIO imaging facility of the Charité Berlin for support in acquisition of the chromatography, mass spectrometry, RNA-seq, and real-time fluorescence data. We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of the Charité Universitätsmedizin Berlin. We thank all current and former members of the Kramer and Herzel laboratories for technical and intellectual assistance. All sketches were created with BioRender.com. Funding: This work was funded by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation, grants 278001972–TRR 186 and KR1989/12 to A.K.) and the Joachim Herz Stiftung (Add-On Fellowship for Interdisciplinary Life Sciences to A.-M.F.). A.E.G.’s research was supported by the German Federal Ministry for Education and Research (BMBF) through the Junior Network in Systems Medicine, under the auspices of the e:Med Programme (grant 01ZX1917C). Author contributions: Study design and conceptualization: A.K., A.-M.F., and S.J. Methodology: A.K., A.-M.F., S.J., H.H., R.H., and A.E.G. Investigation, A.-M.F., S.J., and M.d.O. Writing (original draft and editing): A.-M.F. and A.K. Writing (review): A.K., A.-M.F., H.H., A.E.G., and M.d.O. Funding acquisition: A.K., H.H., A.-M.F., and A.E.G.. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Requests for additional data related to this paper or on resources and reagents should be directed to and will be fulfilled by the corresponding authors. Raw and processed RNA-seq data reported in this paper are available at GEO under the accession number GSE158577. Software for recording and visualization of luminometer data, as well as for extraction of rhythm parameters, were generated in-house and can be obtained online from achim-kramer-lab.de. All other software used is openly available. Mathematical algorithms used in this study are described or referenced in their respective method section.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/30/eabg5174/DC1
REFERENCES AND NOTES
- 1.Woelfle M. A., Ouyang Y., Phanvijhitsiri K., Johnson C. H., The adaptive value of circadian clocks: An experimental assessment in cyanobacteria. Curr. Biol. 14, 1481–1486 (2004). [DOI] [PubMed] [Google Scholar]
- 2.DeCoursey P. J., Walker J. K., Smith S. A., A circadian pacemaker in free-living chipmunks: Essential for survival? J. Comp. Physiol. A 186, 169–180 (2000). [DOI] [PubMed] [Google Scholar]
- 3.J. S. Takahashi, Molecular architecture of the circadian clock in mammals, in Research and Perspectives in Endocrine Interactions, P. Sassone-Corsi, Y. Christen, Eds. (Springer, 2016), pp. 13–24; 10.1007/978-3-319-27069-2_2. [DOI] [PubMed]
- 4.Ukai-Tadenuma M., Kasukawa T., Ueda H. R., Proof-by-synthesis of the transcriptional logic of mammalian circadian clocks. Nat. Cell Biol. 10, 1154–1163 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Gerber A., Esnault C., Aubert G., Treisman R., Pralong F., Schibler U., Blood-borne circadian signal stimulates daily oscillations in actin dynamics and SRF activity. Cell 152, 492–503 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Travnickova-Bendova Z., Cermakian N., Reppert S. M., Sassone-Corsi P., Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc. Natl. Acad. Sci. U.S.A. 99, 7728–7733 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welsh D. K., Yoo S.-H., Liu A. C., Takahashi J. S., Kay S. A., Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr. Biol. 14, 2289–2295 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Welsh D. K., Logothetis D. E., Meister M., Reppert S. M., Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron 14, 697–706 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Leise T. L., Wang C. W., Gitis P. J., Welsh D. K., Persistent cell-autonomous circadian oscillations in fibroblasts revealed by six-week single-cell imaging of PER2::LUC bioluminescence. PLOS ONE 7, e33334 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoo S.-H., Yamazaki S., Lowrey P. L., Shimomura K., Ko C. H., Buhr E. D., Siepka S. M., Hong H.-K., Oh W. J., Yoo O. J., Menaker M., Takahashi J. S., PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. U.S.A. 101, 5339–5346 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saini C., Liani A., Curie T., Gos P., Kreppel F., Emmenegger Y., Bonacina L., Wolf J.-P., Poget Y.-A., Franken P., Schibler U., Real-time recording of circadian liver gene expression in freely moving mice reveals the phase-setting behavior of hepatocyte clocks. Genes Dev. 27, 1526–1536 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tahara Y., Kuroda H., Saito K., Nakajima Y., Kubo Y., Ohnishi N., Seo Y., Otsuka M., Fuse Y., Ohura Y., Komatsu T., Moriya Y., Okada S., Furutani N., Hirao A., Horikawa K., Kudo T., Shibata S., In vivo monitoring of peripheral circadian clocks in the mouse. Curr. Biol. 22, 1029–1034 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Sinturel F., Gos P., Petrenko V., Hagedorn C., Kreppel F., Storch K.-F., Knutti D., Liani A., Weitz C., Emmenegger Y., Franken P., Bonacina L., Dibner C., Schibler U., Circadian hepatocyte clocks keep synchrony in the absence of a master pacemaker in the suprachiasmatic nucleus or other extrahepatic clocks. Genes Dev. 35, 329–334 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landgraf D., Achten C., Dallmann F., Oster H., Embryonic development and maternal regulation of murine circadian clock function. Chronobiol. Int. 32, 416–427 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Moore S. R., Pruszka J., Vallance J., Aihara E., Matsuura T., Montrose M. H., Shroyer N. F., Hong C. I., Robust circadian rhythms in organoid cultures from PERIOD2::LUCIFERASE mouse small intestine. Dis. Model. Mech. 7, 1123–1130 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noguchi T., Wang L. L., Welsh D. K., Fibroblast PER2 circadian rhythmicity depends on cell density. J. Biol. Rhythms 28, 183–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Neill J. S., Hastings M. H., Increased coherence of circadian rhythms in mature fibroblast cultures. J. Biol. Rhythms 23, 483–488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guenthner C. J., Luitje M. E., Pyle L. A., Molyneux P. C., Yu J. K., Li A. S., Leise T. L., Harrington M. E., Circadian rhythms of PER2::LUC in Individual primary mouse hepatocytes and cultures. PLOS ONE 9, e87573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rougemont J., Naef F., Dynamical signatures of cellular fluctuations and oscillator stability in peripheral circadian clocks. Mol. Syst. Biol. 3, 93 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akhtar R. A., Reddy A. B., Maywood E. S., Clayton J. D., King V. M., Smith A. G., Gant T. W., Hastings M. H., Kyriacou C. P., Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr. Biol. 12, 540–550 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Koronowski K. B., Kinouchi K., Welz P.-S., Smith J. G., Zinna V. M., Shi J., Samad M., Chen S., Magnan C. N., Kinchen J. M., Li W., Baldi P., Benitah S. A., Sassone-Corsi P., Defining the independence of the liver circadian clock. Cell 177, 1448–1462.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noguchi T., Ikeda M., Ohmiya Y., Nakajima Y., Simultaneous monitoring of independent gene expression patterns in two types of cocultured fibroblasts with different color-emitting luciferases. BMC Biotechnol. 8, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagoshi E., Saini C., Bauer C., Laroche T., Naef F., Schibler U., Circadian gene expression in individual fibroblasts: Cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell 119, 693–705 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Schmal C., Herzog E. D., Herzel H., Measuring relative coupling strength in circadian systems. J. Biol. Rhythms 33, 84–98 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abraham U., Granada A. E., Westermark P. O., Heine M., Kramer A., Herzel H., Coupling governs entrainment range of circadian clocks. Mol. Syst. Biol. 6, 438 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aton S. J., Colwell C. S., Harmar A. J., Waschek J., Herzog E. D., Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat. Neurosci. 8, 476–483 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi S., Isejima H., Matsuo T., Okura R., Yagita K., Kobayashi M., Okamura H., Synchronization of cellular clocks in the suprachiasmatic nucleus. Science 302, 1408–1412 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Jiang Z.-G., Yang Y.-Q., Allen C. N., Tracer and electrical coupling of rat suprachiasmatic nucleus neurons. Neuroscience 77, 1059–1066 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Maier B., Wendt S., Vanselow J. T., Wallach T., Reischl S., Oehmke S., Schlosser A., Kramer A., A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes Dev. 23, 708–718 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang E. E., Liu A. C., Hirota T., Miraglia L. J., Welch G., Pongsawakul P. Y., Liu X., Atwood A., Huss J. W., Janes J., Su A. I., Hogenesch J. B., Kay S. A., A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell 139, 199–210 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Börding T., Abdo A. N., Maier B., Gabriel C., Kramer A., Generation of human CRY1 and CRY2 knockout cells using duplex CRISPR/Cas9 technology. Front. Physiol. 10, 577 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korge S., Maier B., Brüning F., Ehrhardt L., Korte T., Mann M., Herrmann A., Robles M. S., Kramer A., The non-classical nuclear import carrier transportin 1 modulates circadian rhythms through its effect on PER1 nuclear localization. PLOS Genet. 14, e1007189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welsh D. K., Takahashi J. S., Kay S. A., Suprachiasmatic nucleus: Cell autonomy and network properties. Annu. Rev. Physiol. 72, 551–577 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueda H. R., Hirose K., Iino M., Intercellular coupling mechanism for synchronized and noise-resistant circadian oscillators. J. Theor. Biol. 216, 501–512 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Ko C. H., Yamada Y. R., Welsh D. K., Buhr E. D., Liu A. C., Zhang E. E., Ralph M. R., Kay S. A., Forger D. B., Takahashi J. S., Emergence of noise-induced oscillations in the central circadian pacemaker. PLOS Biol. 8, e1000513 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stockis J., Dedobbeleer O., Lucas S., Role of GARP in the activation of latent TGF-β1. Mol. Biosyst. 13, 1925–1935 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Deheuninck J., Luo K., Ski and SnoN, potent negative regulators of TGF-β signaling. Cell Res. 19, 47–57 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J., Weinberg M. S., Zerbini L., Prince S., The oncogenic TBX3 is a downstream target and mediator of the TGF-β1 signaling pathway. Mol. Biol. Cell 24, 3569–3576 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verrecchia F., Tacheau C., Schorpp-Kistner M., Angel P., Mauviel A., Induction of the AP-1 members c-Jun and JunB by TGF-β/Smad suppresses early Smad-driven gene activation. Oncogene 20, 2205–2211 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Chen S.-J., Ning H., Ishida W., Sodin-Semrl S., Takagawa S., Mori Y., Varga J., The early-immediate gene EGR-1 is induced by transforming growth factor-β and mediates stimulation of collagen gene expression. J. Biol. Chem. 281, 21183–21197 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Tao W., Wu J., Zhang Q., Lai S.-S., Jiang S., Jiang C., Xu Y., Xue B., Du J., Li C.-J., EGR1 regulates hepatic clock gene amplitude by activating Per1 transcription. Sci. Rep. 5, 15212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kornhauser J., Nelson D., Mayo K., Takahashi J., Regulation of jun-B messenger RNA and AP-1 activity by light and a circadian clock. Science 255, 1581–1584 (1992). [DOI] [PubMed] [Google Scholar]
- 43.Warren J., Im M., Ballesteros A., Ha C., Moore T., Lambert F., Lucas S., Hinz B., Dveksler G., Activation of latent transforming growth factor-β1, a conserved function for pregnancy-specific beta 1-glycoproteins. Mol. Hum. Reprod. 24, 602–612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Awwad K., Hu J., Shi L., Mangels N., Abdel Malik R., Zippel N., Fisslthaler B., Eble J. A., Pfeilschifter J., Popp R., Fleming I., Role of secreted modular calcium-binding protein 1 (SMOC1) in transforming growth factor β signalling and angiogenesis. Cardiovasc. Res. 106, 284–294 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Su L., Lei X., Ma H., Feng C., Jiang J., Jiao J., PRDM16 orchestrates angiogenesis via neural differentiation in the developing brain. Cell Death Differ. 27, 2313–2329 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gauger K. J., Chenausky K. L., Murray M. E., Schneider S. S., SFRP1 reduction results in an increased sensitivity to TGF-β signaling. BMC Cancer 11, 59 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gauger K. J., Shimono A., Crisi G. M., Schneider S., Loss of SFRP1 promotes ductal branching in the murine mammary gland. BMC Dev. Biol. 12, 25 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flanders K. C., Yang Y.-A., Herrmann M., Chen J., Mendoza N., Mirza A. M., Wakefield L. M., Quantitation of TGF-β proteins in mouse tissues shows reciprocal changes in TGF-β1 and TGF-β3 in normal vs neoplastic mammary epithelium. Oncotarget 7, 38164–38179 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu H.-S., Li Y. F., Chou C.-I., Yuan C. C., Hung M. W., Tsai L. C., The concentration of serum transforming growth factor beta-1 (TGF-β1) is decreased in cervical carcinoma patients. Cancer Invest. 20, 55–59 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Khan S. A., Joyce J., Tsuda T., Quantification of active and total transforming growth factor-β levels in serum and solid organ tissues by bioassay. BMC. Res. Notes 5, 636 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu G., Anafi R. C., Hughes M. E., Kornacker K., Hogenesch J. B., MetaCycle: An integrated R package to evaluate periodicity in large scale data. Bioinformatics 32, 3351–3353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.G. Mönke, F. A. Sorgenfrei, C. Schmal, A. E. Granada, Optimal time frequency analysis for biological data - pyBOAT. bioRxiv 2020.04.29.067744 [Preprint]. 5 June 2020. 10.1101/2020.04.29.067744. [DOI]
- 53.Mullen A. C., Orlando D. A., Newman J. J., Lovén J., Kumar R. M., Bilodeau S., Reddy J., Guenther M. G., DeKoter R. P., Young R. A., Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 147, 565–576 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Massagué J., TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Topper J. N., DiChiara M. R., Brown J. D., Williams A. J., Falb D., Collins T., Gimbrone M. A., CREB binding protein is a required coactivator for Smad-dependent, transforming growth factor β transcriptional responses in endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 95, 9506–9511 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen W., Lam S. S., Srinath H., Schiffer C. A., Royer W. E., Lin K., Competition between Ski and CREB-binding protein for binding to Smad proteins in transforming growth factor-β signaling. J. Biol. Chem. 282, 11365–11376 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Kon N., Hirota T., Kawamoto T., Kato Y., Tsubota T., Fukada Y., Activation of TGF-β/activin signalling resets the circadian clock through rapid induction of Dec1 transcripts. Nat. Cell Biol. 10, 1463–1469 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Sloin H. E., Ruggiero G., Rubinstein A., Smadja Storz S., Foulkes N. S., Gothilf Y., Interactions between the circadian clock and TGF-β signaling pathway in zebrafish. PLOS ONE 13, e0199777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonze D., Bernard S., Waltermann C., Kramer A., Herzel H., Spontaneous synchronization of coupled circadian oscillators. Biophys. J. 89, 120–129 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen W.-D., Yeh J.-K., Peng M.-T., Shie S.-S., Lin S.-L., Yang C.-H., Chen T.-H., Hung K.-C., Wang C.-C., Hsieh I.-C., Wen M.-S., Wang C.-Y., Circadian CLOCK mediates activation of transforming growth factor-β signaling and renal fibrosis through cyclooxygenase 2. Am. J. Pathol. 185, 3152–3163 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Sato F., Sato H., Jin D., Bhawal U. K., Wu Y., Noshiro M., Kawamoto T., Fujimoto K., Seino H., Morohashi S., Kato Y., Kijima H., Smad3 and snail show circadian expression in human gingival fibroblasts, human mesenchymal stem cell, and in mouse liver. Biochem. Biophys. Res. Commun. 419, 441–446 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Brancaccio M., Edwards M. D., Patton A. P., Smyllie N. J., Chesham J. E., Maywood E. S., Hastings M. H., Cell-autonomous clock of astrocytes drives circadian behavior in mammals. Science 363, 187–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Myung J., Schmal C., Hong S., Tsukizawa Y., Rose P., Zhang Y., Holtzman M. J., De Schutter E., Herzel H., Bordyugov G., Takumi T., The choroid plexus is an important circadian clock component. Nat. Commun. 9, 1062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crosby P., Hamnett R., Putker M., Hoyle N. P., Reed M., Karam C. J., Maywood E. S., Stangherlin A., Chesham J. E., Hayter E. A., Rosenbrier-Ribeiro L., Newham P., Clevers H., Bechtold D. A., O’Neill J. S., Insulin/IGF-1 drives PERIOD synthesis to entrain circadian rhythms with feeding time. Cell 177, 896–909.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown S. A., Zumbrunn G., Fleury-Olela F., Preitner N., Schibler U., Rhythms of mammalian body temperature can sustain peripheral circadian clocks. Curr. Biol. 12, 1574–1583 (2002). [DOI] [PubMed] [Google Scholar]
- 66.Balsalobre A., Brown S. A., Marcacci L., Tronche F., Kellendonk C., Reichardt H. M., Schütz G., Schibler U., Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 289, 2344–2347 (2000). [DOI] [PubMed] [Google Scholar]
- 67.Yamajuku D., Inagaki T., Haruma T., Okubo S., Kataoka Y., Kobayashi S., Ikegami K., Laurent T., Kojima T., Noutomi K., Hashimoto S., Oda H., Real-time monitoring in three-dimensional hepatocytes reveals that insulin acts as a synchronizer for liver clock. Sci. Rep. 2, 439 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colak S., ten Dijke P., Targeting TGF-β signaling in cancer. Trends Cancer 3, 56–71 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Akhurst R. J., TGFβ signaling in health and disease. Nat. Genet. 36, 790–792 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Shafi A. A., Knudsen K. E., Cancer and the circadian clock. Cancer Res. 79, 3806–3814 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sulli G., Lam M. T. Y., Panda S., Interplay between circadian clock and cancer: New frontiers for cancer treatment. Trends Cancer 5, 475–494 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z., GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.B. Maier, S. Lorenzen, A.-M. Finger, H. Herzel, A. Kramer, Searching novel clock genes using RNAi-based screening, in Methods in Molecular Biology (Clifton, N.J.), S. A. Brown, Ed. (Humana, 2021), pp. 103–114; 10.1007/978-1-0716-0381-9_8. [DOI] [PubMed]
- 74.Reyes J., Chen J.-Y., Stewart-Ornstein J., Karhohs K. W., Mock C. S., Lahav G., Fluctuations in p53 signaling allow escape from cell-cycle arrest. Mol. Cell 71, 581–591.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Broutier L., Andersson-Rolf A., Hindley C. J., Boj S. F., Clevers H., Koo B.-K., Huch M., Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 11, 1724–1743 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Zhang R., Lahens N. F., Ballance H. I., Hughes M. E., Hogenesch J. B., A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. U.S.A. 111, 16219–16224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hughes M. E., DiTacchio L., Hayes K. R., Vollmers C., Pulivarthy S., Baggs J. E., Panda S., Hogenesch J. B., Harmonics of circadian gene transcription in mammals. PLOS Genet. 5, e1000442 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weger B. D., Gobet C., David F. P. A., Atger F., Martin E., Phillips N. E., Charpagne A., Weger M., Naef F., Gachon F., Systematic analysis of differential rhythmic liver gene expression mediated by the circadian clock and feeding rhythms. Proc. Natl. Acad. Sci. U.S.A. 118, e2015803118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Y., Song L., Liu M., Ge R., Zhou Q., Liu W., Li R., Qie J., Zhen B., Wang Y., He F., Qin J., Ding C., A proteomics landscape of circadian clock in mouse liver. Nat. Commun. 9, 1553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mure L. S., Le H. D., Benegiamo G., Chang M. W., Rios L., Jillani N., Ngotho M., Kariuki T., Dkhissi-Benyahya O., Cooper H. M., Panda S., Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 359, eaao0318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.M. D. Ruben, G. Wu, D. F. Smith, R. E. Schmidt, L. J. Francey, R. C. Anafi, J. B. Hogenesch, A population-based human enCYCLOPedia for circadian medicine. bioRxiv 301580 [Preprint]. 14 April 2018. 10.1101/301580. [DOI]
- 82.Li Y., Li G., Wang H., Du J., Yan J., Analysis of a gene regulatory cascade mediating circadian rhythm in zebrafish. PLOS Comput. Biol. 9, e1002940 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Robles M. S., Cox J., Mann M., In-Vivo quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. PLOS Genet. 10, e1004047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mauvoisin D., Wang J., Jouffe C., Martin E., Atger F., Waridel P., Quadroni M., Gachon F., Naef F., Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. Proc. Natl. Acad. Sci. U.S.A. 111, 167–172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang J., Mauvoisin D., Martin E., Atger F., Galindo A. N., Dayon L., Sizzano F., Palini A., Kussmann M., Waridel P., Quadroni M., Dulić V., Naef F., Gachon F., Nuclear proteomics uncovers diurnal regulatory landscapes in mouse liver. Cell Metab. 25, 102–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Robles M. S., Humphrey S. J., Mann M., Phosphorylation is a central mechanism for circadian control of metabolism and physiology. Cell Metab. 25, 118–127 (2017). [DOI] [PubMed] [Google Scholar]
- 87.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/30/eabg5174/DC1