

Abstract
Physiologically based pharmacokinetic (PBPK) models have been proposed as a tool for more accurate individual pharmacokinetic (PK) predictions and model‐informed precision dosing, but their application in clinical practice is still rare. This study systematically assesses the benefit of using individual patient information to improve PK predictions. A PBPK model of caffeine was stepwise personalized by using individual data on (1) demography, (2) physiology, and (3) cytochrome P450 (CYP) 1A2 phenotype of 48 healthy volunteers participating in a single‐dose clinical study. Model performance was benchmarked against a caffeine base model simulated with parameters of an average individual. In the first step, virtual twins were generated based on the study subjects' demography (height, weight, age, sex), which implicated the rescaling of average organ volumes and blood flows. The accuracy of PK simulations improved compared with the base model. The percentage of predictions within 0.8‐fold to 1.25‐fold of the observed values increased from 45.8% (base model) to 57.8% (Step 1). However, setting physiological parameters (liver blood flow determined by magnetic resonance imaging, glomerular filtration rate, hematocrit) to measured values in the second step did not further improve the simulation result (59.1% in the 1.25‐fold range). In the third step, virtual twins matching individual demography, physiology, and CYP1A2 activity considerably improved the simulation results. The percentage of data within the 1.25‐fold range was 66.15%. This case study shows that individual PK profiles can be predicted more accurately by considering individual attributes and that personalized PBPK models could be a valuable tool for model‐informed precision dosing approaches in the future.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Physiologically based pharmacokinetic (PBPK) models have been proposed as a tool for more accurate individual pharmacokinetic predictions and model‐informed precision dosing, but a systematic investigation is still missing.
WHAT QUESTION DID THIS STUDY ADDRESS?
Using caffeine as an exemplary use case, we analyzed to which extent individual data on demography, physiology, and enzyme activity improved the accuracy of personalized PBPK predictions.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Personalized pharmacokinetic predictions for caffeine improved if PBPK models were generated based on individual demography and cytochrome P450 1A2 phenotyping.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
In the future, PBPK modeling may provide a platform for model‐informed precision dosing to improve personalized drug treatment.
INTRODUCTION
Although everyone is different, therapeutic drug treatment still mostly follows the one‐size‐fits‐all paradigm. 1 , 2 Treatment regimens are developed for average individuals, but obviously only a small fraction of the population is well represented by the assumption of such an average individual. 3
Precision dosing is a novel concept in clinical pharmacology that aims to achieve the best drug response by finding the best dose for every patient. To optimally modulate target activity, the right amount of drug molecules has to reach the drug target at the right time. Numerous factors influence drug pharmacokinetics (PK) in patients and thus their pharmacodynamic (PD) response. The most important factors influencing drug PK are demography, physiology, genetic makeup, lifestyle, comorbidities, comedication, and patient compliance. Understanding the contribution of individual patient characteristics to absorption, distribution, metabolism, and excretion (ADME) processes to PK is essential to achieving an optimal dosing design.
Model‐informed precision dosing (MIPD) is a pharmacometric concept that employs mathematical models to predict optimal doses for individual patients. MIPD is currently only applied to a few drugs that are subject to therapeutic drug monitoring and for which appropriate PK models are available. 4 It is a reasonable expectation that physiologically based PK (PBPK) models may contribute to MIPD in the future because PBPK models have a mechanistic basis and describe ADME processes in the body at a large level of physiological detail. 5 , 6 , 7 , 8 , 9 To date, PBPK models are already routinely applied in drug development to study PK in special populations 10 , 11 , 12 or drug–drug interactions in virtual clinical trials. 13 , 14
The structure of PBPK models is ideally suited for the integration of heterogeneous patient data because most parameters correspond to observable quantities such as blood flow rates or organ volumes. Generic PBPK models of average individuals rely on population average data for the organism, but individual measurements can easily replace these parameters. Therefore, PBPK models provide an excellent framework for personalized PK predictions. 15
In this study, we exemplarily employed a PBPK model of caffeine to assess the benefit of individual data on the accuracy of personalized PK simulations in a cohort of healthy volunteers. Caffeine is a widely consumed neurostimulant, and its PK is well understood. 16 , 17 The compound is almost completely bioavailable and is mainly metabolized by cytochrome P450 (CYP) 1A2 in the liver, with paraxanthine being the major metabolite. Only about 0.4%–3.7% of the dose are excreted unchanged to urine. 18 , 19 There are no clinically relevant CYP1A2 genotypic differences, but CYP1A2 is inducible by lifestyle factors such as oral contraception or smoking. 20 It has been proposed to use the paraxanthine/caffeine ratio ( as a measure for CYP1A2 activity. 6 , 21 , 22
We here systematically evaluated to what extent individual data about demography, physiology, and CYP phenotype improve the accuracy of personalized PBPK predictions of caffeine PK. A specifically designed clinical study had been conducted to acquire relevant attributes of study participants along with PK measurements for PBPK model personalization. 23
METHODS
Data collection
Caffeine PK was investigated in an open‐label, single‐dose PK study conducted in healthy volunteers. The study was approved by the ethics committee of the University of Tübingen and the German Federal Drug Administration. It was registered at ClinicalTrials.gov (NCT01788254) and in the European clinical trials database (EudraCT 2011‐002291‐16). All volunteers gave their written informed consent before participation in the study. The study participants received a single dose of 50 mg (trimethyl‐13C3) caffeine (99% 13C; Cambridge Isotope Laboratories) provided in a gelatin capsule. The 13C caffeine was administered orally in a cocktail together with 1 mg midazolam, 0.25 mg torsemide, 2.5 mg talinolol, 5 mg pravastatin, and 5 mg codeine. 24 13C3‐caffeine and 13C2‐paraxanthine in plasma were quantified by liquid chromatography‐tandem mass spectrometry as described. 24 Additional blood samples were taken to determine the hematocrit and the glomerular filtration rate (GFR).
Hepatic blood flows were determined by magnetic resonance imaging (MRI). Electrocardiographically gated MRI was performed on a 1.5‐T Siemens Aera scanner using commercially available software and dedicated surface coils. Multislice TrueFISP images were acquired in three orthogonal planes to locate the liver and vessels. Subsequently, blood flow measurements were performed in the portal vein and hepatic artery using through‐plane velocity‐encoded (VENC) phase‐contrast MRI (PC‐MRI). VENC limits were set individually to 50–150 cm/s. Each PC‐MRI measurement was acquired during a single 15–20‐second breath hold. For each vessel, a region of interest was drawn to estimate the mean vessel lumen cross‐sectional area and mean velocity across the cardiac cycle. Vessel flow was calculated as the product of the cross‐sectional area and velocity of the respective vessel for each cardiac phase.
The total number of subjects included in the clinical study was 103. However, GFR and MRI measurements of the liver blood flow were only available for 48 subjects. Therefore, the full PBPK model personalization workflow could only be performed for 48 subjects.
PBPK modeling
PBPK modeling was carried out with PK‐Sim, which is part of the Open Systems Pharmacology suite (Version 8.0, www.open‐systems‐pharmacology.org). 25 Partition coefficients were automatically calculated based on physicochemical properties with a previously described distribution model. 26 Base model parameters (specific CYP1A2 clearance [Cl] and intestinal permeability) were optimized with the built‐in Levenberg‐Marquardt parameter identification method (randomized start values, 20 runs, default settings). Details of the caffeine base model development are described in the supplement.
Local sensitivities of the model to personalized parameters (e.g., liver blood flow) were calculated with the built‐in sensitivity analysis in PK‐Sim. The sensitivity of the model toward height, weight, and sex was calculated with customized MATLAB scripts (MathWorks, Version 2018b) using the MoBi® Toolbox for MATLAB. The variation range was 0.1 for all parameters except for sex. The base model was also simulated for a female individual with the same demography to calculate changes of PK parameters of the female in relation to the male reference individual.
PBPK model evaluation
Different metrics were calculated to evaluate the performance of the model personalization: the root mean square error (RMSE), the percentage of predicted concentrations that fell within the 1.25‐fold and twofold range of the observed data, and the geometric mean fold error (GMFE) of PK parameters.
Geometric mean fold error (GMFE)
The GMFE is a measure to compare observed and predicted PK parameters. The PK parameters of Cl, volume of distribution (V d), and area under the curve (AUC) were calculated by noncompartmental analysis (NCA). Cl and V d depend on the terminal rate constant lambda estimated by the “Rsquare” method. A set of linear regressions was performed on log‐transformed concentration data for the last n (n = 3, 4, 5, …) data points before the maximum observed concentration (Cmax). Lambda was calculated from the regression with the highest adjusted R 2.
The GMFE is calculated as follows:
| (1) |
where PK parameter refers to the AUC until the last measurement (AUCt0−last) or the V d, n denotes the number of observations, and i the i th observation and corresponding model prediction. A GMFE below 2 indicates a good prediction of the PK parameters.
PBPK model personalization
Caffeine PBPK models were personalized by setting the organism properties of the PBPK model to the observed volunteer attribute in the clinical study. These PBPK model instances are called virtual twins because they represent the respective study subject in the model simulation. PBPK model personalization was carried out in MATLAB (The MathWorks, Version 2018b) using the MoBi® Toolbox for MATLAB.
The base model simulated the caffeine PK for an average individual (male, 30 years old, 1.76 m tall, and weighing 73 kg) and had been fitted to the mean caffeine plasma concentration of the entire study population (n = 103).
PK‐relevant virtual twin attributes were grouped into 3 different classes: (1) demography, (2) physiology, and (3) CYP activity. Figure 1 shows the workflow for the PBPK model personalization. Starting from a base model, information on study subject demography (Step 1), physiology (Step 2), and CYP activity (Step 3) was successively included. In addition to the workflow, the effect of integrating either only physiological information (Additional Analysis 1) or only CYP phenotyping (Additional Analysis 2) was simulated. Furthermore, specific Cl of virtual twins from Step 1 was fitted to the data to benchmark how good the individual model predictions could maximally get (Additional Analysis 3).
FIGURE 1.
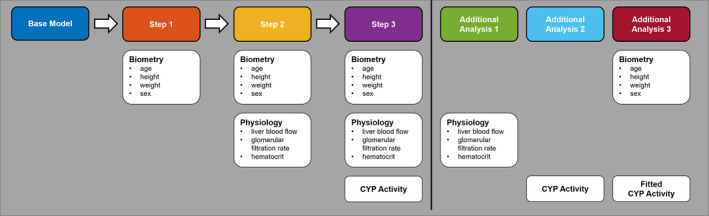
Physiologically based pharmacokinetic model personalization workflow. Colored boxes indicate the names of the analyses. White boxes below indicate the respective information that was used to generate a personalized prediction. CYP, cytochrome P450
Step 1: personalization of demography
Virtual twins with matching ethnicity, age, weight, height, and sex of the study subjects were generated with the function “PKSimCreateIndividual(),” which is part of the MATLAB toolbox for MoBi and PK‐Sim. In consequence, organ volumes and blood flows in the PBPK model were accordingly scaled based on the height and weight of the respective individuals, as described previously. 27 Ethnicity was Caucasian for all subjects.
Step 2: personalization of physiology
Virtual twin physiology was matched to the observed physiological data by adapting the liver blood flows, the GFR, and the hematocrit level.
Personalization of liver blood flow
Blood flows (Q) of virtual twins were rescaled to match the measured liver blood flows. The specific liver blood flow parameter was adjusted so that the liver blood flow rate equaled the measured blood flow in the hepatic artery. In the PBPK model, the portal vein blood flow is defined as a formula and equals the sum of the blood flows through the gastrointestinal (GI) organs (stomach, small intestine, large intestine, pancreas, and spleen). The ratio of the portal vein blood flow in the data and the model (Equation 2) was used as a scaling factor to adjust the blood flows of the GI organs (Equation 3) and the portal vein (Equation 4). Blood flows of the other organs were not changed except for the lung blood flow, which equals the cardiac output (Equation 5).
| (2) |
| (3) |
| (4) |
| (5) |
Personalization of GFR
The PK‐Sim PBPK model required a specific GFR (ml/min/g kidney tissue) as an input. The observed GFR was calculated from the cystatin C plasma concentration with the caucasian asian pediatric adult equation. 28 The observed kidney weight was unknown. Therefore, the respective virtual twin's kidney weight was used for the calculation of the specific GFR as shown in Equation (6). A few individuals had GFRs outside the reference range, but measurements were considered clinically not relevant and included in the study.
| (6) |
Step 3: CYP1A2 phenotyping
There is no method to measure each volunteer's CYP1A2 activity for model parameterization directly, but the in blood plasma correlates with CYP1A2 activity. 21 We here used the blood plasma (Equation 7) of a single point concentration (C) measurement 4 h after drug administration to phenotype CYP1A2 activity.
| (7) |
The observed was subsequently translated to the model CYP1A2 activity parameter‐specific Cl (Clspec). First, the study population mean of the was calculated, which corresponds to the Clspec of the base model. Then a Cl scale factor was calculated for every individual i based on the (Equation 8) and multiplied with the base model enzyme activity to get the individualized Cl (Equation 9).
| (8) |
| (9) |
RESULTS
To systematically test how the accuracy of PBPK predictions improved by adding knowledge about individuals, a workflow for PBPK model personalization was developed (Figure 1).
The workflow started with a base model for caffeine, which represented an average individual. In three consecutive steps, the caffeine PBPK model was then informed with individual data on demography, physiology, and CYP1A2 activity.
Base model
The base model served as a benchmark for evaluating the PBPK personalization workflow, and all individual caffeine PK measurements were compared with the base model prediction. Most of the observed data fell within the twofold range (92.71%), whereas only a smaller fraction of the data fell within the 1.25‐fold range (45.83%). The RMSE was 1.28 (Table 1 ). In consequence, the visual predictive check (Figure 2a) shows that some individual PK profiles were not described very well with the base model. In the following, we investigated whether the integration of study subject data in personalized PBPK models improved the accuracy of the PK simulations.
TABLE 1.
Caffeine physiologically based pharmacokinetic model evaluation metrics
| Model Scenario | Biometry | Physiology | CYP activity | Percentage in 1.25‐fold range | Percentage in twofold range | GMFE AUC | GMFE V d | RMSE |
|---|---|---|---|---|---|---|---|---|
| Benchmark | ||||||||
| Base model | 45.83 | 92.71 | 1.31 | 1.21 | 1.28 | |||
| Workflow | ||||||||
| Step 1 | X | 57.81 | 93.75 | 1.24 | 1.14 | 1.08 | ||
| Step 2 | X | X | 59.11 | 93.49 | 1.24 | 1.12 | 1.07 | |
| Step 3 | X | X | X | 66.15 | 97.40 | 1.16 | 1.12 | 0.97 |
| Additional analysis | ||||||||
| Additional Analysis 1 | X | 47.14 | 92.45 | 1.31 | 1.21 | 1.30 | ||
| Additional Analysis 2 | X | 64.84 | 97.66 | 1.17 | 1.18 | 1.05 | ||
| Additional Analysis 3 | X | Fit | 89.06 | 98.44 | 1.04 | 1.11 | 0.79 | |
X indicates whether the information was used in the respective step or additional analysis (see also Figure 1).
ABBREVIATIONS: AUC, area under the curve; CYP, cytochrome P450; GMFE, geometric mean fold error; RMSE, root mean square error; V d, volume of distribution.
FIGURE 2.
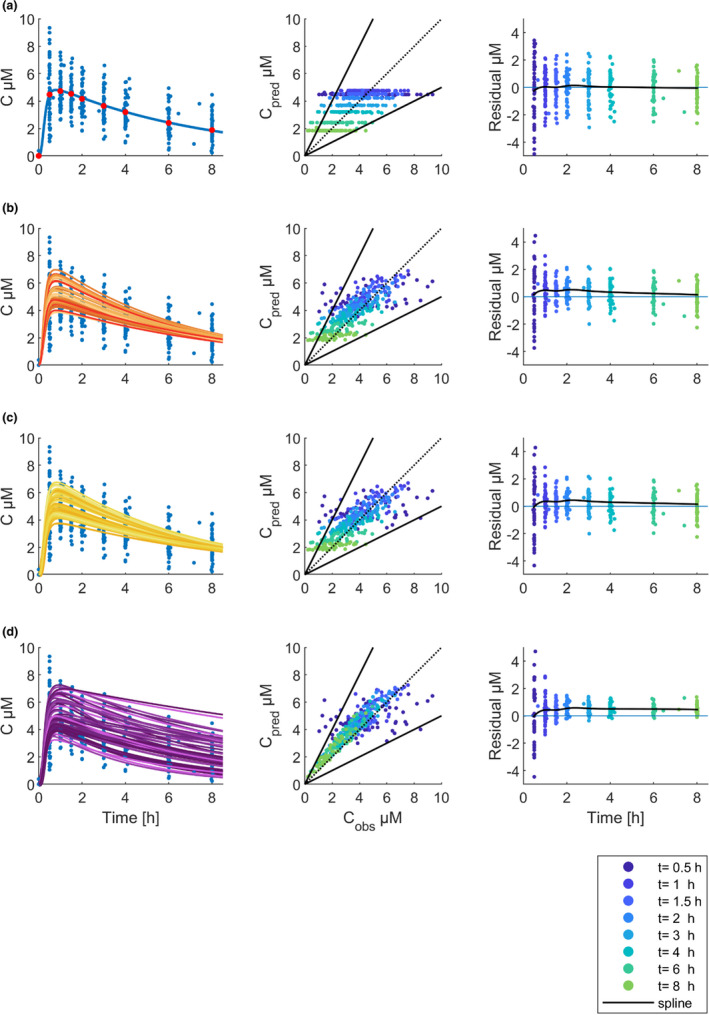
Basic goodness‐of‐fit plots. Rows show the result of the execution of the workflow steps: (a) Base model, (b) Step 1, (c) Step 2, (d) Step 3. Visual predictive check (left column): blue dots show observed caffeine plasma concentrations (C µM), and red dots show the mean of the observed plasma concentrations. The lines display the caffeine physiologically based pharmacokinetic model simulations for the respective cohort of virtual twins. Predicted versus observed concentrations (middle column): dots show the predicted concentrations that were plotted against the observed concentrations. The color of the dots corresponds to the timepoint of the measurement. The dashed black line is the line of unity; solid black lines indicate the twofold range. Residuals versus time (right column): dots show the residuals. The blue line marks the zero line. The black line is a cubic spline through the mean of the residuals
Simulation of virtual twins
Step 1: demography
Demographic information was included in the first step of the PBPK model personalization. The generation of virtual twins based on demographic information implied that organ volumes and blood flows were allometrically scaled and adjusted accordingly in the PBPK model. The mean of the observed demographic data (31.5 years old, 1.76 m, 73.4 kg) was equivalent to the demographics of the PK‐Sim European reference individual in the base model (30 years old, 1.76 m, 73 kg; Figure 3).
FIGURE 3.
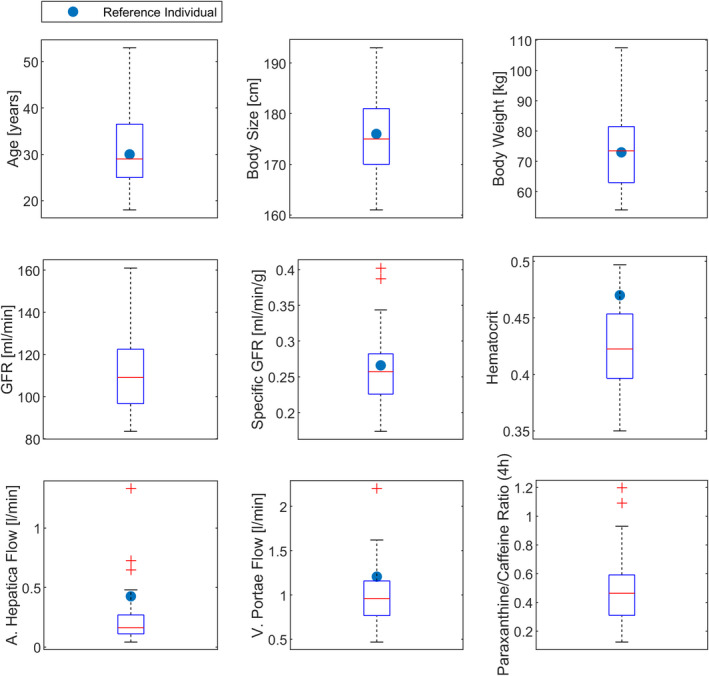
Boxplots of observed parameter values for demographic parameters, physiological parameters, and the paraxanthine/caffeine ratio. Blue dots depict the parameter values of the reference individual if applicable. GFR, glomerular filtration rate
Compared with the base model, the percentage of data in the twofold range increased from 92.71% to 93.75%, the percentage of data in the 1.25‐fold range increased from 45.83% to 57.81%, and the RMSE decreased from 1.28 to 1.08 (Table 1 ). Particularly, Cmax and the distribution phase were influenced by the changes introduced in Step 1 (Figure 2b). The GMFE of the V d was reduced from 1.21 to 1.14, and the boxplots of the PK parameter ratios show that the prediction of V d mainly improved in Step 1 by the inclusion of demographic data (Figure 4a). This agreed with the sensitivity analysis, which showed that Cmax in plasma was sensitive to body weight (−0.58) and height (−0.57). Sex affected the body composition in the PBPK model, and therefore exposure (AUCt0−last) and Cmax both increased by 16% when the sex of the reference individual was changed from male to female (Table S5). Age was not relevant for this cohort of healthy adults.
FIGURE 4.
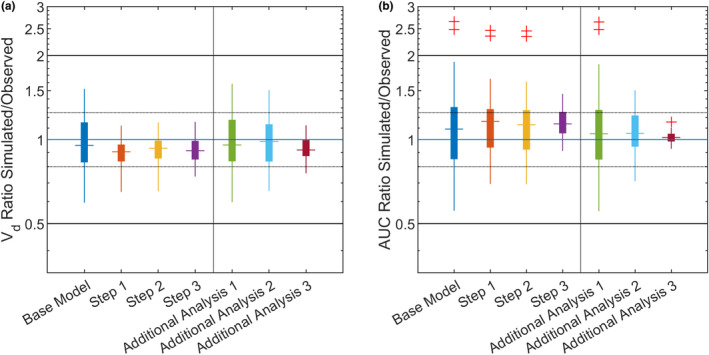
Predicted versus observed pharmacokinetic parameter ratios. Boxplots show the pharmacokinetic parameter ratios for the respective modeling step. Horizontal solid black lines indicate the twofold range; dotted lines indicate the 1.25‐fold range, and the blue line marks a ratio of 1. The vertical line separates the workflow from the additional analyses. (a) Volume of distribution, (b) area under the curve from the start until the last measurement (8 h). AUC, area under the curve
Step 2: physiology
In the second workflow step, physiological parameters (liver blood flow, GFR, and hematocrit) were personalized in addition to the demographic parameters. Although the median specific GFR of the study cohort was similar to the base model parameter, the median of the measured liver blood flows and of the hematocrit were lower (Figure 3).
The physiological information hardly changed the model prediction of the caffeine PBPK model (Figure 2c), likewise the model evaluation metrics were highly similar to the first step (Table 1 , Figure 2). The caffeine PBPK model was not sensitive to the physiological parameters hematocrit, GFR, and liver blood flow (Table S5). Simulation of virtual twins informed with physiological data but not with demographic data confirmed that liver blood flow, GFR, and hematocrit had only a minor influence on the model simulation (Additional Analysis 1; Figure S2a).
Step 3: CYP phenotyping
The third and final step was the personalization of the CYP1A2 activity by the . Compared with Step 2, the percentage of the data inside the twofold range increased from 93.49% to 97.40%, and the percentage of data in the 1.25‐fold range increased from 59.11% to 66.15%. The RMSE decreased from 1.07 to 0.97 (Table 1). Boxplots of the PK parameter ratios (Figure 4b) show that CYP1A2 phenotyping improved the exposure (AUCt0−last) prediction, including two subjects with a much higher Cl than the other study subjects. CYP1A2‐specific Cl was the most sensitive PBPK model parameter with respect to AUC (−0.97; Table S4). Simulation of virtual twins informed with CYP1A2 phenotype data alone also improved the PBPK model prediction (Additional Analysis 2; Table 1, Figure S2b).
Time courses of the paraxanthine/caffeine plasma ratios were calculated for the entire study cohort (n = 48; Figure 5b) to analyze CYP1A2 phenotyping by the metabolite/parent drug ratio. Notably, the ratios increased linearly over time, and the slope was characteristic for the study subjects: individuals with a high metabolite/parent drug ratio in the beginning also had a high metabolite/parent drug ratio in the end. A linear regression model revealed a strong correlation (R 2 = 0.86) of observed caffeine Cl (calculated by NCA) and the measured in blood plasma at 4 h (see Figure 5a). The correlations for the ratios at 2 h, 6 h, and 8 h were equally high for the ratio at 4 h (data not shown). The mean ratio of 48 subjects at 4 h was 0.48 (standard deviation, 0.23; minimum, 0.12; maximum, 1.20; Figure 3).
FIGURE 5.
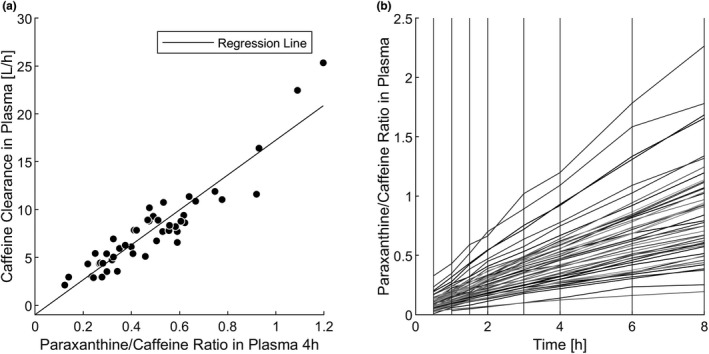
Correlation of observed clearances and paraxanthine/caffeine ratio. (a) Linear regression model with the paraxanthine/caffeine ratio in plasma at 4 h as the explanatory variable and the observed caffeine clearance in plasma as the response variable. (b) Individual time courses of paraxanthine/caffeine ratios in plasma
Best model predictions on individual level
For every study subject, the workflow step that predicted the observed plasma profile best was determined based on the RMSE (Figure S3). For 12 individuals, the base model was still considered the best prediction; an exemplary PK profile is shown in Figure 6a.
FIGURE 6.
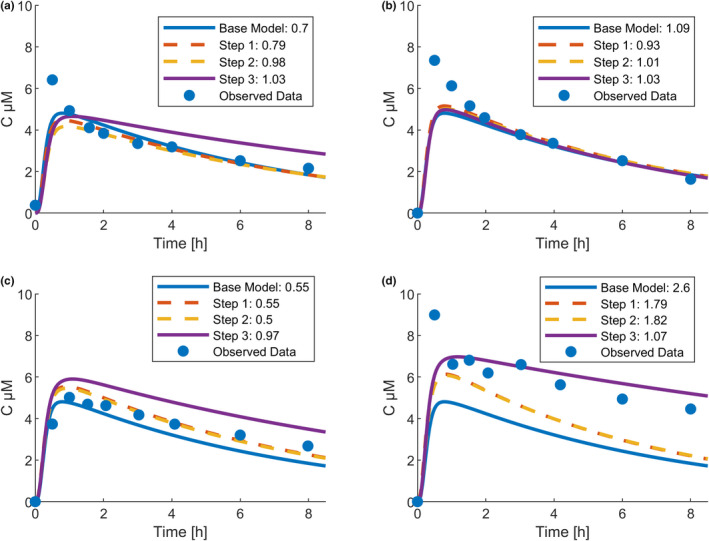
Representative individual caffeine pharmacokinetics. Line colors indicate the respective workflow step, and numeric values in the legend display the corresponding root mean square error: (a) best prediction by base model (male, 42 years old, 179 cm, 83 kg), (b) best prediction by Step 1 (male, 31 years old, 170 cm, 67 kg), (c) best prediction by Step 2 (female, 31 years old, 163 cm, 82 kg), and (d) best prediction by Step 3 (female, 47 years old, 167 cm, 72 kg)
Including only demographic information in Step 1 yielded the best prediction for three individuals, and the combination with physiological information in Step 2 performed best for 10 individuals. Exemplary PK profiles are shown in Figure 6b,c. The time courses of Step 1 and Step 2 are similar, which is in line with the finding that the model metrics hardly changed after the inclusion of physiological information (Table 1).
Model personalization Step 3, which included demographic, physiological, and CYP phenotyping data, yielded the best prediction for 23 of 48 individuals (48%). An exemplary profile is shown in Figure 6d.
DISCUSSION
PBPK models provide a detailed description of ADME processes in the human body and can accommodate patient data from different levels of biological organization. Therefore, personalized PBPK models can be expected to be ideally suited for MIPD. 5 , 6 , 7 , 8 , 9 We here systematically assessed the contribution of demographic, physiological, and CYP1A2 phenotype data to personalized PBPK predictions for caffeine.
Our analysis's key outcome was that consideration of subject‐specific data in personalized PBPK models might increase the accuracy of the PBPK predictions. Informing the caffeine PBPK model with demographic data (age, sex, height, weight) and CYP1A2 phenotype improved the model performance (Table 1). For the base model, 45.83% of the model predictions fell within the 1.25‐fold range. For Step 3 of the workflow, which included demographic, physiological, and CYP1A2 phenotype data, the percentage increased to 66.15%. Individual optimization of the drug Cl for every subject in the Additional Analysis 3 reached 89.06%. This benchmark demonstrates what is theoretically possible with the given model structure.
The second important finding of our study was that the extent of model improvement varied with the type of information included, that is, pharmacology of the compound determines the relevant patient attributes for model personalization. Because hepatic Cl of caffeine is enzyme limited and renal Cl is low, 16 , 17 it is not surprising that liver blood flow and kidney function had a negligible influence on the model simulation in our case. Still, this result also provides guidance for study planning in future clinical trials with different drugs because it highlights the relevance of pharmacology for data generation.
Complementary to an in‐depth analysis of a drug's pharmacology, sensitivity analysis can be used to identify the model parameters that have the biggest influence on the PBPK simulation result and for the personalization of the PBPK model. For caffeine, results of a sensitivity analysis (Table S4) agreed with the observations that demographic and CYP phenotyping information improved the model predictions, whereas physiological information did not.
In practice, the potential improvement of personalized PBPK predictions needs to be weighed against the clinical effort to gather the relevant patient‐specific data. Measurements of liver blood flow and the determination of the CYP1A2 phenotype are not routinely executed and come with economic as well as practical burdens. Therefore, their use for PBPK personalization is only indicated if a substantial improvement of the model accuracy can be expected given the pharmacology of the compound investigated.
Plasma protein binding was not considered for model personalization here because variations within the physiological range do not affect the fraction unbound of caffeine. 29 However, variation in plasma protein concentrations may influence PK for other compounds with a small fraction unbound and for patients with plasma protein concentrations outside the physiological range. 30 Under such conditions, it is important to consider plasma protein concentrations for personalized PK modeling.
Interindividual variability of absorption‐related processes was not considered in our study because all volunteers received the drug cocktail under controlled conditions. Nevertheless, absorption varied between volunteers and in consequence, model predictions missed the observed Cmax for volunteers with an early Cmax around 30 min after administration (Figure 6a,b,d). Late caffeine concentrations are well described by the model predictions even if the Cmax was not. The PBPK model structure would allow for personalization of absorption processes, but informing relevant parameters requires PK data from a crossover study with intravenous and oral administrations.
Quantification of the enzymatic Cl activity was essential for our model personalization approach. However, using a metabolite/drug ratio for enzyme activity phenotyping cannot readily be transferred to other substances because it is only established for a few other compounds. 31 Estimating enzyme activity based on hepatic expression is not suitable for personalized PBPK models because it would require liver biopsies. Endogenous markers of ADME enzyme and transporter activities for personalized PBPK models have been discussed, and plasma exosomes seem to be especially promising. 9 , 32 , 33 , 34
In addition, the intraindividual variability of the measured parameters should be considered. The height of the study subjects or organ weights are relatively stable over long periods, whereas metabolic enzyme activity or organ blood flows are subject to adaptations. When the drug cocktail was administered a second time to a subset of the study subjects, the at 4 h showed a good correlation with the ratio of the first administration (100% of the data within the twofold range, 44.4% of the data within the 1.25‐fold range; Figure S4). Liver blood flows were measured at an additional visit at the study center, not during the day of drug cocktail administration. Similar to most other organ blood flows, liver blood flows are regulated by different mechanisms and vary depending on, for example, posture or exercise. 35 If liver perfusion is of interest in future studies, blood flows and drug plasma concentrations should be measured simultaneously.
The potential power of personalized PBPK models lies in scenarios where extrapolation to unknown conditions is required, for example, for patients with a rare drug‐metabolizing enzyme genotype in combination with a disease. Although population PK models are applied for therapeutic drug monitoring and dose adjustments, their utility is limited to the conditions for which they were established. Such empirical approaches rely on correlations of covariates with PK parameters, which makes the prediction for diseases that affect multiple PK processes at the same time very difficult due to the interactions of covariates.
In addition, PBPK models can be applied to learn about ADME processes for a drug and physiological characteristics of a specific individual. This knowledge can be applied to predict pharmacokinetics for another drug for the same individual. The applicability of such efforts has been successfully demonstrated. 36 This is attributed to the unique PBPK framework that allows such translational concepts in contrast to compartmental approaches.
Single point measurements of probe drugs such as caffeine could be used to specify the phenotype and to translate this knowledge to PBPK models of drugs with more complex metabolism. For example, the in saliva has been applied together with the CYP2C8 genotype to suggest individual dose adjustments for olanzapine. 6
A limitation of the presented approach regarding clinical applications is that the study cohort of healthy Caucasian volunteers aged 18–56 years old represented only a small part of the total population. PBPK models that account for young or old age, 37 impaired liver or kidney function, or pregnancy have been published. 10 , 11 , 12 , 36 A PBPK modeling framework has also been applied to translate PK in a diseased population from one drug to another. 36 Most of these PBPK models perform well for special populations but have not been applied to predict PK in individual patients. To expand personalized PBPK approaches from healthy volunteers to patients, a robust understanding of how diseases affect individual PK is needed. Finally, our work underlines the need for quantitative raw data for PBPK model building and evaluation.
We here showed that personalized PBPK predictions performed better than PBPK predictions, which assume an average person. Therefore, virtual twins that reflect the patient characteristics are a promising concept for MIPD. A future application of personalized PBPK models could be the generation of virtual twins from the patients’ electronic health records and subsequent model‐guided optimization of medication plans. Besides PK, MIPD strategies should also consider PD processes. The integration of personalized PBPK and PD models would be a big step toward the clinical pharmacology paradigm of treating every patient with the right drug and the right dose at the right time.
CONFLICT OF INTEREST
R.F., A.R.P.S., J.F.S., J.L., R.B., and L.K. were employed by Bayer AG during the manuscript's preparation and are potential stockholders of Bayer AG. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
R.F., L.K., and J.F.S. wrote the manuscript. R.F., U.H., R.K., J.L., J.F.S., M.S., and L.K. designed the research. R.F., U.H., E.S., A.Y., J.F.S., and L.K. performed the research. R.F., A.R.P.S., R.B., L.M.B., J.F.S., and L.K. analyzed the data.
Supporting information
Supinfo S1
Supinfo S2
ACKNOWLEDGEMENT
Open Access funding enabled and organized by Projekt DEAL.
Fendt R, Hofmann U, Schneider ARP, et al. Data‐driven personalization of a physiologically based pharmacokinetic model for caffeine: A systematic assessment. CPT Pharmacometrics Syst Pharmacol. 2021;10:782–793. 10.1002/psp4.12646
Matthias Schwab and Lars Kuepfer contributed equally to this article.
Funding information
This research was funded by the German Federal Ministry of Education and Research “Liver Systems Medicine (LiSyM)” Grants 031L0039 and 031L0037, the Horizon 2020‐PHC‐2015 Grant U‐PGx 668353, the Horizon 2020 Framework Programme EU‐STANDS4PM Grant 825843 and the Robert Bosch Stiftung (Stuttgart, Germany).
Contributor Information
Matthias Schwab, Email: matthias.schwab@ikp-stuttgart.de.
Lars Kuepfer, Email: lars.kuepfer@rwth-aachen.de.
REFERENCES
- 1. Lesko LJ, Schmidt S. Individualization of drug therapy: history, present state, and opportunities for the future. Clin Pharmacol Therap. 2012;92(4):458‐466. [DOI] [PubMed] [Google Scholar]
- 2. Schlender JF, Vozmediano V, Golden AG, et al. Current strategies to streamline pharmacotherapy for older adults. Eur J Pharmaceut Sci. 2018;111:432‐442. [DOI] [PubMed] [Google Scholar]
- 3. Leeder JS. Who believes they are "just average": informing the treatment of individual patients using population data. Clin Pharmacol Ther. 2019;106(5):939‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kantasiripitak W, Van Daele R, Gijsen M, Ferrante M, Spriet I, Dreesen E. Software tools for model‐informed precision dosing: how well do they satisfy the needs? Front Pharmacol. 2020;11:620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rowland M, Peck C, Tucker G. Physiologically‐based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45‐73. [DOI] [PubMed] [Google Scholar]
- 6. Polasek TM, Tucker GT, Sorich MJ, et al. Prediction of olanzapine exposure in individual patients using physiologically based pharmacokinetic modelling and simulation. Br J Clin Pharmacol. 2018;84(3):462‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Terrier J, Daali Y, Fontana P, Csajka C, Reny JL. Towards personalized antithrombotic treatments: focus on P2Y12 inhibitors and direct oral anticoagulants. Clin Pharmacokinet. 2019;58(12):1517‐1532. [DOI] [PubMed] [Google Scholar]
- 8. Tucker GT. Personalized drug dosage – closing the loop. Pharm Res. 2016;34(8):1539‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Polasek TM, Rostami‐Hodjegan A. Virtual twins: understanding the data required for model‐informed precision dosing. Clin Pharmacol Ther. 2020;107(4):742‐745. [DOI] [PubMed] [Google Scholar]
- 10. Edginton AN, Willmann S. Physiology‐based simulations of a pathological condition: prediction of pharmacokinetics in patients with liver cirrhosis. Clin Pharmacokinet. 2008;47(11):743‐752. [DOI] [PubMed] [Google Scholar]
- 11. Dallmann A, Ince I, Solodenko J, et al. Physiologically based pharmacokinetic modeling of renally cleared drugs in pregnant women. Clin Pharmacokinet. 2017;56(12):1525‐1541. [DOI] [PubMed] [Google Scholar]
- 12. Ince I, Solodenko J, Frechen S, et al. Predictive pediatric modeling and simulation using ontogeny information. J Clin Pharmacol. 2019;59(Suppl 1):S95‐S103. [DOI] [PubMed] [Google Scholar]
- 13. Britz H, Hanke N, Volz AK, et al. Physiologically‐based pharmacokinetic models for CYP1A2 drug‐drug interaction prediction: a modeling network of fluvoxamine, theophylline, affeine, rifampicin, and midazolam. CPT Pharmacomet Syst Pharmacol. 2019; 8(5): 296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanke N, Frechen S, Moj D, et al. PBPK models for CYP3A4 and P‐gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacomet Syst Pharmacol. 2018; 7(10): 647‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuepfer L, Niederalt C, Wendl T et al. Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacomet Syst Pharmacol 2016;5(10):516‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fredholm BB, Bättig K, Holmén J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. 1999;51(1):83‐133. [PubMed] [Google Scholar]
- 17. Nehlig A. Interindividual differences in caffeine metabolism and factors driving caffeine consumption. Pharmacol Rev. 2018;70(2):384‐411. [DOI] [PubMed] [Google Scholar]
- 18. Birkett DJ, Miners JO. Caffeine renal clearance and urine caffeine concentrations during steady state dosing. Implications for monitoring caffeine intake during sports events. Br J clin Pharmacol. 1991;31:405‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang‐Liu DD, Williams RL, Riegelman S. Disposition of caffeine and its metabolites in man. J Pharmacol Exp Therap. 1983;224(1):180‐185. [PubMed] [Google Scholar]
- 20. Tian DD, Natesan S, White JR Jr, Paine MF Effects of common CYP1A2 genotypes and other key factors on intraindividual variation in the caffeine metabolic ratio: an exploratory analysis. Clin Translat Sci 2019;12(1):39‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fuhr U, Rost KL. Simple and reliable CYP1A2 phenotyping by the paraxanthine/caffeine ratio in plasma and in saliva. Pharmacogenetics. 1994;4(3):109‐116. [DOI] [PubMed] [Google Scholar]
- 22. Fuhr U, Rost KL, Engelhardt R, et al. Evaluation of caffeine as a test drug for CYP1A2, NAT2 and CYP2E1 phenotyping in man by in vivo versus in vitro correlations. Pharmacogenetics. 1996;6(2):159‐176. [DOI] [PubMed] [Google Scholar]
- 23. Kuepfer L, Kerb R, Henney AM. Clinical translation in the virtual liver network. CPT Pharmacomet Syst Pharmacol 2014;3:e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matthaei J, Tzvetkov MV, Strube J, et al. Heritability of caffeine metabolism: environmental effects masking genetic effects on CYP1A2 activity but not on NAT2. Clin Pharmacol Ther. 2016;100(6):606‐616. [DOI] [PubMed] [Google Scholar]
- 25. Lippert J, Burghaus R, Edginton A et al. Open systems pharmacology community‐an open access, open source, open science approach to modeling and simulation in pharmaceutical sciences. CPT Pharmacomet Syst Pharmacol. 2019;8(12):878‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmitt W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol In Vitro. 2008;22(2):457‐467. [DOI] [PubMed] [Google Scholar]
- 27. Willmann S, Hohn K, Edginton A, et al. Development of a physiology‐based whole‐body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34(3):401‐431. [DOI] [PubMed] [Google Scholar]
- 28. Grubb A, Horio M, Hansson LO, et al. Generation of a new cystatin C‐based estimating equation for glomerular filtration rate by use of 7 assays standardized to the international calibrator. Clin Chem. 2014;60(7):974‐986. [DOI] [PubMed] [Google Scholar]
- 29. Blanchard J. Protein binding of caffeine in young and elderly males. J Pharm Sci. 1982;71(12):1415‐1418. [DOI] [PubMed] [Google Scholar]
- 30. Haouala A, Widmer N, Guidi M, et al. Prediction of free imatinib concentrations based on total plasma concentrations in patients with gastrointestinal stromal tumours. Br J Clin Pharmacol. 2013;75(4):1007‐1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fuhr U, Jetter A, Kirchheiner J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the "cocktail" approach. Clin Pharmacol Ther. 2007;81(2):270‐283. [DOI] [PubMed] [Google Scholar]
- 32. Rodrigues AD, Taskar KS, Kusuhara H, Sugiyama Y. Endogenous probes for drug transporters: balancing vision with reality. Clin Pharmacol Ther. 2018;103(3):434‐448. [DOI] [PubMed] [Google Scholar]
- 33. Rodrigues D, Rowland A. From endogenous compounds as biomarkers to plasma‐derived nanovesicles as liquid biopsy; has the golden age of translational pharmacokinetics‐absorption, distribution, metabolism, excretion‐drug–drug interaction science finally arrived? Clin Pharmacol Ther. 2019;105(6):1407‐1420. [DOI] [PubMed] [Google Scholar]
- 34. Rowland A, Ruanglertboon W, van Dyk M, et al. Plasma extracellular nanovesicle (exosome)‐derived biomarkers for drug metabolism pathways: a novel approach to characterize variability in drug exposure. Br J Clin Pharmacol. 2019;85(1):216‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Daneshmend TK, Jackson L, Roberts CJ. Physiological and pharmacological variability in estimated hepatic blood flow in man. Br J Clin Pharmacol. 1981;11(5):491‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krauss M, Hofmann U, Schafmayer C, et al. Translational learning from clinical studies predicts drug pharmacokinetics across patient populations. NPJ Syst Biol Applicat. 2017;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schlender J‐F, Meyer M, Thelen K, et al. Development of a whole‐body physiologically based pharmacokinetic approach to assess the pharmacokinetics of drugs in elderly individuals. Clin Pharmacokinet. 2016;55(12):1573‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supinfo S1
Supinfo S2