

Abstract
Staphylococcus epidermidis is a leading cause of hospital-acquired infections. Traditional antibiotics have significantly reduced efficacy against this pathogen due to its ability to form biofilms on abiotic surfaces and drug resistance. The accessory gene regulator (agr) quorum sensing system is directly involved in S. epidermidis pathogenesis. Activation of agr is achieved via binding of the autoinducing peptide (AIP) signal to the extracellular sensor domain of its cognate receptor, AgrC. Divergent evolution has given rise to four agr specificity groups in S. epidermidis defined by the unique AIP sequence used by each group (AIPs-I–IV), with observed cross-group activities. As agr agonism has been shown to reduce biofilm growth in S. epidermidis, the development of pan-group activators of the agr system is of interest as a potential anti-virulence strategy. To date, no synthetic compounds have been identified that are capable of appreciably activating the agr system of more than one specificity group of S. epidermidis or, to our knowledge, of any other of the Staphylococci. Here, we report the characterization of the structure-activity relationships (SARs) for agr agonism by S. epidermidis AIP-II and AIP-III, and the application of these new SAR data and those previously reported for AIP-I for the design and synthesis of the first multi-group agr agonists. These non-native peptides were capable of inducing the expression of critical biofilm dispersal agents (i.e., phenol soluble modulins) in cell-culture and represent new tools to study the role of quorum sensing in S. epidermidis infections.
Graphical Abstract

INTRODUCTION
The bacterium Staphylococcus epidermidis, previously thought to be an innocuous commensal that colonizes the skin and mucosal tissues, has been recognized recently as a leading cause of hospital-acquired, or nosocomial, infections.1–3 Coagulase-negative Staphylococci account for over a third of all hospital-acquired bloodstream infections in the US,4, 5 and S. epidermidis represents the dominant pathogen within this group due to its propensity to form robust biofilms on abiotic surfaces and its prevalence in the human microbiome.5, 6 These traits, in combination with the rise of antibiotic resistance, make treating S. epidermidis infections increasingly difficult.7, 8 Alternative approaches to combat the virulence of this pathogen would be advantageous to public health.
Pathogenesis in S. epidermidis is largely under the regulation of quorum sensing (QS), a chemical-based cell-cell communication system, which makes QS an attractive target to potentially attenuate virulence phenotypes in this bacterium.9–13 QS activation can increase S. epidermidis’ capability for skin colonization,6 evasion of the host immune system,14 and tissue infiltration.15, 16 In contrast, QS inhibition in S. epidermidis has been shown to allow for more resilient biofilm formation, shielding S. epidermidis from antibiotics and the immune system.6, 15, 17 Non-native small molecules and peptides capable of either inhibiting or activating QS have been developed in other bacteria, and have shown significant value as research tools and as potential anti-virulence agents.18–21 While there are merits to either intentionally activating or inhibiting QS in order to curb S. epidermidis infections, given that the primary mechanism by which S. epidermidis causes device-associated infections is through biofilm formation,1, 3, 22 this connection suggests that the development of chemical tools to agonize QS and thereby promote biofilm dispersion may provide the most benefit in reducing S. epidermidis growth on surfaces and rendering them more vulnerable to treatment with antibiotics or clearance via the host immune system.15, 23 Studies in S. aureus, which utilizes a similar QS system to regulate biofilm formation, have demonstrated that activation of QS through addition of agonists dispersed biofilms, significantly increasing their susceptibility to antibiotics relative to untreated S. aureus biofilms.24 Identifying such QS agonists to combat S. epidermidis biofilms was a motivation for the current study.
Like other Staphylococci, S. epidermidis uses the accessory gene regulator (agr) system for QS (Figure 1), which consists of four protein components (AgrA–D) and a signaling molecule known as the autoinducing peptide (AIP).12, 25 The AIP sequence is contained within the propeptide AgrD that is processed in part by the membrane peptidase AgrB.26 In a manner still not well-understood, the processed AgrD is further modified and exported out of the cell as the mature AIP signal.11, 27 The AIP concentration in the local extracellular matrix increases with cell density; upon reaching a threshold concentration, the AIP effectively binds to its receptor, the transmembrane histidine kinase AgrC, and induces trans-autophosphorylation.28 Phosphorylated AgrC next activates the intracellular response regulator AgrA through phosphorelay. Thereafter, the activated AgrA dimerizes and binds to various promoters to regulate QS gene expression.28, 29 Binding to the P2 promoter upregulates the agr operon and completes the auto-induction circuit;30 binding to the P3 promoter upregulates RNAIII, which is the main effector molecule of agr;31 and binding to the PSM promoters upregulates genes for phenol-soluble modulins (PSMs), peptides that promote biofilm dispersal among other functions.32, 33
Figure 1.
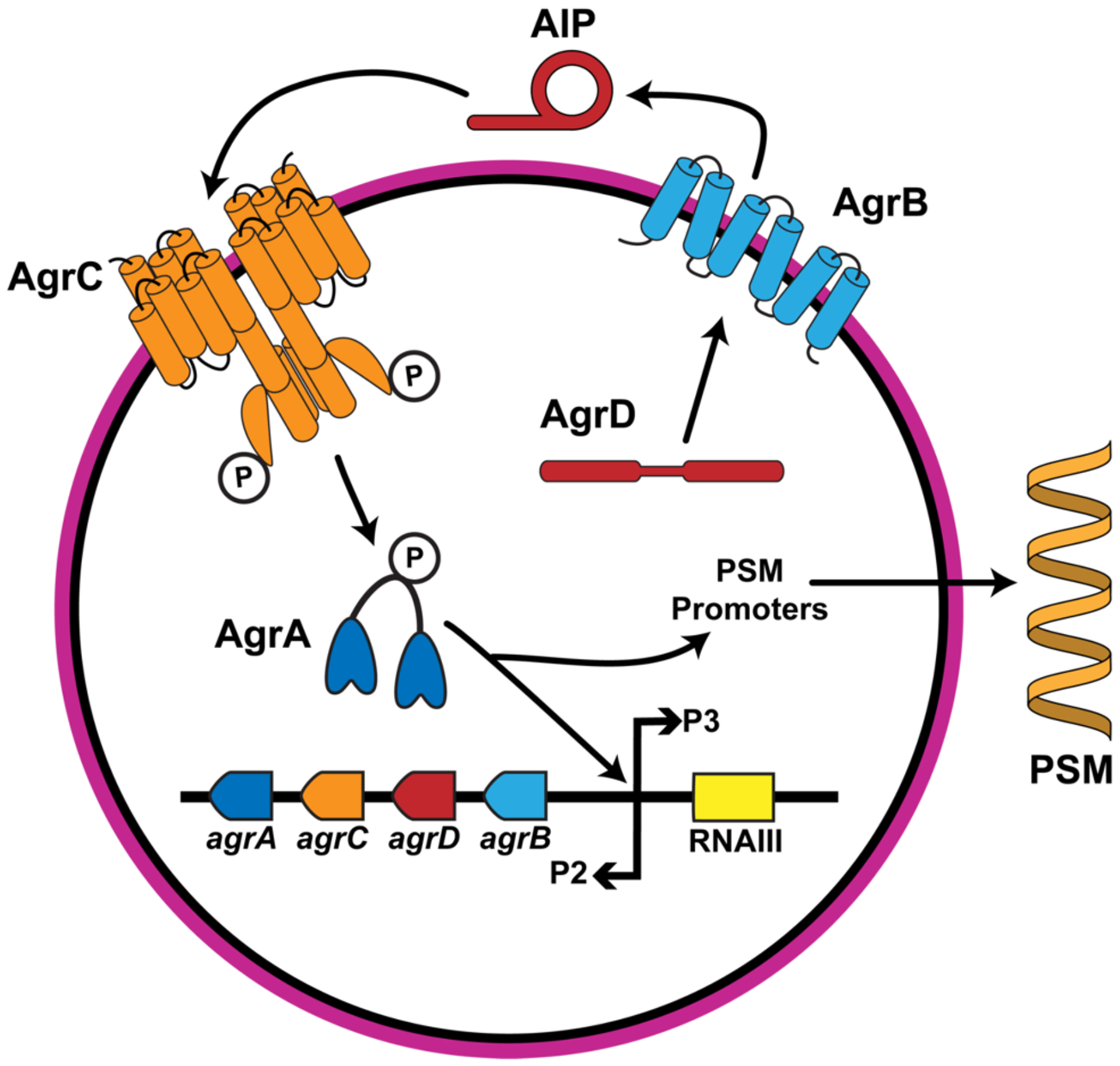
A simplified diagram of the agr QS system in S. epidermidis. The AIP signal is produced through processing of the AgrD polypeptide in part by AgrB and exported out of cell. AIPs bind to the AgrC receptor and induce phosphorylation and activation of the AgrA response regulator, upregulating expression of the agr operon, the RNAIII effector molecule, and production of secreted PSMs that lead to biofilm dispersal. Divergent evolution has led to four agr specificity groups with unique AIPs and agr machinery.
Similar to S. aureus,34 S. epidermidis has evolved into divergent agr specificity groups, where allelic variations of the agr machinery are tuned to respond to their own unique AIP signals. Despite attempts by multiple laboratories to quantify the abundance of different agr groups in human infections, consensus has not yet been reached because the distribution of groups varies substantially in these studies.2, 35–39 The S. epidermidis agr groups-I–III were identified over a decade ago,35 and very recently a new agr group (group-IV) was reported.38 As the latter group still awaits significant further characterization (such as confirmation of its native AIP structure) and appears to be rare, the work presented herein focuses on agr groups-I–III.
Interestingly, and again similar to S. aureus, the AIP signal from one S. epidermidis agr group can have cross-activity in another, “non-self” agr group, resulting in a network of possible agr interference (e.g., AIP-I activates its cognate receptor, AgrC-I, but inhibits both AgrC-II and -III).6, 35, 38 These cross-activity profiles raise interesting questions about the role of agr QS in mixed microbial environments, allowing one group to potentially outcompete another by blocking the non-self group QS system while simultaneously activating its own.40 However, in the development of chemical tools to combat S. epidermidis biofilms in native environments, the cross-activity among agr specificity groups complicates the use of any single native AIP or even a cocktail of the native AIPs to block biofilms without a priori knowledge of the agr specificity groups present in the sample/isolate.21 This challenge could be overcome with a “pan-group” agonist capable of activating all S. epidermidis agr groups. To our knowledge, no such pan-group agonist has been reported in the Staphylococci.11, 41
Our laboratory performed the first study of the structure-activity relationships (SARs) for agr agonism by S. epidermidis AIP-I,42 as well as a NMR structural study of key agonists and antagonists that we identified side-by-side with the native AIPs-I–III.43 These past studies suggested that a multi-group agonist could be designed based on two observations. First, agr activation in group-I S. epidermidis was found to be largely dependent on a single exocyclic residue in AIP-I that likely could be incorporated into new scaffolds.43 Second, AIP-II and AIP-III do not exhibit cross-group inhibition.6 In order to design multi-group agonists, however, we require further information beyond these observations; namely, we need to identify the key agonizing features for each cognate AIP–AgrC interaction in groups-I–III so we can attempt to combine these features to target all three major S. epidermidis groups effectively using one peptide scaffold.
Herein, we report the characterization of SARs for agr agonism by the native AIP-II and AIP-III signals and the combination of these SARs with those for AIP-I to design, synthesize, and identify the first multi-group agr agonists in S. epidermidis. These new peptides were active in cell-based reporters of agr activity and capable of modulating a key phenotype associated with biofilms, PSM production. This study provides powerful chemical tools to investigate the role of QS in S. epidermidis infections, examine interference between groups, and assess pan-group agr activation as a possible approach to prevent or clear biofilm.
RESULTS AND DISCUSSION
Overview of methods and analysis of reported SARs for agr agonism by group-I S. epidermidis AIP.
In our prior study, we determined key SARs for group-I agr agonism by AIP-I through synthesizing a set of analogs and assaying their agonism and antagonism profiles in a group-I S. epidermidis reporter strain (assumed to act via competitively binding AgrC-I due to their very close structural similarity to AIP-I).42, 43 The experimental methods and AIP-I features most pertinent to the current study are highlighted here (shown in Figure 2A; select assay data in Table 1). Briefly, we performed systematic alanine and D-amino acid scans of the AIP-I structure to ascertain the importance of each residue to agonistic activity (Cys4 was not mutated to Ala as it is required for macrocycle formation). We also examined truncated analogs that lacked the exocyclic tail. Peptide activity was measured using a previously reported group-I S. epidermidis strain (AH3408) harboring a reporter plasmid encoding a gfp gene fused to the P3 promoter.6 Activation of the AgrC-I receptor in this strain leads to GFP production, which can be quantitated by fluorescence. AgrC-I agonism by AIP-I analogs was measured in the presence of a known AgrC-I inhibitor, the native S. epidermidis AIP-II. AgrC-I antagonism was measured by testing AIP-I analogs alone.
Figure 2.

Summary of SAR trends for activation of the three S. epidermidis AgrC receptors by their cognate AIPs. SAR trends for (A) AIP-I as determined in our previous report.42 SAR trends for (B) AIP-II and (C) AIP-III determined in the current study. Red: essential for receptor binding. Green: crucial for receptor activation. Blue: either inconsequential or detrimental to receptor binding and activation. Unshaded residues/motifs contribute to receptor activation but are tolerant to changes. (D) Cross-activity network of AIPs-I–III for three S. epidermidis agr specificity groups (groups-I–III) as determined in the current study; ⟞ indicates inhibition and ⟶ indicates activation.
Table 1.
Selected activity data for native S. epidermidis AIPs and key analogs from SAR studies in corresponding reporter strains.
| agr group tested | Compound | EC50 [nM]a | IC50 [nM]b |
|---|---|---|---|
| agr-I | AIP-I | 170 | |
| AIP-II | 9.64c | ||
| AIP-III | 34.3c | ||
| AIP-I V3A | 51.9c | ||
| AIP-I D1AS6A | 10.3c | ||
| AAA | 2.84c | ||
| agr-II | AIP-I | 13.9 | |
| AIP-II | 226 | ||
| AIP-III | 648 | ||
| AAA | 0.36c | ||
| AIP-II K4A | >2000 | ||
| AIP-II D-K4 | >2000 | ||
| AIP-II S9A | >2000 | ||
| AIP-II D-S9 | Inactived | ||
| AIP-II N10A | >2000 | ||
| AIP-II D-N10 | Inactived | ||
| AIP-II 9aa | 83.5 | ||
| agr-III | AIP-I | 2.13 | |
| AIP-II | n/ce | ||
| AIP-III | 71.8 | ||
| AAA | 0.87c | ||
| AIP-III K4A | >2000 | ||
| AIP-III D-K4 | 26.1 | ||
| AIP-III D-A9 | Inactived | ||
| AIP-III S10A | Inactived | ||
| AIP-III D-S10 | 145 | ||
| AIP-III 9aa | 28.9 | ||
Agonism and antagonism assay data obtained in S. epidermidis group-I–III reporter strains. See SI for assay details and full data sets.
Agonism and antagonism assay data obtained in S. epidermidis group-I–III reporter strains. See SI for assay details and full data sets.
Data reproduced from reference42.
Dose-response assays revealed no substantial agonism nor antagonism activity over concentration range tested.
Agonism dose-response curve did not converge over concentration range tested.
Using these assays, we identified the first exocyclic residue of AIP-I (Val3) to be absolutely critical to agonizing AgrC-I because any substitutions at this position eliminated all agonistic activity (Figure 2A, Table 1). Interestingly, an alanine substitution at this position did not affect the overall 3D conformation of the peptide relative to AIP-I, as determined by solution-phase NMR;43 however, this substitution converted the peptide into a potent antagonist, which implicated the branched side chain of Val3 as the principal structural element required for AgrC-I activation. Similarly, removal of the entire exocyclic tail yielded an analog with antagonistic activity. Analogous to the SARs ascertained for AIPs in S. aureus,44 AIP-I binding to AgrC-I appears to be mediated largely by endocyclic hydrophobic residues (i.e., Tyr7 and Phe8). The solution-phase NMR structures of AIP-I reveal a hydrophobic face formed by these residues that is oriented in the opposite direction of Val3, suggestive that the initial binding/recognition site on AgrC-I is relatively distanced from the contact site that engenders receptor activation. Finally, replacing either of the hydrophilic residues Asp1 and Ser6 with alanine increased the agonistic potency of the resulting peptides. Replacement of Ser6 with alanine enforced a β-turn conformation in the macrocycle not present in the native AIP (as revealed by NMR),43 which appears to play a key role in the observed increase in agonism potency. Using these SARs, we developed a second-generation agonist containing two non-native alanine residues (AIP-I D1AS6A) that displayed a ten-fold improvement in potency in AgrC-I relative to AIP-I (Table 1), as well as a potent multi-group antagonist containing three non-native alanine residues (AIP-I D1AV3AS6A, hereby referred to as AAA; Table 1).
SARs for agr agonism by group-II and group-III S. epidermidis AIPs.
We next gathered corresponding SAR data for AIP-II and AIP-III using similar residue scanning methods as for AIP-I. These sets of AIP-II and AIP-III analogs were synthesized using solid-phase peptide chemistry and purified by HPLC using procedures based on our established protocols (see SI).42, 45, 46 Each analog was then tested for its effects on the cognate AgrC receptors using previously reported S. epidermidis group-II and group-III fluorescence reporter strains (AH2673 and AH3409, respectively)6 and methods analogous to those for group-I introduced above. To measure AgrC agonism, we used our multi-group antagonist AAA to block the activation of AgrC-II and AgrC-III by the endogenously produced AIPs.
To start, we first evaluated the activities of the native S. epidermidis AIPs (AIP-I–III) in the group-II and group-III reporters (Table 1, Tables S1–S2), to compare our methods to a prior report by Olson et al.6 While the inhibitory activity of AIP-I against AgrC-II and AgrC-III (~70% and 90% inhibition at 10 μM, respectively) was in agreement with that past study, the group-II and group-III cross-activity differed (trends summarized in Figure 2D). Instead of showing inactivity,6 cross-group activation was observed by AIP-II in AgrC-III and AIP-III in AgrC-II (~40% and 180% activation at 10 μM, respectively). This difference is most likely the result of variances in the agonism assay protocol used in each study. The original report examined whether spent media from one S. epidermidis agr specificity group would affect agr activity in another agr specificity group using a fluorescent reporter as compared to controls without spent media.6 While compounds with strong agonistic or antagonistic activity could be characterized with this assay, compounds with partial agonism activity could be misinterpreted as inactive. Our agonism assay assesses the ability of compounds to agonize agr activity in the presence of a strong antagonist. Inactive or inhibitory compounds will result in no observable activity, while partial or full agonists will restore agr activity. As our assay can more clearly distinguish between inactive compounds and partial agonists relative to the prior study, we believe the observed cross-group activation more accurately defines the interactions between group-II and group-III. This cross-group activation is intriguing, as it suggests the possibility of QS cooperativity between group-II and group-III in a mixed environment; further studies are needed to better understand the mechanisms and extent by which agr cross-group activity could shape native S. epidermidis communities.
Although the sequences of AIP-II and AIP-III differ at only three residues, our previous NMR studies showed substantial differences in their solution-phase conformations.43 While the macrocycle conformations were modestly similar despite differences at positions 9 and 10, the seven residue exocyclic tails exhibited remarkably different conformations in solution (presumably due to the single residue difference at position 3).43 However, in the current study we observed largely comparable trends in the SARs for agonism by these two signals in the reporter assays (Figure 2B–C; see Tables S1–S2 for full efficacy and potency data), which suggests that the exocyclic tails may reorganize to more similar conformations upon binding to their cognate AgrC receptors. We summarize key trends here.
Beginning at the C-terminus of the peptides, we found that substitutions at the endocyclic hydrophobic residues (Tyr11 and Leu12) of AIP-II and AIP-III drastically impaired activity. These observations align well with our findings in AIP-I (vide supra),42, 43 suggesting that these endocyclic hydrophobic residues likely share a common role in initial binding to their respective AgrC receptors. The residues at positions 9 and 10 differ between AIP-II and AIP-III (Figure 2B–C), and we hypothesized that these two positions could dictate receptor specificity. This reasoning was supported by the consequences of substitution at position 10 differing between AIP-II and AIP-III (Table 1). The alanine substitution at position 10 in AIP-II reduced potency but the same substitution in AIP-III completely inactivated the analog; meanwhile, stereochemical inversion of position 10 in AIP-II obliterated activity, while the corresponding inversion in AIP-III had almost no effect on activity (Table 1). Position 9 was more similar between the two AIPs, with substitutions generally being poorly tolerated in terms of agonism efficacy and potency. Taken together, the activity profiles of the AIP-II and AIP-III analogs exploring positions 9 and 10 suggest that these two positions each make significant and distinct contributions to agonistic activity, likely driving receptor specificity.
The final endocyclic residue in AIP-II and AIP-III is Cys8, which forms the thioester linkage with Leu12 to generate the macrocycle. The incorporation of D-cysteine fully inactivated both AIP-II and AIP-III, similar to our result for D-cysteine replacement in AIP-I.42 This loss of activity could be attributed to a stringent requirement for the local orientation of the thioester bridge for binding, or perhaps this inversion has more global ramifications on peptide conformation. Further structural studies are needed to distinguish these effects and are ongoing in our laboratory.
Turning to the exocyclic tail, we found that both AIP-II and AIP-III, analogous to AIP-I,42 require specific exocyclic residues for AgrC activation. Alanine and D-amino acid substitutions of Tyr5, Asn6, or Pro7 converted the peptides into antagonists or removed any observable activity. These findings are interesting because while AIP-I depends on Val3 alone for nearly all agonistic activity,42 AIP-II and AIP-III appear to depend on the simultaneous presence of multiple residues in this region of the exocyclic tail to activate their cognate receptors. The subsequent residue Lys4 also appears essential for AgrC agonism, but its role differs between AIP-II and AIP-III. In AIP-II, altering Lys4 appears to mainly affect potency without affecting agonism efficacy, while the same changes to AIP-III at Lys4 affect both potency and efficacy (Table 1). These divergent outcomes for substitution at an identical residue and position suggests Lys4 may play unique roles when these AIP analogs interact with AgrC-II versus AgrC-III.
Substitutions at the remaining three N-terminal residues of AIP-II and AIP-III had little impact on their activities. Again, this result contrasts with their NMR solution structures that show the end of the exocyclic tails of AIP-II and AIP-III to be in drastically different conformations.43 In fact, removal of these residues through tail truncation actually increased the potency of the peptides, as the AIP analogs with all three N-terminal residues removed (AIP-II 9aa and AIP-III 9aa) were two-fold more potent than their parent AIPs (Table 1). These data suggest that the final three N-terminal residues of AIP-II and AIP-III fail to engage in meaningful contacts with AgrC and may possibly hamper optimal contacts. Continued removal of additional residues, however, turned the AIP analogs into potent antagonists (Tables S1–S2). We also explored extending the exocyclic tail by one residue (i.e., AIP-II 13aa and AIP-III 13aa, each containing an N-terminal glycine in accordance with the next residue in the AgrD sequence), but this had no effect on efficacy or potency. These results underscore the minor role of the N-terminal residues in AIP-II and AIP-III for AgrC agonism. These surprising data suggest the possibility that AIP-II and AIP-III could be further tailored to 9-mers outside of the cell (i.e., by some extracellular peptidase), and these shorter and more potent agonists may represent the native forms of AIP-II and AIP-III. Additional studies are warranted to probe this hypothesis.
Design and biological characterization of multi-group agonist scaffolds.
With SARs in hand for agonism of AIPs-I–III in their cognate AgrCs, we used these collected data to design peptides that united various motifs critical for activity in an attempt to generate scaffolds with multi-group agonistic activity. We designed the scaffolds with the following features in mind: 1) the endocyclic, hydrophobic residues of all three AIPs are essential and likely adopt similar orientations for receptor binding; 2) a length of nine amino acids appears optimal for agonistic activity in AgrC-II and AgrC-III; 3) the exocyclic residues immediately neighboring the cysteine (Val3 in AIP-I, and Tyr5, Asn6, and Pro7 in AIP-II and AIP-III) largely control agonism in all three AgrC receptors; and 4) the endocyclic residues neighboring the cysteine likely dictate specificity between AgrC-II and AgrC-III. From these features, we designed and synthesized a series of chimeric peptide scaffolds blending the three native AIPs together (see SI for discussion of all scaffolds explored) and identified three peptides for further study: Chimeras 1, 2, and 3 (Cmr1, Cmr2, and Cmr3; Figure 3).
Figure 3.
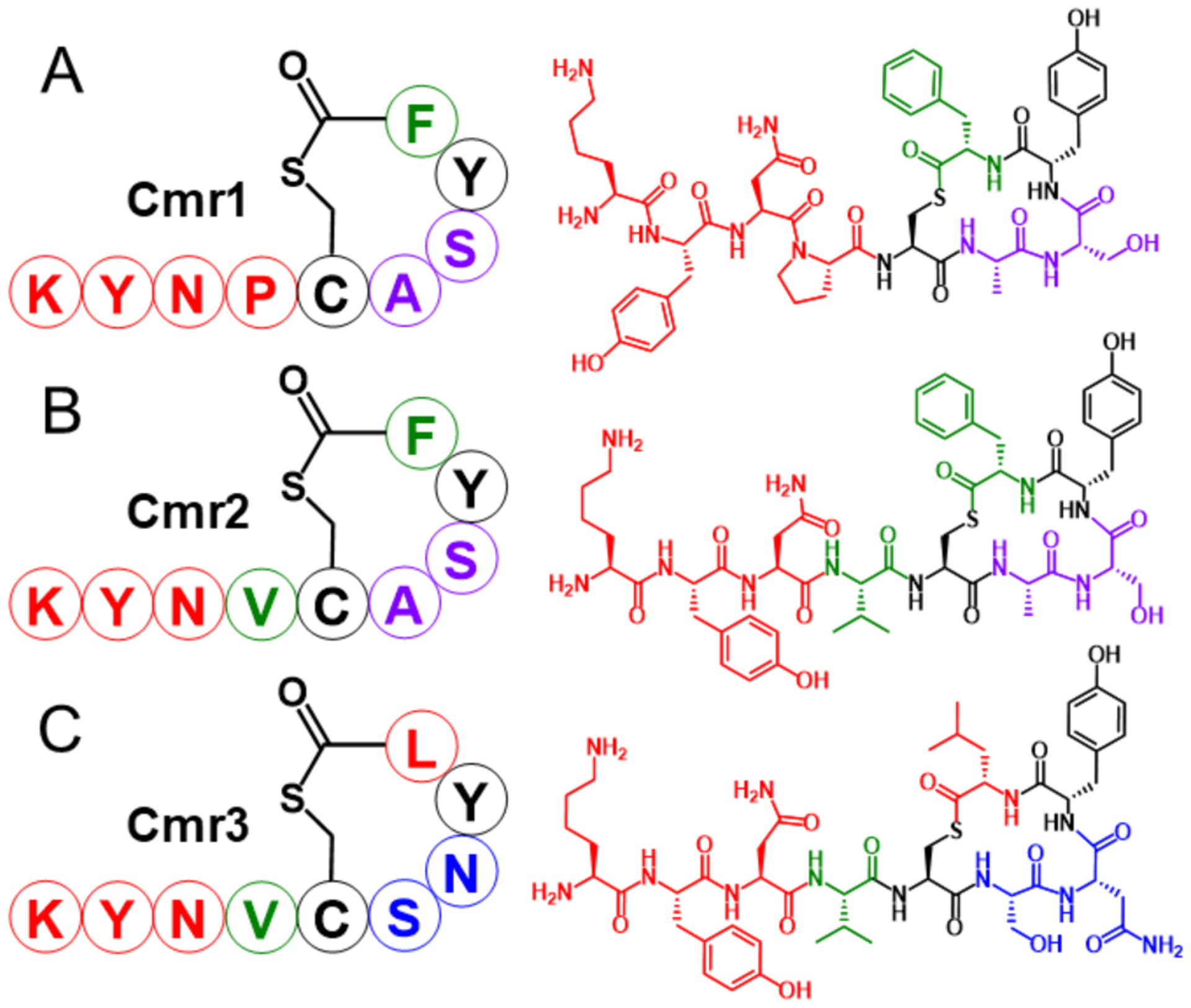
Structures of the three AIP chimeric scaffolds (Cmr1–3). Each scaffold represented with single-letter amino acid abbreviations (left) and chemical structures (right). Black signifies residues shared by AIPs-I–III, green signifies residues solely from AIP-I, blue signifies residues from AIP-II, purple signifies residues shared by AIP-I and AIP-III, and red signifies residues of the shared by AIP-II and AIP-III.
Cmr1 combined the macrocycle of AIP-I with the shared exocyclic tail of AIP-II 9aa and AIP-III 9aa (Figure 3). Given that Cmr1 lacks the exocyclic valine deemed critical for agonism of AgrC-I, we initially expected this scaffold to be an antagonist of AgrC-I, while functioning as an agonist in AgrC-II and AgrC-III. To our surprise, Cmr1 exhibited agonistic activity in all three AgrC receptors, albeit with varying efficacies and potencies (Table 2). Cmr1 was capable of agonizing AgrC-I to approximately 50%, yet displayed low potency. We hypothesize that this reduced activity for Cmr1 is due to the inability of the exocyclic Pro4 to fully replace the agonizing, hydrophobic interactions of the native valine residue in AIP-I with AgrC-I. Cmr1 was similarly able to activate AgrC-II to 60%. In AgrC-III, Cmr1 had an exceptional agonism profile, displaying full activation and sub-nanomolar potency and signifying the most potent AgrC-III agonist we have discovered thus far. Indeed, with activity in all three groups, Cmr1 represents to our knowledge the first known multi-group agonist for the agr systems in S. epidermidis, and in any other related Staphylococci.
Table 2.
Agonism activity data for native AIPs, chimera scaffolds, and identified multi-group agonists (indicated in bold).a
| Peptide Name | agr-I | agr-II | agr-III | |||
|---|---|---|---|---|---|---|
| Max. Activation (%)b | EC50 (nM)c | Max.Activation (%)b | EC50 (nM)c | Max. Activation (%)b | EC50 (nM)c | |
| AIP-I | 147 | 170 | ---d | ---d | ---d | ---d |
| AIP-II | ---d | ---d | 421 | 226 | 39.1 | n/ce |
| AIP-III | ---d | ---d | 182 | 648 | 220 | 71.8 |
| Cmr1 | 51.1 | >2000 | 60.4 | 61.3 | 223 | 0.929 |
| Cmr2 | 146 | 71.8 | ---d | ---d | 46.8 | 40.6 |
| Cmr3 | ---d | ---d | 290 | >2000 | ---d | ---d |
| Cmr1 S7A | 38.4 | 844 | 136 | 63.7 | 398 | 1.47 |
Agonism assay data obtained in S. epidermidis group-I–III reporter strains. See SI for methods.
100% activity corresponds to level of activity produced by endogenously-produced AIP from vehicle controls in absence of 25 nM AAA, 0% activity corresponds to media controls.
As determined by dose-response curves with compound competing against 25 nM AAA.
Only antagonism activity observed.
Dose-response analysis did not converge over concentration range tested.
While Cmr2 also incorporates the macrocycle of AIP-I, Cmr2 differs from Cmr1 in its tail sequence that unites the critical valine residue of AIP-I with the remaining tail residues of AIP-II 9aa and AIP-III 9aa (Figure 3). With the valine residue in place, Cmr2 performed substantially better as an AgrC-I agonist, with mid-nanomolar potency and full efficacy (Table 2). However, the presence of this valine reduced its activity in AgrC-II and AgrC-III; Cmr2 had no observable agonism activity in AgrC-II and had greatly reduced agonistic activity in AgrC-III relative to Cmr1. Lastly, Cmr3 incorporates the hybrid tail of Cmr2 but instead trades the AIP-I macrocycle for the AIP-II macrocycle. While Cmr3 could strongly agonize AgrC-II (Table 2), it showed no agonism activity in either AgrC-I or AgrC-III. Although Cmr3 does not demonstrate multi-group agonism, it did provide new insights into the importance of the macrocycle on AgrC agonism. The inability of Cmr3 to agonize AgrC-I—even with the critical exocyclic valine present—suggests that the macrocycle likely aids in presenting this side chain properly to AgrC-I for activation and that improper orientation by the AIP-II macrocycle thereby hinders agonism. Conversely, Cmr3 was able to strongly agonize AgrC-II, which suggests the AIP-II macrocycle orients the valine to contact and agonize AgrC-II in a functionally similar fashion to the native proline in AIP-II. These new SARs and the differential activities of the three chimeras in the AgrC receptors guided our design of second-generation chimeras; we return to those below.
Multi-group agonists promote PSM production in S. epidermidis.
We sought to demonstrate that the multi-group agr agonist Cmr1 was capable of modulating a phenotype in S. epidermidis relevant to virulence. We selected to investigate PSM production, as PSMs have been shown to facilitate biofilm dispersal and are positively regulated by the agr system in S. epidermidis.15, 16, 23, 47–49 PSMs also can be readily isolated from bacterial culture and quantitated using RP-HPLC.48, 50, 51 As wild-type S. epidermidis cultures can produce a substantial amount of PSMs without exogenous agr agonists added (Figure 4A–C, black trace; see SI for details), we first inhibited PSM production in group I–III strains by adding one of our in-house agr antagonists. When added alone, the antagonists either eliminated or substantially reduced the production of PSMs for each of the three agr groups (Figure 4A–C, red trace); however, addition of both an antagonist and the multi-group agonist Cmr1 was observed to fully or partially restore the production of PSMs for the three agr groups (as determined by comparison to an internal standard; Figure 4A–C, blue trace). PSM peaks were restored to 47% of the baseline value in group-I, and to 11% and 263% in group-II and group-III, respectively. MALDI-MS and ESI-MS confirmed the peak with a RT of 46 minutes contained PSMα, PSMβ, and PSMγ (see SI Table D). The restoration of PSM production by Cmr1 signifies activation of agr within each group and serves to validate the use of reporter strains to identify compounds capable of modulating agr controlled phenotypes in S. epidermidis. Moreover, the ability of Cmr1 to activate PSM production in all three groups underscore the potential of this chemical approach to combat virulence via the pan-group or “global” targeting of the agr system.
Figure 4.

Representative portions of HPLC traces of samples of S. epidermidis cultures showing the PSM peaks. A) Group-I PSM production inhibited with 1 μM AIP-II 9aa and restored with 9 μM Cmr1. B) Group-II PSM production inhibited with 250 nM AAA and restored with 9 μM Cmr1. C) Group-III PSM production inhibited with 1 μM AAA and restored with 9 μM Cmr1. See SI for strain and assays details.
Design and characterization of second-generation multi-group agonist scaffolds.
From the assay data accrued for the three chimera scaffolds above, we designed and synthesized a set of second-generation analogs in an attempt to identify peptide scaffolds with improved agonistic activity in the three S. epidermidis agr groups (see Figures S1–S6, Tables S3–S10 for full list of compounds and activity data). We largely focused on altering the macrocycle of our chimera scaffolds, guided by the hypothesis that we could improve activity in specific groups by tailoring the macrocycle to be more similar to that of the native AIP of the desired group. For example, starting with Cmr1, we aimed to enhance its activity towards AgrC-II by making substitutions at positions 6 and 7 to more resemble AIP-II. As expected, the more closely these analogs resembled the AIP-II macrocycle, the more active they were at agonizing AgrC-II, but unfortunately often at the cost of losing activity toward AgrC-I (Figure S7). Corresponding strategies in Cmr2 and Cmr3 met with similar results (see SI for further discussion). The only analog resulting from these follow-up studies that displayed multi-group agonism activity was Cmr1 S7A, which maintained activity in group-I and group-III while substantially improving group-II activity when compared to Cmr1 (Table 2, Figure S7). This compound was also active in the S. epidermidis agr phenotypic assay, stimulating PSM production levels comparable to or surpassing Cmr1 in the three agr groups (Figure S8).
Beyond the discovery of Cmr1 S7A, the activity survey of the second-generation analogs had an additional positive outcome as it revealed a new SAR for high potency in the three AgrC receptors; namely, the preferred C-terminal residue for high multi-group potency by AIP analogs appears to be a phenylalanine (native to AIP-I) over leucine (native to AIP-II and AIP-III). In most cases where two peptide analogs differ only at the C-terminal residue, the C-terminal phenylalanine analog is at least as potent as the corresponding C-terminal leucine analog and often substantially more potent across the three agr groups, with few exceptions, namely AIP-II 9aa and AIP-II 9aa L9F in group-II (Table 3). This activity trend holds regardless of whether the peptide is an agonist or antagonist. This result is intriguing and strongly supports the installation of phenylalanine as the C-terminal residue in design of future pan-group modulators of S. epidermidis AgrC receptors.
Table 3.
Potency data for pairs of AIP analogs where the only sequence difference is a C-terminal phenylalanine or leucine.
| agr group tested | Peptide containing Phe | EC50 [nM]a | IC50 [nM]b | Peptide containing Leu | EC50 [nM]a | IC50 [nM]b | Fold change |
|---|---|---|---|---|---|---|---|
| agr-I | AIP-I | 170 | I-tail, III-ring | 1870 | 11.0 | ||
| AIP-II 9aa L9F | 0.470 | AIP-II 9aa | 2.84 | 6.04 | |||
| Cmr2 | 71.8 | Cmr2 F9L | 1020 | 14.2 | |||
| Cmr3 L9F | 2.15 | Cmr3 | 75.8 | 35.2 | |||
| agr-II | AIP-I | 13.9 | I-tail, III-ring | 50.9 | 3.66 | ||
| AIP-II 9aa L9F | 203 | AIP-II 9aa | 83.5 | 0.41 | |||
| Cmr3 L9F | 210 | Cmr3 | >2000 | ---c | |||
| agr-III | AIP-I | 2.13 | I-tail, III-ring | 31.8 | 14.9 | ||
| AIP-II 9aa L9F | >2000 | AIP-II 9aa | >2000 | ---c | |||
| Cmr1 | 0.929 | AIP-III 9aa | 28.9 | 31.1 | |||
| Cmr2 | 40.6 | Cmr2 F9L | >2000 | ---c | |||
| Cmr3 L9F | 34.9 | Cmr3 | >2000 | ---c |
Collected from agonism assay data or antagonism assay data obtained in S. epidermidis group-I–III reporter strains. See SI for methods.
Collected from agonism assay data or antagonism assay data obtained in S. epidermidis group-I–III reporter strains. See SI for methods.
Not calculated due to low potency.
Summary and conclusions.
The agr system plays a critical role in the biofilm life cycle and virulence of S. epidermidis, making chemical or biological agents capable of agr modulation valuable tools to probe and potentially limit infectivity.2, 15, 16, 22, 47 The intentional activation of agr could reduce biofilm accumulation and thereby render the dispersed bacteria more susceptible to antimicrobial agents.8 In this study, we report the design, synthesis, and biological characterization of AIP signal analogs capable of agonizing three specificity groups of S. epidermidis (I–III). Through systematic amino acid scans, we determined the key SARs that engendered AgrC agonism by AIP-II and AIP-III, and combined these SARs strategically with those we previously reported for AIP-I42, 43 to design chimeric peptides scaffolds. These investigations revealed two chimeric peptides (Cmr1 and Cmr1 S7A) that were capable of activating the agr systems in groups-I–III of S. epidermidis. In addition, we demonstrated that these multi-group agonists could stimulate production of PSMs, which plays a key role in the dispersal of biofilms, serving to validate their use as probes to study infection relevant phenotypes in S. epidermidis.
The results of this study are significant as they provide peptide chimeras that represent, to our knowledge, the first reported non-native multi-group activators of the agr QS system in any bacterium. This study also provided a deeper understanding of the signal–receptor interactions for the S. epidermidis agr systems. For example, we discovered that the two endocyclic residues adjacent to the conserved cysteine likely contribute towards group specificity and facilitate AIP recognition by the S. epidermidis AgrC receptors. In addition, we observed that the inclusion of a C-terminal phenylalanine can enhance potency of AIP analogs in all three AgrC receptors. Lastly, and perhaps with the most implications for future ligand design, we identified that the exocyclic residue adjacent to the conserved cysteine is likely the critical residue to explore in future work aimed at increasing the multi-group agonistic activity of AIP analogs. While the current study only examined two amino acids at that position (valine and proline), introducing other natural and non-canonical amino acids (e.g., leucine, pipecolic acid, or thioproline) at this position could reveal side chains with optimal sterics and/or hydrophobicity for agonizing the three AgrC receptors. Ongoing studies focus on exploring these motifs in new peptides to target not only groups-I–III S. epidermidis, but also the newly reported group-IV.38
Supplementary Material
ACKNOWLEDGEMENT
This work was supported by the NSF (CHE-1708714 to H.E.B.), the NIH (AI153185 to A.R.H.), and by a Merit Award from the Department of Veteran Affairs (I01 BX002711 to A.R.H.). K. West and J. Vasquez were supported in part by the UW-Madison NIH Chemistry-Biology Interface Training Program (T32 GM008505). T. Yang was supported in part by the UW-Madison NIH Biotechnology Training Program (T32 GM008349). J. Vasquez was supported in part by an NSF Graduate Research Fellowship (DGE-1747503). MS facilities in the Department of Chemistry were supported by the NIH (1S10 OD020022-1) and a gift from P. Bender. We thank S. Ruderman for her early experimental contributions to this project.
Footnotes
ASSOCIATED CONTENT
Supplemental information (full experimental details, additional discussion, reporter assay data, PSM production assay data, and MS and HPLC data for all new AIP analogs) is available free online.
REFERENCES
- 1.Otto M (2009) Staphylococcus epidermidis--the ‘accidental’ pathogen, Nat. Rev. Microbiol 7, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mertens A, and Ghebremedhin B (2013) Genetic determinants and biofilm formation of clinical Staphylococcus epidermidis isolates from blood cultures and indwelling devises, Eur. J. Microbiol. Immunol. (Bp) 3, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabate Bresco M, Harris LG, Thompson K, Stanic B, Morgenstern M, O’Mahony L, Richards RG, and Moriarty TF (2017) Pathogenic Mechanisms and Host Interactions in Staphylococcus epidermidis Device-Related Infection, Front. Microbiol 8, 1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers KL, Fey PD, and Rupp ME (2009) Coagulase-negative staphylococcal infections, Infect. Dis. Clin. North Am 23, 73–98. [DOI] [PubMed] [Google Scholar]
- 5.Uckay I, Pittet D, Vaudaux P, Sax H, Lew D, and Waldvogel F (2009) Foreign body infections due to Staphylococcus epidermidis, Ann. Med 41, 109–119. [DOI] [PubMed] [Google Scholar]
- 6.Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Buttner H, Dunman PM, Rohde H, Cech NB, Fey PD, and Horswill AR (2014) Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization, J. Bacteriol 196, 3482–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin L, Da F, Fisher EL, Tan DC, Nguyen TH, Fu CL, Tan VY, McCausland JW, Sturdevant DE, Joo HS, Queck SY, Cheung GY, and Otto M (2017) Toxin Mediates Sepsis Caused by Methicillin-Resistant Staphylococcus epidermidis, PLoS Pathog. 13, e1006153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies D (2003) Understanding biofilm resistance to antibacterial agents, Nat. Rev. Drug Discov 2, 114–122. [DOI] [PubMed] [Google Scholar]
- 9.Fuqua C, and Greenberg EP (2002) Listening in on bacteria: acyl-homoserine lactone signalling, Nat. Rev. Mol. Cell Biol 3, 685–695. [DOI] [PubMed] [Google Scholar]
- 10.Rutherford ST, and Bassler BL (2012) Bacterial quorum sensing: its role in virulence and possibilities for its control, Cold Spring Harb. Perspect. Med 2, a012427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang B, and Muir TW (2016) Regulation of Virulence in Staphylococcus aureus: Molecular Mechanisms and Remaining Puzzles, Cell Chem. Biol 23, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novick RP, and Geisinger E (2008) Quorum sensing in staphylococci, Annu. Rev. Genet 42, 541–564. [DOI] [PubMed] [Google Scholar]
- 13.Amara N, Mashiach R, Amar D, Krief P, Spieser SA, Bottomley MJ, Aharoni A, and Meijler MM (2009) Covalent inhibition of bacterial quorum sensing, J. Am. Chem. Soc 131, 10610–10619. [DOI] [PubMed] [Google Scholar]
- 14.Yao Y, Vuong C, Kocianova S, Villaruz AE, Lai Y, Sturdevant DE, and Otto M (2006) Characterization of the Staphylococcus epidermidis accessory-gene regulator response: quorum-sensing regulation of resistance to human innate host defense, J. Infect. Dis 193, 841–848. [DOI] [PubMed] [Google Scholar]
- 15.Wang R, Khan BA, Cheung GY, Bach TH, Jameson-Lee M, Kong KF, Queck SY, and Otto M (2011) Staphylococcus epidermidis surfactant peptides promote biofilm maturation and dissemination of biofilm-associated infection in mice, J. Clin. Invest 121, 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otto M (2013) Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity, Annu. Rev. Med 64, 175–188. [DOI] [PubMed] [Google Scholar]
- 17.Mah T-FC, and O’Toole GA (2001) Mechanisms of biofilm resistance to antimicrobial agents, Trends Microbiol. 9, 34–39. [DOI] [PubMed] [Google Scholar]
- 18.Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, and Blackwell HE (2013) Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide, J. Am. Chem. Soc 135, 7869–7882. [DOI] [PubMed] [Google Scholar]
- 19.Khan BA, Yeh AJ, Cheung GY, and Otto M (2015) Investigational therapies targeting quorum-sensing for the treatment of Staphylococcus aureus infections, Expert Opin. Investig. Drugs 24, 689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, DeLeo FR, Otto M, Cheung AL, Edwards BS, Sklar LA, Horswill AR, Hall PR, and Gresham HD (2014) Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance, PLoS Pathog. 10, e1004174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim MK, Zhao A, Wang A, Brown ZZ, Muir TW, Stone HA, and Bassler BL (2017) Surface-attached molecules control Staphylococcus aureus quorum sensing and biofilm development, Nat. Microbiol 2, 17080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harris LG, Dudley E, Rohde H, Frommelt L, Siemssen N, Wilkinson TS, and Mack D (2017) Limitations in the use of PSMgamma, agr, RNAIII, and biofilm formation as biomarkers to define invasive Staphylococcus epidermidis from chronic biomedical device-associated infections, Int. J. Med. Microbiol 307, 382–387. [DOI] [PubMed] [Google Scholar]
- 23.Vuong C, Durr M, Carmody AB, Peschel A, Klebanoff SJ, and Otto M (2004) Regulated expression of pathogen-associated molecular pattern molecules in Staphylococcus epidermidis: quorum-sensing determines pro-inflammatory capacity and production of phenol-soluble modulins, Cell. Microbiol 6, 753–759. [DOI] [PubMed] [Google Scholar]
- 24.Boles BR, and Horswill AR (2008) Agr-mediated dispersal of Staphylococcus aureus biofilms, PLoS Pathog. 4, e1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thoendel M, Kavanaugh JS, Flack CE, and Horswill AR (2011) Peptide signaling in the staphylococci, Chem. Rev 111, 117–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Gray L, Novick RP, and Ji G (2002) Transmembrane topology of AgrB, the protein involved in the post-translational modification of AgrD in Staphylococcus aureus, J. Biol. Chem 277, 34736–34742. [DOI] [PubMed] [Google Scholar]
- 27.Kavanaugh JS, Thoendel M, and Horswill AR (2007) A role for type I signal peptidase in Staphylococcus aureus quorum sensing, Mol. Microbiol 65, 780–798. [DOI] [PubMed] [Google Scholar]
- 28.Wang B, Zhao A, Novick RP, and Muir TW (2014) Activation and inhibition of the receptor histidine kinase AgrC occurs through opposite helical transduction motions, Mol. Cell 53, 929–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koenig RL, Ray JL, Maleki SJ, Smeltzer MS, and Hurlburt BK (2004) Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region, J. Bacteriol 186, 7549–7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novick RP, Projan SJ, Kornblum J, Ross HF, Ji G, Kreiswirth B, Vandenesch F, and Moghazeh S (1995) The agr P2 operon: an autocatalytic sensory transduction system in Staphylococcus aureus, Mol. Gen. Genet 248, 446–458. [DOI] [PubMed] [Google Scholar]
- 31.Novick RP, Ross HF, Projan SJ, Kornblum J, Kreiswirth B, and Moghazeh S (1993) Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule, EMBO J. 12, 3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, and Otto M (2008) RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus, Mol. Cell 32, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otto M (2014) Phenol-soluble modulins, Int. J. Med. Microbiol 304, 164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji G, Beavis R, and Novick RP (1997) Bacterial interference caused by autoinducing peptide variants, Science 276, 2027–2030. [DOI] [PubMed] [Google Scholar]
- 35.Carmody AB, and Otto M (2004) Specificity grouping of the accessory gene regulator quorum-sensing system of Staphylococcus epidermidis is linked to infection, Arch. Microbiol 181, 250–253. [DOI] [PubMed] [Google Scholar]
- 36.Li M, Guan M, Jiang XF, Yuan FY, Xu M, Zhang WZ, and Lu Y (2004) Genetic polymorphism of the accessory gene regulator (agr) locus in Staphylococcus epidermidis and its association with pathogenicity, J. Med. Microbiol 53, 545–549. [DOI] [PubMed] [Google Scholar]
- 37.Hellmark B, Soderquist B, Unemo M, and Nilsdotter-Augustinsson A (2013) Comparison of Staphylococcus epidermidis isolated from prosthetic joint infections and commensal isolates in regard to antibiotic susceptibility, agr type, biofilm production, and epidemiology, Int. J. Med. Microbiol 303, 32–39. [DOI] [PubMed] [Google Scholar]
- 38.Zhou W, Spoto M, Hardy R, Guan C, Fleming E, Larson PJ, Brown JS, and Oh J (2020) Host-Specific Evolutionary and Transmission Dynamics Shape the Functional Diversification of Staphylococcus epidermidis in Human Skin, Cell 180, 454–470 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinez-Garcia S, Ortiz-Garcia CI, Cruz-Aguilar M, Zenteno JC, Murrieta-Coxca JM, Perez-Tapia SM, Rodriguez-Martinez S, Cancino-Diaz ME, and Cancino-Diaz JC (2019) Competition/antagonism associations of biofilm formation among Staphylococcus epidermidis Agr groups I, II, and III, J. Microbiol 57, 143–153. [DOI] [PubMed] [Google Scholar]
- 40.Brown MM, Kwiecinski JM, Cruz LM, Shahbandi A, Todd DA, Cech NB, and Horswill AR (2020) Novel Peptide from Commensal Staphylococcus simulans Blocks Methicillin-Resistant Staphylococcus aureus Quorum Sensing and Protects Host Skin from Damage, Antimicrob. Agents Chemother 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horswill AR, and Gordon CP (2019) Structure-Activity-Relationship Studies of Small Molecule Modulators of the Staphylococcal Accessory Gene Regulator, J. Med. Chem 10.1021/acs.jmedchem.9b00798. [DOI] [PubMed] [Google Scholar]
- 42.Yang T, Tal-Gan Y, Paharik AE, Horswill AR, and Blackwell HE (2016) Structure-Function Analyses of a Staphylococcus epidermidis Autoinducing Peptide Reveals Motifs Critical for AgrC-type Receptor Modulation, ACS Chem. Biol 11, 1982–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasquez JK, West KHJ, Yang T, Polaske TJ, Cornilescu G, Tonelli M, and Blackwell HE (2020) Conformational Switch to a beta-Turn in a Staphylococcal Quorum Sensing Signal Peptide Causes a Dramatic Increase in Potency, J. Am. Chem. Soc 142, 750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wright JS 3rd, Lyon GJ, George EA, Muir TW, and Novick RP (2004) Hydrophobic interactions drive ligand-receptor recognition for activation and inhibition of staphylococcal quorum sensing, Proc. Natl. Acad. Sci. U. S. A 101, 16168–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blanco-Canosa JB, and Dawson PE (2008) An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation, Angew. Chem. Int. Ed. Engl 47, 6851–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tal-Gan Y, Ivancic M, Cornilescu G, and Blackwell HE (2016) Characterization of structural elements in native autoinducing peptides and non-native analogues that permit the differential modulation of AgrC-type quorum sensing receptors in Staphylococcus aureus, Org. Biomol. Chem 14, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kong KF, Vuong C, and Otto M (2006) Staphylococcus quorum sensing in biofilm formation and infection, Int. J. Med. Microbiol 296, 133–139. [DOI] [PubMed] [Google Scholar]
- 48.Cheung GY, Joo HS, Chatterjee SS, and Otto M (2014) Phenol-soluble modulins--critical determinants of staphylococcal virulence, FEMS Microbiol. Rev 38, 698–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Le KY, Villaruz AE, Zheng Y, He L, Fisher EL, Nguyen TH, Ho TV, Yeh AJ, Joo HS, Cheung GYC, and Otto M (2019) Role of Phenol-Soluble Modulins in Staphylococcus epidermidis Biofilm Formation and Infection of Indwelling Medical Devices, J. Mol. Biol 431, 3015–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yao Y, Sturdevant DE, and Otto M (2005) Genomewide analysis of gene expression in Staphylococcus epidermidis biofilms: insights into the pathophysiology of S. epidermidis biofilms and the role of phenol-soluble modulins in formation of biofilms, J. Infect. Dis 191, 289–298. [DOI] [PubMed] [Google Scholar]
- 51.Mehlin C, Headley CM, and Klebanoff SJ (1999) An inflammatory polypeptide complex from Staphylococcus epidermidis: isolation and characterization, J. Exp. Med 189, 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.