

Abstract
Tick-borne ‘Neoehrlichia (N.) mikurensis’ is the cause of neoehrlichiosis, an infectious vasculitis of humans. This strict intracellular pathogen is a member of the family Anaplasmataceae and has been unculturable until recently. The only available genetic data on this new pathogen are six partially sequenced housekeeping genes. The aim of this study was to advance the knowledge regarding ‘N. mikurensis’ genomic relatedness with other Anaplasmataceae members, intra-species genotypic variability and potential virulence factors explaining its tropism for vascular endothelium. Here, we present the de novo whole-genome sequences of three ‘N. mikurensis’ strains derived from Swedish patients diagnosed with neoehrlichiosis. The genomes were obtained by extraction of DNA from patient plasma, library preparation using 10× Chromium technology, and sequencing by Illumina Hiseq-4500. ‘N. mikurensis’ was found to have the next smallest genome of the Anaplasmataceae family (1.1 Mbp with 27% GC contents) consisting of 845 protein-coding genes, every third of which with unknown function. Comparative genomic analyses revealed that ‘N. mikurensis’ was more closely related to Ehrlichia chaffeensis than to Ehrlichia ruminantium, the opposite of what 16SrRNA sequence-based phylogenetic analyses determined. The genetic variability of the three whole-genome-sequenced ‘N. mikurensis’ strains was extremely low, between 0.14 and 0.22‰, a variation that was associated with geographic origin. No protein-coding genes exclusively shared by N. mikurensis and E. ruminantium were identified to explain their common tropism for vascular endothelium.
Keywords: whole-genome sequencing, de novo sequencing, Anaplasmataceae, Candidatus Neoehrlichia mikurensis, human, neoehrlichiosis
1. Introduction
‘Candidatus (Ca.) Neoehrlichia (N.) mikurensis’ was first isolated in ticks and wild rodents on the Japanese island of Mikura in 2004 [1]. In Europe, it is one of the most common human-pathogenic microbes carried by Ixodes ricinus ticks, after Borrelia burgdorferi sensu lato and Rickettsia spp. [2,3,4]. The bacterium gained attention in 2010, when several case reports revealed its capacity to cause human disease [5,6,7,8], which was later named as neoehrlichiosis [9]. Severe cases of neoehrlichiosis typically feature high fever with thromboembolic or vascular complications, [9] which is a consequence of ‘Ca. N. mikurensis’ tropism for vascular endothelium [10]. Initially, ‘Ca. N. mikurensis’ was considered an opportunistic bacterium that chiefly afflicted immune-suppressed patients with particular hematologic or autoimmune diseases [9]. However, it is increasingly appreciated that persons with normal immune defenses can also become infected by this new pathogen and presented with disease manifestations ranging from asymptomatic infection, skin rash, systemic infection with fever and even suspected death from vascular complications [6,11,12,13]. The bacterium has been considered unculturable until recently, when we were able to cultivate it in tick cell lines and human primary endothelial cell lines [10].
‘Candidatus Neoehrlichia’ is the latest genus to be described out of the 7 genera currently comprising the family Anaplasmataceae. ‘Candidatus Neoehrlichia lotoris’ was the first species to be identified within this genus and is carried by North American raccoons and their associated tick species [14]. Previous studies of the genetic diversity of ‘Ca. N. mikurensis’ based on sequencing of housekeeping genes, particularly the 16S rRNA and groEL genes, indicated that ‘Ca. N. mikurensis’ is closely related to Ehrlichia ruminantium, less so to Anaplasma phagocytophilum and least of all to Wolbachia endosymbiont [6,8,15,16,17].
Three genotypes of ‘Ca. N. mikurensis’ were identified amongst 12 European human isolates analyzed by Multilocus sequence assay [MLSA]: one from the western part of Sweden, one from central Europe and a pan-European type [17]. Overall, there was low genetic diversity in the six analyzed MLSA loci, indicating that the strains infecting humans in Europe were quite similar [17]. In contrast, comparative alignment of the 16S rRNA and groEL gene sequences of European and Asian ‘Ca. N. mikurensis’ strains revealed that they differed considerably from each other [6,15,17].
The main objective of this study was to sequence the entire genome of ‘Ca. N. mikurensis’ to increase the knowledge regarding this emerging pathogen. Specifically, we wanted to shed light on the relatedness of this pathogen with other bacterial species within the Anaplasmataceae family, and possibly unravel shared genes with bacterial species having documented tropism for vascular endothelium, e.g., Ehrlichia ruminantium [18]. Complete sequences of five previously sequenced MLSA genes (16S rRNA, ftsZ, gatB, groEL and lipA) as well as fragments evaluate the degree of intra-species variability between different clinical isolates of ‘Ca. N. mikurensis’. Our strategy was to attempt whole-genome sequencing using two types of material: experimentally infected tick cell lines and plasma samples from neoehrlichiosis patients not yet treated with antibiotics.
2. Materials and Methods
2.1. Clinical Isolates of Ca. N. mikurensis
EDTA-anticoagulated blood samples from three Swedish immunocompromised neoehrlichiosis patients (patient and strain ID SE20, SE24 and SE26) were used. Clinical data pertaining to these patients have been published previously [10]. Neoehrlichiosis was diagnosed by PCR using plasma from EDTA-blood as follows: a real-time PCR against the groEL gene of ‘Ca. N. mikurensis’ was run first, and subsequently confirmed by pan-bacterial PCR reactions with Sanger sequencing of the 16S rRNA gene [11].
2.2. Tick Cell Line Cultivated Isolate of Ca. N. mikurensis
The embryo-derived tick cell line IRE/CTVM20 was inoculated with whole blood from a patient (SE18), diagnosed with neoehrlichiosis, and incubated for 21 weeks according to a published protocol [10]. Successful infection of the tick cells was confirmed as previously described [10,11]. Briefly, visualization of the bacteria inside the tick cells was achieved by image-flow cytometry, as well as by quantification of bacterial DNA in tick cell cultures by the groEL-based real-time PCR.
2.3. Bacterial DNA Extraction
The MagAttract HMW DNA Kit (Qiagen, Hilden, Germany) was used for purification of high-molecular-weight DNA from patient plasma and tick cells, using magnetic beads according to the manufacturer’s protocol. DNA yields and purity were measured by automated electrophoresis using a Tape Station and gDNA kit (Agilent Technologies, Santa Clara, CA, USA).
2.4. 10X Chromium Library and Sequencing
10X Chromium Technology (10X Genomics, Pleasanton, CA, USA) was applied for Gel Bead-In Emulsion (GEM) library preparations (Chromium Genome 10X Library kit, 10X Genomics) using 0.7–1.3 ng/µL of HMW-extracted DNA according to the manufacturer’s recommendations. Final library DNA concentrations and DNA fragment sizes were determined by Tape Station electrophoresis as described above. The libraries were sequenced at Clinical Genomics Stockholm, SciLife Labs, using an Illumina HighSeq-4500 platform in two runs. The first run was based on a fresh blood sample (patient ID SE24) and was selected to be a proof-of-concept sequence run. The subsequent runs were based on two additional frozen plasma preparations from the same patient (SE24-1 and SE24-2) and two additional patient samples, SE20 and SE26. The SE24 libraries were assembled using a combination of Supernova (Supernova v.2.0.1 assembler) [19] using standard parameters, BLAST (GenBank BLAST program http://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 11 April 2019) and CLC Genome Finishing module (CLC bio, Aarhus, Denmark). Genomes were finally annotated with Prokka v1.11 software [20]. The first draft assembly was created by initial mapping of the entire SE24 data set against a human whole-genome reference (HG19) and collecting any reads that did not map to the reference [21]. After assembly, the SE20 and SE26 libraries were mapped against the SE24 assembly using CLC Genomics Workbench software with default settings, followed by a fixed ploidy variant calling (90% probability, 80% minimum frequency, minimum coverage 10, minimum count 8, filter homopolymers >3). Lastly, the genome sequences were annotated with the Prokaryotic Genome Annotation Pipeline [22,23] and submitted to GenBank.
2.5. Genomic Analyses and Comparisons
Bioinformatic analyses to categorize the functions of the proteins encoded by the sequenced Ca. N. mikurensis genomes were done using eggNOG Mapper v2 [24,25].
Pan-genome analyses were also done using the type-strain genomes of Ehrlichia chaffeensis ArkansasT, Ehrlichia ruminantium WelgevondenT, the strain Anaplasma phagocytophilum HZ (the first published genome of A. phagocytophilum [26]) and the genomes of reference strains ‘Ca. N. lotoris’ RAC-413 and ‘Ca. N. mikurensis’ SE24. For normalization purposes, all genomes were annotated using Prokka v1.11. The proteins sequences of the genomes were compared (all vs. all) using the Basic Local Alignment Search Tool for Proteins (BLASTP) [27]. Based on these results, groups of homologous proteins were formed, using the Get Homologues software [28] and based on two different algorithms: Cluster of Orthologous Genes Triangle (COGT) [29] and Orthologous Markov Cluster (OMCL) [30]. The threshold for homology was set to 70% similarity for at least 70% of the respective sequence [31]. Only clusters of homologous proteins detected by both algorithms were considered for further characterizations. A Venn Diagram was constructed based on the presence and absence of the different clusters among the species studied to compare the numbers of shared proteins.
The presence or absence of prophages was determined using the online tool Prophage Hunter [32]. The results are classified by this tool as “Active” (score 0.8–1) or “Ambiguous” (score 0.5–0.8). Functional categorizations of the sets of proteins extracted from the genomes were performed with eggNOG-Mapper v2 [24,25].
2.6. Phylogenetic Analyses
Complete sequences of the16S rRNA genes were extracted from the whole-genome sequences of the strains listed above. Sequences were aligned and similarity matrices were generated. Evolutionary distances were calculated using the Kimura two-parameter model [33]. Clustering analysis was performed and a phylogenetic tree was built based on neighbor-joining [34] using the MEGA v7 software [35]. Bootstrap was set for 1000 replications. Additionally, a core-genome phylogenetic analysis was done, using the same genome annotations employed in the previous pan-genome determination. BLASTP analyses comparing all protein sequence files were made. Based on these results, homologous searches were done using the algorithms COGT, OMCL, Bi-Directional Best Hits (BDBH) [36] and Get_Homologues software. The homology threshold was set as explained previously (70% similarity in at least 70% of the sequences). A core genome composed of proteins encoded by single-copy genes was determined based on the consensus reached by the three algorithms. Each protein group was aligned using Clustal Omega [37]. Alignments were analyzed by GBLOCKS [38] to generate a concatenation of the regions with homologous positions. This final alignment was used to build a core genome tree using the Maximum Likelihood algorithm [39] and the Approximate Likelihood Ratio Test (aLRT) [40] with PhyML software [41].
3. Results and Discussion
Here, we present the complete genome sequence and genomic features of the reference strain Ca. N. mikurensis SE24 (GenBank accession no. CP066557), a clinical isolate from a patient diagnosed with neoehrlichiosis. We also present the genome sequences of two additional Swedish clinical isolates: Ca. N. mikurensis, SE20 (GenBank accession No. CP054597) and SE26 (GenBank accession No. CP060793). We selected clinical isolates derived from immune-suppressed neoehrlichiosis patients because they usually have several 10-log higher concentrations of bacterial DNA in the blood compared with immune-competent subjects [9]. All three Ca. N. mikurensis strains were derived from patients from different geographic locations in Sweden (Figure 1).
Figure 1.
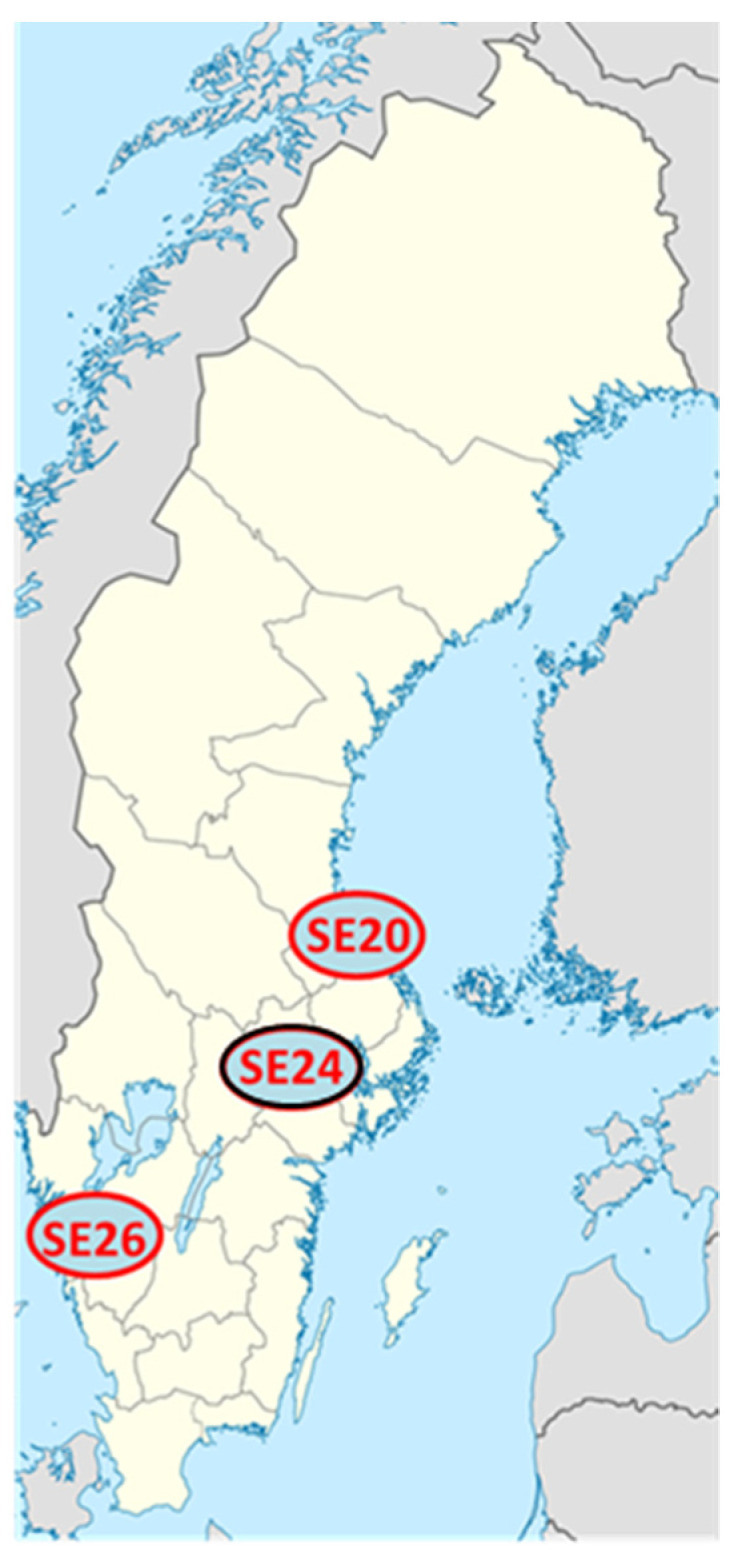
Geographic origin of the sequenced human Ca. N. mikurensis isolates. The sites of residence of the three patients diagnosed with neoehrlichiosis whose blood samples were sequenced are shown. Strain ID SE20 = Gävle, SE24 = Eskilstuna (reference genome, marked in black), SE26 = Kungälv.
The sequencing of these clinical isolates from patient plasma was successful despite the low fractions of bacterial DNA, ranging from 0.1 to 5.1% of the total extracted DNA (Table 1). Meanwhile, enrichment for bacterial DNA by propagating the infection from patient plasma onto tick cells only generated sequence data belonging to Ixodes ricinus. A possible explanation for this failure may be that the bacteria were harvested too late during the infection and, although the tick cells were massively infected, the bacteria may have started to die, thus yielding poor-quality DNA. Moreover, the successful sequencing outcome when using human plasma directly may have depended on the fact that contamination of bacterial DNA with human DNA is advantageous when using 10X technology for barcoding and library preparation because human DNA apparently protects and enhances the recovery and integrity of bacterial DNA by unclear means [42].
Table 1.
Library statistics of Ca. N. mikurensis isolates sequenced from the blood plasma of three neoehrlichiosis patients (SE24, SE20, SE26).
| Patient Sample | Number of Reads | Fraction of Ca. N. mikurensis DNA in Sequenced Plasma Sample (%) | Bacterial Load in Extracted Plasma (c/mL) a |
|---|---|---|---|
| SE24-1 | 775,626,508 | 5.08 | 5.8 × 108 |
| SE24-2 | 729,557,012 | 0.10 | 5.8 × 108 |
| SE24-3 | 673,424,174 | 0.10 | 5.8 × 108 |
| SE20 | 748,875,060 | 1.25 | 1.2 × 108 |
| SE26 | 764,836,740 | 0.57 | 4.6 × 105 |
a Number of groEL gene copies/mL blood by diagnostic PCR. SE24-1, preparation from fresh blood sample. SE24-2 and SE24-3, frozen plasma preparations.
The SE24-1 plasma sample yielded the highest fraction of Ca. N. mikurensis DNA (5.08%), which probably reflects that this sample was extracted from fresh plasma, whereas the other plasma samples from the same patient (SE24-2 and SE24-3) were stored frozen at −120 °C before DNA extraction (Table 1). Moreover, this patient had a high bacterial burden as estimated by a recovery of 5.8 × 108 groEL gene copies/mL blood, as determined by the diagnostic PCR (Table 1). This is equivalent to the number of bacteria/mL blood since the ‘Ca. N. mikurensis’ genome harbors only one copy of the groEL gene (Figure 2).
Figure 2.

Circular representation of the chromosome of the reference genome of Ca. N. mikurensis strain SE24. From inside to outside, the first circle represents the genome size (Mbp), the second circle represents G + C contents (%), the third and fourth circles represent the coding sequences (CDS) on terminus and plus strands, respectively. The location of previously published MLSA genes and the complete ribosomal RNA genes (5S rRNA, 16S rRNA and 23S rRNA) within the genome are shown.
3.1. Genome Assembly and De Novo Annotation
The collected reads that did not map to the human whole-genome reference (HG19) were assembled using the 10× assembler Supernova and contigs were extracted at the megabubble level using limited linkage information whilst not fully collapsing the assembly into a pseudohaplotype. This resulted in 1008 contigs with 7 contigs >50 kb and a N50 (median contig size) 9801 of bp. The contigs were then analyzed by BLAST against the NCBI nucleotide BLAST-database and contigs with a hit against any Ehrlichia species were extracted. In total, 183 contigs were extracted and used as a basis for the draft assembly. Next, the contigs were aligned against themselves using the CLC Genome Finishing module and joined in multiple scaffolding rounds, reducing the total number of contigs to 85. The entire dataset was assembled again using Supernova, this time extracted at the Pseudohap level where the assembler creates a pseudohaplotype scaffold using linkage information and aligned against the clean contigs. This allowed us to join additional contigs and reduce the total number to 50, decreasing the assembly size to 1.14 Mbp. After genome finishing, the raw reads were mapped back to the assembly and the contig sequences were updated according to the mapping information. Initial annotation using Prokka revealed the complete sequences of five previously sequenced MLSA genes: 16S rRNA, ftsZ, gatB, groEL and lipA, as well as fragments of clpB; their spread-out positions within the genome indicated that the assembly was likely to contain the major part of the ‘Ca. N. mikurensis’ genome [17].
An additional sequencing run was performed with the goal of completing the genome of SE24. The new assembly was performed from the three sequenced SE24 samples using Supernova and again extracted at the megabubble level resulting in an assembly of 40,657 contigs. The dataset was reduced by eliminating contigs longer than 30 mbp and shorter than 4000 bp, obtaining 22,913 contigs that were analyzed by BLAST against the previous assembly and an existing reference genome of another Ehrlichia species, Ehrlichia ruminantium [43]. The BLAST results revealed a single contig of approximately 1 Mb containing the majority of the previous assembly sequence and another contig of 900 kb that contained the remaining parts. These two contigs were then joined using contig overlap. Further inspection revealed the new 2 Mb contig to contain two Ca. N. mikurensis genomes that could be split, producing a single contig of 1.11 Mb. The assembly indicated that the edges of the single contig contained a highly repetitive region, which prevented further assembly. Finally, the contig was corrected, first by using contigs from the original assembly, and subsequently by using mapping data and a majority vote where the nucleotide with the highest count at a given position was chosen.
The assembly annotation of the reference genome (SE24) yielded 900 genes in total, of which 860 represented coding sequences (CDS). Every tenth gene encoded a protein of unknown function (Table 2). The annotations contained all 6 MLSA-genes, 34 tRNA, as well as a complete ribosomal RNA operon (5S rRNA, 16S rRNA and 23S rRNA) (Table 2).
Table 2.
Genome properties of Ca. N. mikurensis compared with that of other members of the family Anaplasmataceae.
| Property | Organism | ||||
|---|---|---|---|---|---|
| Ca. Neoehrlichia mikurensis SE24 | Ca. Neoehrlichia lotoris | Ehrlichia ruminantium | Ehrlichia chaffeensis | Anaplasma phagocytophilum | |
| Accession number | CP053279 | NZ_LANX00000000 | NC_005295 | NC_007799 | NC_007797 |
| Size (bp) | 1,112,301 | 1,268,660 | 1,516,355 | 1,176,248 | 1,471,282 |
| GC content (%) | 26.9 | 27.7 | 27.5 | 30.1 | 41.6 |
| Genes, total (n) | 900 | 953 | 987 | 965 | 1152 |
| CDS, total (n) | 860 | 912 | 944 | 922 | 1108 |
| CDS with protein (n) | 845 | 908 | 919 | 886 | 1105 |
| Average CDS length (bp) | 960 | 1016 | 1007 | 995 | 929 |
| Assigned functions (n) | 776 | NR | 758 | 604 | 747 |
| Unknown functions (n) | 90 | NR | NR | 85 | 77 |
| Pseudogenes (n) | 15 | 4 | 25 | 36 | 103 |
| RNA genes (n) | 40 | 41 | 43 | 43 | 44 |
| rRNAs (n) | 3 | 3 | 3 | 3 | 3 |
| tRNAs (n) | 34 | 35 | 36 | 37 | 37 |
| ncRNAs (n) | 3 | 3 | 4 | 3 | 4 |
| Reference | This study | Daugherty, S.C et al. Direct submission | Collins et al. [44] | Dunning Hotopp et al. [26] | Dunning Hotopp et al. [26] |
Bp, base pair; CDS, coding sequence; rRNA, ribosomal ribonucleic acid; tRNA, transfer ribonucleic acid; ncRNA, non-coding ribonucleic acid; NR, not recorded.
3.2. Intra-Species Genomic Comparisons
The SE20 and SE26 datasets were compared to the SE24 reference genome by mapping and variant calling analysis. SE26 differed by a mere 0.22‰ (245 single-nucleotide variants out of 1.1 million) and SE20 differed by 0.138‰ (153/1.1 million) with respect to SE24, respectively. The degree of genetic variation between the strains seemed to be in accordance with their mutual geographic distance, such that the variation was greatest between SE26 and SE24 and lower between SE20 and SE24 (Figure 1).
The proteins encoded by the three Ca. N. mikurensis strains (SE24, SE20 and SE26) were classified into functional Clusters of Orthologous Groups (COG) categories (Table 3). As expected, the majority of the genes’ encoded proteins were essential for bacterial survival, i.e., involved in bacterial biogenesis and replication; nutrient transport and metabolism. It is worth highlighting that close to every tenth protein of Ca. N. mikurensis was classified as “Function unknown” and 20% of the proteins did not assign to any COG category at all (Table 3).
Table 3.
Functional Clusters of Orthologous Groups of protein-coding genes from the three sequenced Ca. N. mikurensis strains.
| Functional Category | Ca. N. mikurensis Strain SE24 | Ca. N. mikurensis Strain SE20 | Ca. N. mikurensis Strain SE26 |
|---|---|---|---|
| Number of Genes (%) | |||
| Translation, ribosomal structure and biogenesis | 112 (13) | 115 (13) | 115 (13) |
| Energy production and conversion | 62 (7) | 62 (7) | 62 (7) |
| Posttranslational modification, protein turnover, chaperones | 58 (7) | 58 (7) | 58 (7) |
| Coenzyme transport and metabolism | 52 (6) | 54 (6) | 54 (6) |
| Replication, recombination and repair | 47 (5) | 47 (5) | 47 (5) |
| Cell wall/membrane/envelope biogenesis | 39 (5) | 39 (5) | 39 (5) |
| Nucleotide transport and metabolism | 37 (4) | 37 (4) | 37 (4) |
| Inorganic ion transport and metabolism | 34 (4) | 34 (4) | 34 (4) |
| Intracellular trafficking, secretion, and vesicular transport | 32 (4) | 32 (4) | 32 (4) |
| Amino acid transport and metabolism | 32 (4) | 32 (4) | 32 (4) |
| Lipid transport and metabolism | 26 (3) | 27 (3) | 27 (3) |
| Transcription | 23 (3) | 23 (3) | 23 (3) |
| Carbohydrate transport and metabolism | 19 (2) | 20 (2) | 20 (2) |
| Cell cycle control, cell division, chromosome partitioning | 11 (1) | 11 (1) | 11 (1) |
| Signal transduction mechanisms | 10 (1) | 10 (1) | 10 (1) |
| Secondary metabolites biosynthesis, transport and catabolism | 9 (1) | 9 (1) | 9 (1) |
| Defense mechanisms | 3 (0.3) | 3 (0.3) | 3 (0.3) |
| General function prediction only | 0 | 0 | 0 |
| Mobilome: prophages, transposons | 0 | 0 | 0 |
| Cell motility | 0 | 0 | 0 |
| Cytoskeleton | 0 | 0 | 0 |
| Extracellular structures | 0 | 0 | 0 |
| RNA processing and modification | 0 | 0 | 0 |
| Chromatin structure and dynamics | 0 | 0 | 0 |
| Nuclear structure | 0 | 0 | 0 |
| Function unknown | 90 (11) | 90 (11) | 90 (11) |
| No category assigned | 170 (20) | 166 (20) | 167 (20) |
Calculations done with eggNOG-mapper v2.
Two of the strains were found to have exclusive proteins not shared by the other two strains: SE24 (4 exclusive proteins) and SE26 (1 exclusive protein) (Figure 3). One of the unique proteins of SE24 belonged to the P44/Msp-family (HL033_02985) and two were classified as hypothetical proteins with unknown function (HL033_02590, HL033_03370). However, the fourth hypothetical protein annotated by Prokka (between positions 53,155 and 63,249) was not annotated by PGAP. The hypothetical protein of strain SE24 that was encoded by HL033_02590 seemed to belong to the TrbC/VirB2 family according to BLASTP analysis. Studies has shown that VirB2 is a major pilus component of T4SS extracellular filaments and may play a critical role in the initial interaction with the host cell for members in the family Anaplasmataceae [45]. The unique protein of SE26 belonged to the thioredoxin-like family (IAH97_01635) and constitutes a small protein with antioxidant properties that is expressed in the periplasm or cytoplasm of certain Gram-negative bacterial species [46].
Figure 3.

Venn diagram based on the consensus clusters of homologous proteins identified by the algorithms COGT and OrthoMCL. The predicted numbers of shared and unique protein-encoding genes derived from the three whole-genome-sequenced strains of Ca. N. mikurensis are shown.
A search for prophages was inconclusive: Although a putative prophage of 13 kb was identified in all three strains, encompassing 6 genes in each case, the confidence score provided by Prophage Hunter software was low (0.56 to 0.63) rendering a verdict of “ambiguous”.
3.3. Comparison of the Ca. N. mikurensis Genome with Other Genomes within the Anaplasmataceae Family
We compared the reference genome of ‘Ca. N. mikurensis SE24′ with other whole-genome-sequenced members of the family Anaplasmataceae (Table 2). The genome of ‘Ca. N. mikurensis’ (1.11 Mb) was found to be the second smallest genome of the Anaplasmataceae family after Neorickettsia sennetsu (0.859 Mb) [26] with low GC contents typical of all members of the family except for A. pagocytophilum (41.6%). Low GC contents is a common trait of the genomes of intracellular bacteria [43]. ‘Ca. N. mikurensis’ and E. ruminantium contain almost the same number of pseudogenes (36 and 32, respectively), whereas Anaplasma phagocytophilum harbors four-fold more pseudogenes. Pseudogenes are genes that have become non-functional due to accumulation of mutations and are more frequent in intracellular bacteria where the loss of gene functions is compensated by bacterial parasitism on the host cell [44].
Phylogenetic analyses based on the complete 16S rRNA gene sequences determined ‘Ca. N. lotoris’ to be the most genetically related relative of ‘Ca. N. mikurensis’ and identified E. ruminantium as the most genetically related relative outside the genus of Neoehrlichia (Figure 4A) supporting earlier MLSA findings [17]. However, a higher-resolution phylogenetic analysis based on 93 sequenced core proteins showed that Ca. N. mikurensis is more closely related to the human pathogen E. chaffeensis than to E. ruminantium (Figure 4B). E. chaffeensis resembles Ca. N. mikurensis by being a human pathogen, in contrast to E. ruminantium, which is pathogenic for ruminants. In contrast, Ca. N. mikurensis and E. ruminantium share tropism for vascular endothelium, unlike E. chaffeensis, which preferentially infects monocytes [47].
Figure 4.

(A) Phylogenetic tree based on comparisons of the complete 16S rRNA gene sequences obtained from the genomes analyzed in this study. The scale bar indicates number of nucleotide subsitutions per site. (B) Core-genome dendrogram based on 93 shared core-proteins identified in this study. Marked in red is the switch in bacterial relatedness seen when a higher resolution phylogenetic analysis was used (B) vs. (A). The scale bar indicates aminoacid substitutions per site.
All earlier sequenced members of the order Rickettsiales have a single rRNA operon in which the 16S rRNA gene is physically separated from the 23S-5S rRNA gene pair [43]. Ca. N. mikurensis also shares this feature, i.e., its 16S rRNA gene was found to be separated from the 23S-5S gene pair (Figure 2). Generally speaking, it is more common for bacteria to have multiple rRNA gene operons composed of genes located one after the other in the order of 16S-23S-5S [48,49]. The phenomenon of unlinked rRNA genes displayed by Ca. N. mikurensis seemed to be more frequent among slow-growing bacterial species and species that contained a single rRNA operon [50].
3.4. Protein Comparisons between Anaplasmataceae Species
Comparisons of the protein sets harbored by Ca. N. mikurensis with those of A. phagocytophilum HZ (GenBank accession no. CP000235), E. chaffeensis ArkansasT, E. ruminatum WelgenvondenT and Ca. N. lotoris RAC-413 were done through a pan-genome approach. All species had 109 proteins in common, and ‘Ca. N. mikurensis’ harbored an additional 83 unique proteins not present in the other species. Further, 31 proteins were uniquely shared by ‘Ca. N. mikurensis’ and Ca. N. lotoris, and 10 proteins were shared by all species except for A. phagocytophilum (Figure 5). No proteins were solely shared by Ca. N. mikurensis and A. phagocytophilum, supporting their more distant relatedness compared with the other species.
Figure 5.

A consensus Venn diagram based on the clusters of homologous proteins detected by the algorithms COGT and OrthoMCL. The numbers of proteins predicted to be unique and shared, respectively, between Ca. N. mikurensis strain SE24 and four other bacterial species that are members of the family Anaplasmataceae, are shown.
A closer look at the ten proteins shared by ‘Ca. N. mikurensis’, Ca. N. lotoris, E. ruminantium and E. chaffeensis, but not by A. phagocytophilum (Table 4), showed that four are involved in translation and DNA repair, two in amino-acid biosynthesis, one in protein secretion, one in cellular detoxification, one in plasmid partitioning (although it is unclear if they possess plasmids) and one of unknown function. Ehrlichia species have a larger number of genes involved in amino acid biosynthesis compared with other members of the Anaplasmataceae-family, and it has been suggested that bacterial production of arginine may counter the host cell’s nitric oxide defense and allow the bacteria to weaken the host immune response [51].
Table 4.
Ten proteins predicted to be shared by Ca. Neoehrlichia mikurensis, Ca. Neoehrlichia lotoris, Ehrlichia chaffeensis and Ehrlichia ruminantium.
| Protein | Function | Gene | Locus Tag |
|---|---|---|---|
| Argininosuccinate lyase | Amino-acid biosynthesis | argH1 | HL033_01080 |
| Argininosuccinate synthase | Amino-acid biosynthesis | argG | HL033_04485 |
| ParA family protein | Partitioning of plasmids | parA | HL033_02250 |
| Type I secretion system permease/ATPase | Protein secretion | prtD | HL033_03355 |
| 50S ribosomal protein L32 | Translation | rpmF | HL033_03630 |
| 50S ribosomal protein L34 | Translation | rpmH | HL033_03600 |
| 50S ribosomal protein L36 | Translation | rpmJ | HL033_04370 |
| DNA repair protein RadA | DNA repair | radA | HL033_02995 |
| DUF2671 domain-containing protein | Protein with domain of unknown function | unknown | HL033_00465 |
| Glutathione S-transferase family protein | Cellular detoxificaion | gstA | HL033_00805 |
All species share a gene (prtD) involved in secretion through the type I secretion system (T1SS), which enables many Gram-negative bacterial species to transport substrates from the bacterial cytosol to the extracellular space and also contributes to their virulence. E.chaffeensis secretes nucleomodulins able to reprogram host cell defense mechanisms and thereby facilitate bacterial invasion of host cells [52]. However, no shared genes were identified for ‘Ca. N. mikurensis’ and E. ruminantium that could explain their tropism for vascular endothelium.
To sum up, we have determined the complete genome sequence of ‘Ca. N. mikurensis’, which we hope will advance our understanding of the pathogenic mechanisms and immune evasion strategies employed by this emerging pathogen. Further, by combining proteomic analyses with the obtained genomic data, it may be possible in the near future to identify candidate outer-membrane proteins for the development of antibody assays to be used for diagnostics and seroepidemiologic studies, helping to determine the prevalence and incidence of this emerging infection in various populations and age groups.
Acknowledgments
Not applicable.
Author Contributions
Conceptualization, C.W. and A.G.; methodology, A.G. and P.S.; software, D.J.-L. and P.S.; validation, A.G.; formal analysis, A.G. and L.W.; investigation, A.G. and L.W.; data curation, D.J.-L.; writing—original draft preparation, A.G.; writing—review and editing, A.G., D.J.-L., L.W., P.S., C.W.; visualization, A.G.; supervision, C.W.; project administration, A.G.; funding acquisition, C.W. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement (ALFGBG-827291), the Västra Götaland Regional Research and Development Fund, the Cancer and Allergy Foundation (19-228), EU Interreg V A project ScandTick Innovation and NorthTick, North Sea Programme of the European Regional Development Fund of European Union, and the Swedish Research Council (2020-01287).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequencing data is available for public use at the sequence read archive of NCBI (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA630882, accessed on 9 July 2021).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kawahara M., Rikihisa Y., Isogai E., Takahashi M., Misumi H., Suto C., Shibata S., Zhang C., Tsuji M. Ultrastructure and phylogenetic analysis of ‘Candidatus Neoehrlichia mikurensis’ in the family Anaplasmataceae, isolated from wild rats and found in Ixodes ovatus ticks. Int. J. Syst. Evol. Microbiol. 2004;54:1837–1843. doi: 10.1099/ijs.0.63260-0. [DOI] [PubMed] [Google Scholar]
- 2.Schouls L.M., Van De Pol I., Rijpkema S.G., Schot C.S. Detection and identification of Ehrlichia, Borrelia burgdorferi sensu lato, and Bartonella species in Dutch Ixodes ricinus ticks. J. Clin. Microbiol. 1999;37:2215–2222. doi: 10.1128/JCM.37.7.2215-2222.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rar V.A., Livanova N.N., Panov V.V., Doroschenko E.K., Pukhovskaya N.M., Vysochina N.P., Ivanov L.I. Genetic diversity of Anaplasma and Ehrlichia in the Asian part of Russia. Ticks Tick Borne Dis. 2010;1:57–65. doi: 10.1016/j.ttbdis.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Andersson M., Raberg L. Wild rodents and novel human pathogen candidatus Neoehrlichia mikurensis, Southern Sweden. Emerg. Infect. Dis. 2011;17:1716–1718. doi: 10.3201/eid1709.101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welinder-Olsson C., Kjellin E., Vaht K., Jacobsson S., Wenneras C. First case of human “Candidatus Neoehrlichia mikurensis” infection in a febrile patient with chronic lymphocytic leukemia. J. Clin. Microbiol. 2010;48:1956–1959. doi: 10.1128/JCM.02423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Von Loewenich F.D., Geissdorfer W., Disque C., Matten J., Schett G., Sakka S.G., Bogdan C. Detection of “Candidatus Neoehrlichia mikurensis” in two patients with severe febrile illnesses: Evidence for a European sequence variant. J. Clin. Microbiol. 2010;48:2630–2635. doi: 10.1128/JCM.00588-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pekova S., Vydra J., Kabickova H., Frankova S., Haugvicova R., Mazal O., Cmejla R., Hardekopf D.W., Jancuskova T., Kozak T. Candidatus Neoehrlichia mikurensis infection identified in 2 hematooncologic patients: Benefit of molecular techniques for rare pathogen detection. Diagn. Microbiol. Infect. Dis. 2011;69:266–270. doi: 10.1016/j.diagmicrobio.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Fehr J.S., Bloemberg G.V., Ritter C., Hombach M., Luscher T.F., Weber R., Keller P.M. Septicemia caused by tick-borne bacterial pathogen Candidatus Neoehrlichia mikurensis. Emerg. Infect. Dis. 2010;16:1127–1129. doi: 10.3201/eid1607.091907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grankvist A., Andersson P.-O., Mattsson M., Sender M., Vaht K., Hoper L., Sakiniene E., Trysberg E., Stenson M., Fehr J., et al. Infections with the Tick-Borne Bacterium “Candidatus Neoehrlichia mikurensis” Mimic Noninfectious Conditions in Patients with B Cell Malignancies or Autoimmune Diseases. Clin. Infect. Dis. 2014;58:1716–1722. doi: 10.1093/cid/ciu189. [DOI] [PubMed] [Google Scholar]
- 10.Wass L., Grankvist A., Bell-Sakyi L., Bergström M., Ulfhammer E., Lingblom C., Wennerås C. Cultivation of the causative agent of human neoehrlichiosis from clinical isolates identifies vascular endothelium as a target of infection. Emerg. Microbes Infect. 2019;8:413–425. doi: 10.1080/22221751.2019.1584017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grankvist A., Sandelin L.L., Andersson J., Fryland L., Wilhelmsson P., Lindgren P.E., Forsberg P., Wenneras C. Infections with Candidatus Neoehrlichia mikurensis and Cytokine Responses in 2 Persons Bitten by Ticks, Sweden. Emerg. Infect. Dis. 2015;21:1462–1465. doi: 10.3201/eid2108.150060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welc-Faleciak R., Sinski E., Kowalec M., Zajkowska J., Pancewicz S.A. Asymptomatic “Candidatus Neoehrlichia mikurensis” infections in immunocompetent humans. J. Clin. Microbiol. 2014;52:3072–3074. doi: 10.1128/JCM.00741-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H., Jiang J.-F., Liu W., Zheng Y.-C., Huo Q.-B., Tang K., Zuo S.-Y., Liu K., Jiang B.-G., Yang H., et al. Human Infection withCandidatusNeoehrlichia mikurensis, China. Emerg. Infect. Dis. 2012;18:1636–1639. doi: 10.3201/eid1810.120594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yabsley M.J., Murphy S.M., Luttrell M.P., Wilcox B.R., Howerth E.W., Munderloh U.G. Characterization of ‘Candidatus Neoehrlichia lotoris’ (family Anaplasmataceae) from raccoons (Procyon lotor) Int. J. Syst. Evol. Microbiol. 2008;58:2794–2798. doi: 10.1099/ijs.0.65836-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Jiang J., Tang F., Sun Y., Li Z., Zhang W., Gong Z., Liu K., Yang H., Liu W., et al. Wide Distribution and Genetic Diversity of “Candidatus Neoehrlichia mikurensis” in Rodents from China. Appl. Environ. Microbiol. 2013;79:1024–1027. doi: 10.1128/AEM.02917-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rar V.A., Epikhina T.I., Livanova N.N., Panov V.V., Doroshenko E.K., Pukhovskaia N.M., Vysochina N.P., Ivanov L.I. Study of the heterogeneity of 16s rRNA gene and groESL operone in the dna samples of Anaplasma phagocytophilum, Ehrlichia muris, and “Candidatus Neoehrlichia mikurensis” determined in the Ixodes persulcatus ticks in the area of Urals, Siberia, and far east of Russia. Mol. Gen. Mikrobiol. Virusol. 2011;2:17–23. [PubMed] [Google Scholar]
- 17.Grankvist A., Moore E.R., Svensson Stadler L., Pekova S., Bogdan C., Geissdorfer W., Grip-Linden J., Brandstrom K., Marsal J., Andreasson K., et al. Multilocus Sequence Analysis of Clinical “Candidatus Neoehrlichia mikurensis” Strains from Europe. J. Clin. Microbiol. 2015;53:3126–3132. doi: 10.1128/JCM.00880-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allsopp B.A. Heartwater--Ehrlichia ruminantium infection. Rev. Sci. Tech. 2015;34:557–568. doi: 10.20506/rst.34.2.2379. [DOI] [PubMed] [Google Scholar]
- 19.Weisenfeld N.I., Kumar V., Shah P., Church D.M., Jaffe D.B. Direct determination of diploid genome sequences. Genome Res. 2017;27:757–767. doi: 10.1101/gr.214874.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seemann T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 21.Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A., et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 22.Tatusova T., DiCuccio M., Badretdin A., Chetvernin V., Nawrocki P., Zaslavsky L., Lomsadze A., Pruitt K.D., Borodovsky M., Ostell J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haft D.H., DiCuccio M., Badretdin A., Brover V., Chetvernin V., O’Neill K., Li W., Chitsaz F., Derbyshire M.K., Gonzales N.R., et al. RefSeq: An update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018;46:D851–D860. doi: 10.1093/nar/gkx1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huerta-Cepas J., Forslund K., Coelho L.P., Szklarczyk D., Jensen L.J., von Mering C., Bork P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017;34:2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huerta-Cepas J., Szklarczyk D., Heller D., Hernández-Plaza A., Forslund S.K., Cook H.V., Mende D.R., Letunic I., Rattei T., Jensen L.J., et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunning Hotopp J.C., Lin M., Madupu R., Crabtree J., Angiuoli S.V., Eisen J.A., Seshadri R., Ren Q., Wu M., Utterback T.R., et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006;2:e21. doi: 10.1371/journal.pgen.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 28.Contreras-Moreira B., Vinuesa P. GET_HOMOLOGUES, a Versatile Software Package for Scalable and Robust Microbial Pangenome Analysis. Appl. Environ. Microbiol. 2013;79:7696–7701. doi: 10.1128/AEM.02411-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kristensen D.M., Kannan L., Coleman M.K., Wolf Y., Sorokin A., Koonin E.V., Mushegian A. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics. 2010;26:1481–1487. doi: 10.1093/bioinformatics/btq229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L., Stoeckert C.J., Roos D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearson W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. Chapter. 2013;3:1–3. doi: 10.1002/0471250953.bi0301s42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song W., Sun H.-X., Zhang C., Cheng L., Peng Y., Deng Z., Wang D., Wang Y., Hu M., Liu W., et al. Prophage Hunter: An integrative hunting tool for active prophages. Nucleic Acids Res. 2019;47:W74–W80. doi: 10.1093/nar/gkz380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 34.Saitou N., Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 35.Kumar S., Stecher G., Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Overbeek R., Fonstein M., D’Souza M., Pusch G.D., Maltsev N. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. USA. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., López R., McWilliam H., Remmert M., Söding J., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castresana J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 39.Tamura K., Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 40.Anisimova M., Gascuel O. Approximate Likelihood-Ratio Test for Branches: A Fast, Accurate, and Powerful Alternative. Syst. Biol. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 41.Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 42.Zheng G.X.Y., Lau B.T., Schnall-Levin M., Jarosz M., Bell J.M., Hindson C.M., Kyriazopoulou-Panagiotopoulou S., Masquelier D.A., Merrill L., Terry J.M., et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016;34:303–311. doi: 10.1038/nbt.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collins N.E., Liebenberg J., de Villiers E.P., Brayton K.A., Louw E., Pretorius A., Faber F.E., van Heerden H., Josemans A., van Kleef M., et al. The genome of the heartwater agent Ehrlichia ruminantium contains multiple tandem repeats of actively variable copy number. Proc. Natl. Acad. Sci. USA. 2005;102:838–843. doi: 10.1073/pnas.0406633102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beare P.A., Sandoz K.M., Omsland A., Rockey D.D., Heinzen R.A. Advances in genetic manipulation of obligate intracellular bacterial pathogens. Front. Microbiol. 2011;2:97. doi: 10.3389/fmicb.2011.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin M., Zhang C., Gibson K., Rikihisa Y. Analysis of complete genome sequence of Neorickettsia risticii: Causative agent of Potomac horse fever. Nucleic Acids Res. 2009;37:6076–6091. doi: 10.1093/nar/gkp642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Achard M.E., Hamilton A.J., Dankowski T., Heras B., Schembri M.S., Edwards J.L., Jennings M.P., McEwan A.G. A periplasmic thioredoxin-like protein plays a role in defense against oxidative stress in Neisseria gonorrhoeae. Infect. Immun. 2009;77:4934–4939. doi: 10.1128/IAI.00714-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cunha B.A., Chandrankunnel J.G., Hage J.E. Ehrlichia chaffeensis human monocytic ehrlichiosis with pancytopenia. Scand. J. Infect. Dis. 2012;44:473–474. doi: 10.3109/00365548.2011.633550. [DOI] [PubMed] [Google Scholar]
- 48.Acinas S.G., Marcelino L.A., Klepac-Ceraj V., Polz M.F. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 2004;186:2629–2635. doi: 10.1128/JB.186.9.2629-2635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krawiec S., Riley M. Organization of the bacterial chromosome. Microbiol. Rev. 1990;54:502–539. doi: 10.1128/mr.54.4.502-539.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klappenbach J.A., Dunbar J.M., Schmidt T.M. rRNA Operon Copy Number Reflects Ecological Strategies of Bacteria. Appl. Environ. Microbiol. 2000;66:1328–1333. doi: 10.1128/AEM.66.4.1328-1333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chakravortty D., Hensel M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect. 2003;5:621–627. doi: 10.1016/S1286-4579(03)00096-0. [DOI] [PubMed] [Google Scholar]
- 52.Patterson L.L., Byerly C.D., McBride J.W. Anaplasmataceae: Dichotomous Autophagic Interplay for Infection. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.642771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequencing data is available for public use at the sequence read archive of NCBI (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA630882, accessed on 9 July 2021).