

Abstract
Background
Lycium Linn. (Solanaceae) is a genus of economically important plants producing fruits and leaves with high nutritional value and medicinal benefits. However, genetic analysis of this plant and molecular breeding for quality improvement are limited by the lack of sufficient molecular markers.
Results
In this study, two parental strains, ‘Ningqi No. 1’ (Lycium barbarum L.) and ‘Yunnan Gouqi’ (Lycium yunnanense Kuang et A.M. Lu), and 200 F1 hybrid individuals were resequenced for genetic analysis. In total, 8,507 well-selected SNPs were developed, and a high-density genetic map (NY map) was constructed with a total genetic distance of 2,122.24 cM. A consensus genetic map was established by integrating the NY map and a previously published genetic map (NC map) containing 15,240 SNPs, with a total genetic distance of 3,058.19 cM and an average map distance of 0.21 cM. The 12 pseudochromosomes of the Lycium reference genome were anchored using this consensus genetic map, with an anchoring rate of 64.3%. Moreover, weak collinearities between the consensus map and the pepper, potato, and tomato genomes were observed. Twenty-five stable QTLs were identified for leaf- and fruit-related phenotypes, including fruit weight, fruit longitude, leaf length, the fruit index, and the leaf index; these stable QTLs were mapped to four different linkage groups, with LOD scores ranging from 2.51 to 19.37 and amounts of phenotypic variance explained from 6.2% to 51.9%. Finally, 82 out of 188 predicted genes underlying stable QTLs for fruit-related traits were differentially expressed according to RNA-seq analysis.
Conclusions
A chromosome-level assembly can provide a foundation for further functional genomics research for wolfberry. The genomic regions of these stably expressed QTLs could be used as targets for further fine mapping and development of molecular markers for marker-assisted selection (MAS). The present study provided valuable information on saturated SNP markers and reliable QTLs for map-based cloning of functional genes related to yield and morphological traits in Lycium spp.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12870-021-03115-1.
Keywords: Genetic map, Resequencing, Leaf- and fruit-related traits, QTL, Genome synteny, Lycium Linn
Background
Lycium Linn. (Solanaceae) is a genus of perennial shrubs with > 80 species worldwide and is mainly distributed in South America, southwestern North America, southern Africa, and eastern Asia [1]. Seven species and three varieties of Lycium occur in China [2]; of these, Lycium barbarum (‘goji berry’ or Chinese wolfberry) has been domesticated and widely cultivated in Northwest China for > 600 years [2, 3]. The edible fruits and leaves of L. barbarum are used as functional foods and traditional Chinese medicinal herbs in China [4, 5]. Many compounds from L. barbarum fruits and leaves, including flavonoids, carotenoids, and polysaccharides, have been reported to be closely associated with the health-enhancing effects of this species [5, 6]. However, it is difficult to improve the quality of Lycium because of the unclear molecular genetic mechanisms underlying Lycium fruit and leaf traits.
Next-generation sequencing (NGS) coupled with the growing number of reference genome sequences presents an opportunity to redesign genotyping strategies for more effective genetic mapping and genome analysis [7], which can result in ultra-high-density linkage map construction and localized quantitative trait loci (QTLs) for multiple traits [8, 9]. Resequencing and high-density genetic mapping in crops with complete genome sequences identified days to heading8 (Dth8) and lax panicle1 (Lax1) as candidate genes in rice [10] and sequence alterations in a novel ion transporter gene (GmCHX1) inducing salt tolerance in wild soybean [11]. Moreover, structural variations were reported in allotetraploid cotton [12]. With the decreasing costs of sequencing technologies, whole-genome sequencing has been applied to an increasing number of plant species; in addition, the numerous single nucleotide polymorphism (SNP) markers developed by aligning resequencing data to the corresponding reference genome can provide a powerful approach for deciphering the genetic basis of complex traits and for large-scale gene discovery [13].
The first sequencing-based linkage map for Lycium was constructed by specific length amplified fragment sequencing (SLAF-seq) using a diploid F1 population derived from a cross between ‘Ningqi No. 1’ (NN) and ‘Chinese gouqi’, and 18 stable leaf and fruit QTLs were mapped onto the resulting genetic map [14]. Recently, a 1,891-Mb Lycium genome sequence (Cao et al., unpublished 2021) provided an opportunity to develop SNP markers for population genotyping. In the present study, we used an F1 population (Fig. 1) of Lycium with the shared parent ‘Ningqi No. 1’ to map QTLs for agronomic traits. Genotyping was performed using resequencing followed by SNP identification. The resulting SNPs were used to construct a high-density linkage map and an integrated consensus map. Using these maps, we were able to map yield-related QTLs in Lycium. Such QTLs and closely linked markers could then be used for molecular breeding to improve Lycium yield and quality.
Fig. 1.

Appearance of fruits and leaves of the two parents (A) and representative F1 individuals (B) of Lycium. Note: A-1 female parent ‘Ningqi No. 1’ fruits; A-2 male parent ‘Yunnan Gouqi’ fruits; A-3 female parent ‘Ningqi No. 1’ leaves; A-4 male parent ‘Yunnan Gouqi’ leaves; B a selection of the hybrid offspring showing differences in fruits and leaves
Results
Leaf and fruit trait variation
Seven leaf- and fruit-related phenotypic traits were measured for three continuous years from 2016 to 2018. The coefficient of variation of most phenotypic traits was > 10%, with the highest value for fruit index (FI) in 2018 (30%) and the lowest (9%) for fruit diameter (FD) in 2016 (Table 1), indicating that all traits showed natural variation in the F1 population. The seven traits were normally or partially normally distributed (Table 1 and Fig. 2A-C). Correlation analysis revealed significant or extremely significant positive correlations between leaf width (LW) and leaf length (LL), leaf index (LI) and LL, fruit longitude (FL) and LL, FI and LL, and LI and FI (P < 0.05) and between single-fruit weight (FW) and FL, FW and FD, and FI and FL (P < 0.01), respectively, whereas a significant or extremely negative correlation was observed between pairs LI and LW and LI and FD (P < 0.05) and between FI and FD (P < 0.01), respectively (Fig. 2A-C).
Table 1.
Data summary for Lycium phenotypes
| Trait | Year | Mean ± SD | Maximum | Minimum | Skewness | Kurtosis | Coefficient of variation (%) | Variance | Shapiro–Wilk P value |
|---|---|---|---|---|---|---|---|---|---|
| Leaf length | 2016 | 45.46 ± 7.69 | 73.13 | 26.26 | 0.48 | 0.67 | 17.00 | 59.19 | 0.010 |
| 2017 | 38.84 ± 7.71 | 60.49 | 21.79 | 0.30 | -0.21 | 20.00 | 59.39 | 0.278 | |
| 2018 | 39.33 ± 9.03 | 62.16 | 21.73 | 0.15 | -0.67 | 23.00 | 81.47 | 0.042 | |
| Leaf width | 2016 | 12.12 ± 2.09 | 17.95 | 7.81 | 0.10 | -0.62 | 17.00 | 4.39 | 0.153 |
| 2017 | 11.78 ± 2.38 | 18.81 | 6.10 | 0.59 | 0.17 | 20.00 | 5.68 | 0.003 | |
| 2018 | 11.41 ± 2.54 | 19.92 | 5.70 | 0.36 | -0.14 | 22.00 | 6.47 | 0.097 | |
| Leaf index | 2016 | 3.60 ± 0.58 | 6.88 | 2.66 | 2.14 | 7.65 | 16.00 | 0.33 | 0.000 |
| 2017 | 3.32 ± 0.48 | 4.80 | 2.53 | 1.05 | 0.86 | 14.00 | 0.23 | 0.000 | |
| 2018 | 3.50 ± 0.52 | 5.32 | 2.21 | 0.58 | 0.74 | 15.00 | 0.27 | 0.007 | |
| Fruit weight | 2016 | 0.79 ± 0.18 | 1.36 | 0.43 | 0.34 | 0.27 | 23.00 | 0.03 | 0.146 |
| 2017 | 0.91 ± 0.22 | 1.68 | 0.40 | 0.20 | 0.15 | 25.00 | 0.05 | 0.436 | |
| 2018 | 0.95 ± 0.24 | 1.66 | 0.51 | 0.76 | 0.30 | 12.00 | 0.06 | 0.000 | |
| Fruit longitude | 2016 | 19.25 ± 2.40 | 26.67 | 11.55 | 0.00 | 1.03 | 14.00 | 5.75 | 0.032 |
| 2017 | 18.68 ± 2.63 | 25.85 | 11.06 | -0.19 | 0.38 | 17.00 | 6.89 | 0.119 | |
| 2018 | 18.67 ± 3.23 | 27.74 | 9.60 | 0.33 | 0.24 | 10.00 | 10.46 | 0.176 | |
| Fruit diameter | 2016 | 9.22 ± 0.89 | 11.22 | 6.55 | -0.62 | 0.20 | 9.00 | 0.80 | 0.003 |
| 2017 | 10.08 ± 0.95 | 13.91 | 7.72 | 0.13 | 0.79 | 14.00 | 0.90 | 0.053 | |
| 2018 | 9.39 ± 1.29 | 12.47 | 2.55 | -0.82 | 3.82 | 12.00 | 1.66 | 0.000 | |
| Fruit index | 2016 | 2.10 ± 0.25 | 2.72 | 1.53 | 0.34 | -0.19 | 13.00 | 0.06 | 0.110 |
| 2017 | 1.87 ± 0.24 | 2.50 | 1.10 | 0.13 | 0.19 | 13.00 | 0.06 | 0.249 | |
| 2018 | 2.05 ± 0.62 | 6.88 | 1.00 | 3.30 | 21.75 | 30.00 | 0.38 | 0.000 |
Fig. 2.
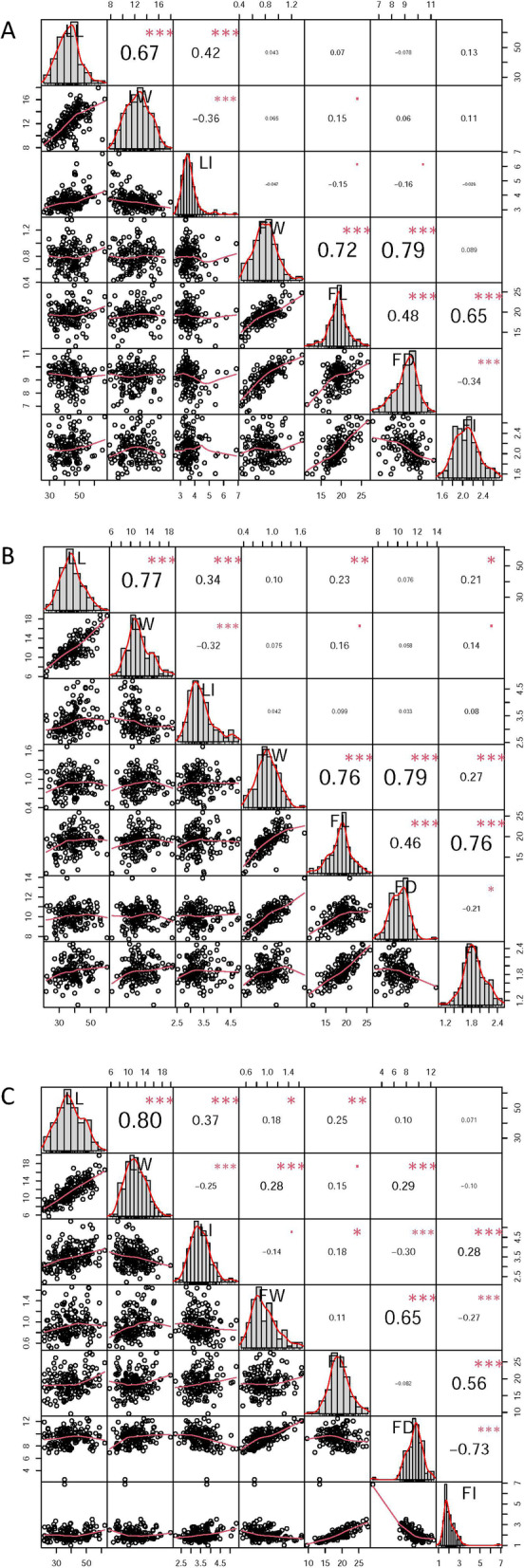
The variation and Pearson pairwise correlation analyses of leaf-related and fruit-related traits of the F1 population. (A), (B) and (C) represent the variation and Pearson pairwise correlations in 2016, 2017 and 2018, respectively. The correlations were calculated with Spearman correlation coefficients, and the P values are indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. The analysis was performed using the R package PerformanceAnalytics. Histograms for LL (leaf length), LW (leaf width), LI (leaf index), FW (fruit weight), FL (fruit length), FD (fruit diameter) and FI (fruit index) are displayed along the diagonal
Variation calling and annotation
Whole-genome resequencing generated a total of 4,549 million clean paired-end reads, 23,968 and 227,408 million in the female and male parents, respectively, with a Q30 value of 92.06%, indicating that high-quality source data were generated. The average depths in the female and male parents were 34 × and 18 × , respectively, and the average depth in the offspring was 2.58 × (Supplementary Table 1). All clean reads were mapped onto the scaffolds of the Lycium reference genome, with 9,015,078 SNPs and 1,317,594 InDels called between the parents. The variation maps of SNPs and InDels are shown in Fig. 3. The SNP density was 4,880 per Mb, and the InDel density was 714 per Mb. Most annotated SNPs (63.74%) were located in intergenic regions, whereas in the coding sequence (CDS) region, most SNPs were nonsynonymous (54.66%). Similar to the SNPs, more than half of the InDels (53.87%) were annotated in intergenic regions, whereas 1.37% were located in the CDS. Of the CDS InDels, 60.77% gave rise to frameshift mutations (Fig. 3). Of all the SNPs, 8,734,495 were successfully classified into eight genotyping patterns, and a set of 3,451,010 SNPs (excluding those with pattern type aa × bb) were used to construct a high-density genetic map of Lycium.
Fig. 3.

Genome variations and annotations. A Circos plot of SNP and InDel distributions. The outer ring indicates the SNP distribution, whereas the inner ring indicates the InDel distribution; B Pie charts of InDel annotations; C Pie charts of SNP annotation information
Construction of an ultradense genetic map and a consensus genetic map
To guarantee a set of high-quality SNP markers, SNPs with a depth in the parents < 10 × , SNPs with an integrity ≤ 70%, and highly significant SNPs with SD (P < 0.01) were filtered out. Finally, a set of 10,446 SNPs was used to construct a high-density genetic map of wolfberry, into which 8,507 SNPs were successfully integrated (Supplementary Fig. 1A). The integrated genetic map included 12 linkage groups (LGs) with a total genetic distance of 2,122.24 cM and an average map distance of 0.25 cM. LG07 included the largest number of markers (1,035), with an average distance of 0.18 cM and a total genetic distance of 182.59 cM. The smallest number of markers (510) was in LG03, spanning 119.69 cM with an average distance of 0.24 cM. The largest gap stretched across 19.41 cM in LG11. The ratios of genetic distance between adjacent markers < 5 cM ranged from 98.19% to 99.83% in LG05 and LG02, respectively (Table 2). To further integrate our published map [14], a consensus genetic map was constructed, which contained 15,240 SNPs with a total genetic distance of 3,058.19 cM and an average map distance of 0.21 cM (Table 2 and Supplementary Fig. 1B). Compared with the NY map, this consensus map harbored 6,733 more SNPs and 0.04 cM less average distance, indicating a higher resolution and likely the genetic map with the largest number of SNP markers for ligneous plants. In addition, the maximum gap was also somewhat narrowed. The consensus map represented a comprehensive improvement.
Table 2.
Summary of the total number of SNP markers in the 12 linkage groups
| Linkage group | Total markers | Total distance (cM) | Average distance (cM) | Maximum gap (cM) | Gaps < 5 cM (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| NY genetic map | Consensus map | NY genetic map | Consensus map | NY genetic map | Consensus map | NY genetic map | Consensus map | NY genetic map | Consensus map | |
| LG01 | 735 | 921 | 218.65 | 278.29 | 0.3 | 0.30 | 15.4 | 6.02 | 99.73 | 99.78 |
| LG02 | 576 | 1262 | 158.03 | 270.61 | 0.27 | 0.21 | 8.41 | 8.41 | 99.83 | 99.92 |
| LG03 | 510 | 1197 | 119.69 | 183.01 | 0.24 | 0.15 | 8.56 | 11.70 | 98.82 | 99.58 |
| LG04 | 518 | 1097 | 159.09 | 229.71 | 0.31 | 0.21 | 11.79 | 8.49 | 99.23 | 99.73 |
| LG05 | 608 | 812 | 193.19 | 261.22 | 0.32 | 0.32 | 17.86 | 17.86 | 98.19 | 98.77 |
| LG06 | 766 | 1436 | 231.03 | 328.13 | 0.3 | 0.23 | 12.16 | 12.16 | 98.95 | 99.37 |
| LG07 | 1,035 | 1725 | 182.59 | 267.50 | 0.18 | 0.16 | 13.7 | 13.70 | 99.61 | 99.65 |
| LG08 | 752 | 1212 | 232.82 | 324.95 | 0.31 | 0.27 | 9.49 | 9.49 | 99.33 | 99.34 |
| LG09 | 819 | 1437 | 190.72 | 250.42 | 0.23 | 0.17 | 9.57 | 9.57 | 99.63 | 99.72 |
| LG10 | 901 | 1395 | 188.58 | 268.11 | 0.21 | 0.19 | 13.07 | 13.07 | 98.89 | 99.5 |
| LG11 | 569 | 1518 | 165.38 | 248.61 | 0.29 | 0.16 | 19.41 | 19.41 | 98.94 | 99.67 |
| LG12 | 718 | 1228 | 82.47 | 147.63 | 0.12 | 0.12 | 14.61 | 6.06 | 99.16 | 99.84 |
| Total | 8,507 | 15,240 | 2,122.24 | 3058.19 | 0.25 | 0.21 | 19.41 | 11.33 | 99.19 | 99.57 |
Genetic map-assisted genome assembly
High-density linkage maps can assist in chromosome-level genome assembly. To assemble the genome of Lycium at the chromosome level on the basis of our high-density consensus genetic map, we used ALLMAPS. Finally, ~ 1.21 Gb of scaffolds were mounted to 12 pseudochromosomes of Lycium, accounting for ~ 64.3% of the genome sequence, 51.3% of which were oriented (Fig. 4; Supplementary Fig. 2; Table 3). The longest pseudochromosome was LG10, with a total length of 132.33 Mb, whereas only 57 Mb and 57.69 Mb were mounted onto LG05 and LG01, respectively, in line with the trends in SNP numbers in the genetic map. More scaffolds were not mounted (783,254), among which scaffolds < 1 kb accounted for 99.3% (777,737/783,254). ALLMAPS scaffolding was performed by inferring and maximizing the collinearity between the genetic map and scaffolds/contigs. By comparing the collinearities between all LGs and pseudochromosomes, we found that certain collinearities occurred between each LG and the corresponding pseudochromosomes. Pseudochromosome 04 showed the lowest P value. Moreover, the P values of collinear pseudochromosomes 01, 02, 08, 09, and 11 were all > 0.8 (Supplementary Fig. 2).
Fig. 4.

Pseudochrome 8 of Lycium anchored by the consensus map. The left panel represents synteny between the consensus map and the reference genome. The right panel represents the correlations between the consensus map and the reference genome. The ρ-values represent the Pearson correlation coefficients
Table 3.
High-density genetic map-assisted genome assembly
| Index | Anchored | Oriented | Unplaced |
|---|---|---|---|
| Unique mapped markers | 12,442 | 10,540 | 2,162 |
| Markers per Mb | 10.3 | 10.9 | 3.2 |
| Scaffolds | 1,643 | 1,044 | 783,254 |
| Scaffolds with 1 anchored marker | 336 | 0 | 104 |
| Scaffolds with 2 anchored markers | 111 | 77 | 187 |
| Scaffolds with 3 anchored markers | 167 | 110 | 70 |
| Scaffolds with ≥ 4 anchored markers | 1,029 | 857 | 181 |
| Total bases (bp) | 1,208,281,949 | 963,551,338 | 670,115,130 |
| Mapping rate (%) | 64.3 | 51.3 | 35.7 |
Synteny analyses
We used the consensus genetic map to perform collinearity analysis with the reference genomes of pepper, potato, and tomato. Weak collinearity was found between wolfberry and the three Solanaceae species, and most regions located at the ends of chromosomes showed high collinearity. The collinear segment pairs between wolfberry and pepper were LG02-chromosome 02, LG04-chromosome 12, LG06-chromosome 03, LG09-chromosome 12, LG10-chromosome 08, and LG11-chromosome 04. The collinear segment pairs of wolfberry and potato were LG02-chromosome 02, LG06-chromosome 03, LG04-chromosome 11, G10-chromosome 01, LG11-chromosome 04, and LG12-chromosome 08. The collinear segment pairs of wolfberry and tomato were LG06-chromosome 03, LG10-chromosome 01, LG11-chromosome 04, and LG12-chromosome 08 (Fig. 5). Among these collinearity pairs, there were overlaps between wolfberry-pepper pairs and wolfberry-tomato pairs on chromosomes 02, 04, 06, 10, and 11 of wolfberry, whereas overlaps between wolfberry-tomato pairs and wolfberry-potato pairs were observed on chromosomes 06, 10, 11, and 12 of wolfberry. Furthermore, there was a potential chromosomal inversion between wolfberry-tomato pairs and wolfberry-potato pairs on chromosome 10 of wolfberry.
Fig. 5.

Synteny analyses between the genetic map of Lycium and the genomes of pepper (A), potato (B), and tomato (C)
QTL mapping
Using the resequencing genetic map and continuous phenotypic data from 2016 to 2018, a large number of QTLs responsible for seven agronomic traits were mapped. The QTLs were distributed in all the LGs of wolfberry except LG08, with phenotypic variance explained (PVE) values ranging from 6% to 73.6%, and the maximum LOD value was 21.39. The fruit-related QTLs were mapped mainly to LG06, LG07, LG09, LG10, and LG12, whereas the leaf-related QTLs were located mainly in LG01, LG06, LG09, and LG10 (Supplementary Table 2).
We further screened stable QTLs detected in at least two years, and a total of 25 such QTLs were identified (Supplementary Table 3). We identified 13 QTLs for the leaf index (LI), and these QTLs were located in LG10 from 45.967 cM to 73.823 cM (27.856 cM), including 44 SNPs. The largest LOD (13.13) and PVE (50.5%) values were observed for qLI10-4 and qLI10-12, respectively (Fig. 6). Some LI QTLs in LG10 were gathered tightly with an average interval of less than 0.63 cM per marker, indicating that these QTLs might belong to the same QTL (Fig. 6). For LL, two QTLs (qLL9-1 and qLL9-2) were mapped to LG09 and supported by 16 SNPs. One stable FW QTL was anchored to LG10 with a PVE value of 59.2% but supported by one marker. Two stable FL QTLs were anchored to LG10 and LG12 (qFL10 and qFL12) with nine significantly linked SNPs, explaining 7.3% to 36.9% of the phenotypic variation (Supplementary Table 3). Five QTLs (qFI10-1 ~ qFI10-5) at 37.344 ~ 66.309 cM in LG10 accounted for 15.8% to 51.9% of the PVE, with the highest LOD value (more than 5.0) in these QTL regions. Notably, qFI10-4 was detected repeatedly in 2017 and 2018 (Fig. 6). Two stable QTLs (qFI7-1 and qFI7-2) for the FI at 60.634 ~ 61.916 cM in LG07 accounted for 8.5% and 19.3% of the PVE, respectively, with an average intermarker distance of 0.18 cM. We extracted predicted genes within 150 kb upstream and downstream of the markers in stable QTLs. To further explore the expression of stable QTLs for fruit-related traits, we performed RNA-seq and found that 82 out of 188 predicted genes showed differential expression (Supplementary Table 4). These genes represent valuable resources for further gene cloning and marker-assisted selection (MAS).
Fig. 6.

Stable QTLs for the fruit index (2017, 2018) and leaf index (2016, 2017) with higher confidence intervals on chromosome 10
Discussion
A high-density genetic map can provide valuable information for deciphering the genetic basis of important and complex agronomic traits. With the rapid development of sequencing technology, SNPs and InDels have been mined for high-density linkage map construction, QTL dissection, candidate gene discovery and breeding applications in crops [10, 15, 16]. Whole-genome resequencing for SNP genotyping in biparental segregation populations has been successfully applied to plants [17–19]. Because of our complete de novo assembled genome sequence of Lycium (Cao et al., unpublished 2021), it has become possible to use whole-genome resequencing for genome-wide SNP discovery and high-density genetic map construction in Lycium. In this study, whole-genome resequencing resulted in 9,015,078 SNPs and 1,646,131 InDels, and a high-density genetic map containing 8,507 SNPs spanning 2,122.24 cM with an average distance of 0.25 cM between adjacent markers was constructed based on a hybrid F1 population (NY map). However, the maximum gap in the integrated map was 19.41 cM, in LG 11. This indicated that there was recombination or undeveloped markers in this area [20]. Compared with that in the first sequencing-based genetic map of Lycium that we constructed [14], the number of F1 individuals was reduced by 100 (the number in the first population containing 302 individuals), whereas the average distance was very similar to that of the published genetic map. Nevertheless, the total number of SNPs and total map distance of this resequencing-based linkage map were higher, indicating higher resolution for resequencing than for reduced-representation sequencing. The consensus map developed by integrating these two high-density maps indicated that a saturated genetic map had been obtained for Lycium.
High-density genetic maps can improve contig/scaffold assembly at the chromosome level [21–23]. Generally, a genetic map can distribute 60–90% of all the assembled contigs/scaffolds on pseudochromosomes [24–26]. Given the increased marker density, the distribution rate of the consensus genetic map will increase accordingly [23, 27]. In this study, a high-density genetic map of Lycium was constructed on the basis of resequencing, and a consensus map was produced by integrating the high-density genetic map and a previously published map [14]. The final genetic map contained 15,240 markers in 12 LGs. Based on this consensus genetic map, the scaffold Lycium genome (Cao et al., unpublished 2021) was mounted onto 12 pseudochromosomes with a distribution rate of 64.30%, which is relatively low. There were 783,254 unmounted scaffolds, which was 476.72 times the number of anchored scaffolds. In addition, the average length of the unplaced scaffolds was 0.86 kb. Therefore, we speculate that a fragmented genome assembly is the cause of the low distribution rate. For the Lycium genome used in this study, the distribution rate could benefit from the use of PacBio long-read sequencing [28], a BioNano optical map [24], or Hi-C technology [29].
To gain insights into the evolutionary history of wolfberry, we conducted collinearity analyses on the genomes of wolfberry, tomato, pepper, and potato. Similar to previous results [14], the genetic map of wolfberry showed low collinearity with these three genomes overall, suggesting greater differentiation of wolfberry. However, high collinearity was observed in some cases, such as segments on chromosomes 02, 04, 06, 10, and 11 of wolfberry for wolfberry-pepper pairs and wolfberry-tomato pairs, suggesting that these intervals are highly conserved among different species of Solanaceae. In addition, marker density was associated with collinearity as more markers meant a greater probability of markers aligning to the corresponding reference genome sequences. The markers on a high-density genetic map were typically developed by reduced-representation sequencing [14, 30] or re-sequencing [17–19, 31], which could yield thousands of randomly distributed markers. But the reference genomes of plants always harbor hundreds of Mbp at least. So only the collinearity of conserved regions could be detected using a sequencing-based high density genetic map and we believe that a comprehensive genomic synteny could be realized using a genome assembly of wolfberry.
Fruits and leaves are the main medicinal and edible parts of Lycium spp. [32]. In our study, many QTLs (Supplementary Table 2) were identified for fruit and leaf traits based on phenotypic data collected over 3 years. Most of the measured traits were located at more than two QTLs, with QTLs for the FI mapped to LG04, LG09, LG11, and LG12 at different positions. In addition, we found a large number of QTLs linked to two or three traits, with those for the FW, FL and FI traits mapped to 37.344 cM in LG10 with LOD and PVE values up to 16.02 and 62.5%, respectively. These results indicated that fruit and leaf traits in Lycium are controlled by multiple loci and single loci with pleiotropic effects [33].
Stable QTLs are valuable and useful for MAS-based breeding programs and have been identified in perennials [34]. In this study, 25 stable QTLs associated with two leaf and three fruit traits were identified in LG07, LG09, LG10, and LG11 (Supplementary Table 3), four of which were identified in two LG10 regions (38.6 cM and 66.3 cM) with a high LOD value (15.7– 45.3) and were related to three traits (LI, FW, and FI). In addition, in a previous study [14], two stable QTLs for FW were located in the same two LG regions (133.6 cM and 146.4 cM) as two stable QTLs for FD, which might indicate that these stable QTLs (qLI10-10, qFW10, qFI10-2, and qFI10-5) show high reliability in different environments and should be considered major QTLs. Of note, given these major QTLs, there is a genetic basis for the phenotypic correlation between FW and the FI, which is consistent with the strong correlation observed between the two traits in phenotypic analyses (Fig. 1A-C). Therefore, we speculate that two regions (38.6 cM and 66.3 cM) in LG10 play crucial roles in regulating Lycium fruit growth and development. The SNPs underlying these QTLs could be verified using the 100 remaining F1 individuals in the future, and the tightly linked SNPs could be converted into kompetitive allele-specific PCR (KASP) markers and potentially used as early selection markers.
Methods
Mapping population construction and phenotyping
A hybrid wolfberry population derived from a cross between ‘Ningqi No. 1’ (NN) (L. barbarum L.) and ‘Yunnan Gouqi’ (YG) (Lycium yunnanense Kuang et A.M. Lu) was generated in August 2014. The female parent NN is a major artificial breeding cultivar in Northwest China. Its fruit is bright red and elliptical with a sweet taste, and its leaves are lanceolate. The male parent YG is a wild-type wolfberry with dark red, long, oval, bitter-tasting fruit (Fig. 1). Seeds of the F1 hybrid and the two parents were collected and sown in the Ningxia Academy of Agriculture and Forestry Sciences National Wolfberry Germplasm Resources Garden (38°38 N, 106°90 E), Yinchuan City, Hui Autonomous Region, Ningxia, China, in May 2015. In total, 300 F1 individuals were grown, 200 of which were randomly selected to establish the mapping population. Water and fertilizer management was the same as that used in the production field. Weeds were managed manually.
The leaf- and fruit-related traits were measured in the F1 population (NY) and the two parents. LL was the maximum distance between the leaf base and tip. LW was the widest distance across the leaf. FW was the weight of one mature fruit. FL was the maximum distance between the top and bottom of the fruit. FD was the widest distance across a fruit. LL, LW, FW, FL, and FD were measured according to methods described elsewhere [35]. LL, LW, FL, and FD were measured using Vernier calipers, whereas FW was acquired using an electronic balance (SE602F, Ohaus, NJ, USA). LI and FI were calculated according to Equations (i) and (ii): (i) LI = LL/LW [35] and (ii) FI = FL/FD [35]. In total, 30 leaves and fruits collected from each tree for 3 consecutive years (from 2016 to 2018) were used to obtain phenotypic data. The average values of each trait per individual were used for QTL analysis. Complex variance analysis, variance analysis, and correlation analysis were carried out using SPSS V17.0 software (SPSS Inc., Chicago, IL, USA).
Population resequencing and genotyping
Genomic DNA was extracted from the young leaves of both parents and 200 F1 plants using a Super Plant Genomic DNA DP360 Kit (Tiangen Biotech, Beijing, China). DNA concentration was measured using a NanoDrop spectrophotometer (ND2000, Thermo Fisher Scientific, USA), and DNA quality was monitored by electrophoresis on 0.85% agarose gels. The genomic DNA was sheared into 350-bp fragments using an S2/E210 Ultrasonicator (Covaris, USA). The purified products were then ligated for end repair, subjected to 3’A and adaptor addition, and selected according to fragment size on a 1% agarose gel. The final library was constructed by PCR. Library quantification was performed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), and the fragments of the libraries were paired-end sequenced (PE125) using the Illumina XTen platform (Illumina, San Diego, CA, USA) according to the manufacturer’s recommendations.The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0001536.
Raw reads were filtered to generate high-quality clean reads by (i) removing adaptor sequences, (ii) filtering reads with > 10% unidentified nucleotides, and (iii) removing reads with > 50% bases with a low Phred quality score (≤ 10). Burrows-Wheeler Aligner [36] was used to align the clean reads to the Lycium genome (Cao et al., unpublished 2021), and duplicates were identified using Picard (Picard: http://sourceforge.net/projects/picard/). SNPs and insertions and deletions (InDels; 1–5 bp) were called using GATK software [37] and then annotated by SnpEff [38]. Genome variation maps of SNPs and InDels were drawn by Circos [39]. SNP genotypes were encoded using biallelic coding rules and eight genotyping patterns (aa × bb, ab × cd, ef × eg, hk × hk, lm × ll, nn × np, ab × cc, and cc × ab). All patterns except aa × bb were selected to construct a high-density genetic map for a cross-pollinated (CP) population.
Genetic linkage map construction and QTL mapping
The resulting SNPs were further screened. Specifically, hk × hk and nn × np segregation-type SNPs with a depth < 6 × in the parents, an integrity < 60%, and significant segregation distortion (SD) (Chi-square P < 0.05) were filtered out, whereas the remaining segregation types with a depth < 10 × in the parents, an integrity < 70%, and highly significant SD (chi-square P < 0.01) were filtered out. SNP markers were arranged into linkage groups (LGs) based on pairwise modified logarithm of odds (MLOD) scores. Markers with MLOD scores > 5 were assigned to a single LG. SMOOTH algorithms [40] were used to correct genotypes, and then the k-nearest neighbor approach [41] was used for genotype imputation. The JoinMap software (V4.1) mapping function for the CP population type was applied for linear arrangement within LGs. Map distances were estimated using the Kosambi mapping function [42]. A heat map of adjacent SNP relationships was generated using R (www.r-project.org/). MapQTL V6.0 with the interval mapping (IM) model was used for QTL analyses [43], and the LOD threshold value was set to 2.5. QTLs with a threshold LOD value > 3.0 and PVE > 10% were considered major QTLs.
Genetic map-assisted genome scaffold assembly and genome synteny analyses
SLAF markers of the published genetic map (NC map) [14] and SNPs identified in this study were aligned to the Lycium scaffold-level genome assembly (Cao et al., unpublished 2021) using BLAT software [44]. The corresponding relationship and shared markers of these two LGs were extracted according to the locations of SLAF and SNP markers. An integrated genetic map was constructed using BioMercator v4.2 [45]. This integrated genetic map was used to anchor the scaffolds of the Lycium genome at the chromosome level using ALLMAPS software [46]. The SNPs integrated genetic map were aligned to the genomes of solanaceous relatives, namely, pepper (http://peppersequence.genomics.cn/page/species/download.jsp #5), tomato (ftp://ftp.solgenomics.net/tomato_genome/assembly/build_2.50/), and potato (http://solanaceae.plantbiology. msu.edu /pgsc_download.shtml), using BLAT [44] and the physical positions of the homologous sequence were used to generate a collinearity diagram in R.
RNA-seq analysis
The fruits of the two parents were sampled in triplicate 36–40 days after pollination for total RNA extraction [RNAprep Pure Plant Kit (Tiangen Biotech, Beijing, China)]. An RNA-seq library was constructed according to [47]; the library was then loaded into Cbot for cluster generation and for 150-bp paired-end read sequencing on the Illumina NovaSeq platform (Illumina, San Diego, CA, USA). Low-quality reads [> 20% of bases with a Q value ≤ 20 or an ambiguous sequence content (‘N’) exceeding 5%] were removed by an in-house Perl script. The clean reads were then mapped to the reference genome of wolfberry (Cao et al., unpublished 2021) using STAR with default settings [48]. StringTie was used to assemble transcripts [49]. The fragments per kilobase of transcript per million mapped reads (FPKM) method was used to quantify transcript expression levels, and the DESeq R package was used to detect differentially expressed genes (DEGs). Significant DEGs were identified using an adjusted P value < 0.05 and a fold change ≥ 2.
Supplementary Information
Additional file 1: Figure 1. Two genetic map. (A) The high-density genetic map by resequencing; (B) The consensus genetic map.
Additional file 2: Figure 2. Genetic map-assisted genome assembly from LG01 to LG12 except LG08.
Additional file 3: Table 1. Data generated during resequencing.
Additional file 4: Table 2. QTLs detected in the F1 population.
Additional file 5: Table 3. Stable QTLs.
Additional file 6: Table 4. Annotation of differentially expressed genes underlying QTLs for fruit-related traits.
Abbreviations
- SNPs
Single-nucleotide polymorphisms
- QTLs
Quantitative trait loci
- LOD
Logarithm of odds
- NN
‘Ningqi No. 1’
- YG
‘Yunnan Gouqi’
- NC
‘Ningqi No. 1’ × ‘Chinese Gouqi’
- NY
‘Ningqi No. 1’ × ‘Yunnan Gouqi’
- MAS
Marker-assisted selection
- NGS
Next-generation sequencing
- LL
Leaf length
- LW
Leaf width
- LI
Leaf index
- FW
Single-fruit weight
- FL
Fruit longitude
- FD
Fruit diameter
- FI
Fruit index
- CDS
Coding sequence
- LGs
Linkage groups
- SD
Segregation distortion
- PVE
Phenotypic variance explained
- KASP
Kompetitive allele-specific PCR
- CNGBdb
China National GeneBank DataBase
- CP
Cross-pollinated
- MLOD
Modified logarithm of odds
- FPKM
Fragments per kilobase of transcript per million mapped reads
- DEGs
Differentially expressed genes
Authors’ contributions
ZJ and AW conceived and designed the experiments; LH, YY, HT and ZB performed F1 population construction and phenotying; WY and LY performed the RNA isolation and RNA-seq data analysis; XY performed the genetic analysis of the plant populations, genetic map analysis and QTL mapping; ZJ and CY wrote the manuscript and ZJ revised the manuscript. All authors have read and approved the final manuscript.
Funding
This work was sponsored by the National Natural Science Foundation of China (32060359, 31660220); Ningxia Hui Autonomous Region Science and Technology Innovation Leading Talents Project (KJT2017004); Special Foundation for Agricultural Breeding of the Ningxia Hui Autonomous Region (2013NYYZ0101).
Availability of data and materials
All the sequencing clean data were uploaded to the China National GeneBank DataBase (CNP0001536). However, the data will be made public on 1 April 2022. Before 1 April 2022, the datasets are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jianhua Zhao, Email: zhaojianhua0943@163.com.
Haoxia Li, Email: lihaoxia0943@163.com.
Yuhui Xu, Email: genetics_2010@163.com.
Yue Yin, Email: yueyin0112@aliyun.com.
Ting Huang, Email: ht20180312@163.com.
Bo Zhang, Email: zhang_bo_0309@126.com.
Yajun Wang, Email: yajun@163.com.
Yanlong Li, Email: lylsxfp@163.com.
Youlong Cao, Email: youlongchk@163.com.
Wei An, Email: angouqi@163.com.
References
- 1.Fukuda T, Yokoyama J, Ohashi H. Phylogeny and biogeography of the genus Lycium (Solanaceae): inferences from chloroplast DNA sequences. Mol Phylogenet Evol. 2001;19:246–258. doi: 10.1006/mpev.2001.0921. [DOI] [PubMed] [Google Scholar]
- 2.Zhao XQ, Guo S, Yan H, Lu YY, Zhang F, Qian DW, et al. Analysis of phenolic acids and flavonoids in leaves of Lycium barbarum from different habitats by ultra-high-performance liquid chromatography coupled with triple quadrupole tandem mass spectrometry. Biomed Chromatogr. 2019;33:e4552. doi: 10.1002/bmc.4552. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Liu X, Zhu L, Wang Y. Nuclear genome size estimation and karyotype analysis of Lycium species (Solanaceae) Sci Hortic. 2013;151:46–50. doi: 10.1016/j.scienta.2012.12.004. [DOI] [Google Scholar]
- 4.Xiao X, Ren W, Zhang N, Bing T, Liu X, Zhao Z, et al. Comparative study of the chemical constituents and bioactivities of the extracts from fruits, leaves and root barks of Lycium barbarum. Molecules. 2019;24:1585. doi: 10.3390/molecules24081585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang CC, Chang SC, Inbaraj BS, Chen BH. Isolation of carotenoids, flavonoids and polysaccharides from Lycium barbarum L. and evaluation of antioxidant activity. Food Chem. 2010;120:184–192. 10.1016/j.foodchem.2009.10.005.
- 6.Pedro AC, Maurer JBB, Zawadzki-Baggio SF, Ávila S, Maciel GM, Haminiuk CWI. Bioactive compounds of organic goji berry (Lycium barbarum L.) prevents oxidative deterioration of soybean oil. Ind Crops Prod. 2018;112:90–97. 10.1016/j.indcrop.2017.10.052.
- 7.Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19:1068–1076. doi: 10.1101/gr.089516.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Q, Miao H, Li S, Zhang S, Wang Y, Weng Y, et al. A sequencing-based linkage map of cucumber. Mol Plant. 2015;8:961–963. doi: 10.1016/j.molp.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Zou G, Zhai G, Feng Q, Yan S, Wang A, Zhao Q, et al. Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J Exp Bot. 2012;63:5451–5462. doi: 10.1093/jxb/ers205. [DOI] [PubMed] [Google Scholar]
- 10.Gao ZY, Zhao SC, He WM, Guo LB, Peng YL, Wang JJ, et al. Dissecting yield-associated loci in super hybrid rice by resequencing recombinant inbred lines and improving parental genome sequences. P Natl Acad Sci USA. 2013;110:14492–14497. doi: 10.1073/pnas.1306579110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qi X, Li MW, Xie M, Liu X, Ni M, Shao G, et al. Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat Commun. 2014;5:4340. doi: 10.1038/ncomms5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S, Chen J, Zhang W, Hu Y, Chang L, Fang L, et al. Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol. 2015;16:108. doi: 10.1186/s13059-015-0678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han K, Lee HY, Ro NY, Hur OS, Lee LJ, Kwon JK, et al. QTL mapping and GWAS reveal candidate genes controlling capsaicinoid content in Capsicum. Plant Biotechnol J. 2018;16:1546–1558. doi: 10.1111/pbi.12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao J, Xu Y, Li H, Yin Y, An W, Li Y, et al. A SNP-based high-density genetic map of leaf and fruit related quantitative trait loci in wolfberry (Lycium Linn.). Front Plant Sci. 2019;10:977. 10.3389/fpls.2019.00977. [DOI] [PMC free article] [PubMed]
- 15.Pandey MK, Monyo E, Ozias-Akins P, Liang X, Guimarães P, Nigam SN, et al. Advances in Arachis genomics for peanut improvement. Biotechnol Adv. 2012;30:639–651. doi: 10.1016/j.biotechadv.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Rothan C, Diouf I, Causse M. Trait discovery and editing in tomato. Plant J. 2019;97:73–90. doi: 10.1111/tpj.14152. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Wei Y, Acharya A, Jiang Q, Kang J, Brummer EC. A saturated genetic linkage map of autotetraploid alfalfa (Medicago sativa L.) developed using genotyping-by-sequencing is highly syntenous with the Medicago truncatula genome. G3-Genes Genom Genet. 2014;4:1971–1979. 10.1534/g3.114.012245. [DOI] [PMC free article] [PubMed]
- 18.Hu Z, Deng G, Mou H, Xu Y, Chen L, Yang J, et al. A re-sequencing-based ultra-dense genetic map reveals a gummy stem blight resistance-associated gene in Cucumis melo. DNA Res. 2018;25:1–10. doi: 10.1093/dnares/dsx033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agarwal G, Clevenger J, Pandey MK, Wang H, Shasidhar Y, Chu Y, et al. High-density genetic map using whole-genome resequencing for fine mapping and candidate gene discovery for disease resistance in peanut. Plant Biotechnol J. 2018;16:1954–1967. doi: 10.1111/pbi.12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Lu J, Yu J, Gibbs RA, F. Yu. An integrative variant analysis pipeline for accurate genotype/haplotype inference in population NGS data. Genome Res. 2013;23:833–842. 10.1101/gr.146084.112. [DOI] [PMC free article] [PubMed]
- 21.Zhao L, Yuanda L, Caiping C, Xiangchao T, Xiangdong C, Wei Z, et al. Toward allotetraploid cotton genome assembly: integration of a high-density molecular genetic linkage map with DNA sequence information. BMC Genom. 2012;13:539. doi: 10.1186/1471-2164-13-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Xia Q, Zhang Y, Zhu X, Zhu X, Li D, et al. Updated sesame genome assembly and fine mapping of plant height and seed coat color QTLs using a new high-density genetic map. BMC Genom. 2016;17:31. doi: 10.1186/s12864-015-2316-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernhardsson C, Vidalis A, Wang X, Scofield DG, Schiffthaler B, Baison J, et al. An ultra-dense haploid genetic map for evaluating the highly fragmented genome assembly of norway spruce (Picea abies). G3-Gene Genom Genet. 2019;9:1623–1632. 10.1534/g3.118.200840. [DOI] [PMC free article] [PubMed]
- 24.Yang J, Liu D, Wang X, Ji C, Cheng F, Liu B, et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat Genet. 2016;48:1225–1232. doi: 10.1038/ng.3657. [DOI] [PubMed] [Google Scholar]
- 25.Hane JK, Ming Y, Kamphuis LG, Nelson MN, Garg G, Atkins CA, et al. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: insights into plant-microbe interactions and legume evolution. Plant Biotechnol J. 2017;15(15):318–330. doi: 10.1111/pbi.12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee BY, Kim MS, Choi BS, Nagano AJ, Au DWT, Wu RSS, et al. Construction of high-resolution RAD-seq based linkage map, anchoring reference genome, and QTL mapping of the sex chromosome in the marine medaka Oryzias melastigma. G3-Gene Genom Genet. 2019;9:3537–3545. 10.1534/g3.119.400708. [DOI] [PMC free article] [PubMed]
- 27.Xue H, Wang S, Yao JL, Deng CH, Wang L, Su Y, et al. Chromosome level high-density integrated genetic maps improve the Pyrus bretschneideri ‘DangshanSuli’ v1.0 genome. BMC Genom. 2018;19:833. 10.1186/s12864-018-5224-6. [DOI] [PMC free article] [PubMed]
- 28.Hazzouri KM, Gros-Balthazard M, Flowers JM, Copetti D, Lemansour A, Lebrun M, et al. Genome-wide association mapping of date palm fruit traits. Nat Commun. 2019;10:4680. doi: 10.1038/s41467-019-12604-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang QJ, Li W, Li K, Nan H, Shi C, Zhang Y, et al. The chromosome-level reference genome of tea tree unveils recent bursts of non-autonomous LTR retrotransposons in driving genome size evolution. Mol Plant. 2020;13:935–938. doi: 10.1016/j.molp.2020.04.009. [DOI] [PubMed] [Google Scholar]
- 30.Geleta M, Gustafsson C, Glaubitz JC, Ortiz R. High-Density Genetic Linkage Mapping of Lepidium Based on Genotyping-by-Sequencing SNPs and Segregating Contig Tag Haplotypes. Front Plant Sci. 2020;11:448. doi: 10.3389/fpls.2020.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tong Z, Zhou J, Xiu Z, Jiao F, Hu Y, Zheng F, et al. Construction of a high-density genetic map with whole genome sequencing in Nicotiana tabacum L. Genomics. 2020;112:2028–2033. doi: 10.1016/j.ygeno.2019.11.015. [DOI] [PubMed] [Google Scholar]
- 32.Dong JZ, Lu DY, Wang Y. Analysis of flavonoids from leaves of cultivated Lycium barbarum L. Plant Foods Hum Nutr. 2009;64:199–204. doi: 10.1007/s11130-009-0128-x. [DOI] [PubMed] [Google Scholar]
- 33.Eduardo I, Pacheco I, Chietera G, Bassi D, Pozzi C, Vecchietti A, et al. QTL analysis of fruit quality traits in two peach intraspecific populations and importance of maturity date pleiotropic effect. Tree Genet Genom. 2011;7:323–335. doi: 10.1007/s11295-010-0334-6. [DOI] [Google Scholar]
- 34.Pan Y, Liang X, Gao M, Liu H, Meng H, Weng Y, et al. Round fruit shape in WI7239 cucumber is controlled by two interacting quantitative trait loci with one putatively encoding a tomato SUN homolog. Theor Appl Genet. 2017;130:573–586. doi: 10.1007/s00122-016-2836-6. [DOI] [PubMed] [Google Scholar]
- 35.Shi Z, Du H, Men H. Goji Germplasm Resource Description Standardization and Data Standards. Beijing: China Forestry Publishing House; 2012. [Google Scholar]
- 36.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20L1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed]
- 38.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Os H, Stam P, Visser RG, van Eck HJ. SMOOTH: a statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor Appl Genet. 2005;112:187–194. doi: 10.1007/s00122-005-0124-y. [DOI] [PubMed] [Google Scholar]
- 41.Huang X, Zhao Y, Wei X, Li C, Wang A, Zhao Q, et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet. 2012;44:32–39. doi: 10.1038/ng.1018. [DOI] [PubMed] [Google Scholar]
- 42.Van Ooijen JW. JoinMap 4: Software for the Calculation of Genetic Linkage Maps in Experimental Populations. Kyazma BV, Wageningen, NL, 2006;33:10–1371.
- 43.Van Ooijen JW. MapQTL 6.0. Software for the Mapping of Quantitative Trait Loci in Experimental Populations of Diploid Species. Kyazma BV, Wageningen, NL, 2009, p 59.
- 44.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, et al. BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics. 2004;20:2324–2326. doi: 10.1093/bioinformatics/bth230. [DOI] [PubMed] [Google Scholar]
- 46.Tang H, Zhang X, Miao C, Zhang J, Ming R, Schnable JC, et al. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 2015;16:3. doi: 10.1186/s13059-014-0573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miao Y, Zhu Z, Guo Q, Zhu Y, Yang X, Sun Y. Transcriptome analysis of differentially expressed genes provides insight into stolon formation in Tulipa edulis. Front Plant Sci. 2016;7:409. doi: 10.3389/fpls.2016.00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure 1. Two genetic map. (A) The high-density genetic map by resequencing; (B) The consensus genetic map.
Additional file 2: Figure 2. Genetic map-assisted genome assembly from LG01 to LG12 except LG08.
Additional file 3: Table 1. Data generated during resequencing.
Additional file 4: Table 2. QTLs detected in the F1 population.
Additional file 5: Table 3. Stable QTLs.
Additional file 6: Table 4. Annotation of differentially expressed genes underlying QTLs for fruit-related traits.
Data Availability Statement
All the sequencing clean data were uploaded to the China National GeneBank DataBase (CNP0001536). However, the data will be made public on 1 April 2022. Before 1 April 2022, the datasets are available from the corresponding author on reasonable request.