

Abstract
The coronavirus pandemic has affected more than 150 million people, while over 3.25 million people have died from the coronavirus disease 2019 (COVID-19). As there are no established therapies for COVID-19 treatment, drugs that inhibit viral replication are a promising target; specifically, the main protease (Mpro) that process CoV-encoded polyproteins serves as an Achilles heel for assembly of replication-transcription machinery as well as down-stream viral replication. In the search for potential antiviral drugs that target Mpro, a series of cembranoid diterpenes from the biologically active soft-coral genus Sarcophyton have been examined as SARS-CoV-2 Mpro inhibitors. Over 360 metabolites from the genus were screened using molecular docking calculations. Promising diterpenes were further characterized by molecular dynamics (MD) simulations based on molecular mechanics-generalized Born surface area (MM-GBSA) binding energy calculations. According to in silico calculations, five cembranoid diterpenes manifested adequate binding affinities as Mpro inhibitors with ΔGbinding < −33.0 kcal/mol. Binding energy and structural analyses of the most potent Sarcophyton inhibitor, bislatumlide A (340), was compared to darunavir, an HIV protease inhibitor that has been recently subjected to clinical-trial as an anti-COVID-19 drug. In silico analysis indicates that 340 has a higher binding affinity against Mpro than darunavir with ΔGbinding values of −43.8 and −34.8 kcal/mol, respectively throughout 100 ns MD simulations. Drug-likeness calculations revealed robust bioavailability and protein-protein interactions were identified for 340; biochemical signaling genes included ACE, MAPK14 and ESR1 as identified based on a STRING database. Pathway enrichment analysis combined with reactome mining revealed that 340 has the capability to re-modulate the p38 MAPK pathway hijacked by SARS-CoV-2 and antagonize injurious effects. These findings justify further in vivo and in vitro testing of 340 as an antiviral agent against SARS-CoV-2.
Keywords: genus Sarcophyton, cembranoid diterpenes metabolites, SARS-CoV-2 main protease, molecular docking, molecular dynamics, reactome
1. Introduction
Coronavirus (CoV) belonging to the Coronaviridae family is one of the largest families of positive-sense, RNA viruses [1]. Based on genomic structure and phylogenetics, CoVs are subdivided into genera alpha through delta with the alpha- and beta-genera responsible for currently known diseases in humans [1]. Unlike subtypes of CoVs that cause mild clinical symptoms, SARS-CoV and MERS-CoV are associated with severe respiratory symptoms [2]. CoVs are recognized to cause infection in the respiratory, hepatic, enteric and neurological systems and pandemic conditions have ensued with high infection rates [3]. Since the World Health Organization (WHO) announced this new beta coronavirus in late 2019, cataloged as SARS-CoV-2 (COVID-19) [4], the organization has authorized several COVID-19 preparations for emergency use immunization including vaccines developed by Pfizer/BioNTech, Astrazeneca/Oxford, Serum Institute of India, Johnson & Johnson, Moderna and Sinopharm. Notwithstanding these advances in controlling the pandemic, with high viral transmission via respiratory droplets from coughing and/or sneezing [5], there is still an urgency to developing effective antiviral drugs for COVID-19 treatment.
In screening for potential COVID-19 drug candidates, metabolite repurposing can be used as a starting point to identify metabolites that already have biological activity against some diseases or infections. Drug repurposing can be achieved by conducting systematic drug-drug target interaction and drug-drug interaction analyses [6]. Another approach is to examine a class or source of natural products with established biological activity; indeed, several in silico studies have been developed to screen for SARS-CoV-2 inhibitors as prospective anti-COVID-19 drug candidates [7,8,9,10,11,12,13]. Natural products play a pivotal role in designing novel and efficient treatments to conquer the current COVID-19 epidemic. Among natural products, alkaloids, flavonoids and terpenoids have enticed considerable attention as potential SARS-CoV-2 drug candidates [14,15,16].
The basis of such studies is that the main protease (Mpro) that cleaves COVID-19 polyproteins is a point of vulnerability for viral replication. The crystal structure of SARS-CoV-2 main protease provides the basis for designing small molecules as inhibitors [17]. As a result, inhibitors can be screened for a suitable molecular structure to bind to the catalytic site of Mpro and interrupt viral replication. Here, we examine cembranoids, a class of diterpenes isolated predominantly from the soft coral genus Sarcophyton that contain a considerable reservoir of bioactive natural products [18]. While screening for marine natural products with pharmacological activity of has led to the discovery of many potent bioactive metabolites [19], the marine pharmacopeia is still a rich source of biological and chemical diversity [20,21,22,23].
In this Sarcophyton specific study, metabolites identified over the last two decades (1998–2019) [18] provided the original database for creating a Sarcophyton chemical library; a subset of those compounds were then virtual screened as Mpro specific inhibitors. According to the anticipated docking scores, the most potent compounds were subjected to molecular dynamics (MD) simulations combined with binding energy calculations utilizing the molecular mechanics-generalized Born surface area (MM-GBSA) approach. The screen identified a promising anti-SARS-CoV-2 agent. Such in silico screening can provide promising inhibitor leads for subsequent in vitro and in vivo studies.
2. Results
The unavailability of approved therapies towards COVID-19 disease indicates a crucial demand to systematically screen and recognize inhibitors that can suppress the SARS-CoV-2 infection and/or replication. Mpro has an indispensable role in the viral reproduction in the host and is considered one of the most promising drug targets. In seeking small molecules to prohibit SARS-CoV-2 replication, molecular docking calculations and molecular dynamics (MD) simulations were applied to scrutinize the potency of a chemical library containing 363 diterpenes as prospective anti-COVID-19 inhibitor candidates.
2.1. Molecular Docking
The molecular docking technique was employed to anticipate the binding affinities for diterpenes towards Mpro. The predicted docking scores of all investigated diterpenes metabolites are tabulated in Table S1. What stands out in Table S1 is the wide range of binding affinities ranging from −4.3 to −10.4 kcal/mol. To evaluate the potency of the studied metabolites, docking scores were compared to darunavir, an antiretroviral therapy that has been clinically tested as a COVID-19 drug candidate [24]. Although darunavir has not shown anti-SARS-CoV-2 activity, darunavir exhibits promising binding affinity against Mpro based on in silico studies [25,26,27].
Interestingly, approximately 23% of the screened metabolites (i.e., 59 compounds) demonstrated higher binding affinity than that of darunavir with a docking score of −8.2 kcal/mol. 2D docking poses of those fifty-nine diterpenes with the key residues inside the Mpro active site are presented in Figure S1 (Supplementary Materials). Most of the investigated metabolites displayed similar Mpro binding modes inside the Mpro’s binding pocket, exhibiting an essential hydrogen bond with GLU166, resulting in high binding affinities (Figure S1). Estimated docking scores, 2D chemical structures, in addition to binding features for the top potent metabolites in complex with Mpro are listed in Table 1.
Table 1.
Calculated binding affinities (in kcal/mol), 2D chemical structures and binding features for darunavir and the top five most promising cembranoid diterpenes against SARS-CoV-2 main protease (Mpro) a.
| Compound Name (Number) | Genus | 2D Chemical Structure | Docking Score (kcal/mol) | Binding Features b |
|---|---|---|---|---|
| Darunavir | --- |
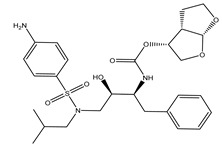
|
−8.2 | LEU167 (1.96 Å), GLU166 (1.94, 2.88 Å) |
| Sarelengan B (363) | S. elegans |
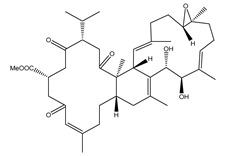
|
−9.8 | GLY143 (2.39 Å), GLU166 (1.94 Å), GLN189 (2.58 Å), THR190 (2.33 Å) |
| Bislatumlide A (340) | S. latum |
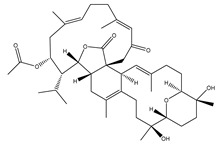
|
−9.6 | GLY143 (1.88 Å), GLU166 (2.68 Å) |
| Dioxanyalolide (347) | S. elegans |
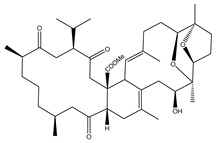
|
−9.5 | GLU166 (2.07 Å) |
| Desacetylnyalolide (345) | S. elegans |
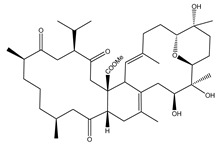
|
−9.1 | GLU166 (1.66, 2.12 Å), THR190 (2.42 Å) |
| Lobophytone W (357) | S. elegans |
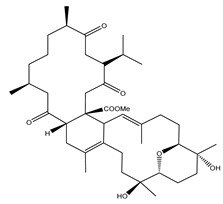
|
−8.7 | HIS41 (2.01 Å), CYS145 (2.34 Å), GLU166 (2.35, 2.86 Å) |
a The potent Sarcophyton cembranoid diterpenes metabolites were selected based on latter MM-GBSA binding energy calculations. b Only hydrogen bonds (in Å) were displayed.
From the data in Table 1, it is apparent that 363, 340, 347, 345 and 357 demonstrated solid binding affinities against Mpro with docking scores in the range of –8.7 to –9.8 kcal/mol. High docking scores are ascribed to the capability of forming hydrogen bonds, van der Waals, hydrophobic and pi-based interactions with the fundamental amino acid residues within the Mpro active site. Sarelengan B (363), separated from S. elegans, demonstrated the highest binding affinity of the diterpenes towards Mpro with a docking score of −9.8 kcal/mol. Investigating the binding mode of 363 inside the Mpro active site revealed that two carbonyl groups of (E)-cyclotetradec-9-ene-1,4,8-trione formed three hydrogen bonds with the imidazole ring backbone of HIS41, SH group of CYS145 and NH group of GLU166, with bond lengths of 2.01, 2.34 and 2.68 Å, respectively (Figure 1 and Table 1). Additionally, a hydroxy group of 363 interacts with the backbone carbonyl group of GLU166 with a bond length of 2.35 Å (Figure 1 and Table 1).
Figure 1.

3D and 2D representations of predicted binding modes of Sarcophyton cembranoid diterpenes metabolites (i) sarelengan B (363), (ii) bislatumlide A (340) and (iii) darunavir against SARS-CoV-2 main protease (Mpro).
Bislatumlide A (340), separated from S. latum, manifested the second-highest binding affinity against Mpro with a docking score of −9.6 kcal/mol. Investigating the binding mode of 340 towards the Mpro showed that the methanol hydroxy group exhibits a hydrogen bond with the backbone NH of GLY143 with a bond length of 1.88 Å (Figure 1 and Table 1). While the carbonyl group of dihydrofuranone shares a hydrogen bond with the backbone NH group of GLU166 with a bond length of 2.68 Å (Figure 1 and Table 1).
It is worth noting that the five potent metabolites containing a cyclotetradecane-trione ring revealed the vital role of the chemical nucleus in the binding affinity of cembranoids with Mpro.
Darunavir demonstrated a satisfactory binding affinity (−8.2 kcal/mol), exhibiting three hydrogen bonds with LEU167 and GLU166 with bond lengths of 1.96, 2.88 and 1.94 Å, respectively (Figure 1 and Table 1). More precisely, the NH2 of the aniline ring forms two hydrogen bonds with the carboxylate group of GLU166 and carbonyl group of LEU167 with bond lengths of 1.94 and 1.96 Å, respectively. Furthermore, the oxygen atom of the tetrahydrofuran ring exhibits a hydrogen bond with the backbone NH of GLU166 with a bond length of 2.88 Å. A docking comparison of darunavir with 363 and 340 disclosed promising binding affinities of the two metabolites as Mpro inhibitors.
2.2. MD Simulations and Binding Energy Calculations
Molecular dynamics (MD) simulations provide structural details, stability of the ligand-enzyme complexes, conformational flexibilities and the reliability of ligand-enzyme binding affinities [28,29]. The most potent diterpenes (59 compounds with docking scores <−8.2 kcal/mol) in complex with Mpro were subjected to MD simulations and binding free energy calculations. To lessen the computational cost and time, the MD simulations were executed in the implicit water solvent for 250 ps in addition to the MM-GBSA approach was used to compute the corresponding binding free energies (see computational methodology section for details). The calculated MM-GBSA binding affinities for the selected metabolites are listed in Table S2. As shown in Table S2, five inhibitors demonstrated lower binding energies (ΔGbinding) than that of darunavir (calc. −31.0 kcal/mol). Generally, the computed MM-GBSA binding energy values were lower than the corresponding docking scores, which might be ascribed to the different evaluation functions of the two employed methods. An over estimation of binding energy using a MM-GBSA approach can occur when not considering the contribution due to entropy [30,31]. To gain more reliable binding affinities of diterpene metabolites in complex with Mpro, those metabolites were further submitted to 10 ns MD simulations in an explicit water solvent. In addition, the corresponding binding energies were calculated (Figure 2).
Figure 2.

Average MM-GBSA binding energies for darunavir and the most potent diterpenes in complex with SARS-CoV-2 (Mpro) over 250 ps MD simulation in an implicit water solvent and 10 ns, 50 ns and 100 ns MD simulations in an explicit water solvent.
What is interesting about the data in Figure 2 is that all investigated metabolites revealed lower binding energies (ΔGbinding) than that of darunavir (calc. −30.4 kcal/mol). Therefore, those potent metabolites were selected and subjected to 50 ns MD simulations in the explicit water solvent, followed by MM-GBSA binding energy calculations (Figure 2). As shown in Figure 2, only 340 demonstrated steady diminution in the binding energies over the simulation times. However, the other investigated molecules exhibited a slight rise in MM-GBSA binding energies throughout the MD simulations. For instance, the evaluated MM-GBSA binding energies for 340 towards Mpro were −45.1, −39.8 and −43.8 kcal/mol throughout 250 ps implicit-solvent MD, 10 ns explicit-solvent MD and 50 ns explicit-solvent MD simulations, respectively. This displays the significance of long MD simulations to anticipate reliable binding affinity of the diterpenes with Mpro. MD simulation for 340 complexed with Mpro was then elongated to 100 ns. In addition, the corresponding MM-GBSA binding energy was evaluated (Figure 2).
What is striking about Figure 2 is that there was no appreciable variation between the estimated MM-GBSA binding energy for 340 complexed with Mpro over the 50 and 100 ns MD simulations. Comparing the binding affinities of 340 and darunavir, 340 demonstrated strong binding affinity throughout the 100 ns MD simulation against Mpro with an average ΔGbinding of −34.8 kcal/mol. The outstanding potency of 340 was attributed to its capability of exhibiting significant hydrogen bonds, hydrophobic, pi-based interactions and van der Waals interactions with the key amino acid residues within the Mpro active site. The average structures for 340 and darunavir inside the active site throughout the 100 ns MD simulations are depicted in Figure 3. The most exciting finding was that 340 preserved three hydrogen bonds with the fundamental amino acid residues of Mpro throughout the 100 ns MD simulation (Figure 3). Darunavir also showed satisfactory binding energy over the 100 ns MD simulation towards Mpro with an average ΔGbinding of −34.8 kcal/mol, exhibiting only two hydrogen bonds with the key amino acid residues of Mpro (Figure 3). In conclusion, the MM-GBSA binding energy calculations displayed a remarkably higher binding affinity of 340 compared to darunavir.
Figure 3.

3D and 2D representations of binding modes of (i) bislatumlide A (340) and (ii) darunavir in complex with SARS-CoV-2 main protease (Mpro) on the basis of the average structure throughout the 100 ns MD simulation.
The MM-GBSA scheme identifies different components the participate in the total binding energy (ΔGbinding), including van der Waals interactions (ΔEvdw), electrostatic interactions (ΔEele), electrostatic solvation free energy evaluated from the generalized Born equation (ΔEGB), the nonpolar component of the solvation energy (ΔESUR), total gas-phase energy (ΔGgas) and solvation free energy (ΔGSolv). All these binding free energy components of 340 and darunavir with SARS-CoV-2 Mpro are summarized in Table 2.
Table 2.
Components of the MM-GBSA binding energies for 340- and darunavir- SARS-CoV-2 main protease (Mpro) complexes over 100 ns MD simulations.
| Compound Name | Estimated MM-GBSA Binding Energy (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|
| ΔEvdw | ΔEele | ΔEGB | ΔESUR | ΔGgas | ΔGSolv | ΔGbinding | |
| Bislatumlide A (340) | −56.1 | −27.7 | 45.6 | −6.6 | −83.8 | 39.0 | −44.8 |
| Darunavir | −47.4 | −15.1 | 33.8 | −6.2 | −62.5 | 27.7 | −34.8 |
Based on the evaluated MM-GBSA binding energies, van der Waals interactions (ΔEvdw) were found to be the prime force inducing molecular complexation with Mpro for both bislatumlide A (ΔEvdw of −56.1 kcal/mol) and darunavir (ΔEvdw of −47.4 kcal/mol). Additionally, electrostatic interactions (Eele) were an appropriate contributor for 340- and darunavir-Mpro binding affinities with an average value of −27.7 and −15.1 kcal/mol, respectively. It is noted that Evdw is approximately two-fold more robust than the ΔEele. Together these results provide quantitative data of the binding affinities of 340 and darunavir as putative Mpro inhibitors.
Moreover, to explore the key residues that exhibit essential contributions to 340- and darunavir-Mpro interactions, the per-residue decomposition of the binding free energy calculations was executed. All the residues with energetic contributions <−0.50 kcal/mol were considered and presented in Figure 4. It is apparent that HIS41, GLY143, MET165, GLU166 and GLN189 amino acid residues contributed to the interactions of 340 and darunavir with Mpro. A considerable contribution of the GLU166 amino acid residue to the total binding free energy was observed with values of −2.9 and −1.2 kcal/mol for 340- and darunavir-Mpro complexes, respectively. In addition, the hydrophobic residues participate in higher binding affinity as a result of hydrophobic interactions between 340 and the hydrophobic residues.
Figure 4.

Per-residue decomposition of the total binding free energy (kcal/mol) of bislatumlide A (340) and darunavir in complex with SARS-CoV-2 Mpro.
Mining of naturally occurring plant-based compounds and marine-derived metabolites has evoked an upsurging interest in discovering potential SARS-CoV-2 Mpro inhibitors. A comparison of the binding affinity of 340 with those previously reported as SARS-CoV-2 Mpro inhibitors would give an informative insight into the preferential inclination of 340 as a prospective drug candidate. Towards an adequate comparison, only natural products and marine metabolites identified by employing a similar in silico technique were considered. Among the explored natural products, salvanolic acid and curcumin accentuated appreciable MM-GBSA binding energies against Mpro over 40 ns MD course with values of −44.8 and −34.2 kcal/mol, respectively [11]. Notably, rutin, a flavonol glycoside molecule, exhibited a debilitated binding affinity against Mpro (ΔGbinding = −28.4 kcal/mol over 150 ns MD simulation) [7]. Two flavone nominees dubbed as PubChem-129-716-607 and PubChem-885-071-27 showed considerable binding affinities against Mpro, over 150 ns MD course, with ΔGbinding values of −69.0 and −68.1 kcal/mol, respectively [7]. Erylosides B, a terpene marine natural product, demonstrated preferential MM-GBSA binding energy with Mpro over 100 ns MD simulation (calc. −51.9 kcal/mol) [8]. Comparing the MM-GBSA binding affinity of 340 to the earlier identified compounds robustly unveiled its competing binding affinity as a prospective Mpro inhibitor. As a consequence, further investigation of the structural and energetic stability of 340 over 100 ns MD simulation is desired.
2.3. Post-MD Analyses
Structural and energetic analyses were conducted throughout the 100 ns MD simulations, as well as compared to those of darunavir to further emphasized the stability and behavior of 340 in complex with Mpro. For structural and energetic analyses, four characteristics were evaluated from respective simulation trajectories: root-mean-square deviation (RMSD), hydrogen bond length, binding energy per frame and center-of-mass (CoM) distance.
2.3.1. Binding Energy per Frame
The correlation between single-trajectory MM-GBSA binding energy and time was used to evaluate the comprehensive structural stability of 340 and darunavir in complex with Mpro throughout the 100 ns MD simulations (Figure 5). An interesting aspect of this graph is the general stabilities for 340 and darunavir with average binding energies (ΔGbinding) of −44.8 and −34.8 kcal/mol, respectively. The most obvious finding to emerge from the analysis is that all complexes maintained stability over 100 ns MD simulations.
Figure 5.

Estimated MM-GBSA binding energies per frame for bislatumlide A (340) (black) and darunavir (red) in complex with SARS-CoV-2 Mpro throughout 100 ns MD simulations.
2.3.2. Hydrogen Bond Length
Hydrogen bonds have a vital role in preserving the binding of investigated compounds with a protein. Consequently, hydrogen bond analysis was carried out for 340 and darunavir in complex with Mpro throughout the 100 ns MD simulations (Table 3). Table 3 demonstrated that the two inhibitors manifested stable hydrogen bonds with GLU166 with H-bond occupancy values of 90.3% and 85.7% for bislatumlide A and darunavir, respectively. The high H-bond occupancy confirms the significant role of GLU166 within the active site of Mpro. A comparison of the data, summarized in Table 3, reveals a higher stability for 340 than darunavir. More exactly, 340 exhibited three stable hydrogen bonds with HIS41, GLU166 and GLN189 with an average H-bond distance of 2.9, 2.8 and 2.6 Å, respectively. However, darunavir formed only a stable hydrogen bond with GLU166 with an average H-bond distance of 2.7 Å. The high stability for the bislatumlide A-Mpro complex compared to darunavir-Mpro complex is clearly supported by the current findings.
Table 3.
Hydrogen bond distance, angle and occupancy for bislatumlide A (340) and darunavir with essential Mpro amino acid residues throughout 100 ns MD simulations.
| Compound Name | Acceptor | Donor | Angle (Degree) a | Distance (Å) a | Occupied (%) b |
|---|---|---|---|---|---|
| Bislatumlide A (340) | HIS41@ND1 | Bislatumlide A@O-H16 | 164 | 2.9 | 67.9 |
| GLU166@O | Bislatumlide A@O2-H25 | 142 | 2.8 | 90.3 | |
| GLN189@O | Bislatumlide A@O3-H47 | 145 | 2.6 | 88.9 | |
| Darunavir | GLU166@O | Darunavir @O5-H36 | 151 | 2.8 | 85.7 |
a The hydrogen bonds are investigated via the acceptor-donor atom distance of <3.5 Å and acceptor-H-donor angle of >120°. b Only hydrogen bonds with occupancy higher than 50% were illustrated.
2.3.3. Center-of-Mass Distance
Center-of-mass (CoM) distances were adopted to allow a deeper insight into the stability of inhibitor-Mpro complexes over the 100 ns MD simulations (Figure 6). From the data, it is apparent that CoM distances were more consistent for 340 complexed with Mpro compared to darunavir with average values of 6.0 and 12.1 Å, respectively. The current results propose that 340 bound more tightly to the Mpro complex compared to darunavir.
Figure 6.

Center-of-mass (CoM) distances (in Å) between bislatumlide A (black) and darunavir (red) and GLU166 of Mpro throughout 100 ns MD simulations.
2.3.4. Root-Mean-Square Deviation
The root-mean-square deviation (RMSD) of the whole complex backbone atoms were plotted as a function of time to examine the structural stability of the 340 and darunavir-Mpro complexes throughout the simulation time (Figure 7). RMSD analysis demonstrated that the scrutinized complexes initiated stabilization after 20 ns and kept their stabilities until the end of the 100 ns MD simulations. The evaluated RMSD values for these complexes stayed beneath 0.26 nm throughout the MD simulation time. Overall, the current results demonstrated that 340 is tightly bonded and does not influence the structural stability of the Mpro, as well as conserved structural integrity.
Figure 7.

Root-mean-square deviation (RMSD) of the backbone atoms from the initial structure of bislatumlide A (black) and darunavir (red) with Mpro over a 100 ns MD simulations.
2.4. In Silico Drug-Likeness
The efficiency of curative drugs fundamentally relies on the molecular property and bioactivity of the molecules [32] was examined. To consider the drug-likeness in addition to bioactivity of 340, in silico molecular features were evaluated using a SwissADME web server and compared to those of darunavir. The predicted properties are shown in Table 4.
Table 4.
Predicted physiochemical parameters and structural descriptors of bislatumlide A (340) and darunavir as prospective SARS-CoV-2 Mpro inhibitors.
| Compound Name | mLogP | TPSA | nON | nOHNH | Nrotb | MWt | %ABS |
|---|---|---|---|---|---|---|---|
| Bislatumlide A (340) | 4.3 | 119.4 | 8 | 2 | 3 | 694.9 | 67.8% |
| Darunavir | 1.2 | 148.8 | 8 | 3 | 13 | 547.7 | 57.7% |
The permeability through the cell membrane, as inspected by the mlogP value, was less than five (4.3 and 1.2 for bislatumlide A and darunavir, respectively), proposing that these inhibitors have adequate membrane permeability. Additionally, the number of hydrogen bond donors (nOHNH) and acceptors (nON) were less than 5 and 10, respectively. Moreover, the molecular weights for bislatumlide A and darunavir were 694.9 and 547.7, respectively, suggesting that these inhibitors are readily transported and/or diffused in the absorption process. Another parameter pointing out the molecular bio-absorption is the topological polar surface area (TPSA). The TPSA of 340 and darunavir were 119.4 and 148.8 Å, respectively, indicating a satisfactory cell membrane permeability and oral bioavailability level.
2.5. Molecular Target Prediction and Network Analysis
One hundred and seventeen genes for severe acute respiratory syndrome diseases (SARS, C1175175) were recorded utilizing DisGeNET online tools. Using Venn diagram comparison analysis, frequently participated genes for 340 involved PRKCA, PRKCB, MAPK1 and MAPK14. A STRING database was utilized to recognize protein-protein interactions for 340 included the biochemical signaling genes AR, MAPK1, MAPK3, MAPK8, LYN, JAK2, SRC, MDM2 PTPN1 and JUN as an in silico natural-product inhibitor towards Mpro and supplies a ligand lead for in vitro enzyme investigations (Figure 8 and Table S3).
Figure 8.

(a) Venn diagram analysis for bislatumlide A (340) towards SARS disease genes and (b) STRING PPI network for the top targets known by network analyzer for bislatumlide A (340) as a SARS-CoV-2 main protease (Mpro) inhibitor.
2.6. Pathway Enrichment Analysis (PEA)
Toward better genome-wide mining for 340, stimulated targets and their interactors, were determined using pathway enrichment analysis (PEA) and Boolean network analysis (BNA). A genome-wide hierarchy map representation of the pathways affected by 340 treatment was constructed (Figure S2). The hierarchy map showed that among the most stimulated pathways was the “disease” pathway, particularly the potential therapeutic for the SARS pathway. In addition, a reacfoam tree of the top targeted/influenced pathways by the top 10 gene targets stimulated by 340 treatment against SARS-CoV-2 infection was constructed (Figure 9). Although the PEA results revealed that the top 10 gene targets stimulated by 340 were identified in 833 reactome pathways (each pathway was hit by at least one of them), the top three significantly enriched major pathways are (A) signal transduction pathway, (B) disease pathway and (C) immune system pathway, with a false discovery rate (FDR) of <0.00001%. Under these pathways, the interaction between the top 10 genes/signals stimulated with 340 treatment was visualized and mapped (Figure S3). Additionally, interactors with these top 10 genes are listed (Table S4).
Figure 9.

FoamTree diagram showing a genome-wide enriched pathways overview based on Voronoi tessellation analysis for the top 10 genes targets stimulated by bislatumlide A (340) as a proposed SARS-CoV-2 inhibitor.
Remarkably, under the most enriched major pathway (signal transduction), it was found that the MAPK family signaling cascades pathway was on the top of the most enriched pathway influenced by 340 within the human biological system (Figure 10). Mining of the PEA analysis outcomes indicated that a set of five genes (MAPK1, MAPK3, JAK2, JUN and SRC) were significantly modulated as biological targets to 340 as potent SARS-CoV-2 inhibitor. Furthermore, the interactome results showed that these five genes were found to interact with other 48 genes/interactors (Table S4).
Figure 10.

A diagrammatic model shows the Signal Transduction Reactome Network.
Previous studies show that various alterations in a few signaling pathways can control many central biological functions of the cell after SARS-CoV-2 infection [33,34]. Recently, several research reports found that the p38 mitogen-activated protein kinase (MAPK), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and epidermal growth factor receptor (EGFR) signaling pathways are altered following coronavirus infection. These pathways play a crucial role in oppressing the host antiviral response and are essential for coronavirus entry, replication and apoptosis [35].
Indeed, accumulated evidence confirmed that the p38 MAPK signaling pathway can operate various essential biological activities depending on the stimuli and the type of stimulated tissue. Therefore, modulation of p38 MAPK signaling was found to raise cell death and survival [36]. Additionally, many reports emphasized the critical role of p38 MAPK signaling in several viral infections targeting the respiratory system; in HCoV-229E, the triggering of p38 MAPK is required to induce cytopathic effect (CPE), as well as the viral replication process. In addition, the upregulation of p38 MAPK may also promote the viral entry through the ACE2 endocytosis and cause subsequent inflammations, thrombosis and could initiate multi-organ failure in COVID-19 patients [37].
Notably, one of the most common consequences of SARS-CoV-2 infection is pulmonary injury. In COVID-19 patients, the p38 MAPK signaling pathway, via p53, or its alternatives TGF-β1, or syntenin, drives apoptosis and ends with lung injury, which means that SARS-CoV-2 is likely to modulate the p38 MAPK signaling pathway to provoke the apoptosis and lung damage. Here, based on the Reactome mining, we found that 340 showed a potential therapeutic effect (Hit:32/144 with FDR:9.17E-1) against SARS-CoV-2 viral infection (Figure S4). This potentiality may be attributed to 340 capability to re-modulate the p38 MAPK signaling pathway hijacked by SARS-CoV-2 viral infection and antagonize its harmful effects. This speculation was supported by recent reports that demonstrated the possibility of inhibiting the p38 MAPK signaling pathway as a promising therapeutic strategy against the SARS-CoV-2 pandemic [38,39].
3. Materials and Methods
3.1. Mpro Preparation
A high-resolution (2.16 Å) biological unit for the X-ray crystallographic structure of SARS-CoV-2 main protease (Mpro) deposited in Protein Data Bank by Jin et al. (PDB ID: 6LU7 [17]) in complex with peptidomimetic inhibitor (N3) was retrieved and utilized as a template for all molecular docking calculations and molecular dynamics (MD) simulations. The protein structure was prepared by eliminating all crystallographic water molecules, heteroatoms and ions, keeping only the amino acid residues. H++ webserver was applied to assign the protonation states of Mpro [40]. Additionally, all missing hydrogen atoms were added. In H++ calculations, physiologic conditions of external dielectric constant = 80, pH = 7, salinity = 0.15 and internal dielectric constant = 10 were used to evaluate the pKa values of Mpro amino acid residues.
3.2. Inhibitor Preparation
A chemical library containing metabolites from the genus Sarcophyton reported over the last twenty years (1998–2019) [18] was assembled. Structures were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov) in SDF format, except for 2D structures of bislatumlides A and B that were taken from the original article [41]. Omega2 software was used to generate three-dimensional (3D) structures of the investigated compounds [42,43]. Geometrical structures were minimized with the assistance of Merck Molecular Force Field 94 (MMFF94S), implemented inside SZYBKI software [44,45]. Undefined stereocenters were enumerated with the help of flipper application inside Omega2 software. A conformational search was performed to generate all conformers within the energy window value of 10 kcal/mol. The lowest energy conformer was subjected to minimization with the assistance of Merck Molecular Force Field 94 (MMFF94S), implemented inside SZYBKI software [44,45]. The protonation state and tautomer enumeration of the compounds were examined by fixpka and tautomer applications, respectively, included in the QUACPAC software [46]. Two-dimensional (2D) structures are presented in Table S1.
3.3. Molecular Docking
In the current study, AutoDock4.2.6 software was applied to perform all molecular docking calculations [47]. According to AutoDock protocol [48], the pdbqt file of SARS-CoV-2 main protease (Mpro) was prepared. In AutoDock4.2.6, the maximum number of energy evaluations (eval) and the number of genetic algorithm (GA) run variables were set to 250 and 25,000,000, respectively. All other docking parameters were preserved at their default settings. The size of the box (60 Å × 60 Å × 60 Å) was specified to encompass the SARS-CoV-2 Mpro active site appropriately. The binding site of SARS-CoV-2 Mpro was precisely located based on the availability of resolved structures of Mpro in complex with inhibitors [49]. An AutoGrid4.2.6 program was applied to generate maps with a grid spacing value of 0.375 Å. The coordinates of the grid center were placed at −13.069, 9.740 and 68.490 (XYZ assignments, respectively). The Gasteiger method was utilized to compute the atomic partial charges of the studied compounds [50]. The anticipated binding modes for each compound were handled using the built-in clustering analysis (1.0 Å RMSD tolerance) and conformation with the lowest energy within the largest cluster was opted as a representative pose.
3.4. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations were carried out for the most potent diterpenes in complex with Mpro using AMBER16 software [51]. An AMBER force field of 14SB was employed to describe the Mpro catalytic site [52], while the general AMBER force field (GAFF2) [53] was used to characterize individual metabolites. Both implicit-solvent and explicit-solvent MD simulations were performed. In the implicit-solvent MD simulations, an AM1-BCC method with the assistance of the Antechamber tool implemented inside AMBER16 software was used to assign the atomic partial charges of the diterpenes [54]. Neither cutoff distance for nonbonded nor periodic boundary conditions were applied. Furthermore, the solvation influence was considered by utilizing igb = 1 solvent model [55]. Energy minimization was initially executed on the docked diterpene in complex with Mpro for 500 steps. Gentle heating was carried out as well on the minimized complexes from 0 K to 300 K over 10 ps NVT MD simulations. The production stage was then performed over 250 ps, in addition to snapshots recorded every 1 ps. Therefore, 250 snapshots were derived from each MD simulation. Herein, the CPU version of pmemd (pmemd.MPI) implemented inside AMBER16 software was employed to conduct all implicit-solvent MD simulations.
In explicit-solvent MD simulations, atomic partial charges of the investigated metabolites were assigned at the HF/6-31G* level with assistance of Gaussian09 software with the restrained electrostatic potential (RESP) fitting approach [56,57]. A water-solvated cubic box was constructed using a TIP3P water model with a minimum distance to the box edge of 15.0 Å (1.5 nm). All solvated systems were neutralized by adding Na+ or Cl− counter-ions [58]. The maximum number of energy minimization steps were adjusted to 5000 utilizing combined steepest and conjugate gradient algorithms. The minimized systems were thereafter heated gradually to 300 K over 50 ps in the NVT ensemble. Additionally, the systems were sufficiently equilibrated for 1 ns under NPT conditions. The production stages were subsequently executed under an NPT ensemble for each studied Mpro-metabolite complex over simulation times of 10 ns, 50 ns and 100 ns. Snapshots were gathered every 10 ps for post-MD analyses. The long-range electrostatic interactions were estimated using the Particle Mesh Ewald (PME) method. A short-range nonbonded interaction cutoff distance of 12 Å was applied [59]. The gamma_ln collision frequency applied was 1.0 ps−1; in addition, the Langevin thermostat was utilized to maintain the temperature at 298 K [60]. The pressure was controlled using a Berendsen barostat with a pressure relaxation time of 2 ps [61]. All bonds, including hydrogen atoms, were constrained using a SHAKE algorithm with a time step of 2 fs [62]. All explicit-solvent MD simulations were executed with the help of a GPU version of pmemd (pmemd.cuda) implemented inside an AMBER16 package. All in silico calculations, including molecular docking calculations, quantum mechanics calculations and MD simulations, were executed on the CompChem GPU/CPU hybrid cluster (hpc.compchem.net). Molecular graphics and analyses were executed with the assistance of BIOVIA DS Visualize 2020 [63].
3.5. MM-GBSA Binding Energy Calculations
The binding free energies for the most potent metabolites that complexed with Mpro were computed using molecular mechanical-generalized Born surface area (MM-GBSA) approach implemented inside AMBER16 software [64]. The MM-GBSA (ΔGbinding) binding free energy calculations were estimated based on the snapshots collected from MD simulations. The average binding free energy (ΔGbinding) was evaluated as follows:
| ΔGbinding = Gcomplex − (GSarcophyton + GMpro) | (1) |
where the energy term (G) is estimated as:
| G = Eele + Evdw + GSA + GGB | (2) |
Eele and Evdw are electrostatic and van der Waals energies, respectively. GSA stands for the nonpolar solvation-free energy, generally estimated via a linear function of the solvent-accessible surface area (SASA) using a LCPO algorithm [65]. GGB is the electrostatic solvation free energy computed from the generalized Born equation. The polar component of the desolvation energy was assigned via Onufriev’s GB [66] (igb = 2). The solvent (exterior) and solute (interior) dielectric constants were set to 80 and 1, respectively. A single-trajectory method was applied, in which the coordinates of each metabolite-Mpro, Mpro and metabolite were obtained from a single trajectory. The configurational entropy (TS) is typically neglected due to the higher computational costs [30,31].
3.6. Drug-Likeness Properties
For the identified cembranoids, physicochemical properties including molecular weight (MW), octanol/water partition coefficient (LogP), hydrogen bond acceptor (HBA), topological polar surface area (TPSA), rotatable bond count (RB) and hydrogen bond donor (HBD) were estimated with assistance of SwissADME web server (http://www.swissadme.ch/).
3.7. Protein Interactions Network and Pathway Enrichment Analysis (PEA)
Cembranoids were screened via an online website-based tool of SwissTargetPredicition (http://www.swisstargetprediction.ch) to recognize the possible targets for each ligand. InteractiVenn online tool was applied to design Venn Diagram [67] for accessible database for severe, acute respiratory syndrome diseases (DisGeNET database; https://www.disgenet.org). The top 100 genes for the most potential metabolite were retrieved. A functional database of STRING for top anticipated targets was then used to generate protein-protein interaction (PPI) [68].
Cytoscape 3.8.2 was used to inspect all probable target-function relations according to a network topology [69]. Furthermore, to explore all potential target-functions relationships for the top 10 targeted genes, pathway enrichment analysis was accomplished using Cytoscape 3.8.2 [69]. In addition, to investigate the impact of bislatumlide A (340) as a COVID-19 inhibitor candidate on human biological pathways computationally, Boolean-network modeling was implemented. The fuzzy logic simulation approach [70] was implemented to mimic Boolean-network dynamic behavior, which consequently transformed from Reactome pathways by extending two-state Boolean variables to variables with values ranging from zero to one. In addition, a FoamTree representation based on Voronoi tessellation analysis for the top 10 gene targets stimulated by 340 was constructed. Finally, a Reactome FIViz plugin tool was utilized for genome-wide visualization of all possible drug-targets interactions [71].
4. Conclusions
COVID-19 is a universal risk for positive human health and economic losses continue to build unabated. A rapid proceed, especially in vaccine and drug development, is substantial to conquer the further outbreak in prevalence and loss of human life from COVID-19 disease. Herein, Sarcophyton cembranoid diterpenes were screened as prospective Mpro inhibitors using combined molecular docking and molecular dynamics approaches. Based on molecular docking calculations, MD simulations, as well as molecular mechanics-generalized born surface area (MM-GBSA) binding energy calculations, 340 manifested a convenient binding affinity with ΔGbinding < −44.0 kcal/mol against Mpro. The energetic and structural analyses throughout 100 ns MD simulations emphasized the high stability of 340. Drug-likeness properties for 340 were evaluated and demonstrated favorable physicochemical properties. The compound has the potential to re-modulate the p38 MAPK signaling pathway. In vitro and in vivo explorations are predicted to further identify the role of 340 as a potential inhibitor therapeutic for COVID-19 treatment.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/md19070391/s1, Figure S1: 2D representations of interactions of darunavir and the top 59 potent Sarcophyton cembranoid diterpenes metabolites with the proximal amino acid residues of SARS-CoV-2 main protease (Mpro), Figure S2: A genome-wide overview of the Reactome Pathway Analysis (RPA) results for the top 20 genes targets stimulated by bislatumlide A (340) against SARS-CoV-2, Figure S3: Pathway digram analysis for enzymes/signals of the top 10 genes products/targets stimulated by bislatumlide A (340), Figure S4: A diagrammatic model shows the potential therapeutic level of bislatumlide A (340) against SARS-CoV-2 viral infection, Table S1: Estimated docking score (in kcal/mol) for darunavir and the investigated Sarcophyton cembranoid diterpenes metabolites towards SARS-CoV-2 main protease (Mpro), Table S2: Computed Autodock and MM/GBSA binding energies (in kcal/mol) for darunavir and the top 58 potent Sarcophyton cembranoid diterpenes metabolites against SARS-CoV-2 main protease (Mpro) over 250 ps implicit solvent MD simulations, Table S3: Network topological analysis for the predicted targets for bislatumlide A (340), Table S4: Interactors list showing the top 10 genes stimulated by bislatumlide A (340), their UniProt Ids and list of genes Ids which Interacts with SARS-CoV-2.
Author Contributions
Conceptualization, M.A.A.I. and M.-E.F.H.; Data curation, A.H.M.A.; Formal analysis, A.H.M.A. and M.A.M.A.; Investigation, A.H.M.A., M.A.M.A. and M.-E.F.H.; Methodology, M.A.A.I.; Project administration, M.A.A.I. and M.-E.F.H.; Resources, M.A.A.I.; Software, M.A.A.I. and M.A.M.A.; Supervision, M.A.A.I.; Visualization, A.H.M.A. and M.A.M.A.; Writing—original draft, A.H.M.A., M.A.M.A. and T.A.M.; Writing—review & editing, M.A.A.I., M.A.M.A., M.F.M., A.R.H., S.A.M.K., F.A.A., F.A., S.H.A., K.S.A., T.E., M.E.S., P.W.P., H.R.E.-S. and M.-E.F.H. All authors have read and agreed to the published version of the manuscript.
Funding
Mahmoud F. Moustafa extends his appreciation to the Deanship of Scientific Research at King Khalid University for funding this work under grant No. (R.G.P.1/143/42). The computational work was completed with resources supported by the Science and Technology Development Fund, STDF, Egypt, Grants No. 5480 & 7972 (Granted to Ass. Mahmoud A. A. Ibrahim). Mohamed-Elamir F. Hegazy gratefully acknowledges the financial support from Alexander von Humboldt Foundation “Georg Foster Research Fellowship for Experienced Researchers”.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article/supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fehr A.R., Perlman S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin Y., Wunderink R.G. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology. 2017;23:130–137. doi: 10.1111/resp.13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y., Liu Q., Guo D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92:418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parrish R.M., Hohenstein E.G., Sherrill D. Tractability gains in symmetry-adapted perturbation theory including coupled double excitations: CCD+ST(CCD) dispersion with natural orbital truncations. J. Chem. Phys. 2013;139:174102. doi: 10.1063/1.4826520. [DOI] [PubMed] [Google Scholar]
- 5.Dhand R., Li J. Coughs and Sneezes: Their Role in Transmission of Respiratory Viral Infections, Including SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020;202:651–659. doi: 10.1164/rccm.202004-1263PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020;60:3277–3286. doi: 10.1021/acs.jcim.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ibrahim M.A., Mohamed E.A., Abdelrahman A.H., Allemailem K.S., Moustafa M.F., Shawky A.M., Mahzari A., Hakami A.R., Abdeljawaad K.A., Atia M.A. Rutin and flavone analogs as prospective SARS-CoV-2 main protease inhibitors: In silico drug discovery study. J. Mol. Graph. Model. 2021;105:107904. doi: 10.1016/j.jmgm.2021.107904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ibrahim M.A., Abdelrahman A.H., Mohamed T.A., Atia M.A., Al-Hammady M.A., Abdeljawaad K.A., Elkady E.M., Moustafa M.F., Alrumaihi F., Allemailem K.S., et al. In Silico Mining of Terpenes from Red-Sea Invertebrates for SARS-CoV-2 Main Protease (M(pro)) Inhibitors. Molecules. 2021;26:2082. doi: 10.3390/molecules26072082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ibrahim M.A., Abdeljawaad K.A., Abdelrahman A.H., Hegazy M.E.F. Natural-like products as potential SARS-CoV-2 M(pro) inhibitors: In-silico drug discovery. J. Biomol. Struct. Dyn. 2020:1–13. doi: 10.1080/07391102.2020.1790037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ibrahim M.A., Abdelrahman A.H., Hegazy M.E.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1791958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ibrahim M.A., Abdelrahman A.H., Hussien T.A., Badr E.A., Mohamed T.A., El-Seedi H.R., Pare P.W., Efferth T., Hegazy M.E.F. In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors. Comput. Biol. Med. 2020;126:104046. doi: 10.1016/j.compbiomed.2020.104046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sencanski M., Perovic V., Pajovic S.B., Adzic M., Paessler S., Glisic S. Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel In Silico Method. Molecules. 2020;25:3830. doi: 10.3390/molecules25173830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keretsu S., Bhujbal S.P., Cho S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020;10:17716. doi: 10.1038/s41598-020-74468-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zakaryan H., Arabyan E., Oo A., Zandi K. Flavonoids: Promising natural compounds against viral infections. Arch. Virol. 2017;162:2539–2551. doi: 10.1007/s00705-017-3417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherrak S.A., Merzouk H., Mokhtari-Soulimane N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS ONE. 2020;15:e0240653. doi: 10.1371/journal.pone.0240653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jo S., Kim S., Kim D.Y., Kim M.-S., Shin D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzym. Inhib. Med. Chem. 2020;35:1539–1544. doi: 10.1080/14756366.2020.1801672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 18.Elkhawas Y.A., Elissawy A.M., El-Naggar M., Mostafa N.M., Al-Sayed E., Bishr M.M., Singab A.N.B., Salama O.M. Chemical Diversity in Species Belonging to Soft Coral Genus Sacrophyton and Its Impact on Biological Activity: A Review. Mar. Drugs. 2020;18:41. doi: 10.3390/md18010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carté B.K. Biomedical Potential of Marine Natural Products: Marine organisms are yielding novel molecules for use in basic research and medical applications. BioScience. 1996;46:271–286. doi: 10.2307/1312834. [DOI] [Google Scholar]
- 20.Martins A., Vieira H., Gaspar H., Santos S. Marketed Marine Natural Products in the Pharmaceutical and Cosmeceutical Industries: Tips for Success. Mar. Drugs. 2014;12:1066–1101. doi: 10.3390/md12021066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atanasov A.G., Zotchev S.B., Dirsch V.M., the International Natural Product Sciences Taskforce. Supuran C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021;20:200–216. doi: 10.1038/s41573-020-00114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hegazy M.-E.F., Mohamed T., Abdel-Latif F.F., Alsaid M.S., Shahat A., Paré P.W. Trochelioid A and B, new cembranoid diterpenes from the Red Sea soft coral Sarcophyton trocheliophorum. Phytochem. Lett. 2013;6:383–386. doi: 10.1016/j.phytol.2013.05.005. [DOI] [Google Scholar]
- 23.Hegazy M.-E.F., El-Beih A.A., Moustafa A.Y., Hamdy A.A., Alhammady M.A., Selim R.M., Abdel-Rehim M., Paré P.W. Cytotoxic Cembranoids from the Red Sea Soft Coral Sarcophyton glaucum. Nat. Prod. Commun. 2011;6:1809–1812. doi: 10.1177/1934578X1100601205. [DOI] [PubMed] [Google Scholar]
- 24.Harrison C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020;38:379–381. doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]
- 25.Ibrahim M.A., Abdelrahman A.H., Allemailem K.S., Almatroudi A., Moustafa M.F., Hegazy M.E.F. In Silico Evaluation of Prospective Anti-COVID-19 Drug Candidates as Potential SARS-CoV-2 Main Protease Inhibitors. Protein J. 2021;40:296–309. doi: 10.1007/s10930-020-09945-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J., Xia L., Liu L., Xu Q., Ling Y., Huang D., Huang W., Song S., Xu S., Shen Y., et al. Open Forum Infectious Diseases. Volume 7. Oxford University Press; New York, NY, USA: 2020. Antiviral Activity and Safety of Darunavir/Cobicistat for the Treatment of COVID-19; p. ofaa241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Vivo M., Masetti M., Bottegoni G., Cavalli A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016;59:4035–4061. doi: 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
- 29.Kerrigan J.E. Molecular dynamics simulations in drug design. In: Kortagere S., editor. In Silico Models for Drug Discovery. Humana Press; Totowa, NJ, USA: 2013. pp. 95–113. [DOI] [PubMed] [Google Scholar]
- 30.Hou T., Wang J., Li Y., Wang W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011;32:866–877. doi: 10.1002/jcc.21666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang E., Sun H., Wang J., Wang Z., Liu H., Zhang J.Z., Hou T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019;119:9478–9508. doi: 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- 32.Shen M., Tian S., Li Y., Li Q., Xu X., Wang J., Hou T. Drug-likeness analysis of traditional Chinese medicines: 1. property distributions of drug-like compounds, non-drug-like compounds and natural compounds from traditional Chinese medicines. J. Cheminform. 2012;4:31. doi: 10.1186/1758-2946-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hemmat N., Asadzadeh Z., Ahangar N.K., Alemohammad H., Najafzadeh B., Derakhshani A., Baghbanzadeh A., Baghi H.B., Javadrashid D., Najafi S., et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021;166:675–696. doi: 10.1007/s00705-021-04958-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Letko M., Marzi A., Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020;5:562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Moller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e1039. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yosimichi G., Nakanishi T., Nishida T., Hattori T., Takano-Yamamoto T., Takigawa M. CTGF/Hcs24 induces chondrocyte differentiation through a p38 mitogen-activated protein kinase (p38MAPK), and proliferation through a p44/42 MAPK/extracellular-signal regulated kinase (ERK) JBIC J. Biol. Inorg. Chem. 2001;268:6058–6065. doi: 10.1046/j.0014-2956.2001.02553.x. [DOI] [PubMed] [Google Scholar]
- 37.Xiao L., Sakagami H., Miwa N. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses. 2020;12:491. doi: 10.3390/v12050491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grimes J.M., Grimes K.V. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020;144:63–65. doi: 10.1016/j.yjmcc.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kono M., Tatsumi K., Imai A.M., Saito K., Kuriyama T., Shirasawa H. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: Involvement of p38 MAPK and ERK. Antivir. Res. 2008;77:150–152. doi: 10.1016/j.antiviral.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon J.C., Myers J.B., Folta T., Shoja V., Heath L.S., Onufriev A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan X.-H., Gavagnin M., Cimino G., Guo Y.-W. Two new biscembranes with unprecedented carbon skeleton and their probable biogenetic precursor from the Hainan soft coral Sarcophyton latum. Tetrahedron Lett. 2007;48:5313–5316. doi: 10.1016/j.tetlet.2007.05.096. [DOI] [Google Scholar]
- 42.Hawkins P.C.D., Skillman A.G., Warren G.L., Ellingson B.A., Stahl M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.OMEGA 2.5.1.4. OpenEye Scientific Software; Santa Fe, NM, USA: 2013. [Google Scholar]
- 44.SZYBKI, 1.9.0.3. OpenEye Scientific Software; Santa Fe, NM, USA: 2016. [Google Scholar]
- 45.Halgren T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999;20:720–729. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 46.QUACPAC, 1.7.0.2. OpenEye Scientific Software; Santa Fe, NM, USA: 2016. [Google Scholar]
- 47.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forli S., Huey R., Pique M.E., Sanner M.F., Goodsell D.S., Olson A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gervasoni S., Vistoli G., Talarico C., Manelfi C., Beccari A.R., Studer G., Tauriello G., Waterhouse A.M., Schwede T., Pedretti A. A Comprehensive Mapping of the Druggable Cavities within the SARS-CoV-2 Therapeutically Relevant Proteins by Combining Pocket and Docking Searches as Implemented in Pockets 2.0. Int. J. Mol. Sci. 2020;21:5152. doi: 10.3390/ijms21145152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gasteiger J., Marsili M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. doi: 10.1016/0040-4020(80)80168-2. [DOI] [Google Scholar]
- 51.Case D.A., Betz R.M., Cerutti D.S., Cheatham T.E., Darden T.A., Duke R.E., Giese T.J., Gohlke H., Goetz A.W., Homeyer N., et al. AMBER16. University of California; San Francisco, CA, USA: 2016. [Google Scholar]
- 52.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K., Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 54.Jakalian A., Jack D.B., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 55.Roux B., Simonson T. Implicit solvent models. Biophys. Chem. 1999;78:1–20. doi: 10.1016/S0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- 56.Bayly C.I., Cieplak P., Cornell W., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993;97:10269–10280. doi: 10.1021/j100142a004. [DOI] [Google Scholar]
- 57.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., et al. Gaussian 09. Gaussian Inc.; Wallingford, CT, USA: 2009. Revision E01. [Google Scholar]
- 58.Jorgensen W.L., Chandrasekhar J., Madura J., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 59.Darden T., York D., Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 60.Izaguirre J.A., Catarello D.P., Wozniak J.M., Skeel R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001;114:2090–2098. doi: 10.1063/1.1332996. [DOI] [Google Scholar]
- 61.Berendsen H.J.C., Postma J.P.M., Van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 62.Miyamoto S., Kollman P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. doi: 10.1002/jcc.540130805. [DOI] [Google Scholar]
- 63.Dassault Systèmes BIOVIA, B.D.S.V. Dassault Systèmes BIOVIA; San Diego, CA, USA: 2019. Version 2019. [Google Scholar]
- 64.Massova I., Kollman P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000;18:113–135. doi: 10.1023/A:1008763014207. [DOI] [Google Scholar]
- 65.Weiser J., Shenkin P.S., Still W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO) J. Comput. Chem. 1999;20:217–230. doi: 10.1002/(SICI)1096-987X(19990130)20:2<217::AID-JCC4>3.0.CO;2-A. [DOI] [Google Scholar]
- 66.Onufriev A., Bashford D., Case D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 67.Heberle H., Meirelles G.V., Da Silva F.R., Telles G.P., Minghim R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015;16:169. doi: 10.1186/s12859-015-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li R., Ma X., Song Y., Zhang Y., Xiong W., Li L., Zhou L. Anti-colorectal cancer targets of resveratrol and biological molecular mechanism: Analyses of network pharmacology, human and experimental data. J. Cell. Biochem. 2019;120:11265–11273. doi: 10.1002/jcb.28404. [DOI] [PubMed] [Google Scholar]
- 69.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morris M.K., Saez-Rodriguez J., Clarke D., Sorger P.K., Lauffenburger U.A. Training Signaling Pathway Maps to Biochemical Data with Constrained Fuzzy Logic: Quantitative Analysis of Liver Cell Responses to Inflammatory Stimuli. PLoS Comput. Biol. 2011;7:e1001099. doi: 10.1371/journal.pcbi.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blucher A., McWeeney S.K., Stein L., Wu G. Visualization of drug target interactions in the contexts of pathways and networks with ReactomeFIViz. F1000Research. 2019;8:908. doi: 10.12688/f1000research.19592.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available in the article/supplementary material.