

Abstract
A novel Enterobacter cloacae phage, EC151, was isolated and characterized. Electron microscopy revealed that EC151 has a siphovirus-like virion morphology. The EC151 nucleotide sequence shows limited similarity to other phage genomes deposited in the NCBI GenBank database. The size of the EC151 genome is 60,753 bp and contains 58 putative genes. Thirty-nine of them encode proteins of predicted function, 18 are defined as hypothetical proteins, and one ORF identifies as the tRNA-Ser-GCT-encoding gene. Six ORFs were predicted to be members of the deazaguanine DNA modification pathway, including the preQ0 transporter. Comparative proteomic phylogenetic analysis revealed that phage EC151 represents a distinct branch within a group of sequences containing clades formed by members of the Seuratvirus, Nonagvirus, and Vidquintavirus genera. In addition, the EC151 genome showed gene synteny typical of the Seuratvirus, Nonagvirus, and Nipunavirus phages. The average genetic distances of EC151/Seuratvirus, EC151/Nonagvirus, and EC151/Vidquintavirus are approximately equal to those between the Seuratvirus, Nonagvirus, and Vidquintavirus genera (~0.7 substitutions per site). Therefore, EC151 may represent a novel genus within the Siphoviridae family. The origin of the deazaguanine DNA modification pathway in the EC151 genome can be traced to Escherichia phages from the Seuratvirus genus.
Keywords: Enterobacter phage, Seuratvirus, Nonagvirus, deazaguanine modification pathway, preQ0 transporter
1. Introduction
The Enterobacteriaceae family includes anaerobic, motile, Gram-negative rods belonging to the Enterobacterales order, Gammaproteobacteria. This family comprises many genera (Citrobacter, Enterobacter, Escherichia, Klebsiella, Proteus, Salmonella, Shigella, Yersinia, etc.) containing both environmental, medical, and agriculturally important bacteria. Currently, the genus Enterobacter contains 22 species [1,2] and is associated with a variety of environmental habitats. These bacteria are found in water, soil and are phytopathogens for various plant species [2]. Enterobacter spp. are also found as a part of the normal animal and human gut microbiota. Among these bacteria, only certain subspecies/species have been associated with hospital-acquired infections and outbreaks. E. cloacae and E. hormaechei represent the most frequently isolated species described in clinical infections [3]. Since many medically important strains of enterobacteria are antibiotic resistant, lytic phages against such bacteria are of great interest.
There are thirty-six prototype Enterobacter phage genomes deposited in the NCBI GenBank database (www.ncbi.nlm.nih.gov/genomes/GenomesGroup accessed on 1 June 2021). Seventeen Enterobacter phages possess the myovirus morphotype: sixteen of them belong to the Myoviridae, and one, to the Ackermannviridae family. Eight and three phages are members of the Autographiviridae and Podoviridae families, respectively. The remaining eight phages possessed the siphovirus morphotype and were defined as Siphoviridae (six phages) and Drexlerviridae (two phages) family members.
It has been previously found that some enterobacteria phages contain genes responsible for the biosynthesis of queuosine [4,5,6,7]. Queuosine biosynthesis pathway produces modified nucleoside derivatives of guanosine (queuosine (Q) in bacteria and archaeosine (G+) in archaea). 7-Cyano-7-deazaguanine (PreQ0), an intermediate in both the Q and G+ pathways, is synthesized from GTP by four enzymes: FolE, QueC, QueD, and QueE. Both Q and G+ were thought to exist exclusively in tRNA molecules. Queuosine is known to be involved in the modification of cognate tRNAs (histidine, asparagine, aspartic acid, and tyrosine) by replacing guanine in the wobble position (position 34), and it was shown to improve decoding accuracy in protein synthesis [8].
Recently, it has been found that queuosine biosynthesis genes in phage genomes are responsible for 7-deazaguanine derivative insertion into phage DNA [8,9,10]. 7-Deazaguanine modifications are believed to protect phage DNA from host restriction enzymes [9,10,11,12]. Phage genomes may possess various sets of queuosine biosynthesis genes. Besides the folE, queD, queC, and queE genes, phage genomes contain dpdA and Gat-queC (instead of queC) genes or encode a preQ0 transporter [10,11,12,13]. Notably, queuosine biosynthesis genes have been found in genomes of phages infecting both enterobacteria and other Gammaproteobacteria [11,14].
Here, we report for the first time the genome sequence and characteristics of a new Enterobacter phage EC151 belonging to the Siphoviridae family and encodes the deazaguanine DNA modification pathway.
2. Materials and Methods
2.1. Bacterial Strains and Cultivation Media
The Enterobacteria strains used in this study were obtained from the Collection of Extremophilic Micro-organisms and Type Cultures (CEMTC) of the Institute of Chemical Biology and Fundamental Medicine Siberian Branch of the Russian Academy of Science (ICBFM SB RAS), Novosibirsk, Russia. These strains were grown in Luria–Bertani medium and on plates containing Luria–Bertani agar (1.5% w/v). All cultures were grown at 37 °C.
Phage host strain CEMTC 2064 was obtained from the same collection. The strain was identified as Enterobacter cloacae by sequencing a 1308-bp fragment of the 16S rRNA gene and a 501 bp internal portion of the rpoB gene, as described previously [15]. Sequences of the 16S rRNA and rpoB of the investigated strain were deposited in the GenBank database under accession numbers MW980939 and MZ062217, respectively.
2.2. Bacteriophage EC151 Isolation, Propagation and Properties
Bacteriophage EC151 was isolated from a human feces sample, and bacteriophage isolation and propagation were performed as described previously [16]. The study was approved by the Local Ethics Committee of the Center for Personalized Medicine in Novosibirsk, Russia; Protocol #2, 12.02.2019.
The bacterial host range for phage EC151 was examined by spotting serial phage dilutions onto freshly prepared lawns of bacteria, as described previously [17]. Eighty bacterial strains from the Collection of Extremophile Microorganisms and Type Cultures of the ICBFM SB RAS were used for host range examination. The list of strains is given in Table S1.
A phage sample was prepared for electron microscopy as described previously [15]. A drop of phage EC151 suspension was adsorbed for 1 min on a copper grid covered with formvar film; the excess liquid was removed, and the grid was contrasted on a drop of 1% uranyl acetate for 5–7 s. An electron micrograph of phage EC151 particles was obtained using a JEM 1400 transmission electron microscope (JEOL, Tokyo, Japan). Digital images were collected using a side-mounted Veleta digital camera (Olympus SIS, Hamburg, Germany).
2.3. Phage DNA Isolation, Genome Sequencing, and Cleavage of EC151 DNA with Type II Restriction Endonucleases
Phage DNA was extracted from the phage preparation, as described previously [18]. Briefly, phage particles were precipitated using a PEG/NaCl solution and dissolved in a STM buffer (10 mM NaCl, 50 mM Tris-HCl, pH 8.0, 10 mM MgCl2). RNase and DNase (Thermo Fisher Scientific, Waltham, MA, USA) were added to the phage preparation with a final concentration of 5 μg/mL, and the mixture was incubated for 1 h at 37 °C. The phage suspension was then supplemented with EDTA, proteinase K (Thermo Fisher Scientific, Waltham, MA, USA), and SDS to final concentrations of 20 mM, 100–200 μg/mL, and 0.5%, respectively; the mixture was incubated for 3 h at 55 °C. Afterward, phage DNA was purified by phenol/chloroform extraction and subsequent ethanol precipitation.
A paired-end library of phage EC151 DNA was prepared using the Nextera DNA Sample Preparation Kit (Illumina Inc., Foster City, CA, USA). Sequencing was conducted using the MiSeq Benchtop Sequencer and MiSeq Reagent Kit v.1 (2 × 150 base reads) (Illumina Inc.). The genome was assembled de novo by the CLC Genomics Workbench software v.6.0.1 (Qiagen, Venlo, Netherlands) and resulted in one genomic contig with an average coverage of 150.
Recognition sequences for type II restriction endonucleases were found in the EC151 genome using Vector NTI software [19]. EC151 DNA hydrolysis was performed using endonucleases Acc65I, ApaI, DraI, KpnI, SalI, and XmaI (SibEnzyme, Novosibirsk, Russia), 0.3 μg of DNA was incubated with 2 U of endonuclease in the appropriate buffer at 37 °C overnight, and the hydrolyzed DNA profile was revealed using electrophoresis in 1% agarose gel.
2.4. Genome Analysis
Putative open reading frames (ORFs) were annotated using RAST [20] and verified manually by checking all of the predicted proteins against the NCBI GenBank protein database. Screening for t-RNA genes was done using tRNAscan-SE [21]. The CGView server [22] was used for comparative analysis of the EC151 genome and genomes of the Pseudomonas phage NP1 (NC_031058), Escherichia phage 9 g (NC_024146), and Escherichia phage Seurat (NC_027378). MAFFT was used to align genomes [23], and the genetic distance between genomes was calculated using MEGA 7.0 software [24]. A comparative proteomic analysis was performed using a Viral Proteomic tree server [25]. Queuosine pathway components were identified using BLASTx and BLASTp searches against the NCBI GenBank protein database and verified using InterProScan software (https://www.ebi.ac.uk/interpro accessed on 1 June 2021). A phylogenetic analysis of the pathway components with those found in other phages was performed as follows: concatenated protein sequences were prepared using BioEdit 7.2 [26], and the resulting sequences were aligned and analyzed using MEGA 7.0.
3. Results and Discussion
3.1. Phage Characteristics
Phage EC151 forms small plaques with a diameter of about 0.5 mm on the lawn of host strain E. cloacae CEMTC2064 (Figure S1). Electron microscopy revealed that RP180 has a slightly elongated capsid 62 × 76 nm in diameter, which is connected with a long noncontractile tail of approximately 200 nm in length (Figure 1A). Phage particles’ morphology corresponds to that of Siphoviridae family members.
Figure 1.

Phage EC151 characteristics. (A) Electron micrograph of phage EC151 particle. Transmission electron microscopy, negative staining with 1% uranyl acetate; (B) genome map of Enterobacter phage EC151 constructed using the CGView server. For sequence similarity comparison, TBLASTX was used versus Escherichia phage 9 g (light green), Pseudomonas phage NP1 (brown), and Escherichia phage Seurat (cyan).
In order to examine the host range of the EC151, a panel of 44 Enterobacter strains of seven approved species of the genus and 32 other Enterobacteriaceae strains were screened (Table S1). Four Pantoea agglomerans strains were included as well. The host range of bacteriophage EC151 was narrow and included only one E. cloacae strain (host strain CEMTC 2064) of the 80 tested Enterobacterales strains (Table S1). The host strain had been previously isolated from a clinical sample from a patient with a wound infection.
3.2. Genome Characteristics
EC151 genome is a double-stranded DNA molecule with a length of 60,753 bp. The genome contains 58 putative ORFs, 39 of them encoding proteins of predicted function, 18 defined as hypothetical proteins, and one identified as the tRNA-Ser-GCT-encoding gene (Figure 1B). The EC151 nucleotide sequence showed limited similarity to other phage genomes deposited in the NCBI GenBank database, and the BLASTx algorithm revealed the similarity of phage EC151 proteins to proteins of phages belonging to the Seuratvirus, Nonagvirus, and Nipunavirus genera (Figure 1B) [4,5,6,7]. In addition, the investigated proteins possessed similarities to proteins of many unclassified phages, especially unclassified Vibrio phages, and Pantoea phage vB_PagS_Vid5 (NC_042120), which is the only representative of the Vidquintavirus genus [14]. Note that, among related phages, the tRNA-Ser-GCT-encoding gene was revealed only in the genome of the vB_PagS_Vid5 phage; no tRNA genes were found in the Nipunavirus, Seuratvirus, and Nonagvirus genomes.
The comparative proteomic analysis of the EC151 with similar bacteriophages suggested that the phage EC151 represented a distinct branch within a group of sequences containing clades formed by members of the Seuratvirus, Nonagvirus, and Vidquintavirus genera (Figure 2). The mean genetic distances in and between these genera and phage EC151 were calculated (Table 1), and it was revealed that the genetic distances between EC151 and the investigated genera were close to the genetic distances between these genera (~0.7 substitutions per site); therefore, EC151 may represent a new genus in the family Siphoviridae. The complete genome sequence of E. cloacae phage EC151 was deposited in the NCBI GenBank database with the accession number MW464860.
Figure 2.

Comparative proteomic analysis of the EC151 with similar bacteriophages prepared using Viral Proteomic tree server. GenBank identifiers (gi) for the sequences are provided in parentheses. The phage EC151 sequence ID is marked by a black box.
Table 1.
Mean genetic distances in and between members of Seuratvirus, Nonagvirus, Vidquintavirus genera, and Enterobacter phage EC151 in substitutions per site.
| Genus | EC151 | Nonagvirus | Seuratvirus | Vidquintavirus |
|---|---|---|---|---|
| EC151 | - | 0.765 | 0.731 | 0.793 |
| Nonagvirus | 0.765 | 0.119 | 0.609 | 0.717 |
| Seuratvirus | 0.731 | 0.609 | 0.069 | 0.697 |
| Vidquintavirus | 0.793 | 0.717 | 0.697 | - |
3.3. Digestion of EC151 DNA with Type II Restriction Endonucleases
In order to estimate phage EC151 DNA modification, the EC151 genome was screened in silico for recognition sequences of type II restriction endonucleases. Acc65I (G^GTACC), KpnI (GGTAC^C), ApaI (GGGCC^C), DraI (TTT^AAA), SalI (G^TCGAC), and XmaI (C^CCGGG) sites were revealed and chosen for further analysis. The calculated EC151 DNA digestion patterns for these restriction enzymes are summarized in Table S2. It was revealed experimentally that EC151 DNA is highly resistant to hydrolysis with the endonucleases used, except DraI, which is specific to the A/T recognition sequence (TTT^AAA) and hydrolyzes DNA efficiently (Figure 3). Low partial digestion was found in the DNA/SalI hydrolysis, where the recognition sequence contained a G/T cleavage site. All the data suggested that EC151 DNA was highly modified and probably contained G- or C-base modifications.
Figure 3.
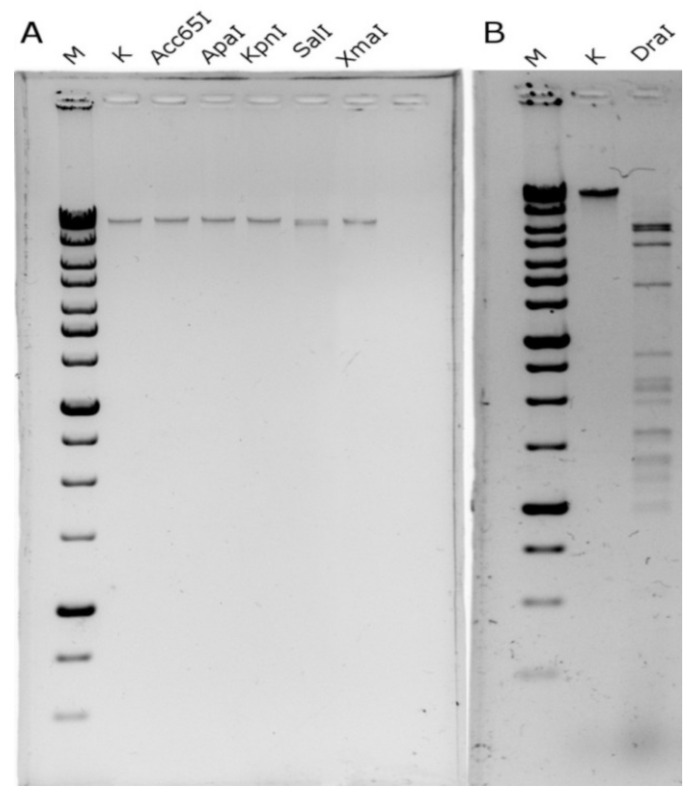
EC151 DNA hydrolysis performed using endonucleases Acc65I, ApaI, KpnI, SalI, and XmaI (A) and DraI (B). The hydrolyzed DNA profile was revealed using electrophoresis in 1% agarose gel. Lanes: M—DNA molecular weight standard 1kb (SibEnzyme, Novosibirsk, Russia), K—EC151 DNA without endonucleases.
3.4. Queuosine Metabolic Pathway
A cluster of genes encoding the deazaguanine DNA modification pathway was revealed in the EC151 genome, similar to that found in the Nipunavirus, Seuratvirus, and Nonagvirus genera’s genomes [4,5,6,7]. The investigated EC151 genome contains four genes (folE, queD, queC, queE), sufficient for the synthesis of a precursor of queuosine (PreQ0) and one more gene, dpdA, which encodes a DNA-modifying protein (Figure 4). Probably, this phage modifies its DNA with dpreQ0, as was shown previously for many phages [11,27]. This cluster contains one more gene, which was annotated using a BLASTx search as a preQ0 transporter, and this function was confirmed using InterProScan software. Such proteins are proposed to salvage the preQ0 or preQ1 precursor from the natural environment [28,29], and phage EC151 probably uses it to increase the preQ0 amount in the host bacterial cell.
Figure 4.
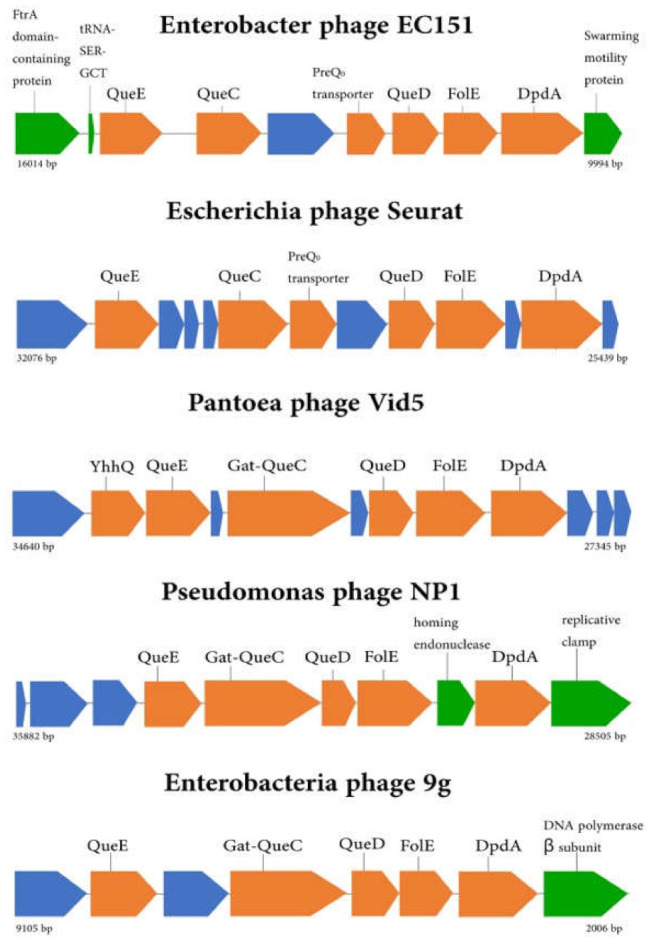
The EC151 deazaguanine DNA modification pathway compared with the corresponding pathways in the most similar phage genomesera. Deazaguanine DNA modification pathway genes are marked with orange boxes, hypothetical proteins are marked with blue, and other proteins are marked with green.
EC151 is the first Enterobacter phage known to contain the full preQ0 synthesis pathway; previously, only the Enterobacter phage phiEM4 (LC373201) (Ackermannviridae; Agtrevirus), containing one component of the pathway, the queC gene (BBD52218), had been revealed [11].
The EC151 deazaguanine DNA modification pathway was compared with the corresponding pathways of the most similar phage genera (Figure 4). It was found that the EC151 pathway possesses a gene synteny typical of the same cluster of Seuratvirus genus members (Figure 4, Escherichia phage Seurat).
In order to evaluate the origin of the deazaguanine DNA modification pathway in the EC151 phage, a phylogenetic analysis of the concatenated QueE, QueC, QueD, FolE, and DpdA proteins of this pathway with the most similar protein sequences was performed. It was found that the investigated cluster of proteins grouped with similar proteins of the Seuratvirus genus (Figure 5). Note that QueC proteins were found in members of the Seuratvirus genus (Figure 4) and some of the unclassified Vibrio phages; meanwhile, members of the Nipunavirus, Vidquintavirus, and Nonagvirus genera encode Gat-QueC proteins (Figure 4), which possess an additional Gat domain required for preQ0 modification into the G+ derivative. Proteins similar to the EC151 preQ0 transporter were found in Seuratvirus genus members and several unclassified phages, especially unclassified Vibrio phages. According to the phylogenetic analysis, the EC151 preQ0 transporter formed a distinct branch within the clade, which included the members of the Seuratvirus genus (Figure 6). Therefore, we can conclude that the deazaguanine DNA modification pathway in the EC151 genome is related to the same pathway in the Seuratvirus genus members, which mostly include Escherichia phages. Phage EC151 and phages of the Seuratvirus genus probably had a common ancestor from which they inherited this operon.
Figure 5.

Phylogenetic analysis of the concatenated QueE, QueC, QueD, FolE, and DpdA proteins of the EC151 deazaguanine DNA modification pathway with the most similar protein sequences. Alignment and analysis were performed using MEGA 7.0, and the maximum-likelihood method was applied; bootstrap value 1000 was used. Bayes branch support values above 80% are provided at nodes. GenBank identifiers (gi) for the sequences are provided in parentheses. The Enterobacter cloacae phage EC151 sequence is indicated by a black circle.
Figure 6.

Phylogenetic analysis of the preQ0 transporter with the most similar protein sequences. Alignment and analysis were performed using MEGA 7.0, and the maximum-likelihood method was applied; bootstrap value 1000 was used. Bayes branch support values above 80% are provided at nodes. GenBank identifiers (gi) for the sequences are provided in parentheses. The preQ0 of Enterobacter cloacae phage EC151 is indicated by a black circle.
4. Conclusions
The novel Enterobacter cloacae phage EC151 was characterized for the first time and is suggested to represent a new genus in the Siphoviridae family. Electron microscopy showed that the EC151 phage possesses a siphovirus-like capsid morphology and EC151 genome organization is typical for the Siphoviridae family. Although increasingly more studies show that the host range of some phages is a function of the environment rather than evolution, EC151 has a very narrow host range. Our results showed that EC151 can only infect E. cloacae CEMTC 2064 strain from 80 tested Enterobacterales strains, including 44 strains of seven Enterobacter species.
Notably, the EC151 genome encodes a complete set of proteins of the deazaguanine DNA modification pathway. The only type II restriction endonuclease able to efficiently hydrolyze the EC151 genome is DraI with a cleavage site TTT^AAA. This fact confirmed that the deazaguanine DNA modification gene cluster in the EC151 genome is active and provides modifications sufficient to protect phage DNA from at least some restriction enzymes.
The EC151 deazaguanine DNA modification gene cluster has a clear gene synteny with corresponding gene clusters of the Seuratvirus genus and a group of unclassified siphoviruses containing mostly Vibrio phages. The trees constructed for the complete protein sequences and proteins encoded by the deazaguanine DNA gene cluster differ in their topology. However, the similarity of phage EC151 proteins to proteins of phages of the Seuratvirus genus containing mostly Escherichia phages was higher than other siphoviruses tested in this study. The obtained data supports the idea that the EC151 deazaguanine DNA modification gene cluster may be inherited from the common enterobacterial phage ancestor and remains active because of its necessity to the EC151 life cycle.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/v13071372/s1, Figure S1. A photograph of the plaques formed by phage EC151 on the lawn of host strain Enterobacter cloacae CEMTC 2064. Table S1: The list of microbial strains tested for sensitivity to phage EC151, the strain host CEMTC 2064 is marked with yellow. Table S2: Calculated digestion patterns of EC151 DNA for type II restriction endonucleases. Recognition sequences for endonucleases were found in the EC151 genome using Vector NTI software.
Author Contributions
Conceptualization, V.M.; methodology, V.M.; investigation, Y.K., A.T., and G.J.; resources, N.T.; data curation, I.B. and G.J.; writing—original draft preparation, V.M. and G.J.; writing—review and editing, N.T.; visualization, V.M. and I.B.; supervision, N.T.; project administration, N.T.; funding acquisition, N.T. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by the Russian Scientific Foundation, Project No. 21-14-00360. The Collection of Extremophile Microorganisms and Type Cultures of ICBFM SB RAS was supported by the Ministry of Education and Science of the Russian Federation, Project No. 121031300043-8.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and was approved by a Local Ethics Committee of the Center for Personalized Medicine in Novosibirsk, Russia; Protocol #2, 12.02.2019.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hormaeche E., Edwards P.R. A Proposed Genus Enterobacter. Int. J. Syst. Bacteriol. 1960;10:71–74. doi: 10.1099/0096266X-10-2-71. [DOI] [Google Scholar]
- 2.Davin-Regli A., Lavigne J.-P., Pagès J.-M. Enterobacter spp.: Update on Taxonomy, Clinical Aspects, and Emerging Antimicrobial Resistance. Clin. Microbiol. Rev. 2019;32:00002–19. doi: 10.1128/CMR.00002-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mezzatesta M.L., Gona F., Stefani S. Enterobacter cloacaecomplex: Clinical impact and emerging antibiotic resistance. Futur. Microbiol. 2012;7:887–902. doi: 10.2217/fmb.12.61. [DOI] [PubMed] [Google Scholar]
- 4.Kulikov E.E., Golomidova A.K., Letarova M.A., Kostryukova E.S., Zelenin A.S., Prokhorov N.S., Letarov A.V. Genomic Sequencing and Biological Characteristics of a Novel Escherichia Coli Bacteriophage 9g, a Putative Representative of a New Siphoviridae Genus. Viruses. 2014;6:5077–5092. doi: 10.3390/v6125077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doan D.P., Lessor L.E., Hernandez A.C., Everett G.F.K. Complete Genome Sequence of Enterotoxigenic Escherichia coli Siphophage Seurat. Genome Announc. 2015;3:e00044-15. doi: 10.1128/genomeA.00044-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carstens A.B., Kot W., Lametsch R., Neve H., Hansen L.H. Characterisation of a novel enterobacteria phage, CAjan, isolated from rat faeces. Arch. Virol. 2016;161:2219–2226. doi: 10.1007/s00705-016-2901-0. [DOI] [PubMed] [Google Scholar]
- 7.Flores V., Sepúlveda-Robles O., Cazares A., Kameyama L., Guarneros G. Comparative genomic analysis of Pseudomonas aeruginosa phage PaMx25 reveals a novel siphovirus group related to phages infecting hosts of different taxonomic classes. Arch. Virol. 2017;162:2345–2355. doi: 10.1007/s00705-017-3366-5. [DOI] [PubMed] [Google Scholar]
- 8.Hutinet G., Swarjo M.A., De Crécy-Lagard V. Deazaguanine derivatives, examples of crosstalk between RNA and DNA modification pathways. RNA Biol. 2017;14:1175–1184. doi: 10.1080/15476286.2016.1265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weigele P., Raleigh E.A. Biosynthesis and Function of Modified Bases in Bacteria and Their Viruses. Chem. Rev. 2016;116:12655–12687. doi: 10.1021/acs.chemrev.6b00114. [DOI] [PubMed] [Google Scholar]
- 10.Tsai R., Corrêa I.R., Xu M.Y., Xu S.-Y. Restriction and modification of deoxyarchaeosine (dG+)-containing phage 9 g DNA. Sci. Rep. 2017;7:1–13. doi: 10.1038/s41598-017-08864-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutinet G., Kot W., Cui L., Hillebrand R., Balamkundu S., Gnanakalai S., Neelakandan R., Carstens A.B., Lui C.F., Tremblay D., et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 2019;10:1–12. doi: 10.1038/s41467-019-13384-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma R., Lai J., Chen X., Wang L., Yang Y., Wei S., Jiao N., Zhang R. A Novel Phage Infecting Alteromonas Represents a Distinct Group of Siphophages Infecting Diverse Aquatic Copiotrophs. mSphere. 2021;6:e0045421. doi: 10.1128/mSphere.00454-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crippen C.S., Lee Y.-J., Hutinet G., Shajahan A., Sacher J., Azadi P., de Crécy-Lagard V., Weigele P.R., Szymanski C.M. Deoxyinosine and 7-Deaza-2-Deoxyguanosine as Carriers of Genetic Information in the DNA of Campylobacter Viruses. J. Virol. 2019;93:e01111-19. doi: 10.1128/JVI.01111-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Šimoliūnas E., Šimoliūnienė M., Kaliniene L., Zajančkauskaitė A., Skapas M., Meškys R., Kaupinis A., Valius M., Truncaitė L. Pantoea Bacteriophage vB_PagS_Vid5: A Low-Temperature Siphovirus That Harbors a Cluster of Genes Involved in the Biosynthesis of Archaeosine. Viruses. 2018;10:583. doi: 10.3390/v10110583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morozova V., Babkin I., Kozlova Y., Baykov I., Bokovaya O., Tikunov A., Ushakova T., Bardasheva A., Ryabchikova E., Zelentsova E., et al. Isolation and Characterization of a Novel Klebsiella pneumoniae N4-like Bacteriophage KP8. Viruses. 2019;11:1115. doi: 10.3390/v11121115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morozova V., Fofanov M., Tikunova N., Babkin I., Morozov V.V., Tikunov A. First crAss-Like Phage Genome Encoding the Diversity-Generating Retroelement (DGR) Viruses. 2020;12:573. doi: 10.3390/v12050573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kutter E. Phage Host Range and Efficiency of Plating. Adv. Struct. Saf. Stud. 2009;501:141–149. doi: 10.1007/978-1-60327-164-6_14. [DOI] [PubMed] [Google Scholar]
- 18.O’Flaherty S., Coffey A., Edwards R., Meaney W., Fitzgerald G.F., Ross R. Genome of Staphylococcal Phage K: A New Lineage of Myoviridae Infecting Gram-Positive Bacteria with a Low G+C Content. J. Bacteriol. 2004;186:2862–2871. doi: 10.1128/JB.186.9.2862-2871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu G. Vector NTI, a balanced all-in-one sequence analysis suite. Brief. Bioinform. 2004;5:378–388. doi: 10.1093/bib/5.4.378. [DOI] [PubMed] [Google Scholar]
- 20.Aziz R.K., Bartels D., Best A.A., DeJongh M., Disz T., Edwards R.A., Formsma K., Gerdes S., Glass E.M., Kubal M., et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008;9:1–15. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowe T.M., Chan P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–W57. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant J.R., Stothard P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36:W181–W184. doi: 10.1093/nar/gkn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katoh K., Rozewicki J., Yamada K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019;20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar S., Stecher G., Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishimura Y., Yoshida T., Kuronishi M., Uehara H., Ogata H., Goto S. ViPTree: The viral proteomic tree server. Bioinformatics. 2017;33:2379–2380. doi: 10.1093/bioinformatics/btx157. [DOI] [PubMed] [Google Scholar]
- 26.Hall T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- 27.Kot W., Olsen N.S., Nielsen T.K., Hutinet G., De Crécy-Lagard V., Cui L., Dedon P.C., Carstens A.B., Moineau S., Swairjo M.A., et al. Detection of preQ0 deazaguanine modifications in bacteriophage CAjan DNA using Nanopore sequencing reveals same hypermodification at two distinct DNA motifs. Nucleic Acids Res. 2020;48:10383–10396. doi: 10.1093/nar/gkaa735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodionov D.A., Hebbeln P., Eudes A., ter Beek J., Rodionova I.A., Erkens G.B., Slotboom D.J., Gelfand M., Osterman A.L., Hanson A.D., et al. A Novel Class of Modular Transporters for Vitamins in Prokaryotes. J. Bacteriol. 2009;191:42–51. doi: 10.1128/JB.01208-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan Y., Zallot R., Grove T.L., Payan D.J., Martin-Verstraete I., Šepić S., Balamkundu S., Neelakandan R., Gadi V.K., Liu C.-F., et al. Discovery of novel bacterial queuine salvage enzymes and pathways in human pathogens. Proc. Natl. Acad. Sci. USA. 2019;116:19126–19135. doi: 10.1073/pnas.1909604116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.