

Abstract
Following the clinical success of cancer immunotherapies such as immune checkpoint inhibitors blocking B7/CTLA-4 or PD-1/PD-L1 signaling and ongoing numerous combination therapies in the clinic,3 bispecific antibodies (BsAbs) are now emerging as a growing class of immunotherapies with the potential to improve clinical efficacy and safety further. Here, we describe four classes of BsAbs: (a) immune effector cell redirectors; (b) tumor-targeted immunomodulators; (c) dual immunomodulators; and (d) dual tumor-targeting BsAbs. This review describes each of these classes of BsAbs and presents examples of BsAbs in development. We reviewed the biological rationales and characteristics of BsAbs and summarized the current status and limitations of clinical development of BsAbs and strategies to overcome limitations. The field of BsAb-based cancer immunotherapy is growing, and more data from clinical trials are accumulating. Thus, BsAbs could be the next generation of new treatment options for cancer patients.
Keywords: bispecific antibody, cancer immunotherapy
1. Introduction
Immunotherapy, including tumor-targeted monoclonal antibodies (mAbs) and immune checkpoint inhibitors (ICIs), has revolutionized anticancer therapy since the late 1990s. Currently, more than 30 different therapeutic mAbs have been developed and approved by the Food and Drug Administration (FDA) for oncology since the approval of rituximab, a B-cell-depleting anti-CD20 mAb used for the treatment of patients with B-cell non-Hodgkin lymphoma (NHL) [1,2]. Ipilimumab was the first FDA-approved ICI in 2011 for melanoma, which blocks cytotoxic T lymphocyte antigen-4 (CTLA-4), a negative regulator of T-cell immune function. Programmed cell death protein 1 (PD-1) is a checkpoint protein on immune cells and regulates the function of T cells by binding to programmed death-ligand 1 (PD-L1) on tumor cells. Anti-PD-(L)1 mAbs, which inhibit PD-1 or PD-L1, can block PD-1 signaling significantly and showed promising antitumor activity in treating certain cancers [3]. PD-1 inhibitors, nivolumab and pembrolizumab, were approved in 2014, and PD-L1 inhibitors, such as atezolizumab, avelumab, and durvalumab, were also approved in 2016 and 2017 for various cancers [4,5,6,7]. Currently, immunotherapies are widely used for the clinical treatment of various tumor types, including melanoma, non-small cell lung cancer, and colorectal cancer, and newly developed antibody-based therapeutics are under clinical investigation [8]. Although immunotherapies have shown significant and long-term efficacy in cancer patients, their clinical benefits in the overall population are still low because of limitations and challenges such as low response rate and resistance development [9,10]. In addition, immune-related adverse events (irAEs) caused by an excessively activated immune system, such as colitis, dermatological toxicity, hepatotoxicity, and pneumonitis, limit the clinical use of immunotherapies. Combination therapies seem to have improved efficacy compared to monotherapies but are also more likely to induce autoimmune-like toxicities, such as increased irAEs, which should be managed [11].
Advances in protein engineering technology have enabled the generation of various bispecific antibodies (BsAbs) that target multiple antigens as a single molecule [12]. BsAb-based immunotherapeutics may have the potential to improve clinical efficacy and safety. Therefore, the interest in the development of BsAbs has grown considerably, and there are various types of BsAbs in clinical and preclinical stages [13]. Since BsAbs can directly target two different antigens on immune cells or tumors (tumor-associated antigens; TAAs), BsAbs have an advantage over monospecific antibodies by synergistic inhibition of two different antigens or increasing cytotoxic activity. Currently, most BsAbs are T cell-redirecting antibodies, including two FDA-approved molecules (Blinatumomab and Catumaxomab) and an increasing number of next-generation bispecific or multi-specific antibodies. This review describes the classes, characteristics of BsAbs, biological rationales for developing BsAbs, current status and issues in the clinic, and strategies to overcome some limitations.
2. Binding Modules and Characteristics of Bispecific Antibodies
The characteristics of BsAbs, including physicochemical properties (size, stability, binding affinity, and valency), pharmacokinetics (PK)/pharmacodynamics (PD) properties, manufacturability, and immunogenicity, impact their efficacy, safety, and clinical success [14,15]. To achieve the desired biological characters, different BsAb formats with various modifications have been developed (Table 1).
Table 1.
Immune modulating bispecific antibody formats in clinical trials for cancer therapy.
| Category | Antibody Format/Platform | Structure | Company | Product | Target |
|---|---|---|---|---|---|
| Single domain antibody (sdAb)-based BsAbs [16,17,18] |
|
Alphamab | KN046 | PD-L1 × CTLA-4 | |
| Inhibrix | INBRX-105 | PD-L1 × 4-1BB | |||
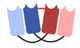
| Single-chain variable fragment (scFv)-based BsAbs [19,20,21,22,23,24,25,26,27] | Bispecific T-cell Engager (BiTE) |

|
Amgen | Blinatumomab (Blincyto) | CD19 × CD3 |
| AMG330 | CD33 × CD3 | ||||
| AMG420 | BCMA × CD3 | ||||
| Bayor | BAY2010112 | PSMA × CD3 | |||
| BiTE-Fc/Half-life extended BiTE (HLE BiTE) |

|
Amgen | AMG701 (Pavurutamab) |
BCMA × CD3 | |
| AMG673 | CD33 × CD3 | ||||
| AMG757 | DLL3 × CD3 | ||||
| Dual affinity retargeting (DART) |
|
Macrogenics | MGD006 (Flotetuzumab) | CD123 × CD3 | |
| DART-Fc |

|
Macrogenics | MGD013 (Tebotelimab) | PD-1 × LAG-3 | |
| MGD019 | PD-1 × CTLA-4 | ||||
| Tandem Diabody (TandAb) |
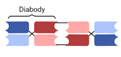
|
Affimed | AFM13 | CD30 × CD16A | |
| Amphivena | AMV564 | CD33 × CD3 | |||
| Trispecific Killer Engager (TriKE) |

|
GT Biopharma | GTB-3550 | CD33 × CD16, IL-15 | |
| IgG-based, heterodimeric bispecifics [28,29,30,31,32,33,34] |
Common light chain (Biclonics, Veloci-Bi) |
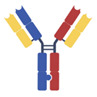
|
Merus | MCLA-145 | PD-L1 × 4-1BB |
| Regeneron | REGN1979 | CD20 × CD3 | |||
| REGN5678 | PSMA × CD28 | ||||
| DuoBody; Controlled Fam-arm exchange |

|
Janssen | Teclistamab (JNJ-64007957) |
BCMA × CD3 | |
| Genmab-Abbvie | Epcoritamab (GEN3013) |
CD20 × CD3 | |||
| Genmab-Biontech | BNT312 (GEN1042) |
CD40 × 4-1BB | |||
| BNT311 (GEN1046) |
PD-L1 × 4-1BB | ||||
| 1 + 1 CrossMAb |

|
Roche | RO7247669 | PD-1 × LAG-3 | |
| Mosunetuzumab | CD20 × CD3 | ||||
| Cevostamab | FcRH5 × CD3 | ||||
| 2 + 1 CrossMAb |
|
Roche | Glofitamab | CD20 × CD3 | |
| RG7802 (RO6958688) |
CEA × CD3 | ||||
| Celgene | CC-93269 | BCMA × CD3 | |||
| IgG-based, homodimeric bispecifics [35,36,37,38,39] |
Grabody, IgG-scFv |
|
ABL Bio | ABL503 | PD-L1 × 4-1BB |
| ABL111 | Claudin 18.2 × 4-1BB | ||||
| IgG-anticalin fusion protein |

|
Pieris | PRS-343 | HER2 × 4-1BB | |
| Fc region with antigen binding (Fcab), mAb2 |

|
F-Star | FS118 | PD-L1 × LAG-3 | |
| Others [40,41,42,43] | XmAb, Fab + scFv + Fc |

|
Xencor | Vibecotamab (XmAb 14045) | CD123 × CD3 |
| XmAb20717 | PD-1 × CTLA-4 | ||||
| Bispecific Engagement by Antibodies based on TCR (BEAT) |

|
Glenmark Pharmaceuticals | GBR1342 | CD38 × CD3 |
2.1. Building Blocks of Bispecific Antibodies
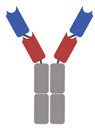
A conventional immunoglobulin (Ig) G consists of two heavy chains (VH-CH1-CH2-CH3) and two light chains (VL-CL) connected by four disulfide bonds. Epitope binding domains consist of two pairs of variable domains of heavy and light chain, (VH + VL)2, with mono-specificity. A single-chain variable fragment (scFv) consists of VH and VL connected by a short amino acid linker, while Fab refers to pairs of VH/CH1 and VL/CL, including the constant domain [19]. Two scFvs with different binding specificity are tandemly linked by a short flexible linker (tandem scFv) in a bispecific T cell engager (BiTE) platform (Amgen, Thousand Oaks, CA, USA) [44]. Diabody refers to two non-covalently associated single chains of tandemly linked VH and VL with different specificity A and B (VHA-VLB and VHB-VLA). Immunoglobulin (IgG2 and IgG3 isotype) from camelids, also called heavy-chain only antibodies, has a unique structure devoid of the light chain component and the first constant domain of the heavy chain (CH1). These epitope binding domains of heavy-chain-only antibody are also called single variable domain, single-domain antibody (sdAb, VHH), or Nanobody [16,17] (Figure 1a).
Figure 1.

The building blocks and formats of bispecific antibodies. (a). The building blocks of BsAbs. Most BsAbs consist of antibody-based fragments (Fv, Fab, Fc, scFv, and sdAb), and some BsAbs include non-antibody-based proteins such as TCR, extracellular domains from receptors or ligands, and antibody-like scaffolds. Fv (VL + VH), Fab (VL-CL + VH-CH1), Fc (CH2-CH3), and scFv (VL-VH) are derived from the conventional antibody. sdAb (VHH) or nanobody is derived from the heavy chain antibody in camelids or cartilaginous fishes. (b). The examples of fragment-based BsAbs. These types of BsAbs are developed by linking the building blocks in a. (c). The example of IgG-based BsAbs. These BsAbs are developed by pairing two different heavy chains and light chains (heterodimeric BsAb) or linking the building blocks to IgG (homodimeric BsAb). Various strategies are used for proper pairing; knob-into-hole, CRIB, and BEAT for heterodimerization of heavy chains, κλ body, Biclonics, DuoBody, CrossMAb, and WuXiBody for heterodimerization of light chains. Abbreviations: BiTE; bispecific T-cell engager; TriKE, trispecific killer cell engager; DART, dual-affinity retargeting; TandAb, tandem diabody; TriTAC, trispecific T cell-activating construct; ImmTAC, immune mobilizing monoclonal TCR against cancer; scMATCH, single-chain multispecific antibody-based therapeutics by cognate heterodimerization; CRIB, charge repulsion induced bispecific; BEAT, bispecific engagement by antibodies based on the TCR; κλ body, kappa lambda body; DVD-Ig, dual-variable-domain immunoglobulin; Fcab, Fc region with an antigen-binding site.
2.2. Single-Chain Variable Fragment (scFv)-Based Bispecific Antibodies
The first scFv-based BsAb is a BiTE, in which two different scFvs are tandemly linked [44]. Blinatumomab, approved for the treatment of B-cell acute lymphoblastic leukemia (B-ALL), binds to CD3 and CD19 and elicits CD19-dependent T-cell activation [20]. Due to low molecular weight (~60 kDa), the half-life of BiTE is generally short, lasting less than several hours. To increase the in vivo half-life, the fragment crystallizable (Fc) domain was linked to BiTE. Other approaches to extend the half-life include ligating human serum albumin (HSA) itself or genetically linking the scFv against HSA [45,46] (Figure 1b).
2.3. IgG-Based Bispecific Antibodies
IgG-based BsAbs generally have a longer half-life compared with scFv- or domain-based antibodies comparable to conventional antibodies. A major challenge in IgG-based BsAbs is the proper pairing of two different heavy and light chains. It is possible to generate 16 different heterodimeric antibodies based on two different heavy chains and two different light chains without modification. The appropriate pairing of light chains with the corresponding heavy chains and the pairing of different heavy chains results in different heterodimeric antibodies. For heterodimerization of heavy chains, the knob-into-hole technology is generally used [32], but other technologies leveraging charge difference (e.g., Charge Repulsion Induces Bispecific; CRIB or substituting the CH3 region with T cell receptor (TCR) interface (used in Bispecific Engagement by Antibodies based on TCR; BEAT, Ichnos Biosciences, New York, NY, USA) are also in use [42,43]. For the pairing of heavy and light chains, diverse technologies have been developed, including (1) sharing the common light chains (Biclonics, Merus), (2) controlled Fab-arm exchange (DuoBody, Genmab), (3) switching the constant regions of heavy (CH1) and light chains (CL1) on one side of the heterodimer (CrossMAb, Roche), and (4) substituting the constant regions of immunoglobulins (CH1/CL) with the constant domains (Cα/Cβ) of TCRs (WuXiBody, WuXi Biologics) [19,42]. Another approach is to maintain a regular IgG structure on one side but substitute the Fab with the scFv on the other side, as in the XmAb from Xencor such as vibecotamab (CD123 × CD3) and XmAb20717 (PD-1 × CTLA-4) [40,41] (Figure 1c).
Homodimeric bispecific antibodies are also frequently found in immunomodulating antibodies, especially in 4-1BB-engaging antibodies or dual immune checkpoint blocking antibodies. To induce TAA-dependent 4-1BB cross-linking and activation, 4-1BB-binding scFvs (ABL503, ABL111) [35,36] or anticalin (PRS-343) are connected to the C-terminal ends of the Fc of TAA-specific antibodies [37]. F-star Therapeutics develops its proprietary modular antibody technology platform, which utilizes some mutations in the Fc region, antigen-binding Fc fragments (Fcab). They are developing multiple pipelines of immuno-oncology therapies such as FS118 (PD-L1 × LAG-3) and FS222 (PD-L1 × 4-1BB) with its innovative BsAb technology, the tetravalent antibodies called mAb2 [47] (Figure 1c).
The Fc domain has effector functions mediated by their binding to the FcγR for IgG or to complement protein C1q [46], including antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC). Fc-engineering technology was introduced to generate versatile BsAb platform technology. Altering certain functions of the Fc domain of an antibody by substituting an amino acid sequence or glycoengineering can enhance or reduce effector functions [46,48]. Fc domains without Fc function or with retained/enhanced Fc function are generally used for immunomodulatory BsAbs and tumor-targeting BsAbs, respectively.
2.4. Affinity and Valency of Bispecific Antibodies
Depending on the immune cell target for agonistic activation, selecting the appropriate valency and affinity for each target is critical for TAA-dependent augmentation of the immune response and securing safety [15]. For CD3-targeting BsAbs, a single CD3-targeting arm is generally used to trigger TAA expression-dependent CD3 activation and T cell-mediated tumor killing while minimizing potential side effects from non-specific T-cell activation. Affinities of T-cell engagers to CD3 are diverse from 2 to 500 nM. Because CD3 is constitutively expressed in T cells, it is generally conceived that a lower binding affinity to CD3 might improve the PK, biodistribution, and safety [49,50]. To increase the TAA-binding activity yet minimizing the CD3 binding activity in the absence of TAA, two Fab regions targeting TAA and monovalent CD3 Fab were incorporated in a 2 + 1 format on the IgG backbone. Currently, both epcoritamab (CD20 × CD3, 1 + 1 format) and glofitamab (CD20 × CD3, 2 + 1 format) are in phase III clinical trials [31,33].
For 4-1BB-engagers, both one-arm and two-arm 4-1BB-engagers have been used to augment TAA-dependent 4-1BB trimerization and T cell activation. PRS-343 (HER2 × 4-1BB) [38], ABL503 (PD-L1 × 4-1BB) [35], and ABL111 (Claudin 18.2 × 4-1BB) [36] are bivalent 4-1BB engagers with a 2 + 2 format, while MCLA-145 (PD-L1 × 4-1BB) is a monovalent 4-1BB activator with a 1 + 1 format [28]. Tetravalent natural killer (NK) cell-engaging BsAbs, including AFM13, adopt the tandem diabody format, which contains a 9-amino acid linker that links variable domains of heavy and light chains to form a single polypeptide, and functionally active homodimers are generated by autonomous assembly of the corresponding VH to VL [25].
2.5. Immunogenicity of Bispecific Antibodies
The molecular structures of BsAbs are not naturally observed, raising the potential risks of immunogenicity. In addition to reducing drug availability and efficacy, the generation of anti-drug antibodies (ADAs) and neutralizing antibodies could also cause severe drug-related toxicities by forming drug/ADA immune complexes and eliciting hypersensitivity reactions [51]. Various factors affect the immunogenicity of antibodies, including product-related impurities, antibody origin (human, chimeric), dosing regimen, and target molecule [51]. Due to the short history of BsAb development in the clinics, reports on immunogenicity are limited: Less than 1% of patients treated with blinatumomab display ADA [51,52]. In the same way, no ADA-related issues were reported with other B cell-targeting BsAbs, such as mosunetuzumab (CD20 × CD3, 1 + 1 IgG format) and glofitamab (CD20 × CD3, 2 + 1 IgG format), regardless of their format [51]. This might be due to mechanism of action (MoA)-based phenomena in which the elimination of antibody-producing B cells by these BsAbs eliminates the possible generation of ADA. ADA generation was reported in other T cell-activating BsAbs such as PRS-343 (HER2 × 4-1BB using anticalin technology), where BsAbs at 2.5 mg/kg or more elicited ADA in 27.8% of cancer patients [53]. The ADAPTIR platform developed by Aptevo Therapeutics contains two different scFvs linked to the N- and C-terminal ends of the Fc domain. APVO-414 (PSMA × CD3) in the ADAPTIR platform elicited ADA in more than 50% of the patients, and further clinical development has been on hold [54,55]. AFM13 (CD30 × CD16A), a first-in-class tetravalent tandem diabody (TandAb)-based BsAb, showed ADA in 15 of the 28 patients treated, and half of the detected ADAs had neutralizing potential due to the chimeric origin of the anti-CD30 scFv [25]. It is not known whether tetravalent TandAb contributed to immunogenicity. Low ADA titers were observed in 42% of the patients treated with FS118. Interestingly, ADA by FS118 was transiently observed at higher dose levels and did not affect drug exposure [39]. VHH domains from camelids have high sequence identity with human type 3 VH domain and are expected to have low immunogenicity.
3. Multiple Types of Bispecific Therapeutics in Immuno-Oncology
Bispecific therapeutics can be divided into four major categories based on the biological types of targets and modes of action: (a) immune effector cell redirectors, (b) tumor-targeted immunomodulators, (c) dual immunomodulators, and (d) dual tumor-targeting BsAbs (Figure 2). Among these categories, both immune effector cell redirectors and tumor-targeted immunomodulators induce tumor-targeted activation of immune cells and are being developed extensively in the clinics. In both approaches, the selection of the optimal TAA is critical. The TAA expression pattern affects the safety and off-tumor toxicity because TAA is highly expressed in tumor cells or the tumor-associated environment, whereas it is absent in normal tissues. In addition, TAA expression will determine the potential indication of BsAbs due to tumor-specific expression. This review will focus on immune effector cell redirectors and tumor-targeted immunomodulators among BsAbs and briefly introduce the biological rationale for developing dual immunomodulators and dual tumor-targeting BsAbs in the last section.
Figure 2.

The types of bispecific therapeutics in immuno-oncology. Bispecific therapeutics are grouped into four major categories based on the biological targets and mode of actions. (a). Immune effector cell redirectors engage tumor cell and immune effector cells by binding to tumor-associated antigen and immune cell receptors such as CD3, CD16, or NKG2D. (b). Tumor-targeted immunomodulators simultaneously bind to cosignaling molecules (CD28 and 4-1BB) on T cells and tumor cells, inducing tumor-specific activation of T cells. (c). Dual immunomodulators regulate the activity of two different immunomodulatory pathways by dual targeting of cosignaling pathways. (d). Dual tumor-targeting BsAbs modulate two functional pathways in the TME by inhibiting angiogenesis, RTK activity, or CD47 binding.
4. Immune Effector Cell Redirectors
Among the four types of BsAbs, immune effector cell redirectors, especially T cell engagers, are the most advanced, with two approved products and several molecules in clinical development. These types of BsAbs include T cell-engagers, NK cell-engagers, and other immune cell-engagers composed of a TAA-targeting moiety and an immune cell-redirecting moiety.
4.1. T Cell-Engagers
T cells recognize target cells by detecting the cognate peptides presented by the major histocompatibility complex (MHC) through their TCRs under physiological conditions. The interaction between TCRs and the cognate peptide/MHC (pMHCs) brings the T cells and target cells in proximity and results in TCR clustering at the center of the immunological synapse. Following TCR clustering, cytolytic granules release cytolytic molecules, such as perforin and granzymes, into the target cells, eventually inducing target cell death. Cancer cells have several mechanisms to escape immunosurveillance, most of which are related to antigen processing and presentation in the context of MHC molecules.
T cell-engagers are BsAbs that engage TAAs and a component of the TCR complex (mostly CD3). They bypass TCR-pMHC interactions and facilitate the formation of cytolytic synapses between T cells and cancer cells, followed by polyclonal activation of T cells, thereby redirecting T cell cytotoxicity to the tumor cells. In this approach, T cell activation by BsAbs is independent of the TCR antigen specificity. Thus, it results in the activation of a large proportion of T cells even with low TAA expression and co-stimulation [56], although co-stimulation further enhances the activity [30]. T-cell-engaging BsAbs have been extensively explored in hematological and solid tumors for many years, and two such FDA-approved BsAbs, blinatumomab (Blincyto) and catumaxomab (Removab), have demonstrated favorable clinical outcomes. Although the latter was voluntarily withdrawn from the market for commercial reasons, there are currently more than 60 T-cell engagers in phase I/II clinical trials for the treatment of hematological and solid tumors.
Blinatumomab is a CD19- and CD3-targeting (CD19 × CD3) BsAb that the FDA approved in 2014 for the treatment of relapsed/refractory B-ALL [20], and is currently undergoing phase I/II clinical trials in other hematological malignancies such as diffuse large B-cell lymphoma and NHL [57]. The complete remission (CR) rate was 32%, although cytokine release syndrome (CRS) and neurotoxicity occurred in some patients [20]. Blinatumomab does not have an Fc region, thus having a short serum half-life of approximately 2 h [20]. Blinatumomab redirects T cell cytotoxicity at sub-picomolar concentrations by inducing immunological synapse formation between T cells and CD19+ target cells, activation and proliferation of T cells, and lysis of target cells [58]. Due to its short half-life, the dosing regimen for blinatumomab has been limited to continuous infusion in a clinical setting. For a prolonged half-life, some T cell engagers consist of the Fc domain, such as half-life extended BiTE and catumaxomab.
Catumaxomab is an EpCAM × CD3 BsAb, which was approved in 2009 for the treatment of malignant ascites in patients with ovarian cancer [59]. In contrast to blinatumomab, catumaxomab has a different molecular format. It consists of an asymmetric Fc-containing full-length Ab comprised of heavy and light chains binding to each target and produced from rat/mouse quadroma. The chimeric Fc domain binds to FcγRs on macrophages, dendritic cells (DCs), and NK cells and activates Fc-mediated functions to tumor cells [59]. However, since its Fc domain is not originated from human Ab, it could trigger immunogenicity and compromise its effectiveness in patients [60]. Other T-cell-redirecting BsAbs in clinical trials are shown in Table 2.
Table 2.
Immune cell-engagers in clinical stages.
| Name | Targets | Developer | Format | Highest Phase | Clinical Trials |
|---|---|---|---|---|---|
| AFM13 | CD30 × CD16A | Affimed GmbH | Fv + (Fv)2 + Fv, 2 + 2 TandAb |
II |
NCT04074746 and 5 studies |
| AFM24 | EGFR × CD16A | Affimed GmbH | Fv + (Fv)2 + Fv, 2 + 2 TandAb |
I/II | NCT04259450 |
| AFM26, RO7297089 |
BCMA × CD16A | Affimed GmbH, Genentech |
Fv + (Fv)2 + Fv, 2 + 2 TandAb |
I | NCT04434469 |
| Solitomab, AMG110, MT110 |
EpCAM × CD3 | Amgen | scFv + scFv, 1 + 1 BiTE |
I | NCT00635596 |
| AMG160 | PSMA × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE |
I |
NCT03792841 NCT04631601 |
| AMG199 | MUC17 × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE |
I | NCT04117958 |
| AMG330 | CD33 × CD3 | Amgen | scFv + scFv, 1 + 1 BiTE |
I |
NCT02520427 NCT04478695 |
| AMG427 | FLT3 × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE |
I | NCT03541369 |
| AMG562 | CD19 × CD3 | Amgen | scFv + scFv, 1 + 1 BiTE |
I | NCT03571828 |
| AMG596 | EGFRvIII × CD3 | Amgen | scFv + scFv, 1 + 1 BiTE |
I | NCT03296696 |
| AMG673 | CD33 × CD3 | Amgen | scFv + scFv + Fc, 1+ 1 HLE BiTE |
I | NCT03224819 |
| Pavurutamab, AMG701 |
BCMA × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE |
I | NCT03287908 |
| AMG757 | DLL3 × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE |
I | NCT03541369 |
| AMG910 | CLDN18.2 × CD3 | Amgen | scFv + scFv + Fc, 1 + 1 HLE BiTE |
I | NCT04260191 |
| Pasotuxizumab, AMG212, BAY2010112 |
PSMA × CD3 | Amgen, Bayer AG | scFv + scFv, 1 + 1 BiTE |
I | NCT01723475 |
| AMG420, BI836909 |
BCMA × CD3 | Amgen, Boehringer Ingelheim |
scFv + scFv, 1 + 1 BiTE |
I | NCT03836053 |
| AMG424 | CD38 × CD3 | Amgen, Xencor | Fab + scFv + Fc, 1 + 1 XmAb |
I | NCT03445663 |
| AMG509 | STEAP1 × CD3 | Amgen, Xencor | (Fab)2 + scFv + Fc, 2 + 1 XmAb |
I | NCT04221542 |
| AMV564 | CD33 × CD3 | Amphivena Therapeutics | Fv + (Fv)2 + Fv, 2 + 2 TandAb |
I | NCT04128423 and 2 studies |
| APVO436 | CD123 × CD3 | Aptevo Therapeutics | (scFv)2 + (scFv)2 + Fc, 2 + 2 ADAPTIR |
I | NCT03647800 |
| CC-93269, EM801 |
BCMA × CD3 | Celgene | Fab + (Fab)2 + Fc, 2 + 1 CrossMAb |
I | NCT03486067 |
| ERY974 | Glypican-3 × CD3 | Chugai Pharmaceutical | Fab + Fab + Fc, 1 + 1 ART-Ig |
I | NCT02748837 |
| A-319 | CD19 × CD3 | Evive Biotech | scFv + Fab, 1 + 1 | I | NCT04056975 |
| GEM333 | CD33 × CD3 | GEMoaB Monoclonals | scFv + scFv, 1 + 1 | I | NCT03516760 |
| GEM3PSCA | PSCA × CD3 | GEMoaB Monoclonals | scFv + scFv, 1 + 1 | I | NCT03927573 |
| RG6160, RO7187797, BFCR4350A |
FcRH5 × CD3 | Genentech | Fab + Fab + Fc, 1 + 1 | I | NCT03275103 |
| RG6194, BTRC4017A |
HER2 × CD3 | Genentech | undisclosed | I | NCT03448042 |
| RG6296, RO7297089 |
BCMA × CD16A | Genentech | Fab + Fab + Fc, 1 + 1 | I | NCT04434469 |
| Mosunetuzumab, RG7828, RO7030816, BTCT4465A |
CD20 × CD3 | Genentech, Roche, Chugai | Fab + Fab + Fc, 1 + 1 | II | NCT03677154 and 8 studies |
| GEN1044 | 5T4 × CD3 | Genmab, Abbvie | Fab + Fab + Fc, 1 + 1 DuoBody |
I | NCT04424641 |
| Epcoritamab, GEN3013 |
CD20 × CD3 | Genmab, Abbvie | Fab + Fab + Fc, 1 + 1 DuoBody |
III | NCT03625037 and 5 studies |
| GTB-3550, OXS-3550 |
CD33 × CD16, IL-15 | GT Biopharma | scFv + ligand + scFv, 1 + 1 + 1 TriKE |
I/II | NCT03214666 |
| HPN424 | PSMA × HSA × CD3 | Harpoon Therapeutics | sdAb + sdAb + scFv, 1 + 1 + 1 TriTAC |
I/II | NCT03577028 |
| ISB1302, GBR1302 |
HER2 × CD3 | Ichnos Sciences, Glenmark Pharmaceuticals | Fab + scFv + Fc, 1 + 1 BEAT |
I/II |
NCT02829372 NCT03983395 |
| ISB1342, GBR1342 |
CD38 × CD3 | Ichnos Sciences, Glenmark Pharmaceuticals | Fab + scFv + Fc, 1 + 1 BEAT |
I/II | NCT03309111 |
| IGM-2323 | CD20 × CD3 | IGM Biosciences | IgM + scFv, 10 + 1 | I | NCT04082936 |
| Tebentafusp, IMCgp100 |
gp100/HLA-A * 02:01 × CD3 | Immunocore | TCR + scFv, 1 + 1 ImmTAC |
III | NCT03070392 and 5 studies |
| IMC-F106C | PRAME/HLA-A * 02:01 × CD3 | Immunocore | TCR + scFv, 1 + 1 ImmTAC |
I/II | NCT04262466 |
| IMC-C103C | MAGE-A4/HLA-A * 02:01 × CD3 | Immunocore, Genentech |
TCR + scFv, 1 + 1 ImmTAC |
I/II | NCT03973333 |
| IMCnyeso, GSK01 |
NY-ESO-1/HLA-A * 02:01 × CD3 | Immunocore, GlaxoSmithKline |
TCR + scFv, 1 + 1 ImmTAC |
I/II | NCT03515551 |
| JNJ-63709178 | CD123 × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody |
I | NCT02715011 |
| JNJ-63898081 | PSMA × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody |
I | NCT03926013 |
| Teclistamab, JNJ-64007957 |
BCMA × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody |
II | NCT04557098 and 5 studies |
| Talquetamab, JNJ-64407564 |
GPRC5D × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody |
II | NCT04634552 and 3 studies |
| JNJ-67571244 | CD33 × CD3 | Janssen Research & Development | Fab + Fab + Fc, 1 + 1 DuoBody |
I | NCT03915379 |
| MGD007 | gpA33 × CD3 | MacroGenics | Fv + Fv + Fc, 1 + 1 DART-Fc |
I |
NCT02248805 NCT03531632 |
| Orlotamab, MGD009 |
B7-H3 × CD3 | MacroGenics | Fv + Fv + Fc, 1 + 1 DART-Fc |
I |
NCT02628535 NCT03406949 |
| Duvortuxizumab, MGD011, JNJ-64052781 |
CD19 × CD3 | MacroGenics, Janssen Research & Development | Fv + Fv, 1 + 1 DART |
I |
NCT02743546 NCT02454270 |
| Flotetuzumab, MGD006, S80880 |
CD123 × CD3 | MacroGenics, Servier | Fv + Fv, 1 + 1 DART |
I | NCT04582864 and 5 studies |
| Tepoditamab, MCLA-117 |
CLEC12A × CD3 | Merus | Fab + Fab + Fc, 1 + 1 Biclonics |
I | NCT03038230 |
| PF-06671008 | P-cadherin × CD3 | Pfizer | Fv + Fv + Fc, 1 + 1 DART-Fc |
I | NCT02659631 |
| PF-06863135 | BCMA × CD3 | Pfizer | Fab + Fab + Fc, 1 + 1 | II | NCT04649359 and 2 studies |
| Odronextamab, REGN1979 |
CD20 × CD3 | Regeneron | Fab + Fab + Fc, 1 + 1 | II | NCT03888105 and 2 studies |
| REGN5458 | BCMA × CD3 | Regeneron | Fab + Fab + Fc, 1 + 1 | I/II | NCT03761108 |
| REGN5459 | BCMA × CD3 | Regeneron | Fab + Fab + Fc, 1 + 1 | I | NCT04083534 |
| REGN4018 | MUC16 × CD3 | Regeneron, Sanofi | Fab + Fab + Fc, 1 + 1 | I/II |
NCT03564340 NCT04590326 |
| Glofitamab, RO7082859, RG6026 |
CD20 × CD3 | Roche | Fab + (Fab)2 + Fc, 2 + 1 CrossMAb |
III | NCT03075696 and 8 studies |
| Cibisatamab, RO6958688, RG7802 |
CEA × CD3 | Roche, Genentech | Fab + (Fab)2 + Fc, 2 + 1 CrossMAb |
I | NCT02650713 and 3 studies |
| SAR440234 | CD123 × CD3 | Sanofi | Fab + Fv + Fc, 1 + 1 | I/II | NCT03594955 |
| TNB-383B | BCMA × CD3 | TeneoBio, AbbVie | sdAb + Fab + Fc, 1 + 1 | I | NCT03933735 |
| M802 | HER2 × CD3 | Wuhan YZY Biopharma | Fab + scFv + Fc, 1 + 1 YBODY |
I | NCT04501770 |
| Plamotamab, Xmab13676 |
CD20 × CD3 | Xencor | Fab + scFv + Fc, 1 + 1 XmAb |
I | NCT02924402 |
| Tidutamab, Xmab18087 |
SSTR2 × CD3 | Xencor | Fab + scFv + Fc, 1 + 1 XmAb |
I/II |
NCT03411915 NCT04590781 |
| Vibecotamab, Xmab14045 |
CD123 × CD3 | Xencor, Novartis | Fab + scFv + Fc, 1 + 1 XmAb |
I | NCT02730312 |
| Nivatrotamab | GD2 × CD3 | Y-mAbs | IgG + (scFv)2, 2 + 2 BiClone |
I/II | NCT04750239 |
Liddy et al. [61] described a different type of BsAbs called ImmTACs (Immune mobilizing monoclonal TCR Against Cancer), consisting of a tumor-associated epitope-specific monoclonal TCR clone with picomolar affinity fused to an anti-CD3 scFv. ImmTAC uses an affinity-matured TCR that recognizes endogenously processed peptides bound to human leukocyte antigens (HLAs) on the cell surface. They generated several ImmTAC clones with the following epitope specificities: gp100280–288, MAGE-A3168–176, Melan-A/MART-126–35, and NY-ESO-1157–165. These ImmTACs efficiently redirect T cells to kill cancer cells in vitro and in vivo in xenograft models [61]. An ImmTAC specific for the gp100 epitope, Tebentafusp (IMCgp100), is the most advanced molecule in the clinic to treat uveal melanoma patients. The overall survival rate at one year in phase I clinical trials was 73–74% [62]. ImmTAC molecules, such as IMC-C103C (anti-MAGE-A4 TCR × CD3), GSK01 (IMCnyeso, anti-NY-ESO-1 TCR × CD3), and IMC-F106C (anti-PRAME TCR × CD3), are under clinical investigation. This strategy suggests the applicability to intracellular antigens, which requires TCR optimization in the context of defined HLA haplotypes.
CD3 BsAb therapy is a form of immunotherapy that enables T cells to recognize and kill tumor cells. To prevent any potential bivalency-driven activation of T cells in the absence of TAAs, most formats for T cell engagers are preferably monovalent for CD3. In addition, T cell-engagers generally have a lower affinity for the CD3-targeting arm than the TAA-targeting arm. Although the binding affinity to CD3 is not thought to be correlated with T cell activation potential [50,63], a lower binding affinity to CD3 may improve the PK properties, biodistribution, and safety profile. Interestingly, CD3 BsAbs with different affinities targeting C-type lectin-like molecule 1 (CLL-1) have been tested: low (50 nM), high (0.5 nM), and very high (0.05 nM). In in vitro cell killing assays, the low-affinity CD3 BsAb showed lower potency than the variants with higher affinity. However, all variants led to a rapid depletion of CLL-1+ cells in transgenic mice, and low-affinity BsAb-treated mice showed a tendency toward a delayed rebound of target cells, suggesting a more sustained activity compared to higher affinity variants. PK analysis showed different clearances for the three variants, with the low-affinity BsAb showing the slowest clearance [64], and the affinity to CD3 also affects the biodistribution of BsAbs. In addition, HER2 × CD3 BsAbs with low-affinity (50 nM) or high-affinity (0.5 nM) CD3-targeting arms were evaluated in human CD3 transgenic mice. Treatment with HER2 × CD3 BsAb having a low-affinity arm led to a higher accumulation in HER2+ tumor tissues and a lower distribution to T-cell rich secondary lymphoid organs such as the spleen and lymph nodes [65].
Although T-cell engagers have shown superior efficacy in clinics, they still face hurdles such as low response rates in solid tumors and significant toxicity, including neurotoxicity and CRS, a systemic inflammatory response [66,67,68]. In a randomized trial, adverse events and serious adverse events, including neutropenia, infection, neurotoxicity, and CRS, were reported in ~99% and 62% of the patients, respectively [69]. Dose fractionation regimens have been suggested to mitigate CRS, which could be clinically manageable by administering steroids or tocilizumab (anti-IL-6R) [68]. In line with the previous affinity issue, the lower affinity of the anti-CD3 arm is related to decreased cytokine release potential. Mice receiving the BsAb variant with a high-affinity anti-CD3 arm showed elevated serum cytokines and proinflammatory mediators and developed vascular shock with fever. However, low-affinity BsAb-treated mice did not exhibit toxicity within 48 h and had variable increases in systemic cytokines that were generally much smaller than those seen in high-affinity BsAb-treated mice [64]. In another example, XmAb13551, which is a CD38 × CD3 BsAb (8 nM affinity for human CD3) against multiple myeloma (MM), showed effective MM cell clearance in mice [70] but triggered CRS at doses >0.2 mg/kg in monkeys [71]. Affinity-engineered XmAb13551 suggested that intermediate and very low-affinity variants were well tolerated at higher doses (0.5–3 mg/kg) and showed enhanced target cell depletion [71].
In addition to modifying the affinity of the anti-CD3 arm, the valency of the BsAb format may also affect its activity. Roche has developed IgG-like T-cell-engaging BsAbs with an asymmetric “2 + 1” structure with two TAA binders and a CD3 binder, and one TAA-binding Fab is linked to the N-terminus of the CD3-binding one. This format increased TAA avidity, enhanced tumor accumulation, and showed superior potency than the “1 + 1” version of the BsAb [72,73]. In clinical trials of a 2:1 CEA × CD3 BsAb (RG7802), 45% of patients showed disease control by the BsAb, and grade ≥ 3 related adverse events were observed in 28% of the patients [34], which was lower than that observed with blinatumomab [69]. The TAA-binding arm may hinder the CD3-binding arm, and TAA binding may unmask the CD3-binding arm. APVO-414 (PSMA × CD3) is an ADAPTIR BsAb format, which has bivalent arms for each target. APVO-414 showed ~30-fold higher cytotoxicity compared to the (scFv)2 format in vitro; however, cytokine production was much lower than that seen with the (scFv)2 format even when the same anti-CD3 Ab was used [74].
To achieve dual antigen-specific activation of CD3, a tandem (scFv)3 was split into two antigen-binding scFvs fused to either the VH or the VL domain of an anti-CD3 scFv, referred to as hemibody. When a complementary pair of hemibodies simultaneously binds to the respective antigens on a single target cell, the VH and VL domains reconstitute the original CD3-binding activity to engage T cells with dual antigen-positive tumor cells [75]. In addition, protease-activation of the anti-CD3 arm can be used for tumor-selective CD3 activation. The CD3-binding arm is masked by a N-terminally fused peptide [76] or anti-idiotypic anti-CD3 scFv [77] and unleashed in the tumor microenvironment (TME), which overexpresses several proteases, including matrix metalloproteinases. These strategies increase the maximum tolerated dose and lower the elevation of systemic cytokine levels compared to unmasked T-cell engagers.
4.2. NK Cell-Engagers
An alternative approach is activating and redirecting NK cells to tumor cells. NK-cell engagers may have a better safety profile than T-cell engagers while showing a similar clinical efficacy [78]. Several activating receptors can induce the cytotoxic functions of NK cells, including CD16 (FcγRIII), natural cytotoxicity receptors (NCRs; NKp30, NKp44, and NKp46), NKG2D (CD314), and DNAM-1 (CD226) [79,80]. CD16, a low-affinity FcγR with two isoforms, CD16A and CD16B, is the most common target for NK-cell engagement. CD16A (FcγRIIIA) is an activating receptor that is mainly expressed on NK cells and macrophages. CD16B (FcγRIIIB) is expressed on granulocytes and is not involved in killing tumor cells. When CD16A binds to the Fc domain of antibodies attached to its cognate antigen, it induces ADCC in antibody-binding cells without costimulation [81]. By activating CD16A, the BsAb can redirect CD16A-expressing effector cells to target cells, even without binding to the Fc region. Most NK-cell engagers have been developed for hematological malignancies.
AFM13 (CD30 × CD16A) is a tetravalent BsAb that targets CD16A and CD30, expressed in Hodgkin and Reed-Sternberg cells of patients with Hodgkin’s lymphoma [25]. AFM13 was well tolerated and showed an overall disease control rate of 61% (partial remission + stable disease) in patients with Hodgkin’s lymphoma, significant NK cell activation, and a decrease in soluble CD30 in peripheral blood during a phase I clinical trial [26] and is currently in a phase II trial. A CD16A-directed tri-specific, tetravalent antibody (BCMA × CD16A × CD200) termed aTriFlex showed dual antigen-selective NK cell-mediated lysis [82]. CD16-targeting BsAbs, which target CD33, CD133, or B7-H3, are further modified by introducing an IL-15 crosslinker, called TriKE, to induce expansion, priming, and survival of NK cells. It showed superior antitumor activity and enhanced survival of human NK cells in vitro compared to non-modified BsAbs [27,83,84].
Other examples of NK-cell engagers are NKp46- or NKG2D-targeting BsAbs, which have been investigated preclinically. NKp46 (NCR1, CD335) is expressed in NK cells, type 1 innate lymphoid cells, and a small population of T cells [79]. CD20 × NKp46 showed tumor regression in a xenograft model. In addition, by introducing an ADCC-enhanced Fc that can bind to CD16, tri-specific antibodies with more efficient ADCC activity than BsAbs were generated [85]. NKG2D is expressed on NK cells, CD8 T cells, and invariant natural killer T (iNKT) cells, and its activation relies on the expression of cognate ligands and induced-self proteins such as MICA and ULBP [80]. MM cell-targeting BsAbs were designed by targeting NKG2D and CS1 (CD319, SLAMF7). A CS1 × NKG2D BsAb induced cytotoxicity against CS1+ MM cells, IFN-γ production, and prolonged survival in a xenograft model of MM [86].
4.3. Other Immune Cell-Engagers
CD64 (FcγRI), a high-affinity FcγR, is expressed on hematopoietic cells such as monocytes, macrophages, activated neutrophils, and immature DCs [81]. CD64-engaging BsAbs were constructed via cross-linking of the anti-CD64 mAb (clone H22) Fab’ fragment with a TAA-targeting Fab’ fragment. The H22 clone reacts with an epitope distinct from the Fc region binding domain. However, no consistent antitumor activity [66,87] or toxicity [88] has been observed in clinical trials of CD64-engaging BsAbs. Additionally, iNKT cell-redirecting strategies using α-galactosylceramide-loaded CD1d extracellular domain fused with tumor-targeting scFv are also at an early preclinical stage [89].
5. Tumor-Targeted Immunomodulators
Similar to T cell-engagers, BsAbs targeting both TAAs and cosignaling molecules on T cells have also been extensively investigated. CD3 is expressed on all T cells, including resting T cells; however, costimulatory receptors, such as 4-1BB, are upregulated in tumor-specific tumor-infiltrating T cells, making them attractive targets for solid tumors [90]. Most cosignaling molecules are involved in either the immunoglobulin superfamily (IgSF) or tumor necrosis factor (TNF) receptor superfamily (TNFRSF) [91,92]. This section reviews tumor-targeted immunomodulatory BsAbs based on the superfamily to which their target belongs, and BsAbs in clinical trials are summarized in Table 3.
Table 3.
Tumor-targeted or Dual immunomodulators in clinical stages.
| Name | Targets | Developer | Format | Highest Phase | Clinical Trials |
|---|---|---|---|---|---|
| ABBV-428 | MSLN × CD40 | AbbVie | (scFv)2 + (scFv)2 + Fc, 2 + 2 | I | NCT02955251 |
| ABL503 | PD-L1 × 4-1BB | ABL Bio, I-MAB | IgG + (scFv)2, 2 + 2 | I | NCT04762641 |
| AK104 | PD-1 × CTLA-4 | Akeso Biopharma | IgG + (scFv)2, 2 + 2 | II | NCT04172454 and 13 studies |
| ATOR-1015, ADC-1015 |
OX40 × CTLA-4 | Alligator Bioscience | IgG + (ligand)2, 2 + 2 | I | NCT03782467 |
| AMG506, MP0310 |
FAP × 4-1BB × HSA | Molecular Partners AG, Amgen | (DARPin)3, 1 + 1 + 1 | I | NCT04049903 |
| CDX-527 | PD-1 × CD40 | Celldex | IgG + (scFv)2, 2 + 2 | I | NCT04440943 |
| LY3415244 | PD-L1 × Tim-3 | Eli Lilly | Fab + Fab + Fc, 1 + 1 | I | NCT03752177 |
| LY3434172 | PD-1 × PD-L1 | Eli Lilly | Fab + Fab + Fc, 1 + 1 | I | NCT03936959 |
| FS118 | PD-L1 × LAG-3 | F-star | IgG with Fcab, 2 + 2 mAb2 |
I/II | NCT03440437 |
| FS120 | OX40 × 4-1BB | F-star | IgG with Fcab, 2 + 2 mAb2 |
I | NCT04648202 |
| FS222 | PD-L1 × 4-1BB | F-star | IgG with Fcab, 2 + 2 mAb2 |
I | NCT04740424 |
| GEN1042 | CD40 × 4-1BB | Genmab, BioNTech | Fab + Fab + Fc, 1 + 1 DuoBody |
I | NCT04083599 |
| GEN1046, BNT311 |
PD-L1 × 4-1BB | Genmab, BioNTech | Fab + Fab + Fc, 1 + 1 DuoBody |
I/II | NCT03917381 |
| GS-1423, AGEN1423 |
CD73 × TGF-b | Gilead Sciences, Agenus | IgG + TGFβ receptor, 2 + 2 | I | NCT03954704 |
| INBRX-105, ES101 |
PD-L1 × 4-1BB | Inhibrx, Elpiscience Biopharma |
(sdAb)2 + (sdAb)2 + Fc, 2 + 2 | I |
NCT03809624 NCT04009460 |
| IBI318 | PD-1 × PD-L1 | Innovent Biologics | Fab + Fab + Fc, 1 + 1 | II | NCT04777084 and 5 studies |
| KN046 | PD-L1 × CTLA-4 | Jiangsu Alphamab Biopharmaceuticals | (sdAb)2 + (sdAb)2 + Fc, 2 + 2 | III | NCT04040699 and 15 studies |
| SHR-1701 | PD-L1 × TGF-b | Jiangsu HengRui Medicine | IgG + TGFβ receptor, 2 + 2 | II | NCT04650633 and 11 studies |
| MGD019 | PD-1 × CTLA-4 | Macrogenics | (Fv)2 + (Fv)2 + Fc, 2 + 2 DART-Fc |
I | NCT03761017 |
| Tebotelimab, MGD013 |
PD-1 × LAG-3 | Macrogenics, Zai Lab | (Fv)2 + (Fv)2 + Fc, 2 + 2 DART-Fc |
II/III | NCT04212221 and 6 studies |
| MEDI5752 | PD-1 × CTLA-4 | MedImmune | Fab + Fab + Fc, 1 + 1 | I | NCT04522323 and 2 studies |
| Bintrafusp Alfa, M7824 |
PD-L1 × TGF-b | Merck KGaA | IgG + TGFβ receptor, 2 + 2 | III | NCT03631706 and 38 studies |
| MCLA-145 | PD-L1 × 4-1BB | Merus, Incyte | Fab + Fab + Fc, 1 + 1 Biclonics |
I | NCT03922204 |
| NM21-1480 | PD-L1 × 4-1BB × HSA | Numab | Fv + Fv + Fv, 1 + 1 + 1 scMATCH3 |
I/II | NCT04442126 |
| PRS-343 | HER2 × 4-1BB | Pieris Pharmaceuticals | IgG + (anticalin)2, 2 + 2 | I |
NCT03330561 NCT03650348 |
| REGN5678 | PSMA × CD28 | Regeneron | Fab + Fab + Fc, 1 + 1 | I/II | NCT03972657 |
| RG6139, RO7247669 |
PD-1 × LAG-3 | Roche | Fab + Fab + Fc, 1 + 1 CrossMAb |
II |
NCT04785820 NCT04140500 |
| RG7769, RO7121661 |
PD-1 × Tim-3 | Roche | Fab + Fab + Fc, 1 + 1 CrossMAb |
II | NCT04785820 and 2 studies |
| Xmab20717 | PD-1 × CTLA-4 | Xencor | scFv + Fab + Fc, 1 + 1 Xtend XmAb |
I | NCT03517488 |
| Xmab22841 | CTLA-4 × LAG-3 | Xencor | scFv + Fab + Fc, 1 + 1 Xtend XmAb |
I | NCT03849469 |
| Xmab23104 | PD-1 × ICOS | Xencor | scFv + Fab + Fc, 1 + 1 Xtend XmAb |
I | NCT03752398 |
5.1. Immunoglobulin Superfamily
CD28 was the first identified costimulatory receptor on T cells and has been extensively characterized. Its ligands, B7-1 (CD80) and B7-2 (CD86), which also interact with CTLA-4 (CD152), are preferentially expressed on antigen-presenting cells (APCs). The B7/CD28 interaction probably remains the most potent costimuli for naïve T cells [91]. CD28 agonistic mAbs have been developed for therapeutic stimulation and induce potent T cell activation without TCR engagement [93]. However, one of these antibodies, TGN1412 (theralizumab), induced life-threatening CRS and multiple organ failure in clinical trials [94]. Several BsAbs have been developed for the controlled stimulation of CD28. r2820, a BsAb targeting CD28 and CD20, induces T cell activation in peripheral blood mononuclear cell (PBMC) cultures of healthy donors and CLL patients and kills CD20+ tumor cells. This proof-of-concept study showed the possibility of target cell-restricted activation of CD28 [95]. Two CD28-targeting BsAbs were developed, one for ovarian cancer (MUC16 × CD28) and another for prostate cancer (PSMA × CD28, REGN5678). Both BsAbs have limited activity but potentiate CD3 BsAbs to activate T cell proliferation and cytokine production. Similarly, a MUC16 × CD28 BsAb enhances the antitumor efficacy of CD3 BsAbs in a xenograft model of ovarian cancer [30]. In a different study, PSMA × CD28 and EGFR × CD28 BsAbs enhanced the antitumor efficacy of the PD-1 blockade in syngeneic and xenograft tumor models and did not induce systemic T cell activation [29,30]. Other tumor antigen-targeting BsAbs, including B7-H3 × CD28 and PD-L1 × CD28, showed similar costimulatory activity [96]. Similar to T-cell engagers, CD28-targeting BsAbs might induce systemic toxicity because of the constitutive expression of CD28 on T cells. Tumor-targeted ICIs have been investigated, such as EGFR × PD-L1 and CSPG4 × PD-L1, to overcome irAEs induced by the indiscriminate activation of T cells through the PD-1 blockade. Both BsAbs show a target antigen-directed PD-1 blockade followed by T-cell activation [97,98]. The BsAb-based ICI approach may represent the next step toward improving tumor selectivity, efficacy, and safety of ICIs in target antigen-overexpressing malignancies.
5.2. TNFR Superfamily
Among the members of TNFRSF, 4-1BB (CD137, tnfrsf9) is a well-documented potent costimulatory receptor. 4-1BB is upregulated upon T cell activation, and 4-1BB ligand (4-1BBL, CD137L)-driven clustering of 4-1BB promotes cytotoxic function, enhances survival, and induces the formation of immunological memory. Although 4-1BB is highly upregulated in activated T cells, it is widely expressed on other immune cells such as NK cells, regulatory T cells (Tregs), DCs, and activated monocytes, as well as in endothelial cells, and has been reported to activate NK cells and induce adhesion molecules on vasculature [90,91,99]. Tumor-infiltrating lymphocytes (TILs) highly express 4-1BB compared to lymphocytes in normal tissue or PBMCs [100], and 4-1BB-expressing CD8 TILs include tumor-specific T cells [101,102]. The restricted expression of 4-1BB on TILs and its costimulatory activity make 4-1BB a promising target for cancer immunotherapy. There was an expectation for the potent stimulatory activity of 4-1BB, so two agonistic 4-1BB mAbs, urelumab (BMS-663513), and utomilumab (PF-05082566), were developed. Urelumab is the first human 4-1BB-agonistic mAb in the IgG4 backbone and does not block the interaction of 4-1BB/4-1BBL [103]. Utomilumab is an IgG2-based 4-1BB agonist that blocks 4-1BBL binding [104]. Although both Abs showed potent activity in preclinical studies, their clinical development was unsuccessful due to the fatal hepatotoxicity of urelumab and the low efficacy of utomilumab [90,104].
Tumor-restricted activation of 4-1BB by BsAbs has been intensively studied to maintain optimal therapeutic efficacy and safety. Several companies are developing 4-1BB BsAbs composed of tumor antigen-targeting arm(s) and 4-1BB-agonistic arm(s). The common feature of these BsAbs is the lack of 4-1BB-agonistic activity in the absence of TAAs. One example is PRS-343 (HER2 × 4-1BB), composed of an Fc-silenced HER2 mAb fused with a 4-1BB-agonistic anticalin [38]. This BsAb is in phase I/II clinical development and has shown a clinical benefit and safety in HER2+ cancer patients [105]. Other examples are RG7827 (FAP × 4-1BBL) and RG6076 (CD19 × 4-1BBL), which are fusion proteins composed of trimeric 4-1BBL in one arm, and FAP or CD19-targeting Fab in the other arm. RG7827 showed synergy with CEA × CD3 BsAb in a xenograft model [106]. The third example is ALG.APV-527 (5T4 × 4-1BB) comprising two 4-1BB-agonistic scFvs and two 5T4-targeting scFvs with a modified Fc domain that minimizes the interaction with the FcγR. ALG.APV-527 enhanced CD8 T cells and NK cells in the presence of 5T4+ cells and inhibited tumor growth in a syngeneic bladder cancer model [107]. A novel 4-1BB BsAb (B7-H3 × 4-1BB) was recently developed to activate 4-1BB signaling only in the context of TAA engagement. This is composed of an Fc-silenced anti-B7-H3 mAb fused with 4-1BB-agonistic scFvs to the C-terminus of the heavy chain. B7-H3 × 4-1BB activates terminally differentiated Tim-3+ CD8 T cells in the TME and synergizes with the PD-1 blockade without inducing irAEs [108]. In addition, the novel PD-L1 × 4-1BB and Claudin18.2 × 4-1BB codeveloped by ABL Bio and I-Mab are in phase I clinical trials. These novel 4-1BB BsAbs are anticipated to show strong antitumor efficacy with no liver toxicity due to the localized activation of 4-1BB in tumors.
Glucocorticoid-induced TNF receptor-related protein (GITR, tnfrsf18) belongs to the TNFRSF and transmits a costimulatory signal upon binding its ligand, GITRL, which is expressed on APCs. GITR is upregulated upon T cell activation and is constitutively expressed on Tregs [91]. Although in vivo GITR activation enhances tumor immunity, irAEs have been observed [109]. Similar to 4-1BB BsAbs, a FAP scFv-targeted GITRL fusion protein was developed to stimulate T cells exclusively in the presence of FAP-expressing tumor cells and cancer-associated fibroblasts. This molecule costimulates T cells and reduces the suppressive function of Tregs [110].
Another interesting concept is the tumor-targeted activation of CD40. CD40 is a potent stimulator of APCs and myeloid cells, and its activation by CD40L has been shown to activate and license APCs to prime cytotoxic T cells, resulting in enhanced tumor immunity. Agonistic CD40 mAbs have shown efficacy, but systemic dose-limiting toxicity similar to agonists targeting other members of the TNFRSF has been observed in clinical trials [99,111]. ABBV-428 (MSLN × CD40) is an MSLN-targeted CD40 agonist BsAb composed of a homodimer of identical chains containing two scFvs and a modified Fc region that cannot induce ADCC. The mouse version of MSLN × CD40 activated B cells and monocyte-derived DCs and showed MSLN-dependent antitumor efficacy without liver toxicity in syngeneic tumor models. ABBV-428 also showed MSLN-directed CD40 activation in APCs and antitumor efficacy in a xenograft model [112] and is undergoing phase I clinical development.
In summary, the goal of developing most tumor-targeted BsAb-based immunomodulators is to increase the therapeutic window by overcoming the toxicity limitations by activating in the TME. The molecular formats of most tumor-targeted immunomodulators consist of IgG-like BsAbs that have modified Fc regions, allowing persistent activity in the system and preventing Fc function-mediated target cell depletion and FcR-mediated activation of the agonistic target. Unlike CD3, expressed in all T cells, the target for tumor-targeted immunomodulators, especially 4-1BB, is expressed only in the activated T cells. Thus, 4-1BB BsAbs have been developed as the second generation of T-cell engagers.
6. Dual Immunomodulators
Among the wide range of immune checkpoint signaling pathways, the PD-1/PD-L1 pathway became the most significant regulator of T cell activity and was extensively investigated [8]. Many dual immune-modulators take an add-on approach of second inhibitory pathways on the PD-1/PD-L1 pathway. The rationale for these BsAbs is their induced expression in the TME and their contribution to primary or acquired resistance to PD-(L)1-targeting therapeutics. Most dual immunomodulators contain the Fc domain to extend their half-life and show enhanced efficacy compared to the combination of mAbs in preclinical studies. Currently, some BsAbs are being evaluated in the clinic (Table 3).
6.1. Dual Blockade of Immunosuppressive Targets
The most established dual immunomodulating BsAbs bind simultaneously to PD-(L)1 and other immune inhibitory molecules, such as CTLA-4, LAG-3, Tim-3, TIGIT, TGF-β, and CD73. This type of BsAb is designed to (1) maximize the antitumor activity of T cells by blocking more than one coinhibitory receptor or modifying immunosuppressive TME, and (2) minimize irAEs occasionally observed in the combination of two different ICIs. Although more comprehensive studies are required, dual IC blocking BsAb has shown superior activity in preclinical studies compared to combination therapy. For example, MEDI5752 (PD-1 × CTLA-4) is a monovalent BsAb that inhibits PD-1 and CTLA-4 with reduced Fc function and was designed to block CTLA-4-mediated inhibition on PD-1+ T cells. Upon binding to targets, MEDI5752 is rapidly internalized and leads to the subsequent degradation of PD-1, which is not observed with mAbs [113,114]. BsAbs targeting LAG-3, Tim-3, and TIGIT with PD-(L)1 have been developed actively because of better efficacy than combinations in preclinical studies [115].
Another example of the dual blockade is the combination of ICIs with TME modulators, such as TGF-β and CD73. M7824 (PD-L1 × TGF-β, Bintrafusp Alfa) is a bifunctional fusion protein composed of a human IgG1 mAb against PD-L1 fused with two extracellular domains of TGF-βRII, which functions as a TGF-β “trap” for all three TGF-β isoforms. Because the upregulation of TGF-β signaling-associated genes has been linked to anti-PD-1 resistance in metastatic melanoma, M7824 showed better efficacy in preclinical studies [116]. CD73 catalyzes AMP breakdown to adenosine and suppresses immune activation through the A2A receptor [117]. Thus, PD-(L)1 blockade could be combined with the inhibition of the purinergic signaling pathway regulated by a series of nucleotidases, including CD73.
6.2. Simultaneous Targeting of Coinhibitory and Costimulatory Pathways
ATOR-1015 (OX40 × CTLA-4) is a tetravalent dual immuno-modulator targeting OX40 and CTLA-4. It consists of a clustering-dependent OX40 agonistic human IgG1 antibody fused with the optimized version of the Ig-like V-type domain of CD86, which is a natural CTLA-4 ligand that binds to CTLA-4 with high affinity while having low affinity for CD28, another counter-receptor. This BsAb may preferentially localize to the tumor because both OX40 and CTLA-4 are highly expressed on activated T cells and Tregs in the TME. ATOR-1015 showed costimulatory activity and enhanced the Fc-mediated ADCC effect on Tregs compared to mAbs [118].
6.3. Dual Stimulation of Costimulatory Pathways
Another strategy in immunomodulation is the dual stimulation of costimulatory receptors on T cells. FS120 (OX40 × 4-1BB) is a tetravalent dual agonist, IgG-based BsAb targeting OX40 and 4-1BB through 4-1BB-binding Fab and OX40-binding Fcab. Both OX40 and 4-1BB stimulate T cell proliferation and activation, but OX40 stimulation preferentially activates CD4 T cells, while 4-1BB stimulation preferentially activates CD8 T cells [99]. Several clinical trials have evaluated agonist mAbs to OX40 or 4-1BB; however, both agonists showed limited efficacy or liver toxicity. FS120 activates CD4 and CD8 T cells through dual binding of OX40 and 4-1BB [119]. As the clinical development of tumor-targeted agonistic BsAbs targeting costimulatory pathways progresses, interest in developing such dual costimulators was expanded.
7. Dual Tumor-Targeting BsAbs
In addition to immunomodulating BsAbs, dual tumor-targeting by BsAbs may offer simultaneous modulation of two functional pathways in the TME, improved payload delivery, and tumor-restricted tumor cell lysis.
7.1. Tumor Receptor Tyrosine Kinase Blockade
Cancer involves multiple disease-driving proteins and pathway crosstalk, which supports a complex molecular network. Identification of these factors has led to the clinical development of targeted therapies for various malignancies. Targeted inhibition of oncogenic receptor tyrosine kinases (RTKs), such as members of the ErbB family, including EGFR and HER2, has been successful in the clinic for over two decades [120]. However, the development of acquired drug resistance is a significant limitation of such therapies. This drug resistance often involves bypassing targeted receptor inhibition by activating crosstalking pathways, such as the heregulin/HER3, hepatocyte growth factor (HGF)/MET, or IGF-1R pathways [121,122]. Several BsAbs targeting two RTKs have been developed to overcome drug resistance, several of which are in clinical trials (Table 4). These BsAbs usually contain the Fc domain for sustained blockade with an extended half-life, and some of them show enhanced ADCC activity through Fc modification.
Table 4.
Dual tumor-targeting BsAbs in clinical stages.
| Name | Targets | Developer | Format | Highest Phase | Clinical Trials |
|---|---|---|---|---|---|
| Dilpacimab, ABT-165 |
DLL4 × VEGF | AbbVie | (Fv)2 + IgG, 2 + 2 DVD-Ig |
II |
NCT01946074 NCT03368859 |
| ABL001, NOV1501, TR009 |
DLL4 × VEGF | ABL Bio | IgG + (scFv)2, 2 + 2 | I/II |
NCT03292783 NCT04492033 |
| BI836880 | VEGF × Ang-2 | Boehringer Ingelheim | sdAb + sdAb + albumin, 1 + 1 | II | NCT03861234 and 5 studies |
| BI905677 | LRP5/6 | Boehringer Ingelheim | sdAb + sdAb + albumin, 1 + 1 | I | NCT03604445 |
| AK112 | VEGF × PD-1 | Akesobio | IgG + (scFv)2, 2 + 2 | I/II | NCT04736823 and 2 studies |
| KN026 | HER2 × HER2 (Biparatopic) |
Jiangsu Alphamab Pharmaceuticals | Fab + Fab + Fc, 1 + 1 | II | NCT04521179 and 6 studies |
| MBS301 | HER2 × HER2 (Biparatopic) |
Beijing Mabworks Biotech | Fab + Fab + Fc, 1 + 1 Fab-arm exchange |
I | NCT03842085 |
| LY3164530 | MET × EGFR | Eli Lilly and Co | Fab + Fab + Fc, 1 + 1 orthoFab-IgG |
I | NCT02221882 |
| EMB-01 | cMET × EGFR | Epimab Biotherapeutics | (Fab)2 + IgG, 2 + 2 FIT-Ig |
I/II | NCT03797391 |
| Duligotuzumab, MEHD7945A, RO5541078, RG7597 |
EGFR × HER3 | Genentech | Fab + Fab + Fc, 1 + 1 | II | NCT01986166 and 4 studies |
| Amivantamab, JNJ-61186372 |
cMET × EGFR | Genmab, Janssen | Fab + Fab + Fc, 1 + 1 DuoBody |
III | NCT04077463 and 5 studies |
| HX009 | PD-1 × CD47 | HanxBio | IgG + (ligand)2, 2 + 2 | I | NCT04097769 |
| IMM0306 | CD20 × CD47 | ImmuneOnco | IgG + (ligand)2, 2 + 2 | I | CTR20192612 |
| IBI322 | PD-L1 × CD47 | Innovent | sdAb + Fab + Fc, 1 + 1 | I | NCT04795128 and 2 studies |
| MM-111 | HER2 × HER3 | Merrimack Pharmaceuticals | (scFv)2 + albumin, 1 + 1 | II | NCT01097460 and 3 studies |
| MM-141 | IGF-1R × HER3 | Merrimack Pharmaceuticals | IgG + (scFv)2, 2 + 2 | II | NCT02538627 and 2 studies |
| Zenocutuzumab, MCLA-128,PB4188 |
HER2 × HER3 | Merus | Fab + Fab + Fc, 1 + 1 Biclonics |
II | NCT02912949 and 2 studies |
| MP0250 | HGF × VEGF × HSA | Molecular Partners AG | (DARPin)4, 1 + 1 + 2 | I/II | NCT03136653 and 2 studies |
| MP0274 | HER2 × HER2 (Biparatopic) |
Molecular Partners AG | (DARPin)4, 1 + 1 + 2 | I | NCT03084926 |
| TG-1801, NI-1701 |
CD19 × CD47 | NovImmune, TG Therapeutics |
Fab + Fab + Fc, 1 + 1 κλ body |
I |
NCT03804996 NCT04806035 |
| Navicixizumab, OMP-305B83 |
DLL4 × VEGF | OncoMed | Fab + Fab + Fc, 1 + 1 common LC |
I | NCT03035253 and 2 studies |
| REGN5093 | MET × MET (Biparatopic) |
Regeneron | Fab + Fab + Fc, 1 + 1 | I/II | NCT04077099 |
| Vanucizumab, RG7221, RO5520985 |
Ang-2 × VEGF-A | Roche | Fab + Fab + Fc, 1 + 1 CrossMAb |
II | NCT02665416 and 4 studies |
| Zanidatamab, ZW25 |
HER2 × HER2 (Biparatopic) |
Zymeworks | Fab + scFv + Fc, 1 + 1 | II | NCT04224272 and 6 studies |
7.2. Angiogenesis Inhibition
Cancer is characterized by some unique properties, such as vascular abnormalities, abnormal tumor immune microenvironment, and hypoxia. The inhibition of angiogenesis to normalize tortuous tumor vasculature, which is essential for tumor development and progression, enabling the alleviation of hypoxia and efficient infiltration of immune cells, has been suggested as a promising approach in cancer therapy [123,124]. Tumor angiogenesis is regulated by multiple angiogenic factors, such as VEGF-A/VEGFR2, Ang-2, angiopoietin-2/angiopoietin receptor, and DLL4/Notch signaling. Therefore, targeting redundancy for multiple angiogenic factors by BsAbs represents an area of interest for cancer therapy (Table 4).
7.3. Improved Delivery of Payloads
Antibody-drug conjugates (ADCs) deliver conjugated payloads directly to the tumor by binding to target antigens, internalizing, and exerting their effects [125]. Because there are no apparent tumor-specific antibodies or tumor-selective antigens that do not always internalize well, BsAbs are used for improved payload delivery. This type of BsAbs enhances internalization by targeting TAA and CD63 [126], which is involved in shuttling between the plasma membrane and lysosomal compartment, or non-overlapping epitopes on the same TAA [127]. In addition, ZW49, a HER2-targeted biparatopic ADC for the treatment of HER2-expressing cancers, is in clinical development [128].
7.4. Tumor-Targeted Tumor Cell Lysis
The last strategy for dual tumor-targeting BsAbs is tumor-targeted tumor cell elimination. These BsAbs target TAAs and the CD47/SIRPα pathway. CD47 is often overexpressed on tumor cells to evade ADCP by macrophages and other myeloid cells [129]. To block the activity of CD47 in the TME selectively, dual targeting BsAbs with CD47 and CD19/CD20/mesothelin have been developed [130]. In addition, the apoptotic pathway is triggered by the binding of pro-apoptotic proteins, including FasL (CD95L) and TRAIL, to death receptors (DRs) such as Fas receptor (CD95), DR4 (TRAIL receptor 1, TRAIL-R1), and DR5 (TRAIL receptor 2, TRAIL-R2) [131]. Tumor-targeted apoptosis can be activated by DR agonistic BsAbs [132,133]. These BsAbs are expected to overcome the safety or efficacy issues of the respective mAbs, and some BsAbs have been clinically evaluated (Table 4).
8. Concluding Remarks
To date, many BsAbs in various forms have been developed and evaluated in clinical trials. Although there are two FDA-approved BsAbs, blinatumomab and catumaxomab, most BsAbs are in the preclinical and early clinical stages. For the success of BsAbs in the clinic, they should overcome the limitations of current immunotherapy and provide better efficacy and safety. Immune effector cell-redirecting BsAbs occupy the most considerable portion of the total BsAbs under development, and among them, T cell-engagers are being developed most actively with more than 60 molecules in clinical development. Safety issues, such as CRS and neurotoxicity, and low efficacy in solid tumors remain critical challenges for T-cell engagers. Some strategies, such as affinity modulation of the CD3-targeting arm, alteration of format, and tumor-specific activation of the CD3-targeting arm, have been suggested for reducing adverse events, but clinical validation is required. NK-cell engagers or 4-1BB-based T-cell engagers may have a better safety profile than CD3-based T-cell engagers, but they are still in the early phase of clinical development and should be validated in human patients.
Tumor-targeted immunomodulators are a class of BsAbs that have been developed to overcome the limitations of their respective immunomodulating mAbs, such as agonistic antibodies. A novel strategy of BsAbs with tumor-specific targeting immunomodulators is designed to activate T cells only in the TME. This type of BsAb is designed to show activity only in the presence of TAAs, which may result in a better safety profile than agonistic mAbs by reducing the risk of irAEs, including liver toxicity. Although both strategies have shown the desired efficacy and safety in early development, it is crucial to select appropriate TAAs to ensure tumor-specific activation and reduce irAEs.
Dual immunomodulators have been developed to exhibit synergistic effects by simultaneously regulating the activities of two different immunomodulatory pathways. These BsAbs are dominated by ICIs, targeting the PD-1/PD-L1 pathway and other immunosuppressive targets, such as LAG-3, Tim-3, TGF-β, and purinergic pathways. There is a general interest in ICIs because of the clinical success of the PD-1/PD-L1 blockade. Thus, it is expected that the development of multiple dual immunostimulators will increase. However, the risk of toxicity due to strong immune activation needs to be monitored.
Finally, dual tumor-targeting BsAbs are designed for the following specific purposes: simultaneous blockade of crosstalking pathways, enhanced drug delivery, and tumor-targeted destruction of tumor cells. To overcome the acquired drug resistance and tumor escape from mAb-based targeted therapy, strategies for the simultaneous inhibition of different signaling pathways using BsAb and simultaneous blockade of two key signaling factors have been proposed. For enhanced efficacy in the TME, several BsAb approaches have been evaluated, typically targeting TAA and effector molecules such as CD63, CD47, pro-apoptotic proteins, and complement-regulatory proteins.
To summarize, the field of immunotherapy using BsAbs is growing rapidly, and the cases of preclinical stage and clinical trials are increasing. Future clinical trials will improve our understanding of the potential and safety profiles of BsAbs. Eventually, BsAbs could provide the next generation of new treatment options for cancer patients.
Acknowledgments
The figures presented in this manuscript were created with BioRender.com.
Author Contributions
Conceptualization, G.Y., J.W., S.H.L., and S.-W.L.; writing—original draft preparation, G.Y. and J.W.; writing—review and editing, G.Y., J.W., Y.L., Y.P., S.H.L., and S.-W.L.; visualization, G.Y. and D.M.; supervision, S.-W.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Korea Initiative for fostering the University of Research and Innovation Program of the National Research Foundation (NRF) funded by the Korean government (MIST) (No.2020M3H1A1075314), by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (2017M3A9C8033570) and by grants from ABL Bio Inc.
Conflicts of Interest
J.W., Y.L., and S.H.L. are current employees of ABL Bio. The other authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lu R.-M., Hwang Y.-C., Liu I.J., Lee C.-C., Tsai H.-Z., Li H.-J., Wu H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020;27:1. doi: 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feugier P. A review of rituximab, the first anti-CD20 monoclonal antibody used in the treatment of B non-Hodgkin’s lymphomas. Future Oncol. 2015;11:1327–1342. doi: 10.2217/fon.15.57. [DOI] [PubMed] [Google Scholar]
- 3.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farkona S., Diamandis E.P., Blasutig I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016;14:73. doi: 10.1186/s12916-016-0623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahoney K.M., Rennert P.D., Freeman G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015;14:561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 6.Chen D.S., Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 7.Darvin P., Toor S.M., Sasidharan Nair V., Elkord E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018;50:1–11. doi: 10.1038/s12276-018-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esfahani K., Roudaia L., Buhlaiga N., Del Rincon S.V., Papneja N., Miller W.H. A Review of Cancer Immunotherapy: From the Past, to the Present, to the Future. Curr. Oncol. 2020;27:87–97. doi: 10.3747/co.27.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai R., Chen N., Li L., Du N., Bai L., Lv Z., Tian H., Cui J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.01290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martins F., Sofiya L., Sykiotis G.P., Lamine F., Maillard M., Fraga M., Shabafrouz K., Ribi C., Cairoli A., Guex-Crosier Y., et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019;16:563–580. doi: 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- 12.Godar M., de Haard H., Blanchetot C., Rasser J. Therapeutic bispecific antibody formats: A patent applications review (1994-2017) Expert Opin. Ther. Pat. 2018;28:251–276. doi: 10.1080/13543776.2018.1428307. [DOI] [PubMed] [Google Scholar]
- 13.Labrijn A.F., Janmaat M.L., Reichert J.M., Parren P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019;18:585–608. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 14.Nie S., Wang Z., Moscoso-Castro M., D’Souza P., Lei C., Xu J., Gu J. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib. Ther. 2020;3:18–62. doi: 10.1093/abt/tbaa003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim S.I. Fine-tuning bispecific therapeutics. Pharmacol. Ther. 2020;212:107582. doi: 10.1016/j.pharmthera.2020.107582. [DOI] [PubMed] [Google Scholar]
- 16.Bannas P., Hambach J., Koch-Nolte F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017;8 doi: 10.3389/fimmu.2017.01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Vlieger D., Ballegeer M., Rossey I., Schepens B., Saelens X. Single-Domain Antibodies and Their Formatting to Combat Viral Infections. Antibodies. 2019;8:1. doi: 10.3390/antib8010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao H., Ma Y., Zhang Y., Hong S., Yang Y., Fang W., Xu J., Van H., Kong P., Yang F., et al. The preliminary efficacy and safety data of KN046 in patients failed on prior immune checkpoint inhibitors therapy. J. Clin. Oncol. 2020;38:3020. doi: 10.1200/JCO.2020.38.15_suppl.3020. [DOI] [Google Scholar]
- 19.Suurs F.V., Lub-de Hooge M.N., de Vries E.G.E., de Groot D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019;201:103–119. doi: 10.1016/j.pharmthera.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 20.Przepiorka D., Ko C.-W., Deisseroth A., Yancey C.L., Candau-Chacon R., Chiu H.-J., Gehrke B.J., Gomez-Broughton C., Kane R.C., Kirshner S., et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015;21:4035–4039. doi: 10.1158/1078-0432.CCR-15-0612. [DOI] [PubMed] [Google Scholar]
- 21.Moore P.A., Zhang W., Rainey G.J., Burke S., Li H., Huang L., Gorlatov S., Veri M.C., Aggarwal S., Yang Y., et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood. 2011;117:4542–4551. doi: 10.1182/blood-2010-09-306449. [DOI] [PubMed] [Google Scholar]
- 22.Uy G.L., Aldoss I., Foster M.C., Sayre P.H., Wieduwilt M.J., Advani A.S., Godwin J.E., Arellano M.L., Sweet K.L., Emadi A., et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. 2021;137:751–762. doi: 10.1182/blood.2020007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel M., Luke J., Hamilton E., Chmielowski B., Blumenschein G., Kindler H., Bahadur S., Santa-Maria C., Koucheki J., Sun J., et al. 313 A phase 1 evaluation of tebotelimab, a bispecific PD-1 x LAG-3 DART® molecule, in combination with margetuximab in patients with advanced HER2+ neoplasms. J. Immunother. Cancer. 2020;8:A193. doi: 10.1136/jitc-2020-SITC2020.0313. [DOI] [Google Scholar]
- 24.Sharma M., Sanborn R.E., Cote G.M., Bendell J.C., Kaul S., Chen F., Berezhnoy A., Moore P., Bonvini E., Sumrow B.J., et al. 1020O A phase I, first-in-human, open-label, dose escalation study of MGD019, an investigational bispecific PD-1 x CTLA-4 DART® molecule in patients with advanced solid tumours. Ann. Oncol. 2020;31:S704–S705. doi: 10.1016/j.annonc.2020.08.1140. [DOI] [Google Scholar]
- 25.Wu J., Fu J., Zhang M., Liu D. AFM13: A first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J. Hematol. Oncol. 2015;8:96. doi: 10.1186/s13045-015-0188-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rothe A., Sasse S., Topp M.S., Eichenauer D.A., Hummel H., Reiners K.S., Dietlein M., Kuhnert G., Kessler J., Buerkle C., et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015;125:4024–4031. doi: 10.1182/blood-2014-12-614636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vallera D.A., Felices M., McElmurry R., McCullar V., Zhou X., Schmohl J.U., Zhang B., Lenvik A.J., Panoskaltsis-Mortari A., Verneris M.R., et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016;22:3440–3450. doi: 10.1158/1078-0432.CCR-15-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayes P., Tacken P., Wang S., Loo P.-F.v., Condamine T., Maaden H.v.d., Rovers E., Engels S., Fransen F., Kulkarni A., et al. Abstract 539: A bispecific Fc-silenced IgG1 antibody (MCLA-145) requires PD-L1 binding to activate CD137. Cancer Res. 2019;79:539. doi: 10.1158/1538-7445.Am2019-539. [DOI] [Google Scholar]
- 29.Waite J.C., Wang B., Haber L., Hermann A., Ullman E., Ye X., Dudgeon D., Slim R., Ajithdoss D.K., Godin S.J., et al. Tumor-targeted CD28 bispecific antibodies enhance the antitumor efficacy of PD-1 immunotherapy. Sci. Transl. Med. 2020;12:eaba2325. doi: 10.1126/scitranslmed.aba2325. [DOI] [PubMed] [Google Scholar]
- 30.Skokos D., Waite J.C., Haber L., Crawford A., Hermann A., Ullman E., Slim R., Godin S., Ajithdoss D., Ye X., et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020;12:eaaw7888. doi: 10.1126/scitranslmed.aaw7888. [DOI] [PubMed] [Google Scholar]
- 31.Hutchings M., Lugtenburg P., Mous R., Clausen M.R., Chamuleau M., Linton K., Rule S., Lopez J.S., Oliveri R.S., DeMarco D., et al. Epcoritamab (GEN3013; DuoBody-CD3 × CD20) to induce complete response in patients with relapsed/refractory B-cell non-Hodgkin lymphoma (B-NHL): Complete dose escalation data and efficacy results from a phase I/II trial. J. Clin. Oncol. 2020;38:8009. doi: 10.1200/JCO.2020.38.15_suppl.8009. [DOI] [Google Scholar]
- 32.Liu H., Saxena A., Sidhu S.S., Wu D. Fc Engineering for Developing Therapeutic Bispecific Antibodies and Novel Scaffolds. Front. Immunol. 2017;8 doi: 10.3389/fimmu.2017.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Killock D. Engaging results with glofitamab. Nat. Rev. Clin. Oncol. 2021 doi: 10.1038/s41571-021-00510-3. [DOI] [PubMed] [Google Scholar]
- 34.Tabernero J., Melero I., Ros W., Argiles G., Marabelle A., Rodriguez-Ruiz M.E., Albanell J., Calvo E., Moreno V., Cleary J.M., et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC) J. Clin. Oncol. 2017;35:3002. doi: 10.1200/JCO.2017.35.15_suppl.3002. [DOI] [Google Scholar]
- 35.Park E., Sung E., Chung H., Lee Y., Yoo J., Park M., Kim E., Son Y.-G., Choi H., Jung J., et al. The PD-L1 x 4-1BB bispecific antibody ABL503 shows potent anti-tumor effect through tumor-directed T cell activation. PEGS Boston. 2019 [Google Scholar]
- 36.Jiang W., Fang L., Wang Z., Guo T.B., Park E., Sung E., Jung J. Abstract 5644: Claudin 18.2 × 4-1BB bispecific antibody induced potent tumor inhibition through tumor-specific 4-1BB activation. Cancer Res. 2020;80:5644. doi: 10.1158/1538-7445.Am2020-5644. [DOI] [Google Scholar]
- 37.Rothe C., Skerra A. Anticalin® Proteins as Therapeutic Agents in Human Diseases. BioDrugs. 2018;32:233–243. doi: 10.1007/s40259-018-0278-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hinner M.J., Aiba R.S.B., Jaquin T.J., Berger S., Dürr M.C., Schlosser C., Allersdorfer A., Wiedenmann A., Matschiner G., Schüler J., et al. Tumor-Localized Costimulatory T-Cell Engagement by the 4-1BB/HER2 Bispecific Antibody-Anticalin Fusion PRS-343. Clin. Cancer Res. 2019;25:5878–5889. doi: 10.1158/1078-0432.CCR-18-3654. [DOI] [PubMed] [Google Scholar]
- 39.Yap T., Wong D., Hu-Lieskovan S., Papadopoulos K., Morrow M., Grabowska U., Gliddon D., Holz J.-B., LoRusso P. 395 A first-in-human study of FS118, a tetravalent bispecific antibody targeting LAG-3 and PD-L1, in patients with advanced cancer and resistance to PD-(L)1 therapy. J. Immunother. Cancer. 2020;8:A240. doi: 10.1136/jitc-2020-SITC2020.0395. [DOI] [PubMed] [Google Scholar]
- 40.Ravandi F., Bashey A., Stock W., Foran J.M., Mawad R., Egan D., Blum W., Yang A., Pastore A., Johnson C., et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XmAb14045), a CD123 x CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood. 2020;136:4–5. doi: 10.1182/blood-2020-134746. [DOI] [Google Scholar]
- 41.Hickingbottom B., Clynes R., Desjarlais J., Li C., Ding Y. Preliminary safety and pharmacodynamic (PD) activity of XmAb20717, a PD-1 x CTLA-4 bispecific antibody, in a phase I dose escalation study of patients with selected advanced solid tumors. J. Clin. Oncol. 2020;38:e15001. doi: 10.1200/JCO.2020.38.15_suppl.e15001. [DOI] [Google Scholar]
- 42.Zhang J., Yi J., Zhou P. Development of bispecific antibodies in China: Overview and prospects. Antib. Ther. 2020;3:126–145. doi: 10.1093/abt/tbaa011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowe K.L., Cole D., Kenefeck R., Okelly I., Lepore M., Jakobsen B.K. Novel TCR-based biologics: Mobilising T cells to warm ‘cold’ tumours. Cancer Treat. Rev. 2019;77:35–43. doi: 10.1016/j.ctrv.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Mack M., Riethmüller G., Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA. 1995;92:7021–7025. doi: 10.1073/pnas.92.15.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roopenian D.C., Akilesh S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 46.Saunders K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lakins M.A., Koers A., Giambalvo R., Hughes R., Marshall S., Wydro M., Gradinaru C., Batey S., Gliddon D., Morrow M., et al. Abstract 4547: Clustering CD137 via cell-expressed PD-L1 crosslinking avoided Fc-mediated agonism and resulted in safe and potent conditional lymphocyte activation. Cancer Res. 2020;80:4547. doi: 10.1158/1538-7445.Am2020-4547. [DOI] [Google Scholar]
- 48.Liu R., Oldham R.J., Teal E., Beers S.A., Cragg M.S. Fc-Engineering for Modulated Effector Functions—Improving Antibodies for Cancer Treatment. Antibodies. 2020;9:64. doi: 10.3390/antib9040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellerman D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods. 2019;154:102–117. doi: 10.1016/j.ymeth.2018.10.026. [DOI] [PubMed] [Google Scholar]
- 50.Staflin K., Zuch de Zafra C.L., Schutt L.K., Clark V., Zhong F., Hristopoulos M., Clark R., Li J., Mathieu M., Chen X., et al. Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI Insight. 2020;5:e133757. doi: 10.1172/jci.insight.133757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krishna M., Nadler S.G. Immunogenicity to Biotherapeutics—The Role of Anti-drug Immune Complexes. Front. Immunol. 2016;7 doi: 10.3389/fimmu.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franquiz M.J., Short N.J. Blinatumomab for the Treatment of Adult B-Cell Acute Lymphoblastic Leukemia: Toward a New Era of Targeted Immunotherapy. Biologics. 2020;14:23–34. doi: 10.2147/BTT.S202746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piha-Paul S., Bendell J., Tolcher A., Hurvitz S., Patnaik A., Shroff R., Pohlmann P., Zettl M., Hahn N., Krishnamurthy A., et al. O82 A phase 1 dose escalation study of PRS-343, a HER2/4–1BB bispecific molecule, in patients with HER2-positive malignancies. J. Immunother. Cancer. 2020;8:A1–A2. doi: 10.1136/LBA2019.2. [DOI] [Google Scholar]
- 54.Jurichson J. Aptevo Therapeutics and MorphoSys End Joint Development and Commercialization Agreement for MOR209/ES414. Aptevo Therapeutics Inc.; Seattle, WA, USA: 2017. [Google Scholar]
- 55.Jurchison S. Aptevo Therapeutics Reports Third Quarter 2018 Financial Results. Aptevo Therapeutics Inc.; Seattle, WA, USA: 2018. [Google Scholar]
- 56.Offner S., Hofmeister R., Romaniuk A., Kufer P., Baeuerle P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006;43:763–771. doi: 10.1016/j.molimm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 57.Dufner V., Sayehli C.M., Chatterjee M., Hummel H.D., Gelbrich G., Bargou R.C., Goebeler M.-E. Long-term outcome of patients with relapsed/refractory B-cell non-Hodgkin lymphoma treated with blinatumomab. Blood Adv. 2019;3:2491–2498. doi: 10.1182/bloodadvances.2019000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Löffler A., Kufer P., Lutterbüse R., Zettl F., Daniel P.T., Schwenkenbecher J.M., Riethmüller G., Dörken B., Bargou R.C. A recombinant bispecific single-chain antibody, CD19 × CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95:2098–2103. doi: 10.1182/blood.V95.6.2098. [DOI] [PubMed] [Google Scholar]
- 59.Linke R., Klein A., Seimetz D. Catumaxomab. mAbs. 2010;2:129–136. doi: 10.4161/mabs.2.2.11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ott M.G., Marmé F., Moldenhauer G., Lindhofer H., Hennig M., Spannagl R., Essing M.M., Linke R., Seimetz D. Humoral response to catumaxomab correlates with clinical outcome: Results of the pivotal phase II/III study in patients with malignant ascites. Int. J. Cancer. 2012;130:2195–2203. doi: 10.1002/ijc.26258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liddy N., Bossi G., Adams K.J., Lissina A., Mahon T.M., Hassan N.J., Gavarret J., Bianchi F.C., Pumphrey N.J., Ladell K., et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012;18:980–987. doi: 10.1038/nm.2764. [DOI] [PubMed] [Google Scholar]
- 62.Damato B.E., Dukes J., Goodall H., Carvajal R.D. Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers. 2019;11:971. doi: 10.3390/cancers11070971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bortoletto N., Scotet E., Myamoto Y., D’Oro U., Lanzavecchia A. Optimizing anti-CD3 affinity for effective T cell targeting against tumor cells. Eur. J. Immunol. 2002;32:3102–3107. doi: 10.1002/1521-4141(200211)32:11<3102::AID-IMMU3102>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 64.Leong S.R., Sukumaran S., Hristopoulos M., Totpal K., Stainton S., Lu E., Wong A., Tam L., Newman R., Vuillemenot B.R., et al. An anti-CD3/anti–CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood. 2017;129:609–618. doi: 10.1182/blood-2016-08-735365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mandikian D., Takahashi N., Lo A.A., Li J., Eastham-Anderson J., Slaga D., Ho J., Hristopoulos M., Clark R., Totpal K., et al. Relative Target Affinities of T-Cell–Dependent Bispecific Antibodies Determine Biodistribution in a Solid Tumor Mouse Model. Mol. Cancer Ther. 2018;17:776–785. doi: 10.1158/1535-7163.MCT-17-0657. [DOI] [PubMed] [Google Scholar]
- 66.Thakur A., Huang M., Lum L.G. Bispecific antibody based therapeutics: Strengths and challenges. Blood Rev. 2018;32:339–347. doi: 10.1016/j.blre.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 67.Yu S., Li A., Liu Q., Yuan X., Xu H., Jiao D., Pestell R.G., Han X., Wu K. Recent advances of bispecific antibodies in solid tumors. J. Hematol. Oncol. 2017;10:155. doi: 10.1186/s13045-017-0522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimabukuro-Vornhagen A., Gödel P., Subklewe M., Stemmler H.J., Schlößer H.A., Schlaak M., Kochanek M., Böll B., von Bergwelt-Baildon M.S. Cytokine release syndrome. J. ImmunoTherapy Cancer. 2018;6:56. doi: 10.1186/s40425-018-0343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kantarjian H., Stein A., Gökbuget N., Fielding A.K., Schuh A.C., Ribera J.-M., Wei A., Dombret H., Foà R., Bassan R., et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017;376:836–847. doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chu S.Y., Miranda Y., Phung S., Chen H., Rashid R., Endo N.A., Chan E.W., Pong E., Bonzon C., Muchhal U.S., et al. Immunotherapy with Long-Lived Anti-CD38 × Anti-CD3 Bispecific Antibodies Stimulates Potent T Cell-Mediated Killing of Human Myeloma Cell Lines and CD38+ Cells in Monkeys: A Potential Therapy for Multiple Myeloma. Blood. 2014;124:4727. doi: 10.1182/blood.V124.21.4727.4727. [DOI] [Google Scholar]
- 71.Moore G.L., Lee S.-H., Schubbert S., Miranda Y., Rashid R., Pong E., Phung S., Chan E.W., Chen H., Endo N., et al. Tuning T Cell Affinity Improves Efficacy and Safety of Anti-CD38 × Anti-CD3 Bispecific Antibodies in Monkeys—A Potential Therapy for Multiple Myeloma. Blood. 2015;126:1798. doi: 10.1182/blood.V126.23.1798.1798. [DOI] [Google Scholar]
- 72.Bacac M., Fauti T., Sam J., Colombetti S., Weinzierl T., Ouaret D., Bodmer W., Lehmann S., Hofer T., Hosse R.J., et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016;22:3286–3297. doi: 10.1158/1078-0432.CCR-15-1696. [DOI] [PubMed] [Google Scholar]
- 73.Bacac M., Umaña P., Herter S., Colombetti S., Sam J., Le Clech M., Freimoser-Grundschober A., Richard M., Nicolini V., Gerdes C., et al. CD20 Tcb (RG6026), a Novel “2:1” T Cell Bispecific Antibody for the Treatment of B Cell Malignancies. Blood. 2016;128:1836. doi: 10.1182/blood.V128.22.1836.1836. [DOI] [Google Scholar]
- 74.Hernandez-Hoyos G., Sewell T., Bader R., Bannink J., Chenault R.A., Daugherty M., Dasovich M., Fang H., Gottschalk R., Kumer J., et al. MOR209/ES414, a Novel Bispecific Antibody Targeting PSMA for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2016;15:2155–2165. doi: 10.1158/1535-7163.MCT-15-0242. [DOI] [PubMed] [Google Scholar]
- 75.Banaszek A., Bumm T.G.P., Nowotny B., Geis M., Jacob K., Wölfl M., Trebing J., Kucka K., Kouhestani D., Gogishvili T., et al. On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat. Commun. 2019;10:5387. doi: 10.1038/s41467-019-13196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boustany L.M., Wong L., White C.W., Diep L., Huang Y., Liu S., Richardson J.H., Kavanaugh W.M., Irving B.A. Abstract A164: EGFR-CD3 bispecific Probody™ therapeutic induces tumor regressions and increases maximum tolerated dose >60-fold in preclinical studies. Mol. Cancer Ther. 2018;17:A164. doi: 10.1158/1535-7163.Targ-17-a164. [DOI] [Google Scholar]
- 77.Geiger M., Stubenrauch K.-G., Sam J., Richter W.F., Jordan G., Eckmann J., Hage C., Nicolini V., Freimoser-Grundschober A., Ritter M., et al. Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. Nat. Commun. 2020;11:3196. doi: 10.1038/s41467-020-16838-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chiang S.C.C., Theorell J., Entesarian M., Meeths M., Mastafa M., Al-Herz W., Frisk P., Gilmour K.C., Ifversen M., Langenskiöld C., et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood. 2013;121:1345–1356. doi: 10.1182/blood-2012-07-442558. [DOI] [PubMed] [Google Scholar]
- 79.Horton N.C., Mathew P.A. NKp44 and Natural Cytotoxicity Receptors as Damage-Associated Molecular Pattern Recognition Receptors. Front. Immunol. 2015;6 doi: 10.3389/fimmu.2015.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chretien A.-S., Le Roy A., Vey N., Prebet T., Blaise D., Fauriat C., Olive D. Cancer-Induced Alterations of NK-Mediated Target Recognition: Current and Investigational Pharmacological Strategies Aiming at Restoring NK-Mediated Anti-Tumor Activity. Front. Immunol. 2014;5 doi: 10.3389/fimmu.2014.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hogarth P.M., Pietersz G.A. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat. Rev. Drug Discov. 2012;11:311–331. doi: 10.1038/nrd2909. [DOI] [PubMed] [Google Scholar]
- 82.Gantke T., Weichel M., Herbrecht C., Reusch U., Ellwanger K., Fucek I., Eser M., Müller T., Griep R., Molkenthin V., et al. Trispecific antibodies for CD16A-directed NK cell engagement and dual-targeting of tumor cells. Protein Eng. Des. Sel. 2017;30:673–684. doi: 10.1093/protein/gzx043. [DOI] [PubMed] [Google Scholar]
- 83.Schmohl J.U., Felices M., Oh F., Lenvik A.J., Lebeau A.M., Panyam J., Miller J.S., Vallera D.A. Engineering of Anti-CD133 Trispecific Molecule Capable of Inducing NK Expansion and Driving Antibody-Dependent Cell-Mediated Cytotoxicity. Cancer Res Treat. 2017;49:1140–1152. doi: 10.4143/crt.2016.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kontos F., Kurokawa T., Vallera D.A., Ferrone S., Ferrone C.R. IL-15/B7-H3 TriKEs-Based Immunotherapy for Pancreatic Ductal Adenocarcinoma. J. Am. Coll. Surg. 2019;229:S176. doi: 10.1016/j.jamcollsurg.2019.08.388. [DOI] [Google Scholar]
- 85.Gauthier L., Morel A., Anceriz N., Rossi B., Blanchard-Alvarez A., Grondin G., Trichard S., Cesari C., Sapet M., Bosco F., et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell. 2019;177:1701–1713.e1716. doi: 10.1016/j.cell.2019.04.041. [DOI] [PubMed] [Google Scholar]
- 86.Chan W.K., Kang S., Youssef Y., Glankler E.N., Barrett E.R., Carter A.M., Ahmed E.H., Prasad A., Chen L., Zhang J., et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol. Res. 2018;6:776–787. doi: 10.1158/2326-6066.CIR-17-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fury M.G., Lipton A., Smith K.M., Winston C.B., Pfister D.G. A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors. Cancer Immunol. Immunother. 2008;57:155–163. doi: 10.1007/s00262-007-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sedykh S.E., Prinz V.V., Buneva V.N., Nevinsky G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des Devel. 2018;12:195–208. doi: 10.2147/DDDT.S151282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Corgnac S., Perret R., Derré L., Zhang L., Stirnemann K., Zauderer M., Speiser D.E., Mach J.-P., Romero P., Donda A. CD1d-antibody fusion proteins target iNKT cells to the tumor and trigger long-term therapeutic responses. Cancer Immunol. Immunother. 2013;62:747–760. doi: 10.1007/s00262-012-1381-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chester C., Sanmamed M.F., Wang J., Melero I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood. 2018;131:49–57. doi: 10.1182/blood-2017-06-741041. [DOI] [PubMed] [Google Scholar]
- 91.Chen L., Flies D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jeong S., Park S.-H. Co-Stimulatory Receptors in Cancers and Their Implications for Cancer Immunotherapy. Immune Netw. 2020;20 doi: 10.4110/in.2020.20.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tacke M., Hanke G., Hanke T., Hünig T. CD28-mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: Evidence for functionally distinct forms of CD28. Eur. J. Immunol. 1997;27:239–247. doi: 10.1002/eji.1830270136. [DOI] [PubMed] [Google Scholar]
- 94.Attarwala H. TGN1412: From Discovery to Disaster. J. Young Pharm. 2010;2:332–336. doi: 10.4103/0975-1483.66810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Otz T., Große-Hovest L., Hofmann M., Rammensee H.G., Jung G. A bispecific single-chain antibody that mediates target cell-restricted, supra-agonistic CD28 stimulation and killing of lymphoma cells. Leukemia. 2009;23:71–77. doi: 10.1038/leu.2008.271. [DOI] [PubMed] [Google Scholar]
- 96.Hedvat M., Zeng V., Diaz J., Bonzon C., Avery K., Rashid R., Leung I., Bartlow N., Bakhit C., Dragovich M., et al. 697 Tumor-targeted CD28 costimulatory bispecific antibodies enhance T cell activation in solid tumors. J. Immunother. Cancer. 2020;8:A419. doi: 10.1136/jitc-2020-SITC2020.0697. [DOI] [Google Scholar]
- 97.Koopmans I., Hendriks D., Samplonius D.F., van Ginkel R.J., Heskamp S., Wierstra P.J., Bremer E., Helfrich W. A novel bispecific antibody for EGFR-directed blockade of the PD-1/PD-L1 immune checkpoint. OncoImmunology. 2018;7:e1466016. doi: 10.1080/2162402X.2018.1466016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koopmans I., Hendriks M.A.J.M., van Ginkel R.J., Samplonius D.F., Bremer E., Helfrich W. Bispecific Antibody Approach for Improved Melanoma-Selective PD-L1 Immune Checkpoint Blockade. J. Investig. Dermatol. 2019;139:2343–2351.e2343. doi: 10.1016/j.jid.2019.01.038. [DOI] [PubMed] [Google Scholar]
- 99.Moran A.E., Kovacsovics-Bankowski M., Weinberg A.D. The TNFRs OX40, 4-1BB, and CD40 as targets for cancer immunotherapy. Curr. Opin. Immunol. 2013;25:230–237. doi: 10.1016/j.coi.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim H.-D., Park S., Jeong S., Lee Y.J., Lee H., Kim C.G., Kim K.H., Hong S.-M., Lee J.-Y., Kim S., et al. 4-1BB Delineates Distinct Activation Status of Exhausted Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Hepatology. 2020;71:955–971. doi: 10.1002/hep.30881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wolfl M., Kuball J., Ho W.Y., Nguyen H., Manley T.J., Bleakley M., Greenberg P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110:201–210. doi: 10.1182/blood-2006-11-056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Williams J.B., Horton B.L., Zheng Y., Duan Y., Powell J.D., Gajewski T.F. The EGR2 targets LAG-3 and 4-1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J. Exp. Med. 2017;214:381–400. doi: 10.1084/jem.20160485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jure-Kunkel M.N., Calarota S., Girit E., Abraham R., Balimane P., Price K., Weiner D., Hefta L. Functional characterization of fully human anti-CD137 antibodies. Cancer Res. 2006;66:1117. [Google Scholar]
- 104.Fisher T.S., Kamperschroer C., Oliphant T., Love V.A., Lira P.D., Doyonnas R., Bergqvist S., Baxi S.M., Rohner A., Shen A.C., et al. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer Immunol. Immunother. 2012;61:1721–1733. doi: 10.1007/s00262-012-1237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ku G., Bendell J.C., Tolcher A.W., Hurvitz S.A., Krishnamurthy A., El-Khoueiry A.B., Patnaik A., Shroff R.T., Noonan A., Hahn N.M., et al. 525O A phase I dose escalation study of PRS-343, a HER2/4-1BB bispecific molecule, in patients with HER2-positive malignancies. Ann. Oncol. 2020;31:S462–S463. doi: 10.1016/j.annonc.2020.08.639. [DOI] [Google Scholar]
- 106.Claus C., Ferrara C., Xu W., Sam J., Lang S., Uhlenbrock F., Albrecht R., Herter S., Schlenker R., Hüsser T., et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019;11:eaav5989. doi: 10.1126/scitranslmed.aav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nelson M., Miller R., Nilsson A., Ljung L., Chunyk A., McMahan C., Bienvenue D., Askmyr M., Hernandez-Hoyos G., Fritzell S. 851 Potent tumor-directed T cell activation and in vivo tumor inhibition induced by a 4–1BB x 5T4 ADAPTIR™ bispecific antibody. J. Immunother. Cancer. 2020;8:A507. doi: 10.1136/jitc-2020-SITC2020.0851. [DOI] [Google Scholar]
- 108.You G., Lee Y., Kang Y.-W., Park H.W., Park K., Kim H., Kim Y.-M., Kim S., Kim J.-H., Moon D., et al. B7-H3×4-1BB bispecific antibody augments antitumor immunity by enhancing terminally differentiated CD8+ tumor-infiltrating lymphocytes. Sci. Adv. 2021;7:eaax3160. doi: 10.1126/sciadv.aax3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schaer D.A., Murphy J.T., Wolchok J.D. Modulation of GITR for cancer immunotherapy. Curr. Opin. Immunol. 2012;24:217–224. doi: 10.1016/j.coi.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Burckhart T., Thiel M., Nishikawa H., Wüest T., Müller D., Zippelius A., Ritter G., Old L., Shiku H., Renner C. Tumor-specific Crosslinking of GITR as Costimulation for Immunotherapy. J. Immunother. 2010;33:925–934. doi: 10.1097/CJI.0b013e3181f3cc87. [DOI] [PubMed] [Google Scholar]
- 111.Vonderheide R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020;71:47–58. doi: 10.1146/annurev-med-062518-045435. [DOI] [PubMed] [Google Scholar]
- 112.Ye S., Cohen D., Belmar N.A., Choi D., Tan S.S., Sho M., Akamatsu Y., Kim H., Iyer R., Cabel J., et al. A Bispecific Molecule Targeting CD40 and Tumor Antigen Mesothelin Enhances Tumor-Specific Immunity. Cancer Immunol. Res. 2019;7:1864–1875. doi: 10.1158/2326-6066.CIR-18-0805. [DOI] [PubMed] [Google Scholar]
- 113.Dovedi S.J., Elder M.J., Yang C., Sitnikova S.I., Irving L., Hansen A., Hair J., Jones D.C., Hasani S., Wang B., et al. Design and efficacy of a monovalent bispecific PD-1/CTLA-4 antibody that enhances CTLA-4 blockade on PD-1+ activated T cells. Cancer Discov. 2021 doi: 10.1158/2159-8290.CD-20-1445. [DOI] [PubMed] [Google Scholar]
- 114.Dovedi S.J., Mazor Y., Elder M., Hasani S., Wang B., Mosely S., Jones D., Hansen A., Yang C., Wu Y., et al. Abstract 2776: MEDI5752: A novel bispecific antibody that preferentially targets CTLA-4 on PD-1 expressing T-cells. Cancer Res. 2018;78:2776. doi: 10.1158/1538-7445.Am2018-2776. [DOI] [Google Scholar]
- 115.Anderson Ana C., Joller N., Kuchroo Vijay K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lan Y., Zhang D., Xu C., Hance K.W., Marelli B., Qi J., Yu H., Qin G., Sircar A., Hernández V.M., et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018;10:eaan5488. doi: 10.1126/scitranslmed.aan5488. [DOI] [PubMed] [Google Scholar]
- 117.Zhong T., Huang Z., Pang X., Chen N., Jin X., Xia Y., Wang Z.M., Li B., Xia Y. 702 Dual blockade of the PD-1 checkpoint pathway and the adenosinergic negative feedback signaling pathway with a PD-1/CD73 bispecific antibody for cancer immune therapy. J. Immunother. Cancer. 2020;8:A422. doi: 10.1136/jitc-2020-SITC2020.0702. [DOI] [Google Scholar]
- 118.Kvarnhammar A.M., Veitonmäki N., Hägerbrand K., Dahlman A., Smith K.E., Fritzell S., von Schantz L., Thagesson M., Werchau D., Smedenfors K., et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J. Immunother. Cancer. 2019;7:103. doi: 10.1186/s40425-019-0570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gaspar M., Pravin J., Rodrigues L., Uhlenbroich S., Everett K.L., Wollerton F., Morrow M., Tuna M., Brewis N. CD137/OX40 Bispecific Antibody Induces Potent Antitumor Activity that Is Dependent on Target Coengagement. Cancer Immunol. Res. 2020;8:781–793. doi: 10.1158/2326-6066.CIR-19-0798. [DOI] [PubMed] [Google Scholar]
- 120.Hynes N.E., Lane H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 121.Zhao B., Wang L., Qiu H., Zhang M., Sun L., Peng P., Yu Q., Yuan X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget. 2016;8:3980–4000. doi: 10.18632/oncotarget.14012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pohlmann P.R., Mayer I.A., Mernaugh R. Resistance to Trastuzumab in Breast Cancer. Clin. Cancer Res. 2009;15:7479–7491. doi: 10.1158/1078-0432.CCR-09-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guo F., Cui J. Anti-angiogenesis: Opening a new window for immunotherapy. Life Sci. 2020;258:118163. doi: 10.1016/j.lfs.2020.118163. [DOI] [PubMed] [Google Scholar]
- 124.Khan K.A., Kerbel R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018;15:310–324. doi: 10.1038/nrclinonc.2018.9. [DOI] [PubMed] [Google Scholar]
- 125.Moek K.L., de Groot D.J.A., de Vries E.G.E., Fehrmann R.S.N. The antibody–drug conjugate target landscape across a broad range of tumour types. Ann. Oncol. 2017;28:3083–3091. doi: 10.1093/annonc/mdx541. [DOI] [PubMed] [Google Scholar]
- 126.de Goeij B.E.C.G., Vink T., ten Napel H., Breij E.C.W., Satijn D., Wubbolts R., Miao D., Parren P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody–Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016;15:2688–2697. doi: 10.1158/1535-7163.MCT-16-0364. [DOI] [PubMed] [Google Scholar]
- 127.Li J.Y., Perry S.R., Muniz-Medina V., Wang X., Wetzel L.K., Rebelatto M.C., Hinrichs M.J.M., Bezabeh B.Z., Fleming R.L., Dimasi N., et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell. 2016;29:117–129. doi: 10.1016/j.ccell.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 128.Hamblett K.J., Hammond P.W., Barnscher S.D., Fung V.K., Davies R.H., Wickman G.R., Hernandez A., Ding T., Galey A.S., Winters G.C., et al. Abstract 3914: ZW49, a HER2-targeted biparatopic antibody-drug conjugate for the treatment of HER2-expressing cancers. Cancer Res. 2018;78:3914. doi: 10.1158/1538-7445.Am2018-3914. [DOI] [Google Scholar]
- 129.Zhang W., Huang Q., Xiao W., Zhao Y., Pi J., Xu H., Zhao H., Xu J., Evans C.E., Jin H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dheilly E., Moine V., Broyer L., Salgado-Pires S., Johnson Z., Papaioannou A., Cons L., Calloud S., Majocchi S., Nelson R., et al. Selective Blockade of the Ubiquitous Checkpoint Receptor CD47 Is Enabled by Dual-Targeting Bispecific Antibodies. Mol. Ther. 2017;25:523–533. doi: 10.1016/j.ymthe.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Walczak H. Death Receptor–Ligand Systems in Cancer, Cell Death, and Inflammation. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brünker P., Wartha K., Friess T., Grau-Richards S., Waldhauer I., Koller C.F., Weiser B., Majety M., Runza V., Niu H., et al. RG7386, a Novel Tetravalent FAP-DR5 Antibody, Effectively Triggers FAP-Dependent, Avidity-Driven DR5 Hyperclustering and Tumor Cell Apoptosis. Mol. Cancer Ther. 2016;15:946–957. doi: 10.1158/1535-7163.MCT-15-0647. [DOI] [PubMed] [Google Scholar]
- 133.Bendell J., Blay J.-Y., Cassier P., Bauer T., Terret C., Mueller C., Morel A., Chesne E., Xu Z.-x., Tessier J., et al. Abstract A092: Phase 1 trial of RO6874813, a novel bispecific FAP-DR5 antibody, in patients with solid tumors. Mol. Cancer Ther. 2018;17:A092. doi: 10.1158/1535-7163.Targ-17-a092. [DOI] [Google Scholar]