

Abstract
The monitoring of the progression of non-alcoholic fatty liver disease is hindered by the lack of suitable noninvasive imaging methods. Here, we show that the endogenous pigment lipofuscin displays strong near infrared and shortwave infrared fluorescence when excited at 808 nm, enabling the label-free imaging of liver injury in mice and the discrimination of pathological processes from normal liver processes with high specificity and sensitivity. We also show that lipofuscin near infrared and shortwave infrared fluorescence can be used to monitor the longitudinal progression and regression of liver necroinflammation and fibrosis in mouse models of non-alcoholic fatty liver disease and of advanced fibrosis, and to detect non-alcoholic steatohepatitis and cirrhosis in biopsied samples of human liver tissue.
One-sentence editorial summary:
Label-free imaging of the endogenous pigment lipofuscin at near and shortwave-infrared wavelengths enables the longitudinal monitoring of liver injury in mouse models of non-alcoholic fatty liver disease and of advanced fibrosis, and in biopsied human livers.
Chronic liver diseases, and consequently liver fibrosis, represent a prevalent and growing public health problem. In the Western world specifically, non-alcoholic fatty liver disease (NAFLD) has become the leading cause of chronic liver disease, whereby its prevalence is increasing in parallel with increases in obesity, expected to encompass nearly 1 in 2 U.S. adults by 2030, and increases in type II diabetes.1–3 It is estimated that 25% of U.S. adults have some form of NAFLD, and an estimated 2–3% of the general population have non-alcoholic steatohepatitis (NASH), where steatosis is accompanied by necroinflammation and hepatocellular injury, thus predisposing patients to severe liver fibrosis, referred to as cirrhosis, and hepatocellular carcinoma.4,5
In addressing these chronic but dynamic diseases, researchers and clinicians are faced with both a limited ability to screen for certain liver diseases and challenges in monitoring the progression of the disease once it is discovered. Corroboration of the diagnosis requires verifying the degree of parenchymal inflammation and the extent of fibrosis.6,7 In preclinical animal studies, accurate staging of disease progression or regression requires sacrificing animals at multiple time points. In the clinic, despite recent advances with liver elastography, which is capable of noninvasively estimating liver fibrosis, liver biopsy remains the gold standard in determining both necroinflammation and fibrosis stage.8 These limitations are due to a lack of noninvasive longitudinal methods for monitoring the progression of inflammation and fibrosis.
It is thought that NAFLD progression is subject to multiple pathophysiological disruptions, with oxidative stress one of the key drivers in the progression of the disease.5,9 Liver injury and necroinflammation are mediated by reactive oxidative species (ROS), which drive fibrosis, but liver injury and necroinflammation are thus far only confirmed by liver biopsies.10,11 Here, we describe an imaging technique to assess necroinflammation activity and fibrosis stage noninvasively in a preclinical study to track the evolution of these chronic and highly dynamic diseases.
We investigate the signal of an endogenous autofluorescent pigment, lipofuscin, a byproduct of ROS, that is surprisingly bright when imagined using 808 nm excitation and detected in the near infrared (NIR, 700 – 1000 nm) and the shortwave infrared (SWIR, 1000 – 2000 nm) (NIR/SWIR detection). We show that NIR/SWIR imaging can detect and track the evolution of multiple liver pathologies in mouse models and in human tissue. Endogenous autofluorescent molecules, such as aromatic amino acids, lipopigments, NAD(P)H, and flavin coenzymes, can act as label-free reporters of the morphological and metabolic properties of cells and tissue.12–14. However, to date, most autofluorescence imaging modalities utilize ultraviolet radiation (UVA, 315 – 400 nm) or visible light (400 – 750 nm) for tissue excitation. The choice of 808 nm excitation enables nondestructive, noninvasive, in vivo detection of tissue autofluorescence, and as we demonstrate, high sensitivity to lipofuscin. Excitation with 808 nm light provides greater penetration depths through tissue and reduces the risk of cellular damage.15,16 Detection of NIR/SWIR autofluorescence beyond 850 nm further facilitates deep-tissue measurements due to greater tissue transmissivity, reduced light scattering, and lower interfering background signals.15,17–19 In this study, we first investigate NIR/SWIR autofluorescence in two mouse models of progressive liver fibrosis and determine that lipofuscin is the origin of the signal. Noninvasive monitoring of autofluorescence intensity with disease progression shows that lipofuscin can thus serve as an endogenous NIR/SWIR autofluorescent pigment. We further apply this finding to two additional mouse models of NAFLD and demonstrate that NIR-SWIR autofluorescence from lipofuscin is sensitive to both early and late stages of NAFLD. We finally show that biopsied human samples also exhibit NASH and NASH-cirrhosis related NIR/SWIR autofluorescence.
Results
NIR/SWIR Autofluorescence Induced by Oxidative Stress in CCl4 Model.
We investigated autofluorescence in two models of progressive liver fibrosis with distinct disease mechanisms.20,21 Hepatotoxin-induced liver fibrosis is achieved by administering carbon tetrachloride (CCl4), which is metabolized into free radicals and propagates into reactive oxygen species.22 CCl4 was administered in olive oil by oral gavage to three cohorts of wild-type (WT) mice three times per week for three, six, or eight weeks along with 5% ethanol drinking water ad libitum.23 Age-matched controls were administered olive oil by oral gavage and supplied with 5% ethanol drinking water (see Materials Methods). An obstructive cholestasis procedure was utilized for the second model to induce portal fibrosis.21 Common bile duct ligation (CBDL) surgery was performed in WT mice, which were then sacrificed four weeks after the procedure when secondary biliary cirrhosis is known to develop.24 Liver autofluorescence of each disease cohort was imaged in vivo through intact shaved skin, ex vivo in whole excised livers, and by microscopy imaging of formalin-fixed paraffin-embedded (FFPE) liver tissue slides.
We found that the liver autofluorescence signal of CCl4-treated mice is clearly distinguishable from age-matched control mice at all measured time-points. Using an InGaAs camera (900 nm – 1620 nm at −80ºC), SWIR autofluorescence from the fibrotic liver is sufficiently bright for real-time imaging (9.17 fps) in awake and freely behaving mice (Fig. 1a and Supplementary Video 1 and Material and Methods). Detecting NIR/SWIR autofluorescence wavelengths on a Silicon camera (850 nm – 1050 nm at −70ºC), likewise shows that the in vivo liver autofluorescence signal is approximately two-fold higher than in control mice after three weeks of CCl4 administration and also after six-weeks of CCl4 administration (Fig. 1b,c and Supplementary Table 1). Quantification of ex vivo liver autofluorescence suggests that the average signal intensity increases significantly relative to age-matched controls with a ratio between treated and control groups of 1.8 and 2.2 for three weeks and six weeks, respectively (Fig. 1d,e and Supplementary Table 1).
Fig. 1 |. Real-time and noninvasive imaging of quantifiable autofluorescent pigments in rodent liver with progressive fibrosis.

a, Autofluorescence (AF) image of a representative mouse from the carbon tetrachloride (CCl4) treated progressive fibrosis group (bottom, liver indicated by yellow arrow) with a representative mouse from the olive oil (OO) control group (top, liver indicated by white arrow) obtained noninvasively (abdominal hair removed) and in real time (9.17 frames per second), with mice awake and freely behaving (Movie S1). b, Reflectance image (top) and AF image (bottom) of a representative CCl4-treated mouse (left) and OO-treated mouse (right) after six weeks of their respective treatments with a yellow arrow indicting fibrotic liver and a white arrow indicating control liver. Scale bar, 2 cm. Images taken > 10 per cohort. c, Quantification of AF intensity of the liver region from in vivo images in counts per millisecond (cpms) of mice receiving no treatment (standard chow diet, SC) (white, n=6), control mice given OO by oral gavage (beige, n=4–5), and fibrotic mice given CCl4 in OO by oral gavage (maroon, n=4–5, slight offset, 3 wks: ***P=0.0001, 6 wks: *** P=0.0003). d, Reflectance image (top) and AF image (bottom) of excised livers after six weeks of CCl4 treatment (left) or OO treatment (right). Scale bar, 1 cm. Images taken > 10 per cohort. e, Quantification of excised liver AF with the same color scheme as (c), (n=4–6, 3 wks: ***P=0.00002, 6 wks: ***P<0.001). f, In vivo (n=5, **P<0.0055) and g, ex vivo comparison of liver AF intensity between pathologically normal mice on SC diet (white) and mice four weeks after a common bile duct ligation (CBDL) procedure (green) (n=5, *P<0.022). All graph data average intensities are indicated by a black bar, ± 1 s.d. indicated by scatter points. Statistical analysis: two-way ANOVA with post-hoc Tukey’s test for CCl4 data and one-way ANOVA with post-hoc Tukey’s test for CBDL data. Details in Supplementary Tables 1 and 2.
In the CBDL model of liver fibrosis, we find that liver autofluorescence of both in vivo and ex vivo measurements is on average slightly elevated compared to age-matched control mice that did not undergo the CBDL (Fig. 1f,g and Supplementary Fig.1a – c and Supplementary Table 1). However, the absolute liver autofluorescence signal in CBDL mice is substantially less pronounced than that of the CCl4 mouse model (Supplementary Fig.1a – c). We note further that in both CBDL and CCl4-treated mice, the bile duct and gall bladder were not autofluorescent in the NIR/SWIR imaging window, ruling out autofluorescent components of the bile (i.e. biliverdin/bilirubin) as the origin of the disease-associated NIR/SWIR autofluorescence signal (fig. S1A & S5).
We further investigated the autofluorescence signal through microscopy of liver tissue samples from the CCl4-treated and CBDL models and found that the disease-correlated autofluorescence signal can be detected in unstained, FFPE liver tissue (Fig. 2a,b and Supplementary Fig. 1e,f). All cohorts of the CCl4-treated mice showed significantly greater percent area of autofluorescence relative to their age-matched controls (~15-fold increase by six weeks of treatment in tissue slices), in parallel with increasing fibrosis, which was confirmed by Sirius Red staining (Fig. 2b,c).
Fig. 2 |. Quantification of lipofuscin/ceroid pigment by AF microscopy in progressive liver fibrosis model tissue.
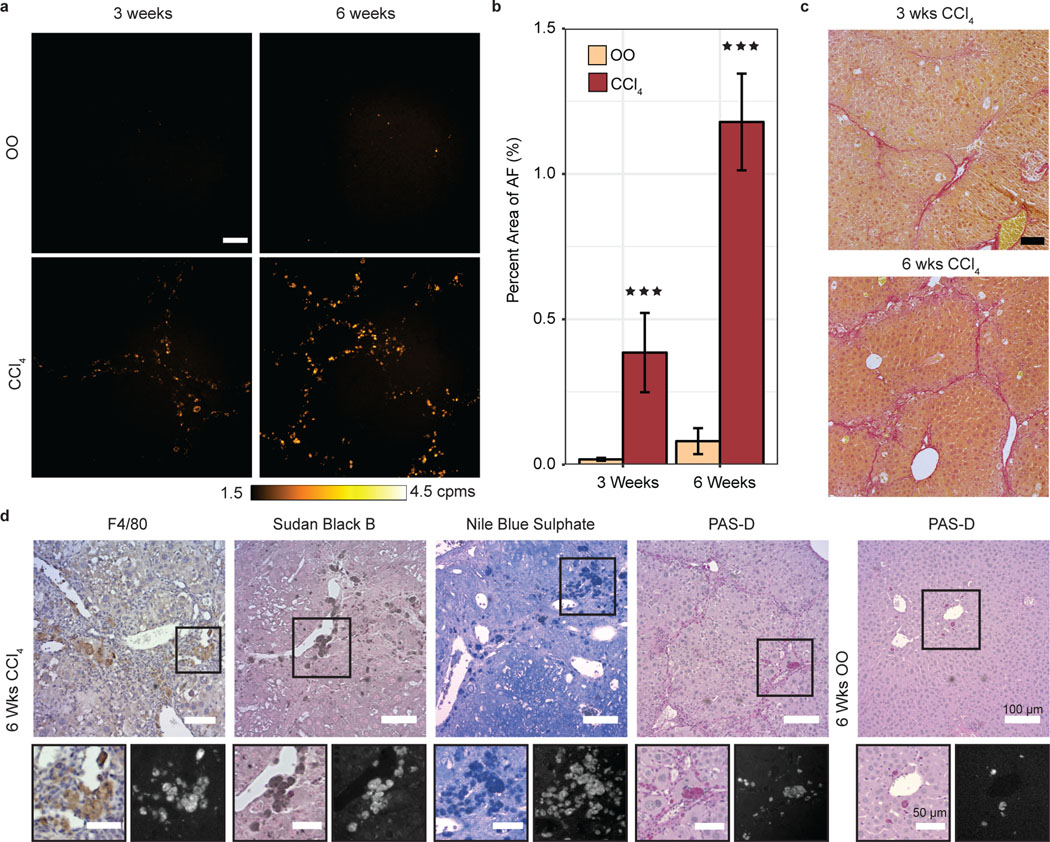
a, AF microscopy images of 10 μm slices of paraffin-embedded olive oil (OO) control liver tissue, and CCl4-treated mice liver tissue after three weeks or six weeks of treatment, represented with false color according to the color bar in counts per millisecond (cpms). Scale bar, 100 μm. b, Percent area of AF in OO-treated (beige, n=4–5) versus CCl4-treated (maroon, n=4–5, 3 wks: ***P=0.0009, 6 wks: *** P<0.001) mice tissue slices. c, Bright-field images of three weeks (top) and six weeks (bottom) CCl4-treated liver tissue, stained with Sirius Red. Scale bar, 100 μm. d, Bright-field and AF images of 5 μm slices of CCl4-treated mice tissue immunohistochemically stained with markers for F4/80, Sudan Black B, Nile Blue Sulfate, and PAS-D. Scale bars, 100 μm (zoomed region scale bars, 50 μm). Bar Plot: mean ± 1 s.d. Statistical analysis: two-way ANOVA with post-hoc Tukey’s test. Details in Supplementary Table 1.
During fibrosis development, the main source of collagen producing myofibroblasts differ between the CBDL model and CCl4-induced fibrosis and in CBDL, while there is some oxidative stress, it is not nearly as pronounced as in CCl4-induced liver fibrosis.20,22,24 RT–PCR analysis indicated high levels of alpha-smooth muscle actin (α–Sma), a marker for activated hepatic stellate cells (HSCs) and fibrosis at three weeks of CCl4 treatment (Supplementary Fig. 2a). Expression of the fibrotic gene Col1a1 was significantly pronounced in the CBDL and CCl4-treated cohorts compared to controls (Supplementary Fig. 2b). The inflammatory genes: Tnf-α, F4/80, Il-1b and CD11b markers were expressed in the CBDL cohort, while the CCl4-treated mice showed only elevated F4/80 and CD11b markers relative to age-matched controls (Supplementary Fig. 2c – f). These results confirm that both models led to the activation of fibrosis and inflammation in the liver.
Consistent with our in vivo and ex vivo imaging, there is a significant difference between CCl4-treated mice and the age-matched controls (Supplementary Table 1). While the CBDL tissue samples did have statistically significant percent area compared with control mice, the absolute signal difference was not as appreciable as in the CCl4 treatment, despite the presence of substantial fibrosis (Supplementary Fig. 1d and Supplementary Table 2). Relative to the CCl4 model, CBDL mice experience far less oxidative stress during the development of fibrosis.20 Hence, these results suggest that the oxidative damage pathway is an important factor in the enhancement of autofluorescence with liver disease progression, and point toward an autofluorescent pigment whose formation is linked with oxidation, such as the lipopigment lipofuscin (or ceroid).25,26
Lipofuscin NIR/SWIR Autofluorescence in CCl4-Induced Cirrhosis.
We then investigated the origin of the observed autofluorescence signal and its relationship with fibrosis and other tissue components involved in fibrosis through histological staining and autofluorescence microscopy of liver tissue from the CCl4 fibrosis model (see Materials and Methods). We found that the autofluorescence signal did not originate from the fibrosis itself but was closely associated with the fibrotic septa. The signal survives deparaffinization and mounting procedures using a variety of mounting media types and stains, indicating a robust complex resilient to both degradation and quenching (Supplementary Fig. 3a). We also stained for the macrophage marker F4/80, α–SMA, and for the ductular reaction marker cytokeratin 19 (CK19). The signal did not correlate with α-SMA or CK19 (Supplementary Fig. 3c,d), but the signal did exhibit a high colocalization with F4/80 (Fig. 2d and Supplementary Fig. 3e). NIR/SWIR positive cells in the F4/80 stains have a light brown appearance of the cell cytoplasm that is reminiscent of ceroid macrophages; this is even more pronounced in the α-SMA – F4/80 double stain, which reconfirmed autofluorescence colocalization with F4/80, but not with α-SMA (Supplementary Fig. 3e).27 As ceroid macrophages are known to be associated with liver injury and liver fibrosis, we subsequently explored the correlation of the autofluorescent pigment with three known stains for lipofuscin/ceroid lipopigments: Sudan Black B (SBB), Nile Blue A Sulphate (NBS) and periodic acid Schiff reaction after diastase (PAS-D).27–31 Our results show that the NIR/SWIR autofluorescence strongly correlates with NBS-positive, PAS-D positive, and SBB-positive regions of the tissue (Fig. 2d and Supplementary Fig. 3f – h). Autophagy is known to be able to affect lipofuscin formation.32,33 However we found that the autofluorescence signal did not overlap with p62 or LC3A/B stain, two markers of autophagy, indicating that the signal may not correlate with autophagy in this model (Supplementary Fig. 3i,j). Thus, we attribute the oxidative stress-correlated autofluorescence in the ex vivo and in vivo measurements of the fibrosis models to lipofuscin/ceroid.
To support this assignment, we further used autofluorescence microscopy to probe the spectral properties of the autofluorescent pigment. Lipofuscin and ceroid are terms often used interchangeably in the literature and refer to a lipopigment complex of undegradable polymers of oxidized proteins and lipids.26,32 The composition of this lipopigment complex is context-dependent and varies significantly, thus the pigment likewise has variable chemical properties.26,32 Consequently, lipofuscin/ceroid is known to have broad excitation and emission spectra. We found in the CCl4-induced model of liver fibrosis that indeed the NIR/SWIR autofluorescent pigment emits in many visible channels as well, consistent with the already known behaviour of lipofuscin/ceroid and lipofuscin-like pigments (Supplementary Fig. 4a,c and Supplementary Table 3).33,34 Moreover, we found that the autofluorescent pigment in the CCl4 model are quenched at visible wavelengths after SBB staining. SBB is a known quencher of tissue autofluorescence caused specifically by lipofuscin among other endogenous fluorophores, but we find that the lipofuscin signal is still detectable at NIR/SWIR wavelengths beyond the strong absorption maximum of SBB (Supplementary Fig. 4b,d).35
Our results demonstrate the feasibility of using NIR to excite lipofuscin autofluorescence for both ex vivo and noninvasive in vivo measurements. Traditionally, UV-Vis excitation is used to induce lipofuscin autofluorescence, however, these measurements are almost exclusively limited to ex vivo or surgically exposed tissue.36 For example, we show that a 660 nm visible excitation source, instead of 808 nm excitation, enhances signal from the gastrointestinal tract, which interferes with the quantification of the liver signal in vivo (Supplementary Fig. 5b). Imaging ex vivo liver samples with 660 nm excitation also produces a highly fluorescent gallbladder (attributed to bilirubin/biliverdin fluorescence), which dominates the signal from lipofuscin in the surrounding liver tissue (Supplementary Fig. 5c,d). Excitation with 808 nm light and NIR/SWIR emission detection, however, minimizes autofluorescence from background structures, resulting in an overall increased specificity to lipofuscin emission relative to using UV-Vis excitation. We note the quantification of the signal is relatively normalized; it is normalized per animal from its initial state prior to disease progression. A method of absolute normalization will require additional data beyond the scope of this study. Thus, NIR/SWIR imaging of lipofuscin can provide a quantitative and specific modality to track liver injury progression noninvasively.
Noninvasive Monitoring of CCl4-Induced Hepatic Injury Regression.
The ability to measure signal that correlates with disease progression has practical implications for diagnosis, disease staging, and monitoring. Additionally, it is desirable to be able to measure disease regression to assess response to treatment. In vivo studies of the CCl4-induced fibrosis model have shown that after discontinuing CCl4 treatment, activated HSCs are both cleared by apoptosis in addition to reverting to an inactive state. Macrophages also play a vital role in the resolution of both cellular and matrix components of the fibrotic response and gradually recede as well.37,38 This suggests that in this fibrosis model, the affiliated autofluorescent ceroid (residing in macrophages) could likewise regress, enabling tracking of the resolution of the fibrosis. We therefore investigated the behavior of the autofluorescence signal in response to fibrosis regression by discontinuing CCl4 treatment and allowing for recovery from CCl4-induced hepatic injury. We administered CCl4 for three weeks, followed by five weeks of recovery, during which time only olive oil was administered. This cohort was compared with mice that received CCl4-treatment for a total of three weeks (presented in Fig. 1) to show the extent of damage with CCl4, and eight-weeks to show the total duration of the disease, along with age-matched controls which received only olive oil (Fig. 3a). All mice additionally received 5% ethanol drinking water ad libitum for the duration of the study.
Fig. 3 |. Regression of liver fibrosis is accompanied by reversal of lipofuscin/ceroid AF signal.

a, Treatment schedule schematic for progression and regression groups, with CCl4 administered three times per week for three, eight, or for three weeks followed by five weeks of recovery. b, Reflectance image (top) and AF image (bottom) of eight weeks of CCl4 (left), eight weeks olive oil (OO) (control, middle), and three weeks of CCl4 followed by five weeks of recovery (regression) (right). Scale bar, 2 cm. Images taken > 10 per cohort. c, AF intensity values of the liver region from in vivo images of OO-treated mice (beige, n=4–8), continuously-treated CCl4 mice at three-weeks and eight-weeks of treatment (maroon, n=5–6, 3 wks: ***P=0.0001, 8 wks: *** P<0.001) and regression mice (yellow, n=8, ***P<0.001). P-values determined by age-matched controls. Average intensities are indicated by a black bar, ± 1 s.d. indicated by scatter points. d, Reflectance image (top) and AF image (bottom) of excised livers from mice receiving eight weeks of continuous CCl4 (top), eight weeks of OO (bottom-right), and regression mice (bottom-left). Scale bar, 2 cm. Images taken > 10 per cohort. e, AF intensity values of excised livers from the three cohorts with the same color scheme as (c), (n=4–8, 3 wks: ***P=0.00002, 8 wks: *** P<0.001) and regression mice (yellow, n=8, ***P<0.001). Average intensities are indicated by a black bar, ± 1 s.d. indicated by scatter points. f, Representative AF microscopy images of 10 μm paraffin-embedded liver tissue slices from OO, continuous CCl4, and regression cohorts. Scale bar, 200 μm. g, Representative liver tissue stained with Sirius Red after OO treatment, CCl4 treatment and regression treatment, respectively. Scale bar, 50 μm. h, Percent area of AF signal in images in (f) from OO treatment (beige, n=4–8), continuous CCl4 treatment (maroon, n=5–8, 3 wks: ***P = 0.0009, 8 wks: *** P<0.001) and regression cohort tissue (yellow, n=8, ***P=0.00001,). Bar Plot: mean ± 1 s.d. i, Percent area quantification of Sirius Red staining in (g) of OO treatment (beige, n = 4), CCl4 treatment (maroon, n=5, *** P<0.001) and regression treatment (yellow, n=8, *** P<0.001). Bar Plot: mean ± 1 s.d. Statistical analysis: two-way ANOVA with post-hoc Tukey’s test. Details in Supplementary Table 1.
We found that the autofluorescence intensity is significantly lower in mice that have undergone regression of liver fibrosis in comparison to three weeks or eight weeks of continuous CCl4-treatment, demonstrating reversal of the autofluorescence signal with disease regression (Fig. 3b,d). The in vivo autofluorescence of the regression cohort was similar to that of the mice receiving only olive oil, while the ex vivo whole liver autofluorescence signal exhibits a slightly higher residual autofluorescence compared to control groups, (Fig. 3c,e and Supplementary Fig. 6a and Supplementary Table 1). This difference is likely due to the attenuation and scattering of relatively low levels of autofluorescence by overlying tissue in the in vivo imaging.
Autofluorescence microscopy of unstained, FFPE liver tissue slides shows a similar trend as in the in vivo and ex vivo measurements. The percent area of autofluorescence signal in the regression cohort was markedly lower than mice receiving continuous three weeks or eight weeks of CCl4-treatment but did not fully decrease to the level of non-fibrotic controls (Fig 3f,h). Regression of the liver fibrosis after cessation of CCl4 was substantiated by hematoxylin and eosin (H&E) and Sirius Red staining, which indicated a significant reduction of collagen relative to mice with continued CCl4 treatment (Fig. 3g,i and Supplementary Fig. 6b,c). The relative decrease of autofluorescence in the regression cohort corresponds with a decrease in the number of fibrosis-associated macrophages, which are the source of the lipofuscin induced autofluorescence, as known from regression experiments in literature (Supplementary Fig. 6d).27,38–40 Furthermore, expression of fibrotic and inflammatory genes, α–Sma, Col1a1, and Il-1b, was decreased after regression compared to continuous CCl4 treatment for 8-weeks (Supplementary Fig. 2g,h,k). Although, we did not observe a significant difference in the transcriptomic expression of F4/80 between the CCl4 treatment group and the regression cohort as we identified at the protein level (Supplementary Fig. 2j and Supplementary Fig. 6d), we did, however, find that macrophage markers, CD11b and Ly6c, were reduced to similar levels as the olive oil control group (Supplementary Fig. 2l,m). Taken together, these results suggest that NIR/SWIR imaging of lipofuscin autofluorescence can provide a quantitative modality to track both the progression and regression of CCl4-induced hepatic injury.
High Sensitivity to Early Stages of CDAHFD-induced NAFLD in Mice.
The striking difference in autofluorescence observed between CCl4 and CBDL-induced fibrosis is likely related to their respective disease pathophysiology, which in the case of CCl4, highly depends on oxidative stress – the main driver of lipofuscin formation.26 Oxidative stress likewise plays an important pathophysiological role in NAFLD and therefore we investigated the applicability of our technique to a mouse model of NAFLD.41 We studied a diet-based NAFLD model, in which mice were fed either a control diet (CD) or a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) for three, six, nine, twelve, or fifteen weeks. After six to nine weeks of diet, CDAHFD induces early non-alcoholic steatohepatitis (NASH) with accumulation of fibrosis.41 We imaged the autofluorescence of the liver in vivo at the end of each diet period, directly followed by excision of the liver tissue for ex vivo imaging, and subsequent tissue processing.
We found that NIR/SWIR autofluorescence is indeed detectable at higher levels for both in vivo and ex vivo imaging in the CDAHFD mice compared with CD mice. After nine weeks of diet, the in vivo and whole liver ex vivo autofluorescence average signal intensity was 1.2 times and 1.6 times higher, respectively, than age-matched controls. By fifteen weeks of diet, the in vivo and whole liver ex vivo average signal intensities increased to 1.8 times and 2.8 times higher than controls, respectively (Fig. 4a – c and Supplementary Table 4). We observed during these experiments that the CDAHFD chow itself is autofluorescent, while CD chow is not, which could affect the observations in the liver due to its near proximity to the gastrointestinal tract (Supplementary Fig. 9a – d). However, quantification of the signal in FFPE liver tissue slices by autofluorescence microscopy, in which interfering chow fluorescence should not be a factor, also shows a steady increase of autofluorescence with time, and with progression of disease (Fig. 4d – f and Supplementary Fig. 7c). Moreover, stained CDAHFD liver tissue showed good correlation between F4/80, PAS-D, and SBB stains with the autofluorescent pigment, similar to the findings in the CCl4-induced fibrosis model (Supplementary Fig. 7d). We also noted in in vivo imaging that the male genitourinary anatomy, known to contain lipofuscin, frequently exhibited measurable autofluorescence as well (Supplementary Fig. 10a,b). Finally, other potential sources of competing signals were also explored or ruled out, such as bilirubin/biliverdin, blood samples, and brown adipose tissue (Supplementary Fig. 5c,d and Supplementary Fig. 10c,d).13,42,43
Fig. 4 |. Tracking lipofuscin AF with the progression of NAFLD in a high-fat diet mouse model.

a, In vivo AF images of mice with progressive NAFLD from three weeks through fifteen weeks of CDAHFD (liver indicated by yellow arrow). Scale bar, 2 cm. Images taken > 10 per cohort. b, Representative ex vivo liver AF images of mice in (a). Scale bar, 1 cm. Images taken > 10 per cohort. c, AF microscopy images of 10 μm paraffin-embedded liver tissue slices from the excised tissue in (b). Scale bar, 100 μm. d, Quantification of liver AF intensity of CDAHFD (maroon, n=4–8, 3 wks – 9 wks: NS, 12 wks: ***P=0.00002, 15 wks: *** P<0.001) measured in vivo through intact skin as a percentage of the age-matched control cohort average at each time point. Average intensities are indicated by a black bar, ± 1 s.d. indicated by scatter points. e, AF intensity values for excised tissue represented as a percentage of age-matched control averages (n=5–8, 3 wks – 15 wks: *** P<0.001). Average intensities are indicated by a black bar, ± 1 s.d. indicated by scatter points. f, Liver tissue percent area quantification relative to control average (n=6, 3 wks – 9 wks: NS, 12 wks: *** P<0.001, 15 wks: *** P<0.001). Bar Plot: mean ± 1 s.d. Statistical analysis: two-way ANOVA with post-hoc Tukey’s test. Details in Supplementary Table 4.
Histological review of liver sections from each diet and control cohort confirmed that CD mice had normal livers, while CDAHFD mice had severe steatosis from three weeks onwards, with the development of steatohepatitis and fibrosis (based on H&E and Sirius Red stains) beginning at twelve weeks of diet (Supplementary Fig. 7e,f and Supplementary Table 9 and Supplementary Table 10). As expected, the overall quantity of the autofluorescence signal, as well as the occurrence of the respective stain, were lower than the CCl4-induced fibrosis model, and the lipofuscin structure and distribution in early stages of NAFLD is unlike the CCl4-induced fibrosis model where ceroid macrophages are abundant. While there is some accumulation of signal in macrophages, lipofuscin accumulation is mainly seen in the hepatocytes in this model, with punctate structure following the build-up of pigment in the hepatocellular cytoplasm (Fig. 4c). This pattern is similar to age-related lipofuscin and the clinical samples shown in a section below.25,44
Analogous to the regression experiments with the CCl4-induced fibrosis model, we studied a regression cohort of NAFLD model mice, which after nine weeks of CDAHFD diet was switched to CD for six additional weeks (Supplementary Fig. 7a – f). Reversal was histologically evidenced in our experiment by the regression of the steatohepatitis in the livers of these mice (Supplementary Fig. 7g – i and Supplementary Table 9 and Supplementary Table 10). We found that the in vivo, whole liver ex vivo, and tissue slices autofluorescence levels are comparable to those of the nine-weeks CDAHFD only cohort (Supplementary Fig. 7a – f).
The lack of full autofluorescence reversal in the NAFLD model can be explained by the hepatocellular localization of lipofuscin in NAFLD, which unlike ceroid macrophages in the CCl4-induced fibrosis model, do not rapidly clear once the injury-inducing treatment is terminated. The tendency for higher levels of autofluorescence (compared to nine-weeks of CDAHFD) can be regarded as the effect of weaning lipofuscinogenesis (i.e. residual but weakening lipofuscin driving processes) once the diet was switched. Hence, in the CDAHFD/CD regression, the signal rather reflects the cumulative amount of oxidative stress, hence the ‘biological age’ experienced thus far by the liver parenchyma in this liver disease (Supplementary Fig. 7h – i).32
Gene expression associated with liver inflammation and fibrosis (α–Sma, Col1a1, and F4/80) remained elevated in CDAHFD mice over the course of study from six weeks to fifteen weeks compared to age-matched CD cohorts (Supplementary Fig. 8a – d). In the regression cohort, marker expression was attenuated to the level of CD mice (Supplementary Fig. 8a – d). Consistent with histological staining, gene expression confirms both progression of liver injury and regression of the liver injury after stopping CDAHFD, consistent with autofluorescence imaging.
Enhanced Signal in Advanced Stages of NAFLD in F-IR/IGFRKO Model.
To further investigate the utility of autofluorescence in monitoring liver disease, we explored a model of NAFLD that develops in the setting of generalized lipodystrophy. In this setting, NAFLD develops as a result of a specific knockout in adipose tissue of the insulin receptor and insulin-like growth factor 1 receptor (F-IR/IGFRKO) independent of diet.45
We imaged H&E stained liver tissue of wild-type (WT) and F-IR/IGFRKO mice at 52-weeks of age under 808 nm excitation and find that F-IR/IGFRKO mice exhibit high levels of autofluorescence in the NIR/SWIR region with respect to age-matched WT mice (Fig. 5a). We quantified the autofluorescence signal by percent area in paraffin-embedded samples of WT and F-IR/IGFRKO mice liver tissue, similar to the method used for CCl4 and CDAHFD experiments, and found a 12-fold increase in percent area of the autofluorescence signal in the F-IR/IGFRKO mice relative to WT controls (Fig. 5b and Supplementary Table 5). The severity of liver fibrosis was assessed by Sirius Red staining (Fig. 5c), and assignment of the autofluorescence to lipofuscin was verified using the same set of stains (F4/80, PAS-D and SBB) as in the previous two mouse models (Supplementary Fig. 11a and Supplementary Table 9 and Supplementary Table 11).
Fig. 5 |. Significant AF signal and fibrosis in a lipodystrophy mouse model.

a, Representative images of H&E stained liver tissue slices (10 μm) from 52-week-old wild type (WT) (left) and F-IR/IGFRKO (right) mice, shown with AF images (bottom) of the same tissue slices. Scale bar, 100 μm. b, Average percent area of AF signal among WT (beige, n=4) and F-IR/IGFRKO (maroon, n=5, ***P=0.0001) mouse liver tissue. Average intensities are indicated by a black bar, ± 1 s.d. indicated by scatter points. c, Comparison of Sirius Red staining for fibrosis in WT and F-IR/IGFRKO mice liver tissue. Scale bar, 200 μm. Statistical analysis: one-way ANOVA with post-hoc Tukey’s test. Details in Supplementary Table 5.
These findings are substantiated by known characteristics of this model, in which hepatocyte balloon degeneration is indicative of progressive NAFLD, and by 12-weeks of age, increased levels of reactive oxygen species and lipid peroxidation are evident, which could account for the increased autofluorescence signal when aged to 52-weeks.26,45 Thus, the lipodystrophy model additionally suggests that increasing autofluorescence with the progression of NAFLD arises from lipopigment molecules, which accumulate due to oxidative stress-related hepatocyte injury as NAFLD progresses. Similar to the CDAHFD study, the accumulation of lipofuscin has punctate structure and is mainly localized in the hepatocytes, contrasting the fibrosis-associated ceroid macrophages as seen in the CCl4-induced fibrosis model (Fig. 4c and Fig. 2a, respectively).
The results of this model, in addition to the lipopigment staining and the CDAHFD-induced NAFLD results, substantiate that indeed the enhanced signal in NAFLD arises as a consequence of the disease progression, with higher levels of autofluorescence with increasing histological severity.
Human NASH Liver Tissue Exhibits NIR/SWIR Autofluorescence.
In patients, NAFLD has a highly variable natural history, exhibiting vastly different rates of progression among individuals, as well as diverse clinical manifestations due to the multiple pathways involved in the pathogenesis.5,46 Moreover, the pathogenic drivers of NAFLD are not likely to be identical between patients, further complicating assessment of steatohepatitis and resulting fibrosis.46,47 We investigated the potential for extending our NIR/SWIR autofluorescence imaging technique to assess human liver samples.
We imaged liver biopsy tissue obtained from 17-patients, with ranging histories and stages of fibrosis (Fig. 6a and Supplementary Table 6). We found that indeed NIR/SWIR autofluorescence is detectable in the human liver biopsy samples (Fig. 6b). Staining for lipofuscin followed the results of the mouse models, with PAS-D and SBB showing correlation with autofluorescence (Supplementary Fig. 12a – c). While some of the signal resides within the macrophages (Kupffer cells), the majority of the signal arises from punctate structures in hepatocytes, as observed in the mouse models (Fig. 4c and 6b).
Fig. 6 |. Liver injury in humans with restructuring of lipofuscin distribution and quantity.

a, Masson’s trichrome strained liver samples from a patient with NASH (top) and NASH-cirrhosis (bottom). Steatosis is widespread in the NASH sample; black arrow indicates remnant steatosis in cirrhotic tissue. Scale bar, 100 μm. b, AF signal detected in NASH patient (top) relative to patient with cirrhosis of the selected area in (a) in an unstained liver tissue (5 μm). Scale bar, 50 μm. Details in Supplementary Table 7.
We then quantified the autofluorescence signal by determining the percent area, using the same methods as in the mouse experiments (Supplementary Fig. 12f,g and Supplementary Table S7). To evaluate how the autofluorescence signal corresponds to the amount of necro-inflammatory activity and the degree of fibrosis, we evaluated the liver biopsy samples using the Brunt criteria for grading and staging (Supplementary Table 6 and Supplementary Table 7)48. Briefly, NASH severity (or NASH grade) was based on the degree of steatosis, extent of hepatocyte ballooning, and the amount of intralobular and portal inflammation. Fibrosis stage was based on the extent and location of fibrosis in the Masson’s Trichrome stained samples (Fig. 6a and Supplementary Fig. 12d,e).
Our preliminary data reveals high autofluorescence in patients with non-cirrhotic NASH and decreased signal in highly fibrotic tissue (Supplementary Fig. 12f). When comparing NASH grade, patients with milder grades of NASH had significant autofluorescence followed by a decrease in signal at moderate and severe grades (Supplementary Fig. 12g). Of note, patients with moderate grade NASH (grade 2) in this study are also cirrhotic (fibrosis grade 4), hindering a clear interpretation of signal to NASH grade and disease severity.
Furthermore, complications arise when comparing to the mouse models of fibrosis, whereby high levels of autofluorescence are exhibited as progression of the liver disease tends toward cirrhosis (Fig. 2a – c and 3f – h). This may be explained by noting that biopsied samples are relatively more fibrotic in the liver parenchyma compared to mouse liver tissue (Fig. 5c and Supplementary Figs. 1d, 6c, 7h, 12d). Additionally, the occurrence of newly formed regeneration nodules is present in human tissue leading to less lipofuscin containing cells and in turn less NIR/SWIR autofluorescence (Supplementary Fig. 12d). In contrast to mouse models, in human biopsy samples, patients with cirrhosis exhibit a larger variety of steatosis and necroinflammation, complicating quantification of the autofluorescence signal due to high variance within our small sample size (Supplementary Fig. 12h – k and Supplementary Table 6 and Supplementary Table 7).
With this human study, we have confirmed the feasibility to detect lipofuscin accurately in human tissue samples with a potential to discern disease stages. However, further studies are required to obtain granularity in distinguishing among intermediate disease stages, as well as determining the pathological drivers of lipofuscin accumulation with respect to steatosis, ballooning, and inflammation in human samples.
Discussion
In conclusion, we demonstrate enhanced NIR/SWIR autofluorescence signal in a variety of animal models of chronic liver disease, sufficiently bright to be detected noninvasively in vivo, and with sufficient contrast to delineate between abnormal and healthy biological structures. We attribute the NIR/SWIR autofluorescence signal in these models to the lipopigment lipofuscin (also known as ceroid). The intracellular accumulation of lipofuscin-like pigments can be found as a consequence of aging, of cumulative oxidative stress, as a response to pathological conditions, or to toxic compounds. Based on our results that NIR/SWIR autofluorescence can measure accumulation of lipofuscin/ceroid, we suggest that this technique can aid investigations into mechanisms of storage diseases that accumulate lipofuscin, mechanisms of senescence and/or serve as a diagnostic/prognostic medical tool. The possibility to monitor samples directly and in real-time is one of the most attractive features of autofluorescence-based techniques. Indeed, longitudinal animal studies can be carried out without the need for sacrificing animals at multiple time points. We show here that indeed we can detect the stage of disease in animal models in real time, and suggest that the NIR/SWIR wavelengths can open a new window for autofluorescence imaging at all length scales, from in vivo imaging down to microscopy of cellular and sub-cellular structures. Furthermore, we have shown that NIR/SWIR autofluorescence can distinguish between non-cirrhotic NASH and patients with NASH-cirrhosis opening the door for further studies of this complex and evolving field.
Methods
Animal care and study approval.
Animal experiments were conducted in accordance with the approved Massachusetts Institute of Technology and Massachusetts General Hospital institutional protocols following the Public Health Service Policy on Humane Care of Laboratory Animals. Human NASH and cirrhosis samples were obtained from Brigham and Women’s Hospital (BWH) under approved institutional protocols (IRB Protocol #2018P000924). F-IR/IGFRKO tissue samples were received from Joslin Center for Diabetes and collected under protocol approved by the Joslin Diabetes Center Institutional Animal Care and Use Committee and were in accordance with National Institutes of Health guidelines.
Animal Treatment Procedures.
CCl4-induced fibrosis progression and regression studies:
Adult wild type, male C3Hf/Sed mice of 8–12 weeks of age, were administered 20% CCl4 (Sigma-Aldrich; 289116) in olive oil via oral gavage (200 μL) three times per week, or were administered just olive oil in the case of the respective controls, for three, six, or eight weeks (Supplementary Table 1). Both groups were supplied with 5% ethanol drinking water ad libitum.22,23 Mice were imaged and sacrificed after 24 or 72 hours of their last CCl4 administration.
For the regression study, mice were administered 20% CCl4 in olive oil via oral gavage (200 μL) three times per week for three weeks then allowed to recover for five weeks receiving only olive oil (200 μL). All mice were supplied with 5% ethanol drinking water ad libitum for the duration of the study. Mice were imaged and sacrificed within 72 hours after their final treatment.
Common Bile Duct Ligation (CBDL) surgery:
The CBDL surgical procedures were performed under sterile conditions. Adult wild type, male C3Hf/Sed mice of 8–12 weeks of age were placed under ketamine-xylazine anesthesia and a midline abdominal incision was made whereby the common bile duct (CBD) was identified. The CBD was occluded with a double ligature using a non-resorbable suture. The first suture was placed beneath the junction of the hepatic ducts and the second suture was made at the entrance of the pancreatic duct, thereafter sectioning the CBD in between the two ligatures.24
Control mice for reference to the CBDL mice cohort were on a standard chow (SC) diet (VAC-PAC RODENT 6/5# IRRADIATED 5066) with no other changes to diet nor supplied any type of treatment.
Non-alcoholic Fatty Liver Disease (NAFLD) progression and regression study:
Adult wild type, male C3Hf/Sed mice of 8–12 weeks of age were fed a control diet (CD) or a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) (Research Diets; A06071314i and A06071302i) 41. CD and CDAHFD treatments were ended at three, six, nine, twelve or fifteen weeks, after which mice were imaged and sacrificed.
For the regression study, similarly, adult wild type, male C3Hf/Sed mice of 8–12 weeks of age were fed a CDAHFD for nine weeks followed by a CD for six weeks. These mice were then imaged and sacrificed.
Lipodystrophy mice study:
Fat-specific insulin receptor and insulin-like growth factor 1 receptor knockout mice (F-IR/IGFRKO) were generated via the Cre-LoxP recombination technique as described in 45. This model leads to generalized lipodystrophy whereby mice exhibit diabetes, hyperlipidemia and NAFLD, including development of NASH and fibrosis by 52-weeks.
All mice had ad libitum access to water and SC. Mice liver tissue samples were donated to address the possibility of enhanced fluorescence with lipodystrophy and NAFLD progression in this model. Tissue sections of liver fixed to glass slides and stained with Haemotoxylin and Eosin (H&E) or Sirus Red were used this study (10 μm).
Animal tissue fixation and staining.
We performed a suite of histological stains on formalin fixed paraffin-embedded (FFPE) tissue sections (5 μm) to determine the correlation of the NIR/SWIR autofluorescent pigment with three known stains for lipofuscin lipopigments: Nile Blue A Sulphate (NBS), periodic acid Schiff reaction after diastase (PAS-D), and Sudan Black B (SBB).28–31,49 We also tested for correction of the autofluorescent signal to fibrosis with Sirius Red, activated hepatic stellate cells (HSC) with immunohistochemistry for α-smooth muscle Actin (α-SMA), macrophages with immunohistochemistry for F4/80, ductular reaction with immunohistochemistry for cytokeratin 19 (CK19) and autophagy with immunohistochemistry for p62 and LC3A/B.
For the NBS stain, deparaffinized sections (using xylenes and ethanol) were stained for 20 minutes in NBS at room temperature, prepared with 0.05% Nile Blue A (Acros Organics; 415690100; Lot number A0377281) in 1% sulfuric acid. Sections were then rinsed in running water for 15 minutes and no counterstain was performed. A liver tissue sections subset was alternatively rinsed in 1% sulfuric acid followed by four changes of absolute acetone to remove most of the NBS, and hence destain the NBS-positive lipofuscin/ceroid in the slide. Finally, another subset was restained with SBB (Sigma-Aldrich; 199664; Lot number MKBX9860V) after NBS-destaining with acetone to confirm specificity of lipofuscin/ceroid positivity.
For PAS-D, deparaffinized sections were incubated in 0.2% malt diastase (MP Biomedicals; ICN10153925) in a phosphate buffer (PBS) for 1 hour at 37˚C. After washing, they were subsequently oxidized with 1% periodic acid for 5 minutes and incubated with Schiffs reagent (Electron Microscopy Sciences; 26052–05) for 20 minutes at room temperature. Counterstain was performed with Gills haematoxylin #2.
For the SBB stain, deparaffinized sections were incubated in 0.7% SBB in 70% ethanol for 6 minutes, then rinsed quickly in 50% ethanol solution and subsequently counterstained with nuclear fast red.
The extent of fibrotic tissue development was determined by staining mouse tissue with Sirius Red. Deparaffinized tissue sections were countered stained with Gill’s Hematoxylin #2, washed with tap water, followed by staining with Scott’s bluing agent and re-rinsed with tap water. The sections were stained with Sirius Red (EMS solution; 26357–02) for one-hour at room temperature, washed with 5mL glacial acetic acid in 1L of water, and then rinsed with 100% ethanol followed by xylenes before mounting.
To verify the extent of fibrosis and determine if the autofluorescence signal regions correlate with the development of activated HSC, deparaffinized sections were incubated with DAKO regular Ag retrieval solution, followed by washing with PBS, and quenching of endogenous peroxidase by placing slides into 3% v/v hydrogen peroxide and water. Sections were washed and incubated in mouse on mouse (M.O.M, Vectastain; BMK-220) blocking reagent for one hour, rinsed and the primary antibody (anti-α-SMA antibody, DAKO, M0851 (Clone 1A4); 1/1000) was applied in dilute M.O.M overnight. The sections were then re-washed with PBS, labeled with anti-mouse polymer-HRP, washed and stained with 3,3’-diaminobenzidine (DAB) under the microscope. The reaction was stopped with distilled water, counter stain was applied, and slides were mounted.
Macrophages/monocytes were detected using an anti-F4/80 antibody. Deparaffinized liver sections were incubated with DAKO regular Ag retrieval solution followed by washing with PBS after cooling, and subsequent quenching of endogenous peroxidase by placing slides into 3% v/v hydrogen peroxide and water. Sections were washed and incubated in 3% v/v bovine serum albumin (BSA) in PBS for one-hour at room temperature before washing and exposing the sections to the primary antibody (Serotec, MCA497BB; 1/100 in BSA) overnight at 4ºC. The processed sections were then re-washed and incubated with avidin–biotin complex followed by washing and stained with DAB. The reactions were stopped with distilled water, counter stain was applied, and slides were mounted. In case of double staining F4/80 and α-SMA, after incubation with DAB, reaction was stopped with distilled water, slides were washed in PBS and incubated with a second primary antibody (Sigma-Aldrich, A569; 1/100 in 3% v/v BSA) for 2 hours at room temperature. The processed sections were re-washed and stained with Perm-Red under the microscope. The wash stopped in with distilled water, counter stain was applied, and slides were mounted.
We checked for ductular reaction using an anti-CK19 antibody. Deparaffinized liver sections were incubated with DAKO regular Ag retrieval solution, cooled, washed with PBS and exposed with 20 uL/mL Proteinase K. The sections were re-washed with PBS, quenched with 3% v/v hydrogen peroxide and water, and rewashed all at room temperature. Five-percent v/v normal donkey serum (NDS) in PBS was incubated with the sections for one-hour at room temperature before washing and exposing the sections to the primary antibody (DSHB; TROMA III:1/1000 in NDS) overnight at 4ºC. The processed sections were then re-washed, received a second application of rat-antibody (1/200 in NDS), washed, then incubated with avidin–biotin complex followed by washing and stained with DAB. The reactions were stopped with distilled water, counter stain was applied, and slides were mounted.
We checked for autophagy using the markers LC3A/B and SQTM1/p62. Deparaffinized liver sections were incubated with DAKO regular Ag retrieval solution followed by washing with PBS and quenching of endogenous peroxidase by placing slides into 3% v/v hydrogen peroxide and water. Sections were washed and incubated in 5% v/v NDS in PBS for one-hour at room temperature before washing and exposing the sections to the primary antibody (Cell Signaling; 12741 (clone D3U4C): 1/100 in NDS; alternatively, Sigma-Aldrich; p0067: 1/200 in NDS) overnight at 4ºC. The processed sections were then re-washed, received a second application of rat-antibody (1/200 in NDS), washed, then incubated with avidin–biotin complex (ABC) followed by washing and stained with DAB. The reactions were stopped with distilled water, counter stain was applied, and slides were mounted.
Real-Time Polymerase Chain Reaction (RT-PCR).
Quantitative RT- PCR was completed for NAFLD for six to fifteen weeks of treatment for selected genes related to fibrosis, and inflammation. Similarly, for four weeks of CBDL along with three, six, eight-weeks of CCl4 treatment and the regression cohort. (Supplementary Fig. 2 and Supplementary Fig. 8). Selected genes include Acta2 (α–Sma), Col1a1, Tnf–α, F4/80, Il-1b, CD11b and Ly6c.
Total RNA was isolated from liver tissue homogenates using the RNeasy minikit (Qiagen; 74106). cDNAs were synthesized using iScript RT Supermix (Bio-Rad; 170–8841). Subsequently, relative gene expression was determined using Real-Time SYBR Green PCR Master Mix (Applied Biosystems) on a Stratagene Mx3000P QPCR System. The fold change in gene expression, compared to zero weeks of diet, was calculated using the comparative threshold cycle method, which was normalized to HPRT as a reference gene. The genes and respective primer sequences can be found in Supplementary Table 8.50
Patient Population and Data Collection.
This study was approved by the Institutional Review Board at the BWH. Human NAFLD liver biopsies were identified through the surgical pathology files at the BWH, Boston, MA, USA. All patient samples were de-identified prior to the study. Patient records at the time of the liver biopsy were reviewed for age, gender, body mass index (BMI), serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), history of metabolic syndrome, alcohol use, and liver disease(s). Exclusionary criteria included excessive alcohol intake (> 30 g/day for males and > 20 g/day for females), other known liver diseases, or decompensated cirrhosis. After application of the exclusion criteria, 17 liver biopsy specimens were included in the study.
Human tissue fixation and staining.
Liver biopsy specimens were fixed in formalin and embedded in paraffin. Sections (5 μm) were stained with H&E, Masson’s trichrome, Sirius Red, Sudan B Black, and PAS-D at BWH following standard hospital protocols.
Human Liver Biopsy Grading and Staging
Liver samples were blindly assessed by two pathologists according to the Brunt scoring.48 Briefly, the degree of macrovesiculair steatosis, necroinflammation were separately scored and a joint score was assigned to express the degree of NASH severity along with the degree of fibrosis.
Steatosis was scored based on the percentage of macrovesicular steatosis: 0 was assigned <5%; 1 point 5–33%; 2 points 33–66%; 3 points >66%. Lobular inflammation was scored as number of foci counted in 20x fields: 0 was assigned for none; 1 point was assigned for <2 foci; 2 points for 2–4 foci; 3 points for >4 foci. Portal inflammation was scored as the density of inflammatory cells in the different portal tracts: 0 was assigned for none; 1 point was assigned for mild; 2 points for moderate; 3 points for severe. Ballooning degeneration was scored by the severity based on numbers of hepatocytes: 0 points assigned as none, 1 point as mild, 2 points as marked.
Each component was used to assign a grade into mild, moderate, and severe NASH categories: mild/grade 1 was up to 66% steatosis in the liver biopsy, none or few ballooning hepatocytes, scattered intralobular inflammation and no or mild portal inflammation; moderate/grade 2 was any degree of steatosis, presence of ballooning hepatocytes, intralobular inflammation and mild/moderate portal inflammation; severe/grade 3 was panacinar steatosis, marked ballooning, and the presence of intralobular and portal inflammation.
Finally, fibrosis grades were defined as the following: stage 1 – zone 3 perisinusoidal/pericellular fibrosis, focal or extensive; stage 2 – zone 3 perisinusoidal/pericellular fibrosis, focal or extensive portal fibrosis; stage 3 – perisinusoidal/pericellular fibrosis, portal fibrosis and bridging fibrosis; stage 4 – cirrhosis.
Autofluorescence Imaging
In vivo imaging:
Prior to imaging, mice were anesthetized via intraperitoneal injection of ketamine/xylazine (100 mg/kg ketamine and 10 mg/kg xylazine in saline) and sufficient depth of anesthesia was maintained, re-dosing with one third the original dose in ketamine only as necessary. Mice were shaved with electric clippers and residual hair was removed using Nair™, applied for approximately 3 minutes.
Animals were then imaged in vivo using two configurations of a single custom-built imaging setup. Coupling a 10 W 808 nm laser (Opto Engine MLL-N-808) with a 910 μm core metal cladded multimode fibre (Thorlabs MHP910L02), the laser output was passed through a ground-glass plate (Thorlabs DG20–220-MD) to provide uniform illimitation over the imaging area. The excitation flux was determined to be roughly 45mW cm−2, for all in vivo imaging experiments. A four-inch square silver mirror (Edmund Optics, 84448) projects the image onto a Navaitar objective, from above, (Navitar MVL50M23, F2.8/50 mm EFL) passing through two-colored glass 850 nm longpass filters (Thorlabs, FGL850S) followed by a single 850 nm dielectric longpass filter (Thorlabs, FELH0850) attached to a cooled Princeton Instruments PIXIS 1024BR camera (−70ºC, 250 nm – 1050 nm).
A single image was taken for each mouse using room light illumination with 500 ms integration time to ensure position of the mouse. The sample was then illuminated with 808 nm excitation as described above and the camera was set to 1000 ms integration time for autofluorescence image acquisition. For movie frame rate imaging, imaged from below, we used an InGaAs camera from Princeton Instruments NIRvana 640 (−80ºC, 10MHz, high gain, 900 – 1620 nm) camera was used with a SWIR corrected 35mm Navaitar objective (Navitar SWIR-35, F1.4/35 mm EFL) along with stacked, two colored glass 850nm and two colored glass 1000 nm, longpass filters. Mice from the CCl4-treated cohort (six weeks of treatment) along with respective control mice (six weeks of olive oil treatment) were imaged awake and freely moving in a plastic cage. The same excitation source was used for movie rate imaging, although the excitation flux was adjusted to 25 mW cm-2.
Ex Vivo imaging:
Subsequently following in vivo imaging, the mouse liver was extirpated and weighed. The liver was imaged in its entirety using the PIXIS camera with the same setup as described above. The left liver lobe was used for histology, while the remaining portion of the liver was snap-frozen in liquid nitrogen and stored at –80˚C until further processing. Finally, the inter-scapular brown adipose tissue (BAT), the gonadal white adipose tissue (as an indicator for visceral fat mass) and the inguinal white adipose tissue (as an indicator for subcutaneous fat mass) were identified, dissected, and snap-frozen.51
A single image of the liver was taken for each mouse using room light illumination with 500 ms integration time to ensure position of the whole liver. The sample was then illuminated with 808 nm excitation as described above and the camera was set to 1000 ms integration time for autofluorescence image acquisition.
FFPE Tissue Slice Microscopy (autofluorescence microscopy):
A Nikon Ti-E inverted microscope equipped with a Stage UP Kit (Nikon) and a backport adaptor was used for tissue imaging. A fiber-coupled 808 nm excitation source (described above) was attached to the backport adaptor along with a laser speckle remover (Edmund Optics, 88–397). Excitation was filtered through a 850-nm dichroic filter (Thorlabs, FEHL0850) to direct the excitation light to the sample and a 900 nm longpass filter (Thorlabs, FELH0900) to select the emission light. Imaging was performed using a 10× objective (Nikon CFI Plan Apo Lambda) magnified by 1.5× by the microscope optics. The PIXIS camera was attached to the side port of the microscope.
Tissue samples were fixed and mounted as described above for all disease and treatment cohorts. Five autofluorescence images with 5000 ms integration time were taken for each sample tissue with 808 nm illumination and used for quantification. Five images with 5000 ms integration time were taken for each sample tissue and used for quantification. Bright field imaging was completed using a Slide scanner on a Zeiss Axioscan.Z1 microscope and using the ZEN2 software package.
Autofluorescence quantification:
16-bit images were processed and quantified using FIJI (ImageJ v.1.5).52 For in vivo quantification, the position of the liver was anterior to that of the stomach, thus a specified region of interest (ROI) was selected and used for all mice. The intensity value was determined by using the ‘Measure’ function on FIJI to calculate the mean grey value for the specified area. Similarly, for the ex vivo quantification, a predefined ROI was selected (avoiding artifacts including reflections), and used on all images, followed by determination of the mean grey value for the selected area. Data points were omitted from the calculated averages in a few instances due to autofluorescent artefacts in the gastrointestinal tract located within close proximity to the liver or due to black skin pigment overlying the liver that inhibited measuring the in vivo liver intensity values (See Supplemental Tables for excluded images).
Autofluorescence quantification of the FFPE tissue slices was completed by first thresholding the entirety of the image converting the 16-bit image to an 8-bit image, followed by binary selection and percent area determination using the “Analyze Particles” function on FIJI. Thresholding was kept consistent through treatment, diet, and control tissue, adjusting only if background counts significantly changed (> 500–1000 counts). Five images, taken at randomly chosen, distinct positions across a given tissue slice, were averaged to obtain a representative value for each sample. For human tissue, averages of three images per sample were used to calculate percent area, due to the limited sample area of the biopsied liver.
Statistics
Two-way or one-way ANOVA was completed for each disease, treatment, or control sample (in vivo, ex vivo, and tissue) to determine statistical significance (P-Value) on RStudio. For two or more factors, Tukey HSD was performed in order to determine the statistical significance of the particular categorical interactions. P-values, equations, and detailed interactions are reported in Excel ANOVA Data Set tabs one through seven and in supporting data Supplementary Tables 1 – 5 and Supplementary Table 7. Data analysis and plots were created using RStudio (v1.1.456).
RT-PCR statistics were calculated using the same procedure as above using SPSS (version 24; IBM SPSS Statistics, Armonk, NY). ANOVA details are reported in Excel ANOVA Data Set tabs eight and nine.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are too big to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request.
Supplementary Material
Table 1 |.
Experimental Summary of In Vivo, Ex Vivo Whole Liver, and Tissue Data for Each Study Group
OO: Olive oil
CCl4/OO: Carbon Tetrachloride/Olive oil
CBDL: Common bile duct ligation
CD: Control diet
CDAHFD: Choline-deficient L-amino acid high-fat diet
WT: Wild type
F-IR/IGFRKO: fat specific -insulin receptor and insulin-like growth factor 1 receptor knock-out
NASH: non-alcoholic steatohepatitis
Acknowledgements
Acknowledgements should be brief, and should not mention anonymous referees or editors, or include effusive comments. Grant or contribution numbers may be included.
We thank Sylvie Roberge for excellent technical support and Thomas Reiberger for early discussions.
Funding: This work received support, in part, from the National Institutes of Health through the Laser Biomedical Research Center, Grant 9-P41-EB015871-26A1 (to M.G.B), and the US Army Research Office through the Institute for Soldier Nanotechnologies at MIT, under Cooperative Agreement W911NF-18-2-0048 (to M.G.B.). M.S. was supported by National Science Foundation Graduate Research Fellowship Program under Award number is #1122374. J.A.C. was supported by a National Defense Science and Engineering Graduate Fellowship 32 CFR 168a. K. vL., Y.Z., and M.G.B. were supported by a Grand Challenges Grant of the MGH/MIT Strategic Initiative. This work was also in part supported by grants from the National Foundation for Cancer Research; the Ludwig Center at Harvard; the U.S. National Cancer Institute grants P01-CA080124, R35-CA197743, R01-CA208205, and U01-CA224173 to R.K.J. J.M.P. was supported by a NIH Training Grant T32HL007627. This work was further supported in part by NIH grants R01 DK031036 and R01 DK033201 to C.R.K and NIH K12 HD000850, P30 DK40561 and NASPGHAN Young Investigator/Nestle Nutrition Award to S.S. M.P. was supported by an Erwin-Schrödinger Fellowship by the Austrian Science Fund (FWF; project number: J 3747-B28). S.C was supported by ABTA Basic Research Fellowship, MGH Tosteon and Fund for Medical Discovery Fellowship and PCRF Emerging Investigator Fellowship Grant. O.T.B. was additionally supported by a European Molecular Biology Organization long-term fellowship.
Footnotes
Competing interests
A competinginterests statement must be included. If you have no competing interests (financial and non-financial) to declare, please write ‘The authors declare no competing interests.’; otherwise, please include the details.
MP is an investigator for Bayer, BMS, and Lilly, he received speaker honoraria from Bayer, BMS, Eisai, and MSD, he is a consultant for Bayer, BMS, Ipsen, Eisai, and Lilly, and he received travel support from Bayer and BMS. R.K.J. received honorarium from Amgen; consultant fees from Chugai, Merck, Ophthotech, Pfizer, SPARC, SynDevRx, XTuit; owns equity in Enlight, Ophthotech, SynDevRx; and serves on the Boards of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund, Tekla World Healthcare Fund. MGB is a consultant and owns equity in Lumicell Inc. Neither any reagent nor any funding from these organizations was used in this study. MIT and MGH have filed a patent based on some of the findings described in this manuscript.
Supplementary information is available for this paper at https://doi.org/10.1038/s41551-01X-XXXX-X.
References
- 1.Younossi ZM et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Younossi Z. et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol 15, 11–20 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Ward ZJ et al. Projected U.S. state-level prevalence of adult obesity and severe obesity. N. Engl. J. Med 381, 2440–2450 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Satapathy SK & Sanyal AJ Epidemiology and natural history of nonalcoholic fatty liver disease. Semin. Liver Dis 35, 221–35 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Brunt EM et al. Nonalcoholic fatty liver disease. Nat. Rev. | Dis. Prim 1–22 (2015) doi: 10.1201/b17616. [DOI] [PubMed] [Google Scholar]
- 6.Kleiner DE On beyond staging and grading: Liver biopsy evaluation in a posttreatment world. Hepatology 65, 1432–1434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rockey DC, Caldwell SH, Goodman ZD, Nelson RC & Smith AD Liver biopsy. Hepatology 49, 1017–1044 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Dietrich CF et al. EFSUMB guidelines and recommendations on the clinical use of liver ultrasound elastography. Ultraschall in Med 2017 vol. 38 (2017). [Google Scholar]
- 9.Masarone M. et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid. Med. Cell. Longev 2018, 9547613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koyama Y. & Brenner DA Liver inflammation and fibrosis Find the latest version : Liver inflammation and fibrosis. 127, 55–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marcellin P. & Kutala BK Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 38, 2–6 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Vishwanath K. & Ramanujam N. Fluorescence Spectroscopy In Vivo. in Encyclopedia of Analytical Chemistry (John Wiley & Sons, Ltd, 2011). [Google Scholar]
- 13.Croce AC, Ferrigno A, Bottiroli G. & Vairetti M. Autofluorescence-based optical biopsy: An effective diagnostic tool in hepatology. Liver Int. 38, 1160–1174 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Monici M. Cell and tissue autofluorescence research and diagnostic applications. Biotechnol. Annu. Rev 11, 227–256 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Frangioni JV In vivo near-infrared fluorescence imaging. Curr. Opin. Chem. Biol 7, 626–34 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Ntziachristos V, Ripoll J. & Weissleder R. Would near-infrared fluorescence signals propagate through large human organs for clinical studies? Opt. Lett 27, 333–335 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Lim YT et al. Selection of quantum dot wavelengths for biomedical assays and imaging. Mol. Imaging 2, 50–64 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Carr JA et al. Shortwave infrared fluorescence imaging with the clinically approved near-infrared dye indocyanine green. Proc. Natl. Acad. Sci. U. S. A 115, 4465–4470 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruns OT et al. Next-generation in vivo optical imaging with short-wave infrared quantum dots. Nat. Biomed. Eng 1, 0056 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwaisako K. et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. U.S.A 111, E3297–E3305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delire B, Stärkel P. & Leclercq I. Animal models for fibrotic liver diseases: what we have, what we need, and what is under development. J. Clin. Transl. Hepatol 3, 53–66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scholten D, Trebicka J, Liedtke C. & Weiskirchen R. The carbon tetrachloride model in mice. Lab. Anim 49, 4–11 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Geerts AM et al. Comparison of three research models of portal hypertension in mice: macroscopic, histological and portal pressure evaluation. Int. J. Exp. Pathol 89, 251–263 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tag C. et al. Induction of experimental obstructive cholestasis in mice. Lab. Anim 49, 70–80 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Majno G. & Joris I. Symptoms of cellular disease: intracellular accumulations. in Cells, Tissues, and Disease: Principles of General Pathology 74–128 (Oxford University Press, 2004). [Google Scholar]
- 26.Terman A. & Brunk UT Lipofuscin. Int. J. Biochem. Cell Biol 36, 1400–1404 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Rantakari P. et al. Stabilin-1 expression defines a subset of macrophages that mediate tissue homeostasis and prevent fibrosis in chronic liver injury. Proc. Natl. Acad. Sci. U.S.A 113, 9298–9303 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barden H. The intragranular location of carboxyl groups in neuromelanin and lipofuscin in human brain and in meningeal melanosomes in mouse brain. J. Histochem. Cytochem 34, 1271–1279 (1986). [DOI] [PubMed] [Google Scholar]
- 29.Lillie RD A nile blue staining technic for the differentiation of melanin and lipofuscins. Stain Technol 31, 151–153 (1956). [DOI] [PubMed] [Google Scholar]
- 30.Evangelou K. & Gorgoulis VG Sudan black B, the specific histochemical stain for lipofuscin: A novel method to detect senescent cells. in Oncogene-Induced Senescence: Methods and Protocols, Methods in Molecular Biology vol. 1534 111–119 (Humana Press, New York, NY, 2017). [DOI] [PubMed] [Google Scholar]
- 31.Everson Pearse AG Pigments and Pigment Precursors. in Histochemistry Theoretical and Applied Vol. 2 898–928 (Churchill Livingstone, 1985). [Google Scholar]
- 32.Terman A, Kurz T, Navratil M, Arriaga EA & Brunk UT Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial–lysosomal axis theory of aging. Antioxid. Redox Signal 12, 503–535 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seehafer SS & Pearce DA You say lipofuscin, we say ceroid: Defining autofluorescent storage material. Neurobiol. Aging 27, 576–588 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Schnell SA, Staines WA & Wessendorf MW Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J. Histochem. Cytochem 47, 719–730 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Erben T, Ossig R, Naim HY & Schnekenburger J. What to do with high autofluorescence background in pancreatic tissues – an efficient Sudan black B quenching method for specific immunofluorescence labelling. Histopathology 69, 406–422 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Nazeer SS, Saraswathy A, Shenoy SJ & Jayasree RS Fluorescence spectroscopy as an efficient tool for staging the degree of liver fibrosis: an in vivo comparison with MRI. Sci. Rep 8, 10967 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kisseleva T. et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. U.S.A 109, 9448–9453 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duffield JS et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest 115, 56–65 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu C. et al. Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab. Invest 90, 1805–1816 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Beljaars L. et al. Hepatic localization of macrophage phenotypes during fibrogenesis and resolution of fibrosis in mice and humans. Front. Immunol 5, 430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsumoto M. et al. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol 94, 93–103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giannessi F, Giambelluca MA, Scavuzzo MC & Ruffoli R. Ultrastructure of testicular macrophages in aging mice. J. Morphol 263, 39–46 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Jara M, Carballada R. & Esponda P. Age-induced apoptosis in the male genital tract of the mouse. Reproduction 127, 359–366 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Brunk UT & Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med 33, 611–619 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Softic S. et al. Lipodystrophy due to adipose tissue-specific insulin receptor knockout results in progressive NAFLD. Diabetes 65, 2187–2200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friedman SL, Neuschwander-Tetri BA, Rinella M. & Sanyal AJ Mechanisms of NAFLD development and therapeutic strategies. Nat. Med 24, 908–922 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alonso C. et al. Metabolomic identification of subtypes of nonalcoholic steatohepatitis. Gastroenterology 152, 1449–1461.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brunt EM, Janney CG, Bisceglie AM Di, Neuschwander-Tetri BA& Bacon BR Nonalcoholic steatohepatitis - a proposal for grading and staging the histological lesions. Am. J. Gastroenterol 94, 2467–2474 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Layton C. & Bancroft JD Connective and other mesenchymal tissues with their stains. in Bancroft’s Theory and Practice of Histological Techniques 215–238 (Elsevier, 2013). [Google Scholar]
- 50.Schmittgen TD & Livak KJ Analyzing real-time PCR data by the comparative CT method. Nat. Protoc 3, 1101–1108 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Casteilla L, Pénicaud L, Cousin B. & Calise D. Choosing an adipose tissue depot for sampling: factors in selection and depot specificity. Methods Mol. Biol 456, 23–38 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biosciences Li-cor. In Vivo Animal Imaging Diet Considerations. (2008). [Google Scholar]
- 54.Bhaumik S, Depuy J. & Klimash J. Strategies to minimize background autofluorescence in live mice during noninvasive fluorescence optical imaging. Lab Anim. (NY) 36, 40–43 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Inoue Y, Izawa K, Kiryu S, Tojo A. & Ohtomo K. Diet and Abdominal Autofluorescence Detected by in Vivo Fluorescence Imaging of Living Mice. Mol. Imaging 7, 21–27 (2008). [PubMed] [Google Scholar]
- 56.Miquel J, Lundgren PR & Johnson JE Spectrophotofluorometric and electron microscopic study of lipofuscin accumulation in the testis of aging mice. J. Gerontol 33, 3–19 (1978). [PubMed] [Google Scholar]
- 57.Paniagua R, Amat P, Nistal M. & Martin A. Ultrastructure of Leydig cells in human ageing testes. J. Anat 146, 173–83 (1986). [PMC free article] [PubMed] [Google Scholar]
- 58.Nygaard MB, Almstrup K, Lindbæk L, Christensen ST & Svingen T. Cell context-specific expression of primary cilia in the human testis and ciliary coordination of Hedgehog signalling in mouse Leydig cells. Sci. Rep 5, 10364 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are too big to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request.