

Abstract
The contribution of mitochondria to oncogenic transformation is a subject of wide interest and active study. As the field of cancer metabolism becomes more complex, the goal of targeting mitochondria using various compounds that inflict mitochondrial damage (so-called mitocans) is becoming quite popular. Unfortunately, many existing cytotoxicity assays, such as those based on tetrazolium salts or resazurin require functional mitochondrial enzymes for their performance. The damage inflicted by compounds that target mitochondria often compromises the accuracy of these assays. Here we describe a modified protocol based on differential staining with two fluorescent dyes, one of which is cell-permeant (Hoechst 33342) and the other of which is not (propidium iodide). The difference in staining allows living and dead cells to be discriminated. The assay is amenable to automated microscopy and image analysis, which increases throughput and reduces bias. This also allows the assay to be used in high-throughput fashion using 96-well plates, making it a viable option for drug discovery efforts, particularly when the drugs in question have some level of mitotoxicity. Importantly, results obtained by Hoechst/PI staining assay show increased consistency, both with trypan blue exclusion results and between biological replicates when the assay is compared to other methods.
Keywords: High-throughput screening, mitochondria-targeted compounds (mitocans), cytotoxicity assay, leukemia, fluorescence microscopy, automated cell counting
SUMMARY:
The protocol describes a rapid, high-throughput, reliable, inexpensive, and unbiased assay for efficiently determining cellular viability. This assay is particularly useful when cells’ mitochondria have been damaged, which interferes with other assays. The assay uses automated counting of cells stained with two nuclear dyes – Hoechst 33342 and propidium iodide.
INTRODUCTION:
The first step to identifying effective cancer treatments is the selection of a robust, unbiased cytotoxicity assay that can be used to examine the effect of treatment. A common choice for low-throughput experiments is the exclusion of trypan blue dye from living cells. This method is favored because it allows a relatively unbiased method for quantifying cell survival. Trypan blue passively diffuses into cells whose membranes are compromised, but it is effectively blocked from entering healthy cells1. The quotient of the living cells and the total cells represents the percent viability, which indicates the efficacy of the treatment. The most significant disadvantage of the trypan blue assay is that it is poorly suited for high-throughput methodologies. It has a relatively low signal-to-noise ratio and prolonged staining can result in artifacts due to the staining of viable cells. Consequently, trypan blue exclusion is typically, but not always2, relegated to manual counting. This makes it too slow and introduces the strong possibility of bias due to subjective judgment of the researcher (unless blinding or independent counts are used, which further reduce laboratory throughput). In general, the throughput of this assay is insufficient for modern drug discovery.
Viability assays, which generally have a much higher throughput, allow researchers to circumvent this limitation, but come with significant caveats (see Table 1). These methods generally fall into two groups. One group is comprised of colorimetric assays that are based on the function of cellular redox enzymes. Colorless or non-fluorescent substrates are converted into vibrant products that can be quantified using a spectrophotometer. Classic examples include tetrazolium salts (MTT, WST-1, XTT, etc.) and resazurin. This category also includes luminescent assays that utilize luciferin to evaluate ATP level. Assays of this type have the underlying limitation that they are measuring cellular metabolism, which is not cellular viability per se. It is quite common for cells to become quiescent under adverse conditions, but still retain the ability to divide3–5. For example, cancer stem cells are often relatively metabolically quiescent6–9, and are likely to be difficult to assay using these techniques. Effectiveness of treatments that damage mitochondrial function, such as most mitocans, is also likely to be significantly overestimated.
Table 1. List of cytotoxicity assays.
Cytotoxicity assays, some of which were used in this study, listed along with the brief description of their key features.
| Assay/dye | Type(s) of cell death detected | Necessary equipment | Key features |
|---|---|---|---|
| MTT, CKK-8, alamarBlue (resazurin) | Apoptosis/Necrosis | Spectrophotometer | Inexpensive, rapid; endpoint assay; dependent on enzymes’ activity (exclusively mitochondrial in case of MTT) and does not discriminate between modes of cell death1,10 |
| LDH release | Necrosis | Spectrophotometer | Rapid, independent of mitochondrial enzymes’ activity; expensive for high-throughput tests; detects necrotic cells with compromised plasma membrane11,12 |
| Trypan blue (TB) | Apoptosis/Necrosis | Microscope | Cell-impermeant; does not discriminate between modes of cell death; laborious and not suitable for high-throughput screening; more difficult to use with adherent cells; prone to subjective judgment of the user, but is considered the standard cell viability measurement method13 |
| Acridine orange (AO) | Apoptosis/Necrosis/Necroptosis | Fluorescence microscope | A nucleic acid dye with unique spectral properties, can distinguish between apoptosis and necrosis/necroptosis14 |
| Hoechst 33342, DAPI | Apoptosis | Fluorescence microscope or flow cytometer | Cell-permeable; inappropriate on its own to monitor cell death; useful for co-staining; can be used to assess chromatin condensation and nuclei fragmentation in early apoptosis; can be paired with propidium iodide to distinguish apoptosis from necrosis15,16 |
| Propidium iodide (PI) | Late apoptosis/Necrosis | Fluorescence microscope or flow cytometer | Cell-impermeant intercalator; detects both late apoptosis and necrosis modes of cell death17. Toxic and permeable after long incubation times18 |
An alternative methodology leverages the chemical properties of various substances that allow them to either cross or not cross biological membranes. One example is nuclear stains such as SYTOX or propidium iodide (PI). This category also includes assays that are similar in concept but different in function, such as the lactate dehydrogenase (LDH) assay, which measures the release of LDH into the extracellular milieu as an indicator of cellular necrosis (Figure 1, Table 1). These assays are more capable of distinguishing between metabolically inactive and dead cells.
Figure 1. Experimental timeline and comparison of existing cytotoxicity assays.
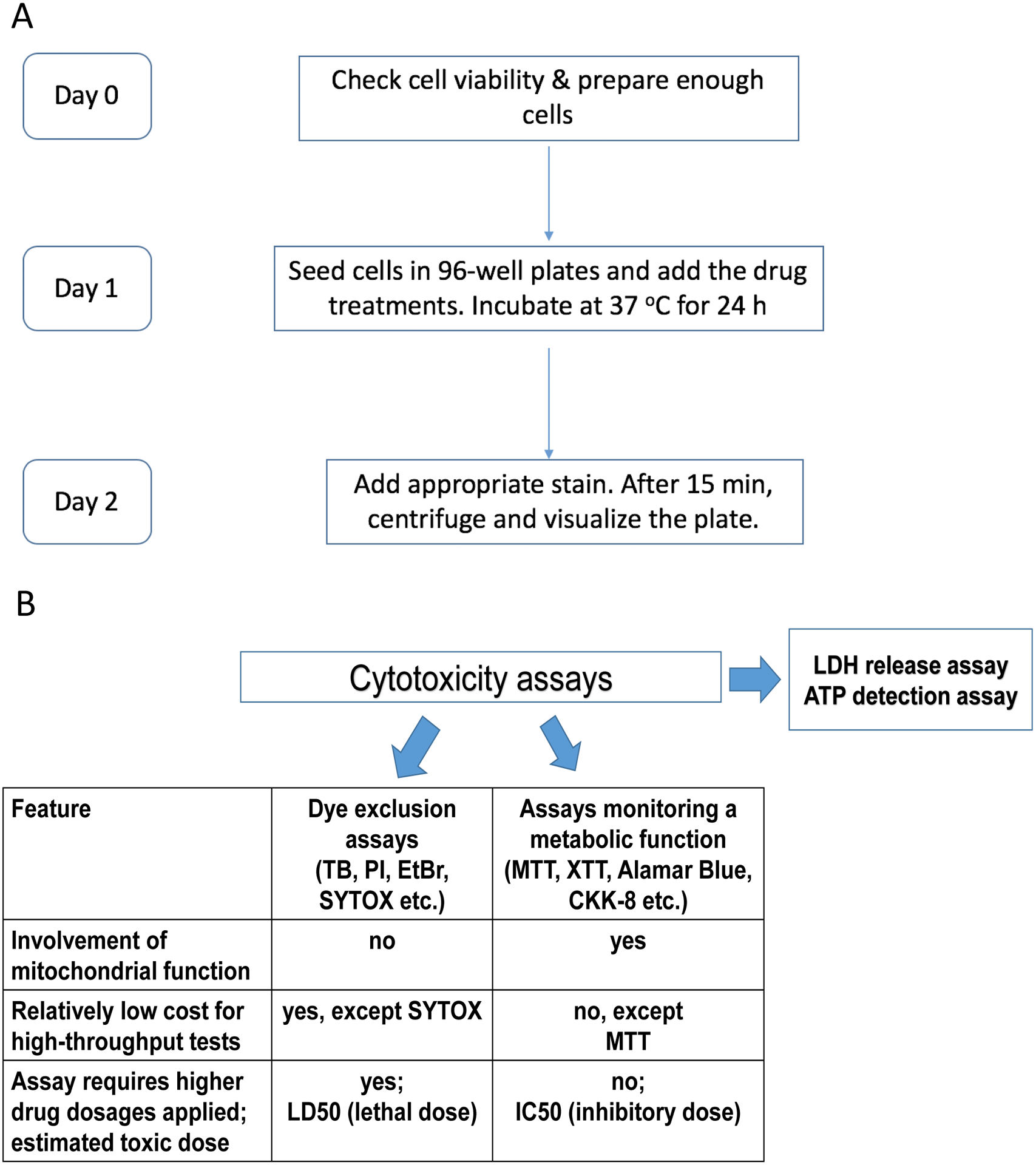
A. Flowchart summarizing the timeline for the experimental procedure, e.g. Hoechst/PI staining. B. Comparison of cytotoxicity assays, some of which were used in this study. Dye exclusion assays involve impermeant nuclear dyes that stain dead cells with compromised plasma membrane: TB – trypan blue, PI – propidium iodide, EtBr – ethidium bromide, and SYTOX. Second group of assays depend on cellular metabolism, for example, tetrazolium salts MTT, XTT, and CKK-8 (WST-8), resazurin-based reagent alamarBlue, etc.
Recent studies have demonstrated that mitochondrial metabolism is altered in some cancers19–25. For example, acute myeloid leukemias have been shown to upregulate their mitochondrial mass, mtDNA content, and mitochondrial respiration to meet their energy demands19,26,27. On the other hand, some solid tumors are characterized by mitochondrial dysfunction, or rather “metabolic reprogramming”, such as downregulation of mitochondrial proteins involved in OXPHOS or decreased mtDNA content, that has been associated with tumor invasiveness, metastatic potential and resistance to apoptosis-inducing drugs28,29. Furthermore, recently, there has been an increased interest in using mechanistically diverse compounds that affect mitochondrial function (generally called mitocans30), as potential therapies for particular cancers. These drugs target ATP synthesis, mitochondrial DNA, OXPHOS, and ROS production, as well as pro- and anti-apoptotic proteins associated with mitochondria30,31. Several studies have shown that this approach has significant promise19,32–34. However, these metabolic deviations in cancer cell biology or mitochondria-targeting treatments may significantly affect conventional viability assays that are based on mitochondrial functionality.
Here, an optimized protocol for a differential nuclear staining assay is described. The protocol allows fast and accurate determination of cytotoxicity of mitocans or their combinations with other compounds. Hoechst 33342 is a cell-permeant nuclear dye that readily crosses cell membranes to stain DNA, allowing the total cell count to be obtained. By co-staining with PI, which only enters the nuclei of dead cells, the proportion of living (Hoechst only) and dead (stained with both) cells can be accurately determined. This protocol refines the published assay35 by adding a step for the optimization of the dye concentration (by cross-referencing results with orthogonal trypan blue method) and centrifugation of the plate prior to imaging. Since many cell lines are semi-adherent or suspended, centrifugation increases the proportion of cells that are imaged and strongly improves accuracy. The assay has several advantages, including that staining does not require removal of media or washing. The dye mixture is also inexpensive, easy to prepare, and compatible with multichannel/robotic pipetting systems.
After cells have been stained, they are imaged with an automated microscope. This has the added advantage of creating a permanent record of the images that can be re-analyzed later and the effects of particular compounds can be re-evaluated by visual inspection of captured images. Once images have been obtained, cells can be counted either manually or by using any of several software packages, including both free (e.g., ImageJ, CellProfiler, etc) and commercial software (e.g., Metamorph, Gen5, etc). Automated cell counting is generally preferable since properly developed automated cell counting pipelines are more accurate and less biased than manual counts. They also more effectively disregard cell debris or insoluble complexes. Development of these pipelines is generally straightforward and is simplified by the efficiency of the stains used. The output is quantitative since the actual number of dead cells is automatically calculated with respect to total cell number, and different thresholds can be applied to increase or decrease the stringency of detection35. For convenience, optimized parameters for counting cells using Gen5 v. 3.00 software compatible with Cytation 5 Cell Imaging Multi-Mode Reader are included.
PROTOCOL:
- Cytotoxicity Assay: Setup
-
1.1.Prepare solutions of compounds of interest at desired concentrations in the appropriate media (serum-free or 1, 2.5, or 5% FBS RPMI-1640).
-
1.1.1.To measure cytotoxicity of a single compound (e.g., to determine effective doses), prepare compounds at 2x final concentration.
-
1.1.2.To measure cytotoxicity of compound combinations, prepare compounds at 4x final concentration.
-
1.1.3.Prepare solvent-only controls by mixing the same amount of solvent with the appropriate medium. For example, if testing compounds dissolved in DMSO and methanol, make solvent-only control for each solvent.
-
1.1.1.
-
1.2.Collect cells from culture dish or flask into a 15 mL conical tube.
-
1.3.Transfer 10 μL of cell suspension into a microcentrifuge tube and stain with 10 μL 0.4% trypan blue. Use a hemocytometer to count viable and non-viable cells for each cell source.
-
1.4.Pellet cells at 200 g for 5 min. Aspirate or decant supernatant.
-
1.5.Resuspend cell pellet in assay-appropriate media (serum-free or 1, 2.5, 5% FBS RPMI-1640) at a cell density of 3*105 cells/mL.Note 1: Cell density of 3*105 cells/mL provides a seeding density of 15,000 cells/well. Seeding density is an important parameter and ideally should be pre-defined prior to the experiment. Seeding density should take into account 1) cell size – usually larger cells are seeded at a lower density; 2) treatment duration – cells are typically seeded at a lower density for experiments that will last longer; and 3) cell division rate – cells with a higher rate of division are seeded at a lower density. Specific examples of optimized seeding densities: K562 cells, larger, 24 h duration – 10,000 cells/well; MOLM-13 cells, moderate size, 24 h treatment – 15,000/well; MOLM-13 cells, 48 h treatment – 8,000/well; small healthy peripheral blood mononuclear cells (PBMCs), 24 h treatment – 50,000/well; primary AML cells, 24 h treatment – 15,000–20,000/well (depending on availability).Note 2: The presence of FBS in media may affect the activity of the compounds. Reducing the concentration of FBS may make assay results simpler to interpret, but it also reduces the physiological accuracy.
-
1.6.Seed 50 μL of cell suspension from step 1.5 into each well of a 96-well plate using a multichannel pipette.
-
1.7.Add compounds as follows:
-
1.7.1.For single compound assays, add 50 μL of 2x compound solution into each well. For solvent-control wells, add 50 μL of test media containing the solvent at the 2x concentration.
-
1.7.2.For combination assays, add 25 μL of each of the compound (4x solutions) into each well. For single compound-control wells, add 25 μL of 4x compound solution and 25 μL of test medium. For solvent-control wells, add 50 μL of test medium or test medium containing the solvent.Note 1: The final concentration of DMSO should not exceed 0.5%.Note 2: It is recommended to add the media containing the compounds with the pipette touching the wall of each well due to low volume.
-
1.7.1.
-
1.8.Gently tap the plate to ensure mixing of the contents of the wells.
-
1.9.Incubate plates at 37 °C in a humidified 5% CO2 atmosphere for an appropriate time, e.g., 24 h.
-
1.1.
- Cytotoxicity Assay: staining with Hoechst 33342 and Propidium Iodide
-
2.1Prepare 10x staining solution. This solution needs to be prepared fresh before each experiment, it cannot be stored. Final dye concentrations should be determined prior to the experiment.
-
2.1.1.For leukemia cell lines and primary leukemia cells, 1 mL of 10x staining buffer contains 10 μL of 20 mM Hoechst 33342 and 50 μL of 1 mg/mL propidium iodide in sterile PBS (final concentrations: Hoechst 33342 20 μM, PI 5 μg/mL).
-
2.1.2.For healthy PBMCs, 1 mL of 10x staining buffer contains 10 μL of 20 mM Hoechst 33342 and 10 μL of 1 mg/mL propidium iodide in sterile PBS (final concentrations: Hoechst 33342 20 μM, PI 1 μg/mL).Note: The final concentration of propidium iodide has to be determined prior to the experiments. Cells should be tested using a range of PI concentrations (1, 2.5, 5 μg/mL), and then Hoechst/PI-calculated viability should be compared with viability measured via trypan blue. The PI concentrations listed above were chosen based on target cell viability in media-control wells (above ~90% for leukemia cell lines, above ~70% for primary normal PBMCs).CAUTION: Hoechst 33342 and propidium iodide are potential carcinogens. Wear appropriate personal protective equipment when handling them.
-
2.1.1.
-
2.2.After incubation, use a multichannel pipette to add 10 μL of 10x staining buffer to each well.Note: To prevent cross-contamination, make sure that the pipette tips do not touch the media.
-
2.3.Gently tap the plate to mix and clear bubbles. Stain at 37 °C for 15 min.
-
2.4.Centrifuge the plate at 200 g for 4 min to bring all of the cells to the bottom of the plate. Carefully wipe the bottom of the plate with a damp kimwipe to remove fibers and/or debris that will interfere with imaging.Note 1: Centrifugation of the plate ensures the highest chances for all of the cells to be captured in the image. Often dead cells will detach and float, providing misleading values of cytotoxicity. Centrifugation mitigates this effect.Note 2: The plate should be imaged as quickly as possible after centrifugation, ideally, within 15 min. Centrifugation can reduce the selectivity of PI staining and may allow cells to slowly accrue propidium iodide. The improvement in accuracy gained by visualizing the dead cells outweighs the slight increase in PI staining. It is recommended to finish imaging within 1 h of centrifugation.
-
2.1
- Cytotoxicity Assay: data acquisition
-
3.1.Set the software for the automated microscope/plate imager to detect fluorescence for both Hoechst 33342 (excitation maximum 350 nm, emission maximum 461 nm) and PI (excitation maximum 493 nm, emission maximum 636 nm). Acquire images for each well in both channels.
-
3.2.Using the software (such as CellProfiler, a free image analysis studio based on MatLab http://cellprofiler.org/, ImageJ, or proprietary software such as Gen5) count the cells in each well in each channel.Note: Since every cell should be stained with Hoechst 33342, and dead cells are stained with PI, the ratio of dead to all represents the fraction of cells that are dead. For example, if the automated count in untreated sample shows 467 cells stained with PI (dead cells) and 2335 cells stained with Hoechst 33342 (total cells), the fraction dead is 0.2 or 20%. This value is then compared to an identically handled sample where treatment was used.
-
3.3.Detailed description of Hoechst/PI staining assay using Cytation5 Cell Imaging Multi-Mode Reader and Gen5 v. 3.00 software:
-
3.3.1.The Cytation5 multimode plate reader/imager was set to image cells in flat bottom, 96-well, generic black plastic plates.
-
3.3.2.An imaging protocol was set to utilize the standard DAPI and Texas Red filter sets. Images were taken at the well center using a 4x magnification objective. No offset (X/Y or Z) was used. The settings used for DAPI filter set were as follows: LED - 10, integration time – 99, gain - 0; for Texas Red filter set: LED - 8, integration time – 950, gain - 18. Autofocusing was performed based on DAPI signal; there was no offset in focusing between channels.
-
3.3.3.Image analysis was performed using Gen5 software v 3.00. In the software settings, cells were defined as shapes between 5 and 25 μm in their size. Primary edge objects were excluded and touching objects were split by turning on special option “Split touching objects”. Next, the image was processed to remove the background (dark background subtraction), a nuclear mask was applied (threshold value DAPI >= 6000 AU), and objects were counted. A subpopulation analysis was performed based on PI staining (threshold value Texas Red >= 5000 AU), and objects were counted. Finally, cell viability %, was defined as .
-
3.3.1.
-
3.1.
REPRESENTATIVE RESULTS:
The aforementioned protocol has been developed using OCI-AML2 cells, which were taken as a representative acute myeloid leukemia (AML) cell line. AML is characterized by abnormal proliferation of undifferentiated and non-functional hematopoietic cells in the bone marrow26. Despite recent developments in AML targeted therapy, the standard of care has remained unchanged for several decades, and consists of induction therapy (typically comprised of three days of anthracycline, e.g., daunorubicin, idarubicin, or anthracenedione mitoxanthrone, and 7 days of cytarabine) followed by consolidation (typically comprised of rounds of cytarabine treatment followed by periods of recovery)36.
Following the protocol outlined in Figure 1A, OCI-AML2 cells were seeded in 96-well plates and treated with the mitochondrial uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP) or the glycolytic inhibitor 2-deoxy-D-glucose (2-DG) at a range of concentrations. Cells were treated for 24 h at 37 °C in serum-free RPMI-1640 media, and then viability was assessed using trypan blue (TB) exclusion or one of four viability assays (Hoechst/PI differential nuclear staining, LDH release assay, MTT, or alamarBlue). Representative images of cells stained with Hoechst/PI are shown in Figure 2A. Several important observations can be made immediately. First, the total number of cells (Hoechst staining) is reasonably high and is markedly greater than the number of dead cells (PI staining). This suggests that the media conditions are not triggering high rates of cell death. Second, since only cells being labeled with both Hoechst and PI are counted as dead (purple cells in the merged image), the probability of counting debris is very low. This image shows a good example of properly stained cells.
Figure 2. Comparison of a panel of cytotoxicity assays with trypan blue exclusion.
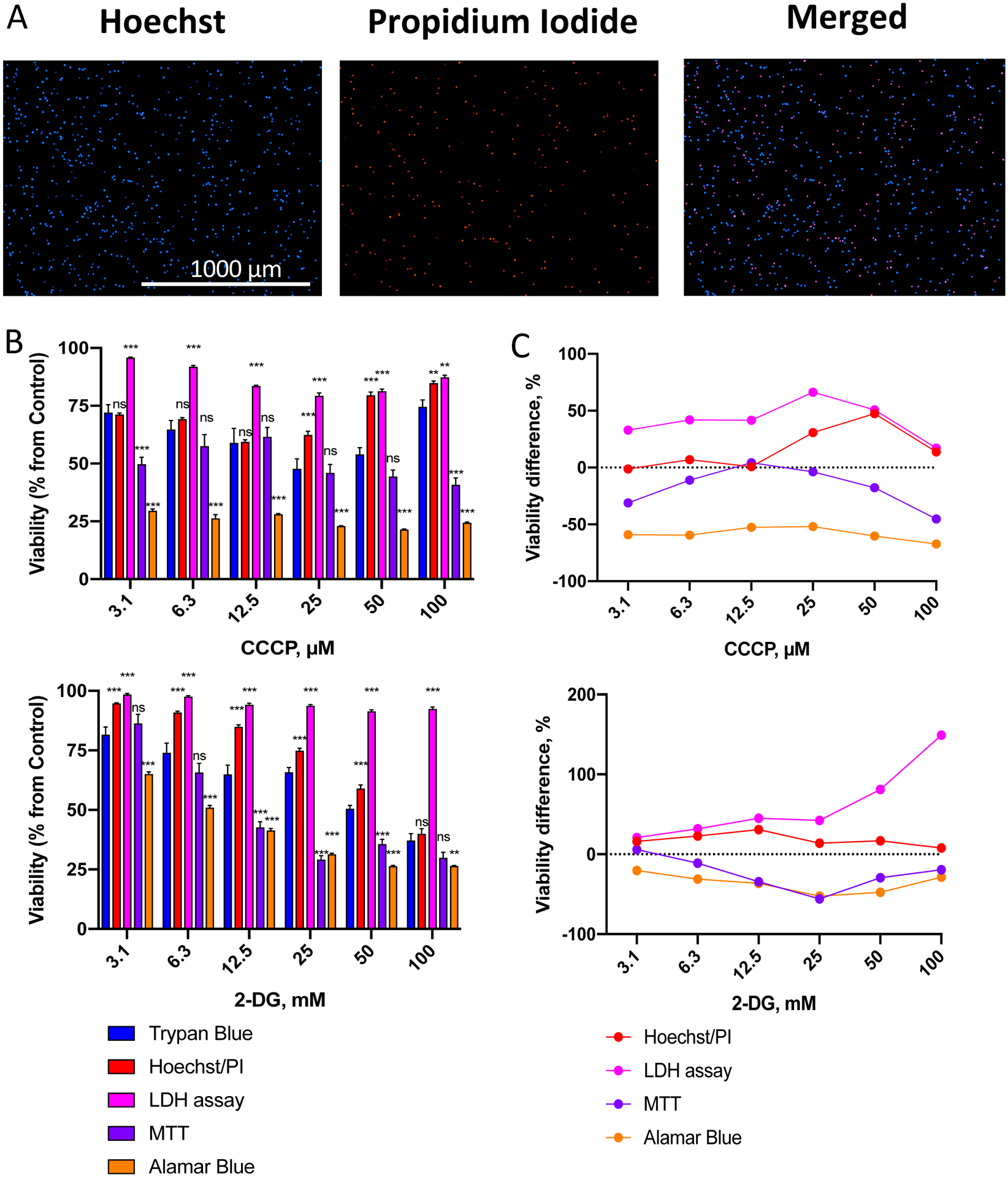
A. Representative images of total (Hoechst 33342) and dead (propidium iodide, PI) OCI-AML2 cells via Hoechst/PI staining. B. Assessment of viability of OCI-AML2 cells using different methods after treatment with a gradient of CCCP (top) or 2-DG (bottom) concentrations. OCI-AML2 were treated with CCCP or 2-DG in serum-free RPMI-1640 for 24 h prior to determination of cell viability. Shown is mean, error bars represent SEM. C. Difference in cell viability between cytotoxicity assays in (B) vs. trypan blue staining (see Supplementary Tables S1–2 for the exact numbers and median difference). Stars indicate significant difference vs. trypan blue staining. ** p < 0.01, *** p < 0.001, ns – non-significant. Group comparison was done via t-test with correction for multiple hypothesis testing. Three independent biological replicates were performed.
As we recently reported19, leukemias are very sensitive to mitotoxic treatments, which indicates that the cells already have underlying mitochondrial damage. On this basis, we predicted that the MTT and alamarBlue assays, which are based on mitochondrial enzyme activity, would inaccurately measure cellular viability. As expected, these assays (especially alamarBlue) showed significantly lower viability compared to Trypan Blue exclusion, see Figure 2B,C, Supplementary Tables S1,2. This is consistent with the fact that doses of these compounds required to induce apoptosis or necrosis are higher than those needed to compromise mitochondrial function.
Amongst the assays that were tested, dual staining with Hoechst 33342 and PI had the best combination of robustness, sensitivity, and consistency with TB staining, with the smallest median deviation from TB exclusion method after CCCP or 2-DG treatment (Supplementary Tables S1–2). Interestingly, at higher doses of CCCP (50 and 100 μM) viability estimated with Hoechst/PI or TB was increased compared to lower doses (Figure 2B, top). This is likely due to the precipitation of CCCP at higher doses due to its hydrophobicity, reducing its effective concentration and impact on cells. Increasing the dose of CCCP further reduced Hoechst/PI-estimated mean viability of OCI-AML2 cells, however: 74% at 150 μM, 62% at 200 μM, 6% at 300 μM (data not shown).
The Hoechst/PI assay was also effective at determining cellular viability after treatment with other mitochondria-targeting molecules. These included rotenone, a poison that compromises Complex I of the mitochondrial electron transport chain37, and 3-bromopyruvate, a glycolytic inhibitor and alkylating agent that impairs mitochondrial respiration and mitochondrial metabolism38 (Figure 3A–D, Supplementary Tables S3–4).
Figure 3. Validation of Hoechst/PI cytotoxicity assay in leukemia cells.
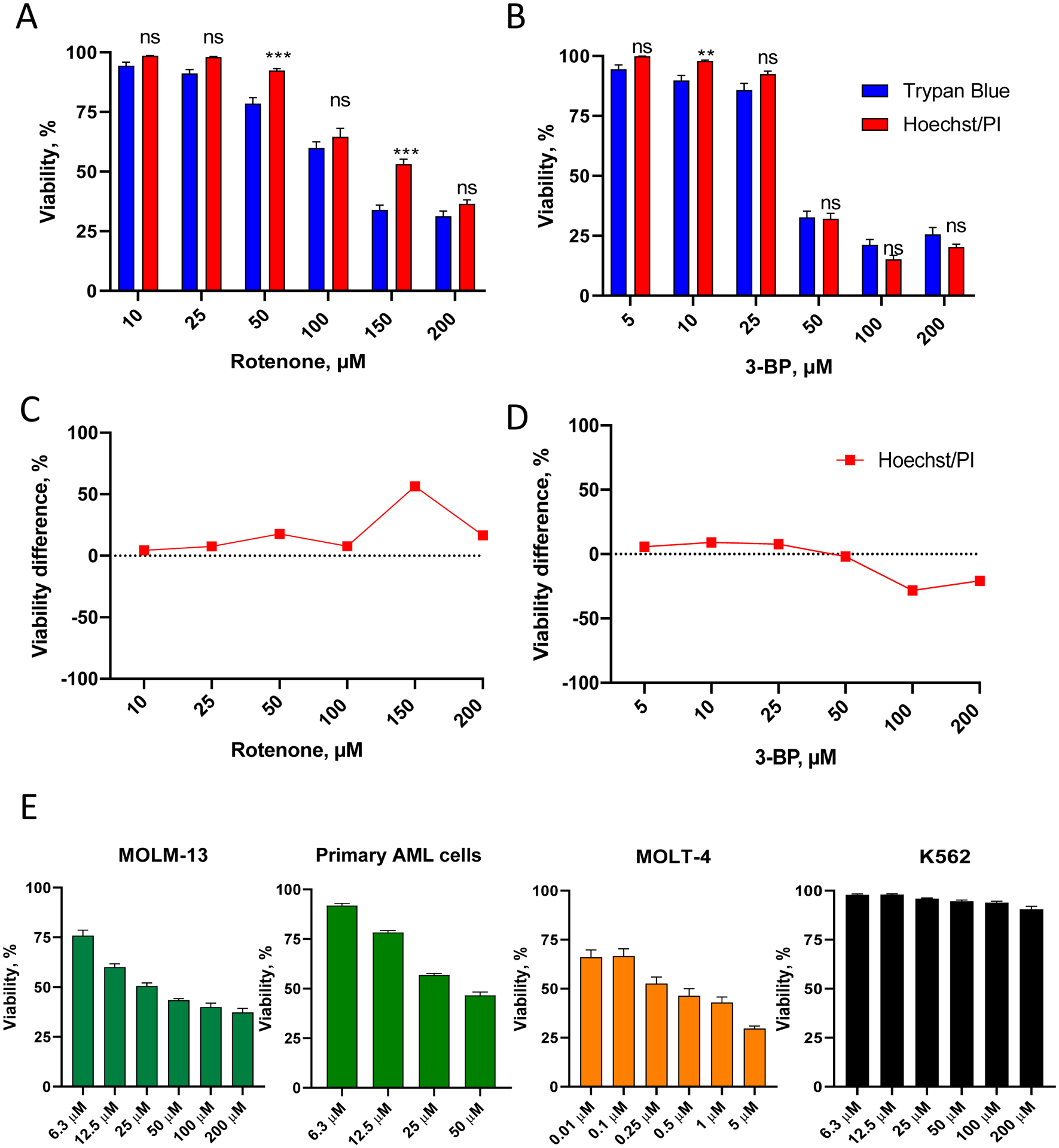
A-B. OCI-AML2 (AML) cells were treated with different concentrations of rotenone (A) or 3-bromopyruvate, 3-BP (B) in serum-free RPMI-1640 media for 24 h, then viability was determined. Shown is mean with SEM. C-D. Comparison of viability difference between Hoechst/PI assay vs. trypan blue staining (for the cells in A-B, see Supplementary Tables S3–4 for quantitation). Stars indicate statistical significance vs. trypan blue staining. ** p < 0.01, *** p < 0.001, ns – non-significant. Group comparison was done via t-test with correction for multiple hypothesis testing. E. MOLM-13 (AML), primary AML cells isolated from a patient, MOLT-4 (ALL), and K562 (CML) cells were treated with the indicated concentrations of rotenone in serum-free RPMI-1640 media for 24 h, prior to viability determination using Hoechst/PI staining. Shown is mean with SEM. Three independent biological replicates were performed.
The Hoechst/PI cytotoxicity assay was further validated using a panel of leukemia cell lines representing multiple types of leukemia, as well as primary cells. They included MOLM-13 (an AML cell line), primary AML cells derived from a representative patient sample, MOLT-4 (an acute lymphoblastic leukemia cell line, ALL), and K562 (a chronic myelogenous leukemia cell line, CML) (Figure 3E, Supplementary Figure S1). Results of the Hoechst/PI assay showed that these cells had profound differences in rotenone sensitivity, ranging from very sensitive (MOLT-4) to resistant (K562) cells.
To demonstrate the robustness of the assay, OCI-AML2 cells were treated with a concentration gradient of 3-bromopyruvate, as described in the protocol above. Cell counts were collected for 6 wells at each concentration and are shown (Figure 4A,B). These counts were used to calculate viability, and showed that as few as 4 wells were sufficient to accurately capture assay outcome (Figure 4C). The resulting dose-response curve is shown (Figure 4D).
Figure 4. Reproducibility of Hoechst/PI staining.
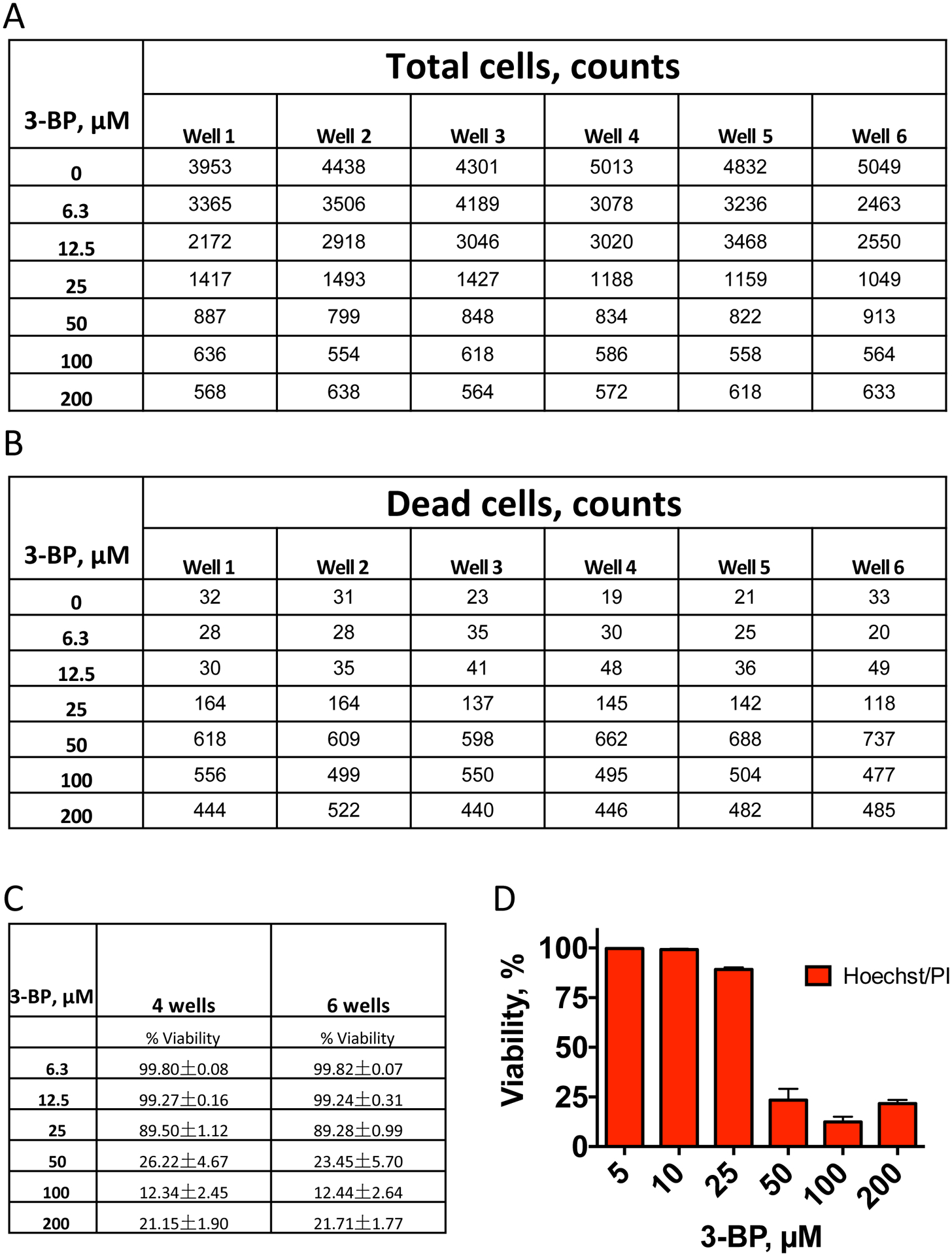
Viability of OCI-AML2 cells after 24 h of treatment with different concentrations of 3-bromopyruvate (3-BP). A. Total cell number counted via Hoechst 33342 staining, B. Dead cell number counted via propidium iodide staining. C. Viability (%) calculated using (A-B). D. Representative dose-response graph. Shown is mean with SEM. Three independent biological replicates were performed.
Although the assay is relatively robust, care must still be taken. Improper performance of the assay may compromise its accuracy. Several compromised outcomes are shown for troubleshooting purposes (Figure 5). The first set of images shows the consequences of over-staining with PI, which may arise from the following sources: using the dye at too high concentration, staining for too long, or using too high LED intensity/integration time in the red channel (Figure 5A). These errors will generate artificially inflated numbers of dead cells. The second issue arises from neglecting the centrifugation step. Often, dead cells begin to lose their attachment from the surface of the dish. Consequently, they will be underrepresented during image acquisition, which generally occurs near the bottom of the well (Figure 5B). Finally, overexposure in the Hoechst channel artificially expands the size of the cells, significantly reducing the total counts per well and limiting the assay power (Figure 5C).
Figure 5. Comparison of optimal and sub-optimal assay parameters.

Comparison of results acquired via optimized parameters (left) or sub-optimal parameters (right). A. Normal PBMCs were stained with propidium iodide at 1 μg/mL (left) or 5 μg/mL (right) for 15 min before imaging. B. Images of OCI-AML2 cells taken with (left) or without (right) plate centrifugation. C. Images of OCI-AML2 cells acquired with optimal (left) or excessive (right) integration time for the Hoechst channel.
DISCUSSION:
Although the protocol for Hoechst/PI cytotoxicity assay is robust and requires comparatively little hands-on time, there are several experimental details that are very important to ensure accurate results. First, it is essential to make sure that the concentration of DMSO remains below 0.5% (v/v). It is generally agreed that exposure to even low doses of DMSO can substantially alter the morphology and attachment of cells and significantly delay cell cycle progression39,40.
Second, the staining should be performed as soon as possible after treatment. Since there are no washing steps, the compound remains in the wells and can continue to inflict damage on the cells. In comparison to the earlier protocol35, the cells were stained for only 15 minutes immediately after treatment. This limits the possibility that viable cells could be inadvertently stained with propidium iodide. For this reason, we suggest staggering treatments and staining if more than two plates will be treated. This allows for imaging time between plates and improves reproducibility.
Third, appropriate dye concentration is dependent on cell type. It is important to empirically determine the optimized propidium iodide concentration, beginning with untreated cells, using trypan blue staining as an orthogonal reference.
Finally, the centrifugation step immediately before visualization is also critical. Based on this protocol, a range of 500–4,000 cells is recorded (depending on seeding density). This is a notable improvement over the 100–400 cells per well used previously35. Considering the variation in the population of cancer cells (especially primary cells), having more cells for analysis is important, and may allow more robust data analysis.
Due to the relative simplicity of the method, a wide variety of modifications can easily be performed. For example, many other cell-impermeant dyes are available from a number of vendors and can be substituted for propidium iodide. While this substitution has relatively little value on its own, the increased palette of colors means that more complex experiments can be performed. For example, the addition of a third color will allow differential survival to be assessed between subpopulations of cells, as is commonly performed with flow cytometry.
A more substantial modification of the protocol is foregoing cell visualization and using a spectrophotometer instead. This approach has the advantage of utilizing a nearly ubiquitous piece of equipment (a standard spectrophotometer with a microplate reader) and is the fastest method to acquire data. However, this method is far less accurate. Fluorescence intensity is representative of cell staining, but stochastic variations in staining intensity, combined with the relatively smaller sample area, introduce significant variability. While further optimization (such as the inclusion of a fixative, additional washing steps, or a larger number of samples) may address these problems, fluorescence microscopy is generally a superior approach.
In conclusion, the modified Hoechst/PI protocol is a fast, accurate, inexpensive, high-throughput cytotoxicity assay that is independent of mitochondrial function. This assay has substantial utility for efficiently screening compounds or compound combinations that target mitochondria.
Supplementary Material
Supplementary Figure S1. Representative images of cells stained with Hoechst/PI, with or without rotenone treatment at indicated concentrations in serum-free RPMI-1640 media for 24 h: A-B. AML: MOLM-13 cell line (A), and primary AML cells (B) derived from a representative patient sample. C. ALL: MOLT-4 cell line. D. CML: K562 cell line.
Supplementary Table S1. Viability differences for a panel of assays in OCI-AML2 cells after CCCP treatment.
Assessment of viability was performed after 24 h of treatment with different concentrations of CCCP. Median viability differences between tested assays (Hoechst/PI, LDH, MTT, or alamarBlue) and trypan blue staining with automated counting was calculated based on viability differences at each drug concentration.
Supplementary Table S2. Viability differences for a panel of assays in OCI-AML2 cells after 2-DG treatment.
Assessment of viability was performed after 24 h of treatment with different concentrations of 2-DG. Median viability differences between tested assays (Hoechst/PI, LDH, MTT, or alamarBlue) and trypan blue staining with automated counting were calculated based on viability differences at each drug concentration.
Supplementary Table S3. Viability difference between Hoechst/PI and TB exclusion method in OCI-AML2 cells after rotenone treatment.
Assessment of viability was performed after 24 h of treatment with different concentrations of rotenone. Median viability difference between Hoechst/PI and trypan blue staining with automated counting was calculated based on viability differences at each drug concentration.
Supplementary Table S4. Viability difference between Hoechst/PI and TB exclusion method in OCI-AML2 cells after 3-bromopyruvate (3-BP) treatment.
Assessment of viability was performed after 24 h of treatment with different concentrations of 3-BP. Median viability difference between Hoechst/PI and trypan blue staining with automated counting was calculated based on viability differences at each drug concentration.
ACKNOWLEDGMENTS:
NVK, a CPRIT scholar in Cancer Research, thanks the Cancer Prevention and Research Institute of Texas (CPRIT) for their generous support, CPRIT grant RR150044. This work was also supported by the Welch Foundation Research Grant C-1930, and by the National Institutes of Health R35 GM129294 awarded to NVK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
DISCLOSURES:
The authors have nothing to disclose.
REFERENCES:
- 1.Ramirez CN, Antczak C & Djaballah H Cell viability assessment: toward content-rich platforms. Expert Opin Drug Discov. 5 (3), 223–233, doi: 10.1517/17460441003596685, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melzer S et al. Trypan blue as an affordable marker for automated live-dead cell analysis in image cytometry. Scanning. 38 (6), 857–863, doi: 10.1002/sca.21335, (2016). [DOI] [PubMed] [Google Scholar]
- 3.Sikora E, Mosieniak G & Sliwinska MA Morphological and Functional Characteristic of Senescent Cancer Cells. Curr Drug Targets. 17 (4), 377–387, doi: 10.2174/1389450116666151019094724, (2016). [DOI] [PubMed] [Google Scholar]
- 4.Coppé JP, Desprez PY, Krtolica A & Campisi J The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 5 99–118, doi: 10.1146/annurev-pathol-121808-102144, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castro-Vega LJ et al. The senescent microenvironment promotes the emergence of heterogeneous cancer stem-like cells. Carcinogenesis. 36 (10), 1180–1192, doi: 10.1093/carcin/bgv101, (2015). [DOI] [PubMed] [Google Scholar]
- 6.Weihua Z, Lin Q, Ramoth AJ, Fan D & Fidler IJ Formation of solid tumors by a single multinucleated cancer cell. Cancer. 117 (17), 4092–4099, doi: 10.1002/cncr.26021, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osisami M & Keller ET Mechanisms of Metastatic Tumor Dormancy. J Clin Med. 2 (3), 136–150, doi: 10.3390/jcm2030136, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S et al. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene. 33 (1), 116–128, doi: 10.1038/onc.2013.96, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mittal K et al. Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate cancer. Br J Cancer. 116 (9), 1186–1194, doi: 10.1038/bjc.2017.78, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKeague AL, Wilson DJ & Nelson J Staurosporine-induced apoptosis and hydrogen peroxide-induced necrosis in two human breast cell lines. Br J Cancer. 88 (1), 125–131, doi: 10.1038/sj.bjc.6600675, (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaja S et al. An optimized lactate dehydrogenase release assay for screening of drug candidates in neuroscience. J Pharmacol Toxicol Methods. 73 1–6, doi: 10.1016/j.vascn.2015.02.001, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan FK, Moriwaki K & De Rosa MJ Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol Biol. 979 65–70, doi: 10.1007/978-1-62703-290-2_7, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piccinini F, Tesei A, Arienti C & Bevilacqua A Cell Counting and Viability Assessment of 2D and 3D Cell Cultures: Expected Reliability of the Trypan Blue Assay. Biol Proced Online. 19 8, doi: 10.1186/s12575-017-0056-3, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plemel JR et al. Unique spectral signatures of the nucleic acid dye acridine orange can distinguish cell death by apoptosis and necroptosis. J Cell Biol. 216 (4), 1163–1181, doi: 10.1083/jcb.201602028, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galluzzi L et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 16 (8), 1093–1107, doi: 10.1038/cdd.2009.44, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cummings BS & Schnellmann RG Measurement of cell death in mammalian cells. Curr Protoc Pharmacol. Chapter 12 Unit 12.18, doi: 10.1002/0471141755.ph1208s25, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brauchle E, Thude S, Brucker SY & Schenke-Layland K Cell death stages in single apoptotic and necrotic cells monitored by Raman microspectroscopy. Sci Rep. 4 4698, doi: 10.1038/srep04698, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiaraviglio L & Kirby JE Evaluation of impermeant, DNA-binding dye fluorescence as a real-time readout of eukaryotic cell toxicity in a high throughput screening format. Assay Drug Dev Technol. 12 (4), 219–228, doi: 10.1089/adt.2014.577, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panina SB, Baran N, Brasil da Costa FH, Konopleva M & Kirienko NV A mechanism for increased sensitivity of acute myeloid leukemia to mitotoxic drugs. Cell Death Dis. 10 (8), 617, doi: 10.1038/s41419-019-1851-3, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caro P et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 22 (4), 547–560, doi: 10.1016/j.ccr.2012.08.014, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagadinou ED et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 12 (3), 329–341, doi: 10.1016/j.stem.2012.12.013, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Senft D & Ronai ZA Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol. 39 43–52, doi: 10.1016/j.ceb.2016.02.001, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vazquez F et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 23 (3), 287–301, doi: 10.1016/j.ccr.2012.11.020, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caino MC & Altieri DC Cancer cells exploit adaptive mitochondrial dynamics to increase tumor cell invasion. Cell Cycle. 14 (20), 3242–3247, doi: 10.1080/15384101.2015.1084448, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ralph SJ, Rodríguez-Enríquez S, Neuzil J, Saavedra E & Moreno-Sánchez R The causes of cancer revisited: “mitochondrial malignancy” and ROS-induced oncogenic transformation - why mitochondria are targets for cancer therapy. Mol Aspects Med. 31 (2), 145–170, doi: 10.1016/j.mam.2010.02.008, (2010). [DOI] [PubMed] [Google Scholar]
- 26.Kreitz J et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells. 8 (8), doi: 10.3390/cells8080805, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sriskanthadevan S et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood. 125 (13), 2120–2130, doi: 10.1182/blood-2014-08-594408, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guerra F et al. Mitochondrial Dysfunction: A Novel Potential Driver of Epithelial-to-Mesenchymal Transition in Cancer. Front Oncol. 7 295, doi: 10.3389/fonc.2017.00295, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guerra F, Arbini AA & Moro L Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. 1858 (8), 686–699, doi: 10.1016/j.bbabio.2017.01.012, (2017). [DOI] [PubMed] [Google Scholar]
- 30.Neuzil J, Dong LF, Rohlena J, Truksa J & Ralph SJ Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion. 13 (3), 199–208, doi: 10.1016/j.mito.2012.07.112, (2013). [DOI] [PubMed] [Google Scholar]
- 31.Ubah OC & Wallace HM Cancer therapy: Targeting mitochondria and other sub-cellular organelles. Curr Pharm Des. 20 (2), 201–222, doi: 10.2174/13816128113199990031, (2014). [DOI] [PubMed] [Google Scholar]
- 32.Yamaguchi R et al. Efficient elimination of cancer cells by deoxyglucose-ABT-263/737 combination therapy. PLoS One. 6 (9), e24102, doi: 10.1371/journal.pone.0024102, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hahn T et al. Use of anti-cancer drugs, mitocans, to enhance the immune responses against tumors. Curr Pharm Biotechnol. 14 (3), 357–376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panina SB, Pei J, Baran N, Konopleva M & Kirienko NV Utilizing Synergistic Potential of Mitochondria-Targeting Drugs for Leukemia Therapy. Front Oncol. 10 435, doi: 10.3389/fonc.2020.00435, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lema C, Varela-Ramirez A & Aguilera RJ Differential nuclear staining assay for high-throughput screening to identify cytotoxic compounds. Curr Cell Biochem. 1 (1), 1–14 (2011). [PMC free article] [PubMed] [Google Scholar]
- 36.Döhner H et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 115 (3), 453–474, doi: 10.1182/blood-2009-07-235358, (2010). [DOI] [PubMed] [Google Scholar]
- 37.Heinz S et al. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci Rep. 7 45465, doi: 10.1038/srep45465, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan T et al. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers (Basel). 11 (3), doi: 10.3390/cancers11030317, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pal R, Mamidi MK, Das AK & Bhonde R Diverse effects of dimethyl sulfoxide (DMSO) on the differentiation potential of human embryonic stem cells. Arch Toxicol. 86 (4), 651–661, doi: 10.1007/s00204-011-0782-2, (2012). [DOI] [PubMed] [Google Scholar]
- 40.Tunçer S et al. Low dose dimethyl sulfoxide driven gross molecular changes have the potential to interfere with various cellular processes. Sci Rep. 8 (1), 14828, doi: 10.1038/s41598-018-33234-z, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Representative images of cells stained with Hoechst/PI, with or without rotenone treatment at indicated concentrations in serum-free RPMI-1640 media for 24 h: A-B. AML: MOLM-13 cell line (A), and primary AML cells (B) derived from a representative patient sample. C. ALL: MOLT-4 cell line. D. CML: K562 cell line.
Supplementary Table S1. Viability differences for a panel of assays in OCI-AML2 cells after CCCP treatment.
Assessment of viability was performed after 24 h of treatment with different concentrations of CCCP. Median viability differences between tested assays (Hoechst/PI, LDH, MTT, or alamarBlue) and trypan blue staining with automated counting was calculated based on viability differences at each drug concentration.
Supplementary Table S2. Viability differences for a panel of assays in OCI-AML2 cells after 2-DG treatment.
Assessment of viability was performed after 24 h of treatment with different concentrations of 2-DG. Median viability differences between tested assays (Hoechst/PI, LDH, MTT, or alamarBlue) and trypan blue staining with automated counting were calculated based on viability differences at each drug concentration.
Supplementary Table S3. Viability difference between Hoechst/PI and TB exclusion method in OCI-AML2 cells after rotenone treatment.
Assessment of viability was performed after 24 h of treatment with different concentrations of rotenone. Median viability difference between Hoechst/PI and trypan blue staining with automated counting was calculated based on viability differences at each drug concentration.
Supplementary Table S4. Viability difference between Hoechst/PI and TB exclusion method in OCI-AML2 cells after 3-bromopyruvate (3-BP) treatment.
Assessment of viability was performed after 24 h of treatment with different concentrations of 3-BP. Median viability difference between Hoechst/PI and trypan blue staining with automated counting was calculated based on viability differences at each drug concentration.