

Abstract
Programmed ribosomal frameshifting (PRF) is a conserved translational recoding mechanism found in all branches of life and viruses. In bacteria, archaea, and eukaryotes PRF is used to downregulate protein production by inducing a premature termination of translation, which triggers messenger RNA (mRNA) decay. In viruses, PRF is used to drive the production of a new protein while downregulating the production of another protein, thus maintaining a stoichiometry optimal for productive infection. Traditionally, PRF motifs have been defined by the characteristics of two cis elements: a slippery heptanucleotide sequence followed by an RNA pseudoknot or stem-loop within the mRNA. Recently, additional cis and new trans elements have been identified that regulate PRF in both host and viral translation. These additional factors suggest PRF is an evolutionarily conserved process whose function and regulation we are just beginning to understand.
Keywords: programmed ribosomal frameshifting, PRF, protein regulators, mRNA regulators, cis-acting elements, trans-acting elements
1. INTRODUCTION
Programmed ribosomal frameshifting (PRF) is a translational recoding mechanism in which the translating ribosome slips into an alternative open reading frame (ORF) during translation. PRF is not a translational mistake or an arbitrary process; it is a regulated, controlled event that results in either the introduction of a premature termination codon or the production of a new protein. It is found in viruses and hosts in all domains of life (bacteria, eukaryotes, archaea) (1, 2). PRF typically arises from the effects of two key motifs within the transcript: a heptanucleotide ribosomal slippery sequence (or slip-site) followed by a downstream RNA structure. However, additional cis factors and new trans factors (both from the virus and from the host) have been discovered to be important for modulating PRF events (Figure 1). In this review, we discuss the mechanism of PRF as well as how the efficiency of PRF can be modified by recently discovered cis and trans modulators of PRF, with an emphasis on the relevance of these mechanisms to viral infection.
Figure 1.
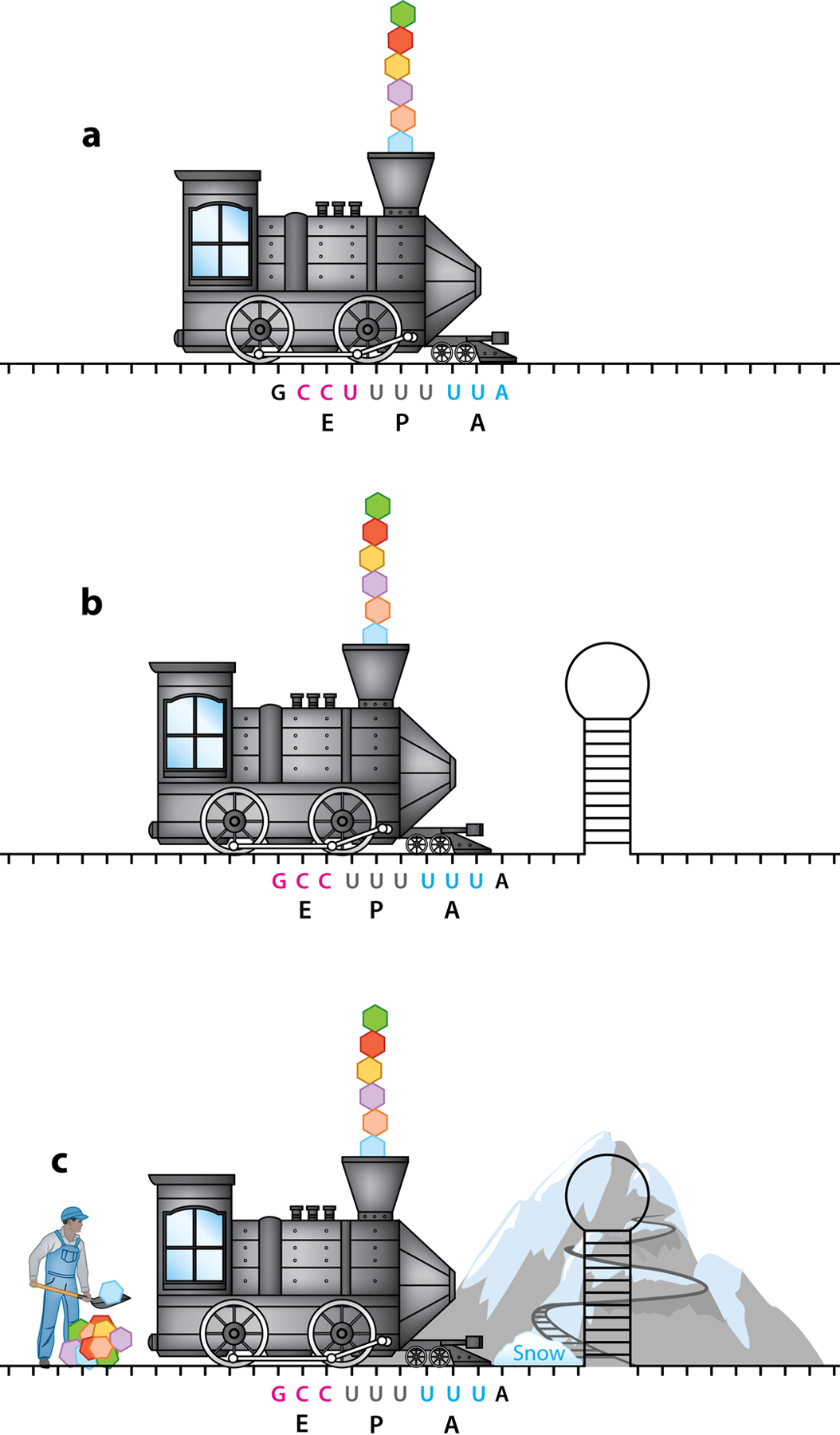
Schematic of how cis and trans elements affect viral and host PRF translation. The train represents the ribosome, and the tracks represent the mRNA. The slippery-site is U UUU UUA with the designated codons in the E, P, and A sites shown in pink, gray, and blue, respectively. The newly formed polyprotein chain is being released from the train’s smokestack. (a) The ribosome chugs along the mRNA, translating a new polyprotein. The E, P, and A sites are labeled along with the corresponding codons. (b) PRF is usually stimulated by an mRNA secondary structure downstream of the slip-site. An encounter with the mRNA stem-loop allowed the train to slip back 1 nucleotide (−1 PRF; shown here), and now new codons are in the EPA sites of the ribosomes. (c) cis and trans regulatory factors have been identified to modulate PRF. These include (but are not limited to) the mountain, which represents proteins or microRNA that bind and stabilize the mRNA stem-loop and forces the train to slow down, enhancing PRF; the snow, which is a host or viral protein that causes the ribosome train to pause, which is quite prevalent; and the concentration of translational factors including tRNAs (depicted by the worker shoveling tRNAs into the train) that influence kinetics and thermodynamics of translation, which is prevalent in +1 PRF. Abbreviations: EPA, exit, peptidyl, and aminoacyl; mRNA, messenger RNA; PRF, programmed ribosomal frameshifting; tRNA, transfer RNA.
2. FRAMESHIFTING IN VIRUSES
The first description of viral PRF was in relation to Rous sarcoma virus (RSV) in 1985 (3), but clues implicating a unique modulator of translation had previously been identified. The RSV genome was sequenced in 1983 by Walter Gilbert and colleagues (4) and allowed for the assignment of RSV proteins to specific genes. Upon assignation it became clear that the pol gene (encoding reverse transcriptase, integrase, and protease) lay 20 nucleotides downstream of the gag gene but appeared to be in a completely different reading frame (4). Given that the gag gene is terminated by an amber stop codon followed by 7 other termination signals in the same reading frame, it was unclear what drove production of the downstream pol gene (4). From previous cellular studies, it was known that the ratio of Gag to Gag-Pol polyprotein was ~20:1 and this stoichiometry was needed for productive infection (5). Therefore, some event was regulating the rate of pol gene translation while simultaneously changing the reading frame (4). One hypothesis was that an RNA splicing event was responsible for a new messenger RNA (mRNA) and translation of the Pol polyprotein. However, there were no donor sites in the RSV genome that would produce the correct change in the reading frame (4). Resolution of this quandary came from Jacks & Varmus (3), who employed a cell-free translation system in order to demonstrate that the product of the pol gene was produced if the ribosome shifted from the 0 to −1 reading frame. From this experiment, it was clear that a single mRNA was responsible for the production of both gene products (3). Furthermore, they found that the ratio of Gag to Gag-Pol polyprotein in their experiments was about 20:1, consistent with the previous observations in infected cells while also providing an explanation for the stoichiometric differences in pol production with relation to gag (3). After discovery of PRF in RSV, other retroviruses were investigated—most notably human immunodeficiency virus type 1 (HIV-1), which contains a similar overlap in the reading frames of the gag and pol genes (6).
The first instance of nonretroviral PRF was found in coronavirus infectious bronchitis virus (IBV) (7). Coronavirus IBV was suspected to undertake PRF because it contains a brief overlapping ORF in two of its genes that was similar to those found in retroviruses (7). Since then, PRF events have been identified in over a dozen viral families (1). During PRF, the ribosome most commonly slips into the −1 reading frame by slipping backward 1 nucleotide, although examples of the ribosome shifting by +1, −2, and +2 nucleotides have also been identified (1, 8–10) (Figure 2). PRF events are mostly found in RNA viruses, although PRFs in DNA viruses have been identified (11–15).
Figure 2.
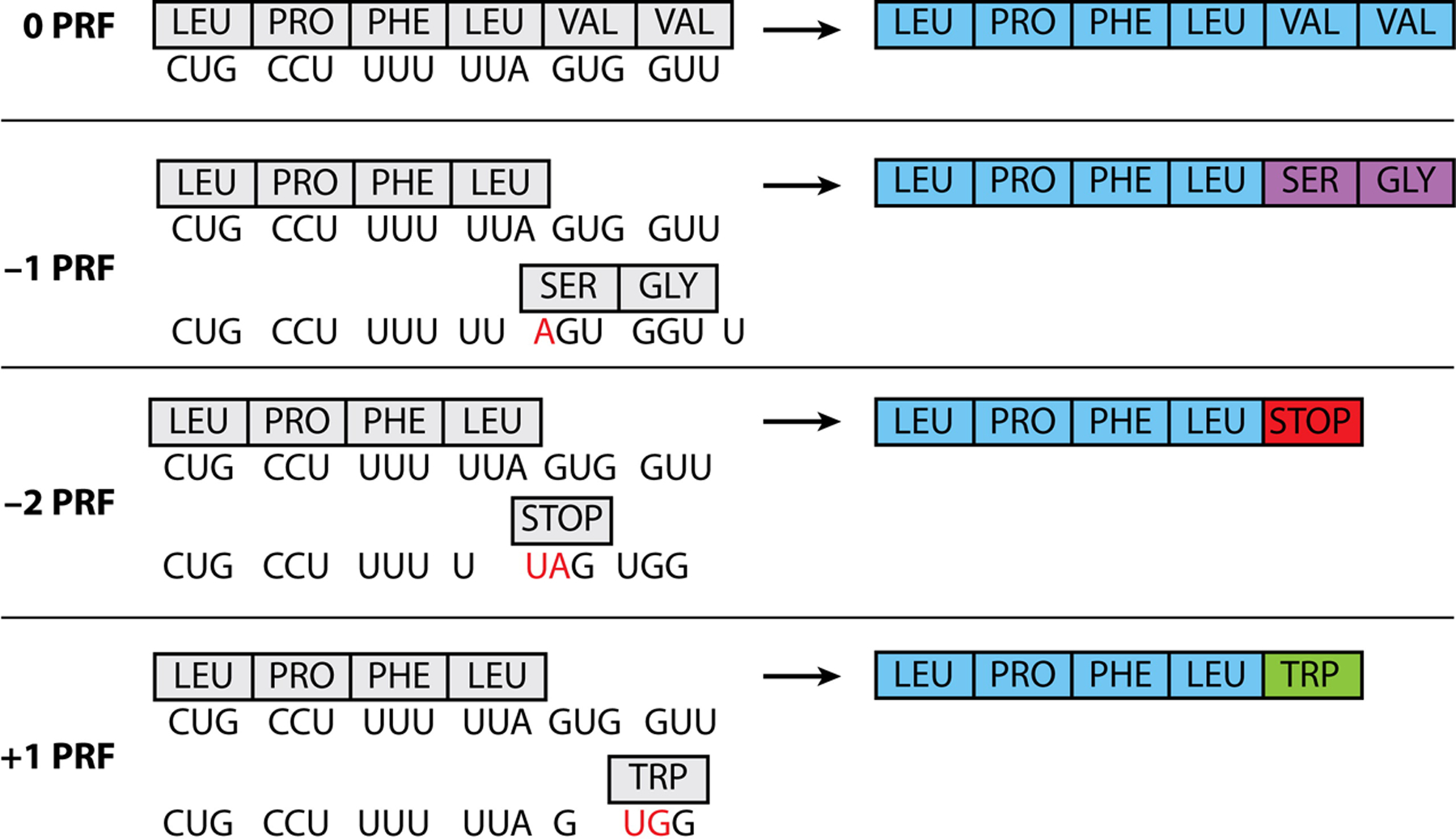
Illustration of different PRFs. The first row depicts no PRF or 0 frame, followed by −1, −2, and +1. (Left) Nucleotide sequences and their corresponding amino acids. The red nucleotide shows the start of the new codon during PRF. (Right) The resulting newly formed polyprotein sequence. Note that the sequence used is for illustration purposes only. While there are characteristic motifs, each virus is slightly unique for their motifs and sequences required for viral PRF events. Abbreviation: PRF, programmed ribosomal frameshifting.
In eukaryotic host cells, most PRF events result in the production of premature termination codons that induce the nonsense-mediated decay (NMD) pathway or generate a truncated protein (1, 2, 16, 17). In contrast, viruses mainly use PRF for the production of new proteins; however, such consistent use of frameshifting potentially makes viral mRNA a target for NMD. While the exact details are beyond the scope of this review [please see Hogg’s excellent review (18)], research has shown a complex, often virus-specific range of effects including mRNA degradation, virus-specific inhibition of NMD, and even global inhibition of NMD (18). PRF also increases the coding capacity of viral genomes, which are often limited in size due to the geometric constraints associated with particle size (19).
Proteins produced by PRF often include virulence factors and polymerases. For example, PRF in viruses is equated with viral proteins whose production is needed for virus replication, or produced late in infection and required for particle assembly (6). Alternatively, or in addition, the PRF products could antagonize host antiviral responses (20–22) that are required for viral spread and pathogenesis. In these cases, PRF allows translational control of their stoichiometry, temporal production, and spatial localization in a manner that is crucial for a successful viral infection (1, 9, 21, 23–29)—too much or too little production at the wrong time or place can hinder a productive infection. Such spatiotemporal regulation of host proteins is typically coordinated by a spectrum of transcription factors and RNA proteins. PRF therefore represents an appealing alternative to complex regulatory schemes that require numerous additional proteins.
3. CIS ELEMENTS THAT MODULATE PROGRAMMED RIBOSOMAL FRAMESHIFTING
3.1. Slippery Sequences
In most contexts, the minimum requirements for PRF are the presence of a heptanucleotide slippery sequence (or slip-site) followed by a downstream mRNA stem-loop or pseudoknot secondary structure. The slip-site itself functionally defines PRF and provides a hardwired position within the transcript where the ribosome can slip or move between alternative reading frames. Traditionally, the slip-site has been defined as X XXY YYZ, where X represents 3 identical nucleotides, Y represents A or U, and Z represents any nucleotide, A, C, or U (30, 31). This slip-site is situated across 3 codons. Prior to frameshifting, the peptidyl (P)-site transfer RNA (tRNA) occupies the XXY nucleotides while the aminoacyl (A)-site tRNA dwells on the YYZ nucleotides. During −1 PRF (the most common event), now the P-site and A-site tRNAs instead occupy the XXX and YYY nucleotides, respectively (Figure 2); this is also referred to as the two-tRNA mechanism of PRF. While rules governing the nucleotide identity at these positions are well established, readers are cautioned against overreliance upon these rules, as bioinformatics and proteomics are identifying exceptions to these rules and deviations are becoming more common.
3.2. Downstream Messenger RNA Structures
Most high-efficiency PRF sites feature a discrete stimulatory mRNA secondary structure positioned 5–9 base pairs downstream from the slip-site (32, 33), although there are some exceptions described in Section 3.2.1. This spacing is critical for the efficiency of PRF (34), as it positions the base of the stimulatory structure at the edge of the mRNA entrance channel while the ribosome dwells on the slip-site (35). Based upon these common features of PRF motifs, Hammell et al. (17) evaluated the relative abundance of putative PRF motifs within various genomes. For example, PRF motifs (slip-site + spacer + RNA structure) are 5.22-fold more common in yeast than would be expected based on random chance. In contrast, PRF motifs appear to be enriched by only 2.67-fold in the human genome. Nevertheless, viruses are clearly more reliant on translational recoding, as PRF motifs are enriched sevenfold within viral genomes, reinforcing the importance of frameshifting in viral infections (2, 17, 27).
Single-molecule investigations have provided considerable insight into the stochastic behavior of individual ribosomes and how the probability of certain transitions changes during PRF. Translation occurs through processive, stepwise translocation events interspersed by kinetic pauses (36). These pauses, and the resulting dwell time, vary considerably in length but tend to increase substantially when the ribosome encounters a region of structured mRNA (36). The ribosome has its own helicase, and RNA viruses also encode additional helicases that unwind these double-stranded RNA and mRNA secondary structures. However, a cryo–electron microscopy structure of a ribosome stalled at the stimulatory pseudoknot from coronavirus shows that these structures appear to block the mRNA entrance channel. Nevertheless, the stimulatory structures that pause ribosomes on viral slip-sites have evolved to test the limits of the ribosomal helicase. The mechanical tension produced by this blockage displaces the ribosomal helicase domain and compresses the P-site tRNA (35). This propagation of mechanical tension suggests that PRF does not arise from the formation of a unique, structurally specific interface between the transcript and ribosome but rather arises from a form of mechanochemical allostery (37). This lack of structural specificity likely accounts for the structural diversity among known stimulatory RNA structures, especially in viruses.
3.2.1. Role of 3′ messenger RNA structures.
Although the distance between the slip-site and stimulatory structure is constrained by the geometry of the ribosome, stimulatory RNA structures themselves come in a variety of shapes and sizes (32). The structural diversity of these stimulatory structures reflects the fact that their effects arise from their mechanical properties rather than from specific structural interactions. While the effect of these structures on the efficiency of PRF does not appear to be strictly related to the thermodynamic stability of the ground-state structure (32, 38), in a few cases, the mechanical force required to globally unfold stimulatory structures does appear to correlate with the observed efficiency of PRF (38, 39). This observation highlights the role of mechanical resistance in the mechanism of PRF. However, such correlations do not appear to be generalizable (40). Subsequent investigations have instead found PRF efficiency to be more closely linked to the mechanical force required to locally unfold the first 3–4 base pairs in the stem-loop of the stimulatory structure (34). Although partial unfolding of these structures may leave much of the remaining downstream structure intact, the rupture of this portion of the structure is sufficient to allow the ribosome to move beyond the slip-site. Once the ribosome passes the slip-site, within which the thermodynamic penalty of recoding is minimal (41), the fidelity of translation is restored.
The local stability at the base of the stem within the ground-state structure appears to be a key feature of the downstream stimulatory regions, at least for those that have an intrinsic thermodynamic propensity to fold into a single unique structure. However, both structural investigations and single-molecule force spectroscopy measurements have provided evidence that the net effects of certain stimulatory regions appear to arise not from the properties of a single structure but instead from an ensemble of competing RNA structures (40, 42, 43). Thus, certain stimulatory regions are likely to sample a spectrum of distinct conformational states between successive rounds of ribosome-mediated unwinding (42). Each of these structures is likely to vary with respect to its intrinsic propensity to stimulate ribosomal frameshifting (44). Based on these considerations, it seems likely that quantitative frameworks to describe the efficiency of ribosomal frameshifting will, in some cases, have to account for the nature of these conformational ensembles as well as the specific effects of each individual structure on ribosomal frameshifting. It should be noted that the discovery of ensemble-based stimulation of PRF has important implications for viral evolution, as mutations within the stimulatory region could disrupt one conformation while preserving another (43). The formation of alternative stimulatory structures may also alleviate certain evolutionary constraints related to the fact that both the mRNA secondary structure and viral proteins are dual encoded. Thus, this structural plasticity likely widens the number of accessible evolutionary pathways that preserve the desired efficiency of ribosomal frameshifting. Additionally, it seems likely that these ensembles are sensitive to changes in both cellular pH and the relative bioavailability of various stabilizing counterions (44–46). Thus, changes in cellular physiology that occur during viral infection could potentially tune these ensembles in a manner that leads to temporal control of PRF.
3.2.2. Kinetic and thermodynamic basis of ribosomal frameshifting.
Efforts to understand how viruses control translational recoding have largely focused on the interplay between the ribosome and the structural features of the transcript. However, it is also clear that recoding efficiencies are dependent upon the abundance of ribosomes (47), tRNAs (discussed more in Section 4.3.1) (48), and elongation factors (49) in relation to the number of viral transcripts. These stoichiometric dependencies highlight the fact that, like most biochemical phenomenon, viral recoding is typically under kinetic control (32). Thus, a fundamental understanding of how the viruses control PRF requires a kinetic framework that accounts for the relative rates of structural transitions involved in decoding and recoding. Recent efforts to probe the mechanism of PRF using single-molecule force spectroscopy and fluorescence resonance energy transfer (both single-molecule and ensemble measurements) have revealed key changes in the kinetics of translation during recoding, and several kinetic models have been proposed based on these observations (50–54).
Current kinetic models of PRF feature some subtle mechanistic differences, which may arise in part from the differences in the abilities of these techniques to detect and resolve certain intermediates. It also seems likely that the kinetic constraints differ across PRF sites and that frameshifting can occur through a spectrum of coexisting pathways (48, 50, 55). Nevertheless, these frameworks contain several common features. PRF occurs during translocation and appears to occur at some point during the transition from the pre- to post-translocation complex (51–54). Importantly, encountering a stimulatory RNA structure significantly increases the dwell time of ribosomes on the slip-site (51–53). During this pause, the ribosome undergoes multiple futile cycles of EF-G binding and GTP hydrolysis that fail to drive translocation (54). The kinetic barrier imposed by the secondary structure effectively decreases the rate of translocation in the 0 frame in relation to the rate of frameshifting (51). Viewed through this lens, it seems that recoding arises from an increase in the kinetic barrier to translocation while the ribosome occupies a site where the tRNA can explore non-native base pairing interactions (41, 54). Given the number of variables that affect translocation and/or the kinetic stability of the downstream stimulatory structure, the interplay between the kinetic and thermodynamic drivers of PRF is likely to vary considerably among PRF sites and perhaps throughout the course of viral infection. Additional insights into the mechanistic basis of PRF are sure to reveal additional manners in which viruses are capable of manipulating recoding events.
3.2.3. Upstream regulatory elements.
Intriguingly, two key Coronaviridae members [severe acute respiratory syndrome-related coronavirus (SARS-CoV) and SARS-CoV-2] utilize upstream hairpin structures to modulate −1 PRF efficiency (56, 57). Termed attenuators by Su and colleagues (56), these elements appear to significantly decrease the amount of frameshifting in both a sequence- and position-dependent manner (58). Conversely, there are examples of PRF being upregulated by RNA sequences far upstream of the slip-site in several viruses and hosts, but the exact mechanism is not well understood (1, 58–61). We refer readers to specific references to learn more about these examples.
3.3. Conformational Transitions in the Nascent Chain
Until recently, the mechanistic basis of PRF was believed to stem from the cis structural features within the mRNA transcript, primarily the slip-site and the downstream mRNA secondary structures. However, a recent investigation of the Sindbis virus structural polyprotein has revealed an unprecedented connection between the conformational state of the nascent polypeptide chain and ribosomal frameshifting. The transmembrane (TM) domain found within the 6K protein (TM3 in Figure 3) is encoded upstream from the slip-site in the mRNA and is therefore present in both 6K (0-frame) and the −1 frameshifted TransFrame (TF) proteins. Interestingly, this TM domain contains an interfacial cluster of conserved cysteine residues that was found to be palmitoylated in TF but not in 6K (25). The topological orientation of these cysteines in relation to the cytosolic localization of palmitoylation machinery provided a key insight—the selective modification of these side chains in the context of the frameshifted TF protein would require a topological inversion of this TM domain. Through an array of biochemical, cellular, and computational techniques, it was shown that the ribosomal frameshifting is stimulated by the less frequent cotranslational membrane integration of an additional, marginally hydrophobic TM domain (62) (TM2 in Figure 3). The membrane integration of TM2 (~20% efficient) coincides with formation of the TF protein and results in topological inversion of TM3, which exposes the interfacial cysteines to the palmitoylation machinery in the cytosol. Moreover, the efficiency of frameshifting was found to be proportional to the mechanical tension generated by the cotranslational membrane integration of TM2. The properties of this TM domain that stimulate PRF were found to be conserved among alphaviruses, suggesting that alphaviruses rely on this mechanism to maintain optimal frameshifting efficiency (63, 64). These observations demonstrate for the first time that pulling forces generated by the cotranslational folding of the nascent polypeptide chain are capable of stimulating PRF (62).
Figure 3.
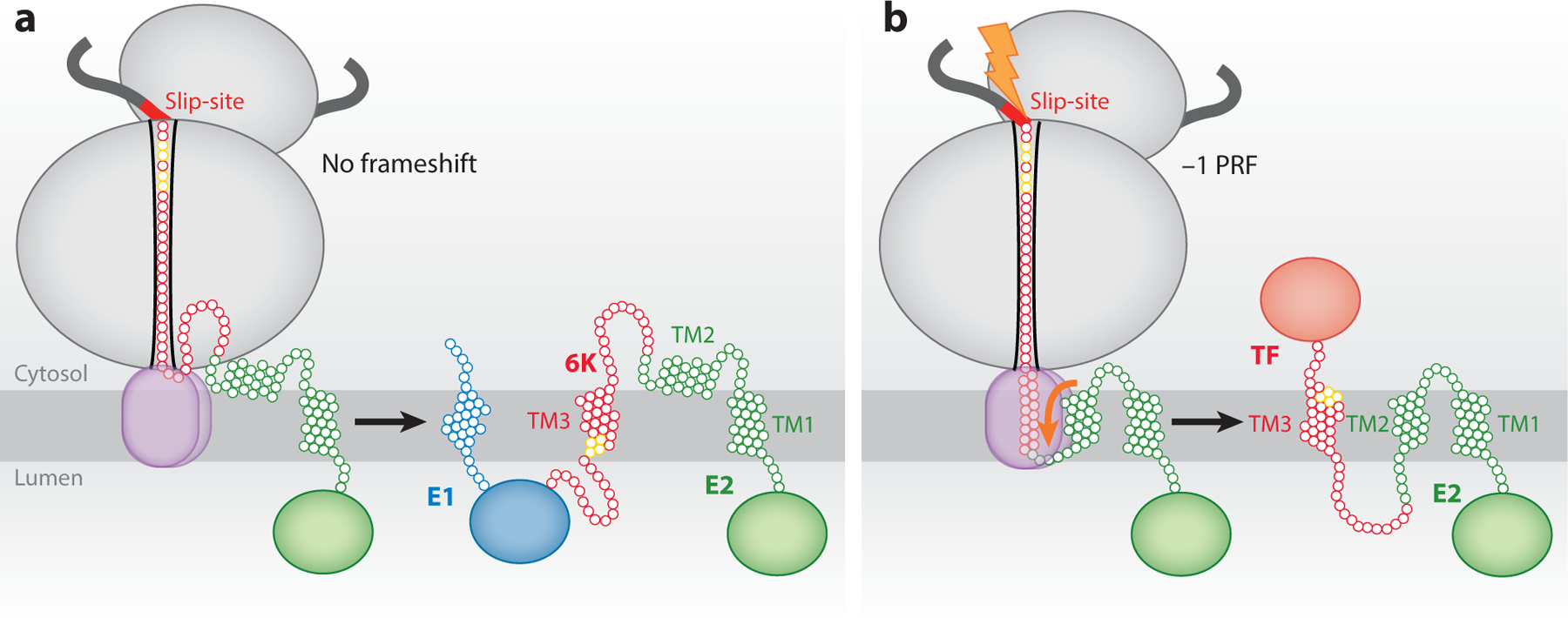
Schematic depicting how conformational transitions in the nascent polyprotein chain affect PRF. The ribosome is shown in gray, and the translocon is outlined in brown. (a) The topological properties of the major form of the nascent alphavirus structural polyprotein of the glycoproteins. In this scenario, the TM2 is too polar to robustly partition into the membrane during translation, no PRF occurs, and the 6K protein is produced. (b) TM2 is hydrophobic enough to occasionally partition into the membrane during translation. When it does, it imposes a tension on the ribosome that stimulates −1 PRF. As a result, the TF protein is translated. In this case, E1 is not produced. Residues in yellow are cysteines that are palmitoylated in TF but not 6K. The residues need to be on the cytoplasmic side for this post-translational event to occur. Abbreviations: E1 and E2, viral envelope glycoproteins; PRF, programmed ribosomal frameshifting; TF, TransFrame protein; TM, transmembrane. Figure adapted from Reference 62.
Emerging research has revealed that a wide array of cotranslational folding and binding reactions can generate tension in the nascent chain. These transitions include the folding of water-soluble protein domains (65), ligand binding reactions in or near the exit tunnel (66), the translocon-mediated membrane integration of nascent TM domains (67–69), and the association of molecular chaperones with the nascent polypeptide chain (70). In principle, it seems likely that any of these events could be exploited to tune the efficiency of PRF, provided they coincide with the ribosome pausing on a slip-site within the transcript. It should also be noted that the mechanical forces generated by folding transitions are typically on the order of 20–40 pN, which is similar in scale to the forces required to mechanically unfold stimulatory RNA structures.
The connection between nascent chain pulling forces and PRF could potentially explain why recoding appears to be sensitive to the expression of ribosome-associated chaperones in yeast (71). These PRF modifiers, and the potential for mechanistic heterogeneity, may also expand the accessible sequence space of viruses that must maintain a specific PRF efficiency. For instance, mutations that weaken stimulatory RNA structures could potentially be compensated for by others that enhance pulling forces on the nascent chain. The diverse nature of these potential effectors may also allow viruses to evolve recoding mechanisms that are active only in certain cell types or that respond to specific changes in cellular conditions. For instance, differences in the composition of the proteostasis network that occur between cells and/or during the course of viral infection may alter the probability associated with the formation of cotranslational chaperone interactions that generate pulling forces in the nascent chain. A link between stimulatory pulling forces and the proteostasis network may therefore allow viruses to adjust their translation products in response to changing cellular conditions during replication and assembly. Additional investigations are needed to evaluate the ways in which viruses exploit this phenomenon to tune ribosomal frameshifting.
Finally, it should be noted that some viral proteins evolve to adopt multiple protein conformations that are associated with distinct functions (19). Thus, the propensity of a protein to switch between distinct conformations during translation could also be hypothetically employed to tune PRF to optimize a productive infection. Differences in the propensity of these conformations to cotranslationally interact with molecular chaperones could also help to tune viral translation in response to changing cellular conditions during replication and assembly. Additional investigations are needed to evaluate the ways in which viruses exploit this emerging mechanism for ribosomal frameshifting.
4. TRANS ELEMENTS THAT MODULATE PROGRAMMED RIBOSOMAL FRAMESHIFTING
Classically, PRF was believed to arise from the interplay between factors intrinsic to the translational process: ribosomes, mRNA, and tRNA. However, a growing body of work has begun to describe a role for factors outside this classical triad. These host- and/or virus-derived trans-acting elements are capable of inducing, modulating, and inhibiting PRF through a variety of mechanisms. The +1 PRF mechanisms in Escherichia coli release factor 2 and human ornithine decarboxylase, the latter of which is stimulated by polyamine, likely represent the earliest description of trans-acting factors that mediate PRF (72, 73). Subsequent to this, Muldoon-Jacobs & Dinman (71) found that the ribosome-associated molecular chaperone complexes Ssz1p/Zuo1p and Ssb1p/Ssb2p are capable of tuning −1 PRF in yeast based on the observation that the deletion of these genes decreased frameshifting efficiency by ~50%. Below we describe trans factors that directly relate to viral infections (Figure 4).
Figure 4.
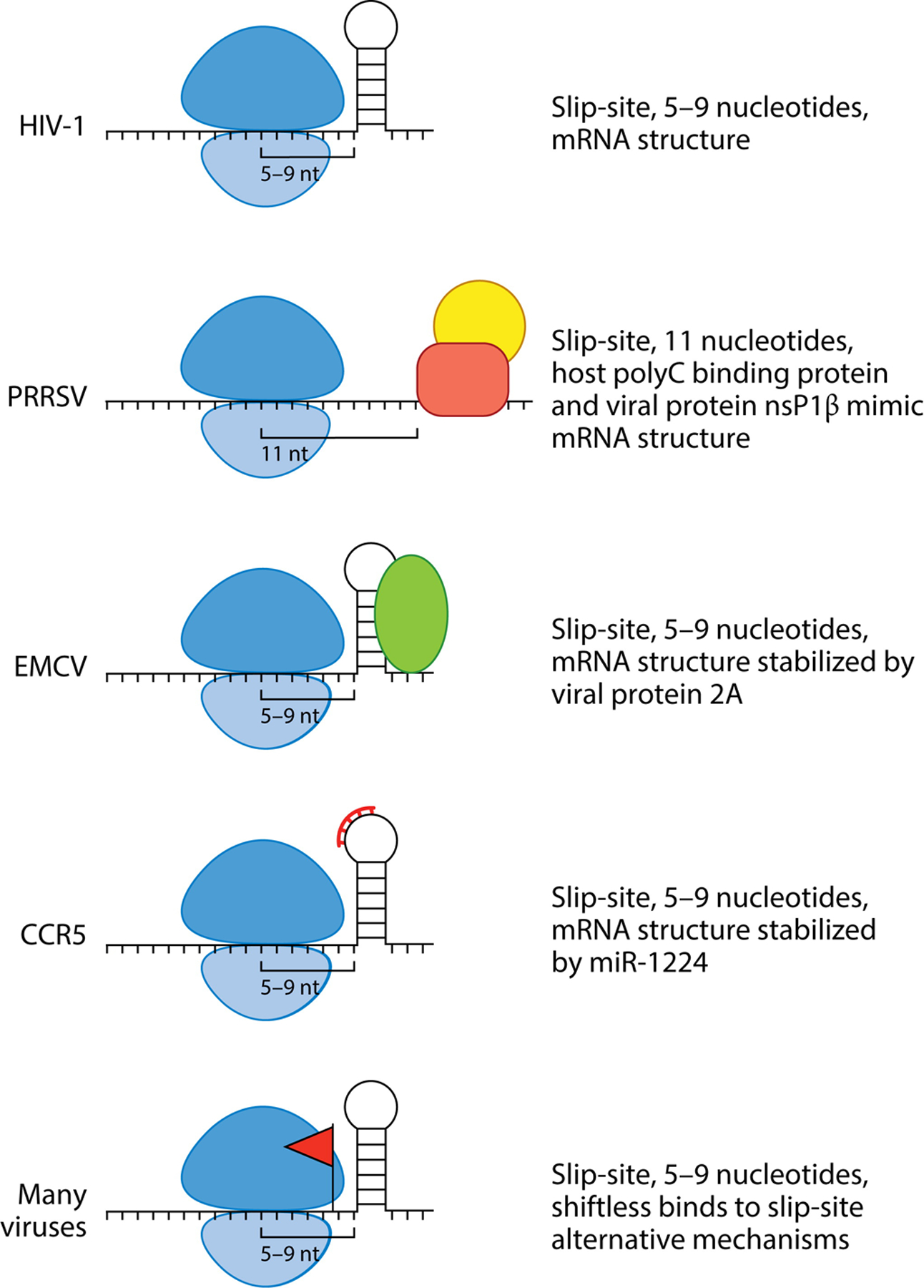
Schematics of different mechanisms of PRF modulation by trans factors, viral and host. The ribosome is depicted in blue. No trans factors that modulate HIV-1 have been identified. PRRSV does not contain a downstream mRNA element but rather two proteins; a viral and a host protein bind together to mimic the structure of the conical mRNA structure. EMCV has a viral protein that is generated late in infection that stabilizes the mRNA loop to promote PRF. CCR5 uses miR-1224 to promote PRF. Shiftless has been found to modulate PRF in many viruses. In some cases, the protein interacts with the slip-site, but in other viruses, PRF has not been identified, so the Shiftless mechanism for protein translation is unknown. Abbreviations: CCR5, cysteine-cysteine chemokine receptor 5; EMCV, encephalomyocarditis virus; HIV-1, human immunodeficiency virus type 1; mRNA, messenger RNA; PRF, programmed ribosomal frameshifting; PRRSV, porcine reproductive and respiratory syndrome virus.
4.1. Nucleic Acids
Various oligonucleotides stimulate frameshifting in trans. Several groups have annealed short DNA and RNA antisense oligonucleotides downstream of −1 PRF slip-sites in experimental frameshifting constructs that lack the pseudoknot/stem-loop stimulatory elements (74–76). These duplex structures appear to induce substantial frameshifting by causing a ribosomal pause on the slip-site. Modifications of the regions downstream of the slip-site are consistent with the work of Mouzakis et al. (34) showing that the thermodynamic stability of the first 3–4 nucleotides of the HIV-1 stem-loop are primary determinants of frameshifting efficiency. In addition, stem-loop plasticity, or the ability to form multiple different pseudoknots, has been shown to mediate PRF in E. coli and West Nile virus (WNV) and may play a role in oligo-mediated frameshifting (42, 77). The mediation of frameshifting by oligonucleotides could potentially play a key role in the immune response. Indeed, Belew and coworkers (78) have shown that miR-1224 induces significant frameshifting of the HIV-1 coreceptor cysteine-cysteine chemokine receptor 5 (CCR5) via NMD. It is hypothesized that miR-1224 binds to the 3′downstream mRNA stem-loop and substantially increases its stability, leading to increased ribosomal pausing and enhanced PRF (78). Importantly, miR-1224 has little effect on HIV-1 frameshifting while small interfering RNA knockdown of argonaute can stimulate or inhibit −1 PRF in interleukin receptors implicating the sequence specificity of this mechanism (78).
4.2. Proteins
The finding that both host and viral proteins are capable of regulating PRF has important implications for the temporal regulation of PRF during viral infection. Indeed, the programmatic expression of infectious viral and immune response host proteins likely underlies the observed variability in PRF induction rates.
4.2.1. Viral proteins.
Protein-stimulated PRF is observed in porcine reproductive and respiratory syndrome virus (PRRSV), a member of the Arteriviridae family. In this virus, two PRF events, −2 and −1, occur in the ORF1a and produce the immune-suppressing nsp2TF and nsp2N proteins, respectively. Interestingly, downstream of the slip-site there appears to be no 3′mRNA structural element, in contrast to the regulatory elements identified in other viral systems. Upstream of the −2 and −1 slip-site is the gene for nsP1β, a replicase protein (20). In the presence of nsP1β and the cellular protein poly (C) binding protein (PCBP), frameshifting of both the nsp2TF and nsp2N proteins is upregulated. nsP1β has been shown to directly interact with the viral RNA, and regions of the protein and regions of the viral RNA that are important for binding have been mapped (20, 79, 80). A complex of nsP1β, PCBP, and viral RNA binds to a C-rich region 11 nucleotides downstream of the slip-site, further away than other PRF motifs; this protein complex is thought to mimic an RNA secondary structure (79, 80).
In encephalomyocarditis virus (EMCV), the 2B protein is produced as a result of −1 PRF. The production of the EMCV 2B protein increases from 0% early in infection to 70% late in infection, in contrast with other viruses where PRF occurs at similar rates throughout the infection cycle (26). Uniquely, the EMCV 2B mRNA secondary structure is 14 nucleotides downstream from its slip-site, more distal than the typical 5–9-nucleotide distance. The viral EMCV 2A protein binds to and stabilizes the downstream mRNA stem-loop similar to what is proposed for CCR5/miR-1124 (26). Consequently, −1 PRF is increased leading to a downregulation of replicase protein translation and promotion of virus assembly. Both the EMCV 2A and 2B proteins have roles in pathogenesis and inhibiting host response (21, 81), so PRF modulation is important for balancing virus assembly and virus infection. Note that the function of 2B protein is not conserved in all picornaviruses, and the regulation described here is specific to EMCV.
4.2.2. Host proteins.
Proper synthetic stoichiometry is critical for viral assembly, packaging, and maturation (82). Thus, processes that perturb these ratios are likely to be inherently antiviral in nature. A novel, interferon (IFN)-stimulated eukaryotic protein that appears to exclusively inhibit −1 PRF has been recently identified (82). Termed Shiftless (SHFL) for its antiframeshifting behavior, the protein was initially described as an inhibitor of dengue virus (DENV) replication as well as hepatitis C virus (HCV), Kunjin virus, Chikungunya virus, Karposi sarcoma–associated herpesvirus (KSHV), and human adenovirus (HA) (83). Mechanistically, SHFL appears to bind the mRNA slip-site in such a way as to induce ribosomal stalling followed by a premature termination of translation, which can be overcome by the eukaryotic release factor (eRF)1-eRF3 complex. Curiously, DENV, HCV, HA, and KSHV have not been shown to undergo −1 PRF, which implies that they must be regulated in a nonslip-site-dependent manner (82). SHFL has been found to affect other viruses including several retroviruses and alphaviruses that do have −1 PRF, and SHFL might be acting on these viruses via different mechanisms. However, by extending their work to show that the human CCR5 and PEG10 genes are also subject to SHFL regulation, Wang and coworkers (82) provided a possible explanation for the non-PRF-associated inhibition of DENV, HCV, HA, and KSHV via modulation of some host factor(s). Furthermore, these effects provide a rationale for speculation as to the healthy-state function of SHFL. While IFN-β has been shown to greatly increase SHFL expression across numerous cell lines, it is important to note that all of these cell lines clearly expressed SHFL absent IFN-β treatment (84). This continuous expression, coupled with the effects of SHFL on CCR5 and PEG10, points to a role for SHFL in the control of normal mammalian translational processes.
4.3. Translational Machinery
The propensity of the ribosome to frameshift is an intrinsic property of the translation machinery. Trans-acting modifiers of translational efficiency largely appear to operate in a concentration-dependent/kinetic manner. Changes in elongation factor abundance and translational machinery composition have been shown to modify frameshifting propensity in trans (85, 86). Relative tRNA abundances appear capable of dictating both the mechanism and type of frameshifting that occurs (48, 87). Below we discuss the contributions of the different components of the translation machinery in PRF.
4.3.1. Transfer RNA abundance.
One often overlooked determinant for regulating PRF is the relative abundance of certain tRNAs. Categorically, tRNAs are trans-acting elements, yet their absolute requirement in translation, let alone PRF, places them in a unique position. Indeed, numerous studies have shown that tRNA is capable of regulating +1/−1 PRF (87–94), and the work of Korniy et al. (48) further demonstrated that tRNA can act as the master regulator of −1 PRF. Unlike the two-tRNA mechanism for −1 PRF (Section 3.1), −1 PRF can also be regulated by tRNA abundance, the one-tRNA or “hungry ribosome” mechanism (48). In this model, −1 PRF is driven by the relative concentration of tRNAs corresponding to the A-site codon in the slippery sequence (or to decode the YYZ position). Take, for example, the well-characterized Gag-Pol slippery sequence from HIV-1: U UUU UUA. Ninety percent of the time this sequence will be read as is, generating the peptide NFLG corresponding to the more common gag polyprotein (95). However, generation of mature, invasion-competent virions requires that 10% of the time, the ribosome slips into the −1 reading frame and the GagPol polyprotein is produced. Intriguingly, previous work (6, 96) established that two unique −1 products (NFLR and NFFR) are produced at a 70:30 ratio during HIV-1 PRF, implying multiple routes to Pol protein production. The hungry model then posits the stoichiometric control of Gag and GagPol polyprotein production is rendered tunable by the abundance of cellular tRNAs. The local tRNA pool could become temporarily depleted in UUA decoding tRNA, pausing the ribosome, and allowing it to slip into the −1 reading frame and produce the NFFR polyprotein. Conversely, if UUA decoding tRNAs are abundant, PRF will occur after accommodation, which will lead to the synthesis NFLR polyprotein. This “hungry ribosome” model is appealing as viral infections very often perturb host cell transcription and translation levels, thereby disrupting the balance of many cellular components, or have evolved to contain sequences for rare codons in their genomes.
4.3.2. Type of programmed ribosomal frameshifting.
While the mechanisms and functional components of −1 PRF are relatively conserved, +1 PRF seems to be mechanistically diverse and surprisingly rare across all domains of life and viruses (97). The lone outlier appears to be ciliates of the genus Euplotes, which utilize +1 PRF in 3,700 genes, some 10% of their genome (97). In comparison, just 3 human and saccharomyces genes are known to use +1 PRF, while influenza A, the Siphoviridae and Listeria phages, and four strains of the Leishmania RNA virus are the only viral examples known to date (98–101). Perhaps the most common mechanisms of +1 PRF derive from the shifty stop mechanism proposed by de Smit and colleagues (87), wherein the slip-site takes the form of a relatively rare codon next to a stop codon. When coupled to the highly selective ribosomal A-site, which efficiently selects for the proper geometry and pairing of A-site tRNAs, this architecture can induce translational pausing, giving rise to kinetic partitioning as the relative abundance of +1/0 frame pairing anticodons can drive the ribosome into the +1 frame (93).
Curran (102) undertook an extensive characterization of factors affecting +1 PRF in prfB synthesis finding that mRNA:tRNA pair stability is a key determinant of +1 PRF. Building upon this, Baranov and colleagues (93) were able to show that while mRNA:tRNA pair stability defines the rate constant of A-site occupancy, the choice between +1 and 0 frame read through is almost entirely dependent upon the concentration of tRNAs corresponding to the A-site 0 frame. In opposition to this model, Hong and coworkers (103) generated a series of crystal structures complexed with the frameshift suppressing tRNAsuf6, which demonstrated how rotation of the 30s ribosomal head and body drives recoding to the +1 frame. Furthermore, as noted above, polysomes have also been shown to induce +1 PRF while others have speculated that it is the overall three-dimensional structure of the mRNA region a ribosome is traversing that drives +1 PRF as opposed to a force/pause interaction (47, 104).
4.3.3. Host specificity and transfer RNAs.
Because frameshifting efficiency is tightly regulated by the stoichiometric needs of viral assembly, one may question whether the tissue/cell specificity of a given virus is tied to its tRNA pool requirements. Poliovirus and foot-and-mouth disease virus, for example, exhibit codon usage patterns similar to those of their host, while other viruses such as influenza A (IAV) and vaccinia virus (VV) are wildly divergent (105, 106). On closer inspection, however, IAV and VV replication complexes appear to associate with unique tRNA pools adapted to their individual needs while not altering the overall cellular tRNA pool (106). Furthermore, tRNA pool composition and modifications can rapidly change under conditions of stress and disease, implying that viral tRNA utilization must be adaptable to both the short- and long-term structure of the tRNA pool (105–108).
4.3.4. Kinetics of translation.
Translational rate is generally thought to be governed by the formation of the initiation complex (109). Reinforcing this notion, Tuller and coworkers (110) have shown that, when averaged across the entire genome, the first 30–50 codons of a given transcript tend to exhibit low translational efficiency. This observation implies that, as the initiation complex forms and elongation begins, translation proceeds slowly. This architecture, which was identified in HCV, PRRSV, and DENV, even appears to occur at multiple points across some viral genomes (111–114). Optimization of translation rates presumably tunes the relative stoichiometry of the corresponding proteins in accordance with the specific needs of virion (115). More recently, numerous groups have shown that elongation is also subject to regulation through mRNA secondary structure and tRNA abundance, which should also allow viruses to fine-tune synthetic rates (116). For example, one group demonstrated that mRNAs with a highly structured coding sequence were more efficiently expressed while others have shown that transcripts enriched in higher abundance tRNA/codons are expressed at higher levels than those containing lower abundance tRNAs (110, 117–120). Effectively, this control is likely the result of proteostatic necessity wherein a given mRNA is translated at a controlled rate to ensure an adequate supply of chaperones or to control the rates of synthesis through regions prone to misfolding (121–124). It should also be noted that the relative load of ribosomes on the viral transcript also affects the propensity of ribosomes to collide, which was recently found to affect the efficiency of both +1 and −1 PRF (47). The relative abundance of viral genomes changes during the course of infection, which should have a direct effect on the density of ribosomes on transcripts. Ribosome collisions could therefore play a key role in the temporal regulation of PRF. Additional investigations are needed to gain insights into the manner in which all of these modifiers are orchestrated in order to tune PRF during the viral life cycle.
5. CONCLUDING REMARKS
Despite the broad relevance of PRF to viral life cycles, there are still many unknown aspects of its regulation. PRF was initially discovered in the context of viruses but was eventually found to occur within all kingdoms of life. Initial investigations of the mechanism of PRF focused on the role of slip-sites and secondary structures within the transcript. Nevertheless, modern advances have provided new insights into the kinetic mechanisms of PRF and how these are modified by various host and viral effectors. A deeper understanding of PRF provides new opportunities to expand on previous efforts to target PRF with antivirals (125–129). The application of new cutting-edge methodologies is likely to result in the identification of additional pieces of the PRF puzzle. For example, deep mutational scanning has recently been employed to identify key residues in viral pathogenesis and evolution (130), as well as new proteins produced by PRF that were previously not detected (131). Using these approaches, we can identify new regulators of PRF and, in conjunction with proteomics, viral and host regulators of PRF.
In a more speculative vein, while PRF is primarily thought of as a mechanism of translational control in viruses, it appears to be a relatively common feature in all domains of life (132). Individual members of these domains are susceptible to infection by viruses that utilize PRF (17). While most of these viruses are RNA based, many of them are also retroviruses. Given that endogenous retroviruses are believed to account for 5–8% of the human genome, it is interesting to speculate whether this mechanism of PRF regulation evolved independently in viruses as well as bacteria, eukaryotes, and archaea, or if it was adapted to use by host cells following viral infection (104, 133–135).
ACKNOWLEDGMENTS
We graciously acknowledge Tamanash Bhattacharya for translating our big-picture ideas into coherent, visually appealing illustrations. We apologize to all colleagues whose contributions were not discussed and cited owing to space constraints. Research on viral frameshifting in the Schlebach and Mukhopadhyay labs is supported by grant NIH R21AI142383.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Atkins JF, Loughran G, Bhatt PR, Firth AE, Baranov PV. 2016. Ribosomal frameshifting and transcriptional slippage: from genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 44:7007–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dinman JD. 2006. Programmed ribosomal frameshifting goes beyond viruses: Organisms from all three kingdoms use frameshifting to regulate gene expression, perhaps signaling a paradigm shift. Microbe Wash. DC 1:521–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacks T, Varmus HE. 1985. Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science 230:1237–42 [DOI] [PubMed] [Google Scholar]
- 4.Schwartz DE, Tizard R, Gilbert W. 1983. Nucleotide sequence of Rous sarcoma virus. Cell 32:853–69 [DOI] [PubMed] [Google Scholar]
- 5.Weiss SR, Hackett PB, Oppermann H, Ullrich A, Levintow L, Bishop JM. 1978. Cell-free translation of avian sarcoma virus RNA: Suppression of the gag termination codon does not augment synthesis of the joint gag/pol product. Cell 15:607–14 [DOI] [PubMed] [Google Scholar]
- 6.Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. 1988. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 331:280–83 [DOI] [PubMed] [Google Scholar]
- 7.Brierley I, Boursnell ME, Binns MM, Bilimoria B, Blok VC, et al. 1987. An efficient ribosomal frameshifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO J. 6:3779–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Firth AE, Jagger BW, Wise HM, Nelson CC, Parsawar K, et al. 2012. Ribosomal frameshifting used in influenza A virus expression occurs within the sequence UCC_UUU_CGU and is in the +1 direction. Open Biol. 2:120109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang Y, Treffers EE, Li Y, Tas A, Sun Z, et al. 2012. Efficient −2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. PNAS 109:E2920–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Firth AE, Brierley I, Cai Y, Napthine S, et al. 2019. Programmed −2/−1 ribosomal frameshifting in simarteriviruses: an evolutionarily conserved mechanism. J. Virol 93:e00370–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xue SA, Jones MD, Lu QL, Middeldorp JM, Griffin BE. 2003. Genetic diversity: Frameshift mechanisms alter coding of a gene (Epstein-Barr virus LF3 gene) that contains multiple 102-base-pair direct sequence repeats. Mol. Cell. Biol 23:2192–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christie GE, Temple LM, Bartlett BA, Goodwin TS. 2002. Programmed translational frameshift in the bacteriophage P2 FETUD tail gene operon. J. Bacteriol 184:6522–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levin ME, Hendrix RW, Casjens SR. 1993. A programmed translational frameshift is required for the synthesis of a bacteriophage λ tail assembly protein. J. Mol. Biol 234:124–39 [DOI] [PubMed] [Google Scholar]
- 14.Condron BG, Atkins JF, Gesteland RF. 1991. Frameshifting in gene 10 of bacteriophage T7. J. Bacteriol 173:6998–7003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwun HJ, Toptan T, Ramos da Silva S, Atkins JF, Moore PS, Chang Y. 2014. Human DNA tumor viruses generate alternative reading frame proteins through repeat sequence recoding. PNAS 111:E4342–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Advani VM, Dinman JD. 2016. Reprogramming the genetic code: the emerging role of ribosomal frameshifting in regulating cellular gene expression. Bioessays 38:21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammell AB, Taylor RC, Peltz SW, Dinman JD. 1999. Identification of putative programmed −1 ribosomal frameshift signals in large DNA databases. Genome Res. 9:417–27 [PMC free article] [PubMed] [Google Scholar]
- 18.Hogg JR. 2016. Viral evasion and manipulation of host RNA quality control pathways. J. Virol 90:7010–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flint SJ, Racaniello VR, Rall GF, Skalka AM, Enquist LW. 2015. Principles of Virology. Washington, DC: ASM [Google Scholar]
- 20.Li Y, Shang P, Shyu D, Carrillo C, Naraghi-Arani P, et al. 2018. Nonstructural proteins nsp2TF and nsp2N of porcine reproductive and respiratory syndrome virus (PRRSV) play important roles in suppressing host innate immune responses. Virology 517:164–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito M, Yanagi Y, Ichinohe T. 2012. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLOS Pathog. 8:e1002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogers KJ, Jones-Burrage S, Maury W, Mukhopadhyay S. 2020. TF protein of Sindbis virus antagonizes host type I interferon responses in a palmitoylation-dependent manner. Virology 542:63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melian EB, Hinzman E, Nagasaki T, Firth AE, Wills NM, et al. 2010. NS1′ of flaviviruses in the Japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J. Virol 84:1641–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder JE, Kulcsar KA, Schultz KL, Riley CP, Neary JT, et al. 2013. Functional characterization of the alphavirus TF protein. J. Virol 87:8511–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramsey J, Renzi EC, Arnold RJ, Trinidad JC, Mukhopadhyay S. 2017. Palmitoylation of Sindbis virus TF protein regulates its plasma membrane localization and subsequent incorporation into virions. J. Virol 91:e02000–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Napthine S, Ling R, Finch LK, Jones JD, Bell S, et al. 2017. Protein-directed ribosomal frameshifting temporally regulates gene expression. Nat. Commun 8:15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kendra JA, de la Fuente C, Brahms A, Woodson C, Bell TM, et al. 2017. Ablation of programmed −1 ribosomal frameshifting in Venezuelan equine encephalitis virus results in attenuated neuropathogenicity. J. Virol 91:e01766–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plant EP, Dinman JD. 2008. The role of programmed −1 ribosomal frameshifting in coronavirus propagation. Front. Biosci 13:4873–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Firth AE, Chung BY, Fleeton MN, Atkins JF. 2008. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J 5:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacks T, Madhani HD, Masiarz FR, Varmus HE. 1988. Signals for ribosomal frameshifting in the Rous sarcoma virus gag-pol region. Cell 55:447–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.ten Dam EB, Verlaan PW, Pleij CW. 1995. Analysis of the role of the pseudoknot component in the SRV-1 gag-pro ribosomal frameshift signal: loop lengths and stability of the stem regions. RNA 1:146–54 [PMC free article] [PubMed] [Google Scholar]
- 32.Giedroc DP, Cornish PV. 2009. Frameshifting RNA pseudoknots: structure and mechanism. Virus Res. 139:193–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chamorro M, Parkin N, Varmus HE. 1992. An RNA pseudoknot and an optimal heptameric shift site are required for highly efficient ribosomal frameshifting on a retroviral messenger RNA. PNAS 89:713–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mouzakis KD, Lang AL, Vander Meulen KA, Easterday PD, Butcher SE. 2013. HIV-1 frameshift efficiency is primarily determined by the stability of base pairs positioned at the mRNA entrance channel of the ribosome. Nucleic Acids Res. 41:1901–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Namy O, Moran SJ, Stuart DI, Gilbert RJ, Brierley I. 2006. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 441:244–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wen JD, Lancaster L, Hodges C, Zeri AC, Yoshimura SH, et al. 2008. Following translation by single ribosomes one codon at a time. Nature 452:598–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leininger SE, Narayan K, Deutsch C, O’Brien EP. 2019. Mechanochemistry in translation. Biochemistry 58:4657–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong Z, Yang L, Zhang H, Shi J, Vandana JJ, et al. 2016. Mechanical unfolding kinetics of the SRV-1 gag-pro mRNA pseudoknot: possible implications for −1 ribosomal frameshifting stimulation. Sci. Rep 6:39549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen G, Chang KY, Chou MY, Bustamante C, Tinoco I Jr. 2009. Triplex structures in an RNA pseudoknot enhance mechanical stability and increase efficiency of −1 ribosomal frameshifting. PNAS 106:12706–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritchie DB, Foster DA, Woodside MT. 2012. Programmed −1 frameshifting efficiency correlates with RNA pseudoknot conformational plasticity, not resistance to mechanical unfolding. PNAS 109:16167–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bock LV, Caliskan N, Korniy N, Peske F, Rodnina MV, Grubmuller H. 2019. Thermodynamic control of −1 programmed ribosomal frameshifting. Nat. Commun 10:4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halma MTJ, Ritchie DB, Cappellano TR, Neupane K, Woodside MT. 2019. Complex dynamics under tension in a high-efficiency frameshift stimulatory structure. PNAS 116:19500–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moomau C, Musalgaonkar S, Khan YA, Jones JE, Dinman JD. 2016. Structural and functional characterization of programmed ribosomal frameshift signals in West Nile virus strains reveals high structural plasticity among cis-acting RNA elements. J. Biol. Chem 291:15788–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Houck-Loomis B, Durney MA, Salguero C, Shankar N, Nagle JM, et al. 2011. An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature 480:561–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nixon PL, Giedroc DP. 2000. Energetics of a strongly pH dependent RNA tertiary structure in a frameshifting pseudoknot. J. Mol. Biol 296:659–71 [DOI] [PubMed] [Google Scholar]
- 46.Nixon PL, Giedroc DP. 1998. Equilibrium unfolding (folding) pathway of a model H-type pseudoknotted RNA: the role of magnesium ions in stability. Biochemistry 37:16116–29 [DOI] [PubMed] [Google Scholar]
- 47.Smith AM, Costello MS, Kettring AH, Wingo RJ, Moore SD. 2019. Ribosome collisions alter frameshifting at translational reprogramming motifs in bacterial mRNAs. PNAS 116:21769–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korniy N, Samatova E, Anokhina MM, Peske F, Rodnina MV. 2019. Mechanisms and biomedical implications of −1 programmed ribosome frameshifting on viral and bacterial mRNAs. FEBS Lett. 593:1468–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng BZ, Bock LV, Belardinelli R, Peske F, Grubmuller H, Rodnina MV. 2019. Active role of elongation factor G in maintaining the mRNA reading frame during translation. Sci. Adv 5:eaax8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liao PY, Choi YS, Dinman JD, Lee KH. 2011. The many paths to frameshifting: kinetic modelling and analysis of the effects of different elongation steps on programmed −1 ribosomal frameshifting. Nucleic Acids Res. 39:300–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caliskan N, Katunin VI, Belardinelli R, Peske F, Rodnina MV. 2014. Programmed −1 frameshifting by kinetic partitioning during impeded translocation. Cell 157:1619–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim HK, Liu F, Fei J, Bustamante C, Gonzalez RL Jr., Tinoco I Jr. 2014. A frameshifting stimulatory stem loop destabilizes the hybrid state and impedes ribosomal translocation. PNAS 111:5538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Petrov A, Johansson M, Tsai A, O’Leary SE, Puglisi JD. 2014. Dynamic pathways of −1 translational frameshifting. Nature 512:328–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi J, O’Loughlin S, Atkins JF, Puglisi JD. 2020. The energy landscape of −1 ribosomal frameshifting. Sci. Adv 6:eaax6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan S, Wen JD, Bustamante C, Tinoco I Jr. 2015. Ribosome excursions during mRNA translocation mediate broad branching of frameshift pathways. Cell 160:870–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Su MC, Chang CT, Chu CH, Tsai CH, Chang KY. 2005. An atypical RNA pseudoknot stimulator and an upstream attenuation signal for −1 ribosomal frameshifting of SARS coronavirus. Nucleic Acids Res. 33(13):4265–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kelly JA, Dinman JD. 2020. Structural and functional conservation of the programmed −1 ribosomal frameshift signal of SARS-CoV-2. bioRxiv 2020.03.13.991083. 10.1101/2020.03.13.991083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cho CP, Lin SC, Chou MY, Hsu HT, Chang KY. 2013. Regulation of programmed ribosomal frameshifting by co-translational refolding RNA hairpins. PLOS ONE 8:e62283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Charbonneau J, Gendron K, Ferbeyre G, Brakier-Gingras L. 2012. The 5′ UTR of HIV-1 full-length mRNA and the Tat viral protein modulate the programmed −1 ribosomal frameshift that generates HIV-1 enzymes. RNA 18:519–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mohan BR, Dinesh-Kumar SP, Miller WA. 1995. Genes and cis-acting sequences involved in replication of barley yellow dwarf virus-PAV RNA. Virology 212:186–95 [DOI] [PubMed] [Google Scholar]
- 61.Hsu HT, Lin YH, Chang KY. 2014. Synergetic regulation of translational reading-frame switch by ligand-responsive RNAs in mammalian cells. Nucleic Acids Res. 42:14070–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harrington HR, Zimmer MH, Chamness LM, Nash V, Penn WD, et al. 2020. Cotranslational folding stimulates programmed ribosomal frameshifting in the alphavirus structural polyprotein. J. Biol. Chem 295:6798–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramsey J, Mukhopadhyay S. 2017. Disentangling the frames, the state of research on the alphavirus 6K and TF proteins. Viruses 9:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chung BY, Firth AE, Atkins JF. 2010. Frameshifting in alphaviruses: a diversity of 3′ stimulatory structures. J. Mol. Biol 397:448–56 [DOI] [PubMed] [Google Scholar]
- 65.Goldman DH, Kaiser CM, Milin A, Righini M, Tinoco I Jr., Bustamante C. 2015. Mechanical force releases nascent chain–mediated ribosome arrest in vitro and in vivo. Science 348:457–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nilsson OB, Hedman R, Marino J, Wickles S, Bischoff L, et al. 2015. Cotranslational protein folding inside the ribosome exit tunnel. Cell Rep. 12:1533–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ismail N, Hedman R, Schiller N, von Heijne G. 2012. A biphasic pulling force acts on transmembrane helices during translocon-mediated membrane integration. Nat. Struct. Mol. Biol 19:1018–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cymer F, Ismail N, von Heijne G. 2014. Weak pulling forces exerted on Nin-orientated transmembrane segments during co-translational insertion into the inner membrane of Escherichia coli. FEBS Lett. 588:1930–34 [DOI] [PubMed] [Google Scholar]
- 69.Niesen MJM, Muller-Lucks A, Hedman R, von Heijne G, Miller TF 3rd. 2018. Forces on nascent polypeptides during membrane insertion and translocation via the Sec translocon. Biophys. J 115:1885–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brodsky JL, Goeckeler J, Schekman R. 1995. BiP and Sec63p are required for both co- and posttranslational protein translocation into the yeast endoplasmic reticulum. PNAS 92:9643–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muldoon-Jacobs KL, Dinman JD. 2006. Specific effects of ribosome-tethered molecular chaperones on programmed −1 ribosomal frameshifting. Eukaryot. Cell 5:762–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adamski FM, Donly BC, Tate WP. 1993. Competition between frameshifting, termination and suppression at the frameshift site in the Escherichia coli release factor-2 mRNA. Nucleic Acids Res. 21:5074–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rom E, Kahana C. 1994. Polyamines regulate the expression of ornithine decarboxylase antizyme in vitro by inducing ribosomal frame-shifting. PNAS 91:3959–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Howard MT, Gesteland RF, Atkins JF. 2004. Efficient stimulation of site-specific ribosome frameshifting by antisense oligonucleotides. RNA 10:1653–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Olsthoorn RC, Laurs M, Sohet F, Hilbers CW, Heus HA, Pleij CW. 2004. Novel application of sRNA: stimulation of ribosomal frameshifting. RNA 10:1702–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu CH, Noteborn MH, Olsthoorn RC. 2010. Stimulation of ribosomal frameshifting by antisense LNA. Nucleic Acids Res. 38:8277–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu B, Zhang H, Sun R, Peng S, Cooperman BS, et al. 2018. Translocation kinetics and structural dynamics of ribosomes are modulated by the conformational plasticity of downstream pseudoknots. Nucleic Acids Res. 46:9736–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Belew AT, Meskauskas A, Musalgaonkar S, Advani VM, Sulima SO, et al. 2014. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature 512:265–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li Y, Treffers EE, Napthine S, Tas A, Zhu L, et al. 2014. Transactivation of programmed ribosomal frameshifting by a viral protein. PNAS 111:E2172–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Napthine S, Treffers EE, Bell S, Goodfellow I, Fang Y, et al. 2016. A novel role for poly(C) binding proteins in programmed ribosomal frameshifting. Nucleic Acids Res. 44:5491–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carocci M, Cordonnier N, Huet H, Romey A, Relmy A, et al. 2011. Encephalomyocarditis virus 2A protein is required for viral pathogenesis and inhibition of apoptosis. J. Virol 85:10741–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang X, Xuan Y, Han Y, Ding X, Ye K, et al. 2019. Regulation of HIV-1 Gag-Pol expression by Shiftless, an inhibitor of programmed −1 ribosomal frameshifting. Cell 176:625–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suzuki Y, Chin WX, Han Q, Ichiyama K, Lee CH, et al. 2016. Characterization of RyDEN (C19orf66) as an interferon-stimulated cellular inhibitor against dengue virus replication. PLOS Pathog. 12:e1005357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Balinsky CA, Schmeisser H, Wells AI, Ganesan S, Jin T, et al. 2017. IRAV (FLJ11286), an interferon-stimulated gene with antiviral activity against dengue virus, interacts with MOV10. J. Virol 91:e01606–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kobayashi Y, Zhuang J, Peltz S, Dougherty J. 2010. Identification of a cellular factor that modulates HIV-1 programmed ribosomal frameshifting. J. Biol. Chem 285:19776–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Inge-Vechtomov S, Zhouravleva G, Philippe M. 2003. Eukaryotic release factors (eRFs) history. Biol. Cell 95:195–209 [DOI] [PubMed] [Google Scholar]
- 87.de Smit MH, van Duin J, van Knippenberg PH, van Eijk HG. 1994. CCC.UGA: a new site of ribosomal frameshifting in Escherichia coli. Gene 143:43–47 [DOI] [PubMed] [Google Scholar]
- 88.Atkins JF, Gesteland RF, Reid BR, Anderson CW. 1979. Normal tRNAs promote ribosomal frameshifting. Cell 18:1119–31 [DOI] [PubMed] [Google Scholar]
- 89.Weiss R, Gallant J. 1983. Mechanism of ribosome frameshifting during translation of the genetic code. Nature 302:389–93 [DOI] [PubMed] [Google Scholar]
- 90.Belcourt MF, Farabaugh PJ. 1990. Ribosomal frameshifting in the yeast retrotransposon Ty: tRNAs induce slippage on a 7 nucleotide minimal site. Cell 62:339–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pande S, Vimaladithan A, Zhao H, Farabaugh PJ. 1995. Pulling the ribosome out of frame by +1 at a programmed frameshift site by cognate binding of aminoacyl-tRNA. Mol. Cell. Biol 15:298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sundararajan A, Michaud WA, Qian Q, Stahl G, Farabaugh PJ. 1999. Near-cognate peptidyl-tRNAs promote +1 programmed translational frameshifting in yeast. Mol. Cell 4:1005–15 [DOI] [PubMed] [Google Scholar]
- 93.Baranov PV, Gesteland RF, Atkins JF. 2004. P-site tRNA is a crucial initiator of ribosomal frameshifting. RNA 10:221–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Korniy N, Goyal A, Hoffmann M, Samatova E, Peske F, et al. 2019. Modulation of HIV-1 Gag/Gag-Pol frameshifting by tRNA abundance. Nucleic Acids Res. 47:5210–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schmalen A, Karius-Fischer J, Rauch P, Setz C, Korn K, et al. 2018. The N-terminus of the HIV-1 p6 Gag protein regulates susceptibility to degradation by IDE. Viruses 10:710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yelverton E, Lindsley D, Yamauchi P, Gallant JA. 1994. The function of a ribosomal frameshifting signal from human immunodeficiency virus-1 in Escherichia coli. Mol. Microbiol 11:303–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Han KZ, Ning J, Zhang BT, Wang YR, Zhang HK, et al. 2016. High power single-frequency Innoslab amplifier. Appl. Opt 55:5341–44 [DOI] [PubMed] [Google Scholar]
- 98.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, et al. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim SN, Choi JH, Park MW, Jeong SJ, Han KS, Kim HJ. 2005. Identification of the +1 ribosomal frameshifting site of LRV1–4 by mutational analysis. Arch. Pharm. Res 28:956–62 [DOI] [PubMed] [Google Scholar]
- 100.Auzat I, Droge A, Weise F, Lurz R, Tavares P. 2008. Origin and function of the two major tail proteins of bacteriophage SPP1. Mol. Microbiol 70:557–69 [DOI] [PubMed] [Google Scholar]
- 101.Zimmer M, Sattelberger E, Inman RB, Calendar R, Loessner MJ. 2003. Genome and proteome of Listeria monocytogenes phage PSA: an unusual case for programmed +1 translational frameshifting in structural protein synthesis. Mol. Microbiol 50:303–17 [DOI] [PubMed] [Google Scholar]
- 102.Curran JF. 1993. Analysis of effects of tRNA:message stability on frameshift frequency at the Escherichia coli RF2 programmed frameshift site. Nucleic Acids Res. 21:1837–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hong S, Sunita S, Maehigashi T, Hoffer ED, Dunkle JA, Dunham CM. 2018. Mechanism of tRNA-mediated +1 ribosomal frameshifting. PNAS 115:11226–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Farabaugh PJ. 1996. Programmed translational frameshifting. Annu. Rev. Genet 30:507–28 [DOI] [PubMed] [Google Scholar]
- 105.Albers S, Czech A. 2016. Exploiting tRNAs to boost virulence. Life 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pavon-Eternod M, David A, Dittmar K, Berglund P, Pan T, et al. 2013. Vaccinia and influenza A viruses select rather than adjust tRNAs to optimize translation. Nucleic Acids Res. 41:1914–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pang YL, Abo R, Levine SS, Dedon PC. 2014. Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: tRNA-seq for quantifying changes in tRNA copy number. Nucleic Acids Res. 42:e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Torrent M, Chalancon G, de Groot NS, Wuster A, Babu MM. 2018. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci. Signal 11:eaat6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Riba A, Di Nanni N, Mittal N, Arhne E, Schmidt A, Zavolan M. 2019. Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. PNAS 116:15023–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, et al. 2010. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141:344–54 [DOI] [PubMed] [Google Scholar]
- 111.Sanchez G, Bosch A, Pinto RM. 2003. Genome variability and capsid structural constraints of hepatitis A virus. J. Virol 77:452–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhou JH, Zhang J, Sun DJ, Ma Q, Chen HT, et al. 2013. The distribution of synonymous codon choice in the translation initiation region of dengue virus. PLOS ONE 8:e77239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ding YZ, You YN, Sun DJ, Chen HT, Wang YL, et al. 2014. The effects of the context-dependent codon usage bias on the structure of the nsp1α of porcine reproductive and respiratory syndrome virus. Biomed. Res. Int 2014:765320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gerresheim GK, Bathke J, Michel AM, Andreev DE, Shalamova LA, et al. 2019. Cellular gene expression during hepatitis C virus replication as revealed by ribosome profiling. Int. J. Mol. Sci 20:1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dinman JD, Wickner RB. 1992. Ribosomal frameshifting efficiency and gag/gag-pol ratio are critical for yeast M1 double-stranded RNA virus propagation. J. Virol 66:3669–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rodnina MV. 2016. The ribosome in action: tuning of translational efficiency and protein folding. Protein Sci. 25:1390–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mauger DM, Cabral BJ, Presnyak V, Su SV, Reid DW, et al. 2019. mRNA structure regulates protein expression through changes in functional half-life. PNAS 116:24075–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324:255–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gingold H, Dahan O, Pilpel Y. 2012. Dynamic changes in translational efficiency are deduced from codon usage of the transcriptome. Nucleic Acids Res. 40:10053–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ben-Yehezkel T, Atar S, Zur H, Diament A, Goz E, et al. 2015. Rationally designed, heterologous S. cerevisiae transcripts expose novel expression determinants. RNA Biol. 12:972–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Calloni G, Chen T, Schermann SM, Chang HC, Genevaux P, et al. 2012. DnaK functions as a central hub in the E. coli chaperone network. Cell Rep. 1:251–64 [DOI] [PubMed] [Google Scholar]
- 122.Waudby CA, Dobson CM, Christodoulou J. 2019. Nature and regulation of protein folding on the ribosome. Trends Biochem. Sci 44:914–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bartoszewski R, Kroliczewski J, Piotrowski A, Jasiecka AJ, Bartoszewska S, et al. 2016. Codon bias and the folding dynamics of the cystic fibrosis transmembrane conductance regulator. Cell. Mol. Biol. Lett 21:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.O’Brien EP, Ciryam P, Vendruscolo M, Dobson CM. 2014. Understanding the influence of codon translation rates on cotranslational protein folding. Acc. Chem. Res 47:1536–44 [DOI] [PubMed] [Google Scholar]
- 125.Neuman BW, Stein DA, Kroeker AD, Churchill MJ, Kim AM, et al. 2005. Inhibition, escape, and attenuated growth of severe acute respiratory syndrome coronavirus treated with antisense morpholino oligomers. J. Virol 79:9665–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Brierley I, Dos Ramos FJ. 2006. Programmed ribosomal frameshifting in HIV-1 and the SARS-CoV. Virus Res. 119:29–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brakier-Gingras L, Charbonneau J, Butcher SE. 2012. Targeting frameshifting in the human immunodeficiency virus. Expert Opin. Ther. Targets 16:249–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hu HT, Cho CP, Lin YH, Chang KY. 2016. A general strategy to inhibiting viral −1 frameshifting based on upstream attenuation duplex formation. Nucleic Acids Res. 44:256–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dinman JD, Ruiz-Echevarria MJ, Peltz SW. 1998. Translating old drugs into new treatments: ribosomal frameshifting as a target for antiviral agents. Trends Biotechnol. 16:190–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dolan PT, Whitfield ZJ, Andino R. 2018. Mapping the evolutionary potential of RNA viruses. Cell Host Microbe 23:435–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ogden PJ, Kelsic ED, Sinai S, Church GM. 2019. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 366:1139–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jacobs JL, Belew AT, Rakauskaite R, Dinman JD. 2007. Identification of functional, endogenous programmed −1 ribosomal frameshift signals in the genome of Saccharomyces cerevisiae. Nucleic Acids Res. 35:165–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Manktelow E, Shigemoto K, Brierley I. 2005. Characterization of the frameshift signal of Edr, a mammalian example of programmed −1 ribosomal frameshifting. Nucleic Acids Res. 33:1553–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Willis SL, Tennstedt SL, Marsiske M, Ball K, Elias J, et al. 2006. Long-term effects of cognitive training on everyday functional outcomes in older adults. JAMA 296:2805–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Clark MB, Janicke M, Gottesbuhren U, Kleffmann T, Legge M, et al. 2007. Mammalian gene PEG10 expresses two reading frames by high efficiency −1 frameshifting in embryonic-associated tissues. J. Biol. Chem 282:37359–69 [DOI] [PubMed] [Google Scholar]