

Abstract
Background
Infantile hypercalcaemia (IH) is a vitamin D3 metabolism disorder. The molecular basis for IH is biallelic mutations in the CYP24A1 or SLC34A1 gene. These changes lead to catabolism disorders (CYP24A1 mutations) or excessive generation of 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] (SLC34A1 mutations). The incidence rate of IH in children and the risk level for developing end-stage renal disease (ESRD) are still unknown. The aim of this study was to analyse the long-term outcome of adolescents and young adults who suffered from IH in infancy.
Design
Forty-two children (23 girls; average age 10.7 ± 6.3 years) and 26 adults (14 women; average age 24.2 ± 4.4 years) with a personal history of hypercalcaemia with elevated 1,25(OH)2D3 levels were included in the analysis. In all patients, a genetic analysis of possible IH mutations was conducted, as well as laboratory tests and renal ultrasonography.
Results
IH was confirmed in 20 studied patients (10 females). CYP24A1 mutations were found in 16 patients (8 females) and SLC34A1 in 4 patients (2 females). The long-term outcome was assessed in 18 patients with an average age of 23.8 years (age range 2–34). The average glomerular filtration rate (GFR) was 72 mL/min/1.73 m2 (range 15–105). Two patients with a CYP24A1 mutation developed ESRD and underwent renal transplantation. A GFR <90 mL/min/1.73 m2 was found in 14 patients (77%), whereas a GFR <60 mL/min/1.73 m2 was seen in 5 patients (28%), including 2 adults after renal transplantation. Three of 18 patients still had serum calcium levels >2.6 mmol/L. A renal ultrasound revealed nephrocalcinosis in 16 of 18 (88%) patients, however, mild hypercalciuria was detected in only one subject.
Conclusions
Subjects who suffered from IH have a greater risk of progressive chronic kidney disease and nephrocalcinosis. This indicates that all survivors of IH should be closely monitored, with early implementation of preventive measures, e.g. inhibition of active metabolites of vitamin D3 synthesis.
Keywords: chronic kidney disease, infantile hypercalcaemia, parathormone, vitamin D
KEY LEARNING POINTS
What is already known about this subject?
• infantile hypercalcaemia (IH) is caused by variants in the CYP24A1 or SLC34A1 gene and the long-term outcome of survivors of IH is not well described.
What this study adds?
• subjects who suffered from IH may develop progressive chronic kidney disease and have a greater risk of end-stage renal disease.
What impact this may have on practice or policy?
• routine recommendations, on the basis of avoidance of sun and the use of vitamin D supplements, are not sufficient, thus other preventive measures and early referral to nephrological care should be considered.
INTRODUCTION
Infantile hypercalcaemia (IH), formerly known as idiopathic IH and also called hypersensitivity to vitamin D3, is a vitamin D3 metabolism disorder due to a disability of enzymatic breakdown of active vitamin D3 or oversynthesis of 1,25-dihydroxyvitamin D3 [1,25(OH)2D3]. This results in high serum levels of 25-hydroxyvitamin D [25(OH)D3] and/or 1,25(OH)2D3, symptomatic hypercalcaemia, including emesis, anorexia, polyuria, dehydration, constipation, generalized hypotonia, arterial hypertension, unexplained fever and clinical signs suggesting urinary tract infection with leucocyturia and erythrocyturia on a general urine test, as well as acute kidney injury. IH is believed to be an infant disease that develops after supplementation of prophylactic doses of vitamin D. The molecular basis of IH is biallelic variants of the CYP24A1 (*126065) or SLC34A1 (*182309) gene, which leads to IH Subtype 1 (143880) or IH Subtype 2 (616963), respectively. The CYP24A1 gene encodes 25-hydroxyvitamin D-24-hydroxylase, whereas SLC34A1 encodes sodium–phosphate cotransporter (NaPi-IIa), which controls proximal tubule phosphate reabsorption. Mutations of these genes result in disturbances to enzymatic breakdown of 25(OH)D3 and 1,25(OH)2D3 in the case of CYP24A1 mutations [1–3] or excessive generation of 1,25(OH)2D3 in the case of SLC34A1 mutations [2, 3] (Figure 1). The clinical symptoms of hypercalcaemia disappear after normalization of serum calcium levels. Although acute consequences of IH are well described, the exact long-term risk to subjects who suffered from IH is not known. There are only a few reports of single cases of chronic kidney disease (CKD) as a long-term outcome of IH. The aim of our study was to analyse the long-term outcome of survivors of IH with a confirmed molecular defect. Our observations suggest that biochemical and structural abnormalities may last longer and may lead to CKD as well as end-stage renal disease (ESRD).
FIGURE 1.

Scheme of vitamin D metabolism.
MATERIALS AND METHODS
All patients (or their parents in case of children) gave written consent for participation in the study. We analysed the data of 42 children (23 girls; average age 10.7 ± 6.3 years) and 26 adults (14 women; average age 24.2 ± 4.4 years) with a personal history of hypercalcaemia with elevated 1,25(OH)2D3 levels who were hospitalized due to hypercalcaemia (calcium serum concentration >2.69 mmol/L) diagnosed in infancy and developed after prophylactic supplementation of vitamin D, with elevated 25(OH)D3/1,25(OH)2D3 levels during the first 14 months of life, in whom molecular analysis of CYP24A1 and SLC34A1 was conducted (Figure 2). All subjects were under the care of the metabolic clinic from childhood until adulthood and everyone was advised to avoid sun exposure, vitamin D and calcium supplements and to drink large amounts of fluids. The data on serum creatinine levels as well as glomerular filtration rates (GFRs) measured at discharge after an IH episode were obtained retrospectively from medical charts. Renal function, calcaemia, vitamin D levels, calciuria and serum parathyroid hormone (PTH) levels were reassessed at the average age of 24 years. In all patients, a genetic analysis of possible IH mutations was conducted, as well as clinical, ultrasonographic and laboratory examinations. Survivors of IH, in whom CYP24A1 or SLC34A1 mutations were found, were included in further analyses. Part of this analysed cohort of patients was already described [3–5].
FIGURE 2.
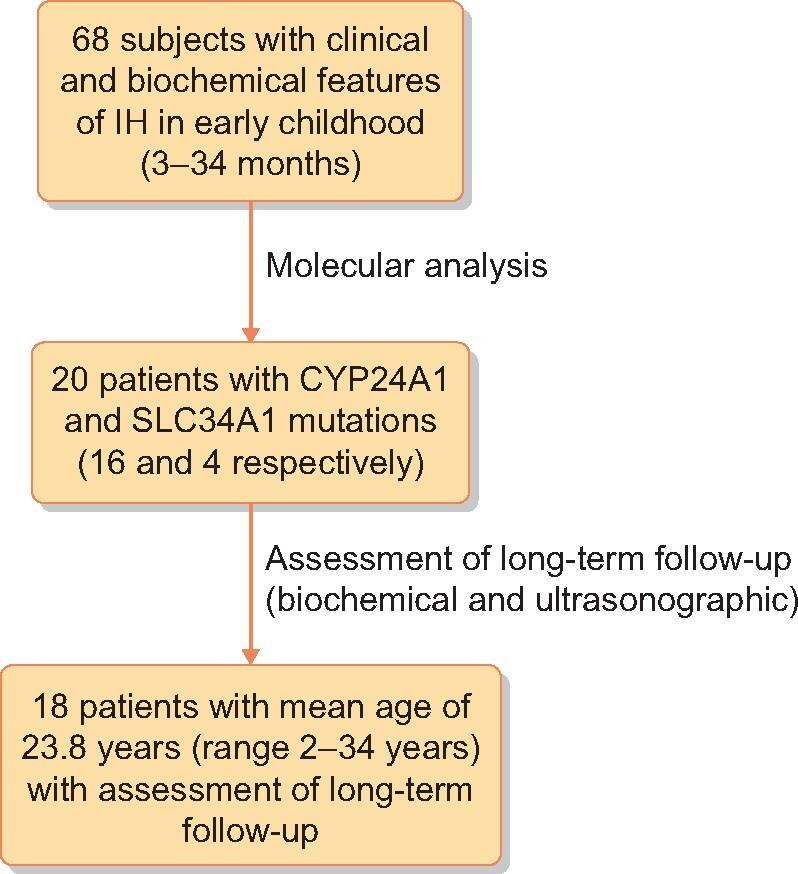
Scheme of the study.
Ultrasonographic, laboratory and molecular analysis
In all subjects, renal ultrasound and laboratory tests were carried out. Laboratory tests involved serum creatinine, cystatin C, 25(OH)D3, 1,25(OH)2D3, PTH, urinary calcium and creatinine in spot urine samples (first morning urine). A biochemical analysis was conducted using traditional methods. Blood (3 mL) was drawn at the medical laboratory after 12 h of fasting and it was centrifuged at 1465 g for 10 min at room temperature, using test tubes. The laboratory tests, including serum creatinine levels, were conducted on the same day by means of routine enzyme assays with commercial kits and with the use of a Cobas 501 Chemistry System; Roche, Basel, Switzerland). Serum cystatin C measurements were performed by a particle-enhanced immune nephelometry assay using the BN ProspecNephelometer system (DadeBehring, Newark, DE, USA). The 25(OH)D3 and 1,25(OH)2D3 were measured by chemiluminescence immunoassay, Liaison system (DiaSorin, Saluggia, Italy) and PTH with an immunoradiometric assay. The estimated GFR (eGFR) was calculated with the Schwartz or Modification of Diet in Renal Disease (MDRD) formula (where appropriate). Serum calcium >2.6 mmol/L was considered elevated and PTH levels <10 pg/mL were considered decreased. The upper limits of normal for 25(OH)D3 and 1,25(OH)2D3 were 50 ng/mL and 109 pg/mL, respectively.
Genomic DNA was extracted from peripheral blood samples of probands and available family members by means of an automated (MagnaPure, Roche) or manual (phenol–chloroform) method. Sanger sequencing analysis was carried out in seven probands and the results were previously described [1–3]. Next-generation sequencing (NGS) using the TruSight One Sequencing Panel (Illumina, San Diego, CA, USA) for simultaneous sequencing of 4813 clinically relevant genes was used as a genetic target in 11 other probands. NGS was performed on a HiSeq 1500 platform using the Exome Enrichment Kit (Illumina) according to a published protocol [6]. Generated reads were first merged and low-quality reads were removed. Then the reads were aligned to the reference human genome hg19/hg38 (GRCh37/GRCh38). Potential polymerase chain reaction duplicates and reads mapping to multiple genomic regions were removed. In the process of variant calling, any call with the ratio <0.2 was assumed to be homozygous, while the rest were heterozygous. Alignments were viewed with Integrative Genomics Viewer version 2.3.82 [7]. The detected variants were annotated using ANNOVAR and converted to Microsoft Access format for a final manual analysis.
All the non-coding and common variants [minor allele frequency (MAF) >0.01 in the general population] were discarded. The rare variants affecting coding regions were filtered on the basis of an autosomal recessive mode of inheritance and predicted consequences at the transcription level. The variants were prioritized according to the population frequency and the predicted effect on protein. The potential consequences were defined in accordance with the conservation of the affected amino acids and in silico predictions by using different algorithms.
To identify the molecular basis for the disease, first we analysed potentially related infantile idiopathic hypercalcaemia (IIH) genes. Furthermore, in order to detect potentially causative variants from the TruSight One target panel, we applied a computational algorithm called the Phenotypic Interpretation of eXomes [8]. The software evaluates and ranks detected gene variants on the basis of pathogenicity and semantic similarity of idiopathic IH (or other disease that causes similar symptoms and needs differential diagnosis) described by Human Phenotype Ontology providing terms to known Mendelian disorders.
The candidate pathogenic variants were verified in the probands and available family members by means of Sanger sequencing using BigDye Chemistry (Applied Biosystems, Waltham, MA, USA).
RESULTS
Of 68 subjects who developed hypercalcaemia in infancy, CYP24A1 or SLC34A1 mutations were found in 20 cases and those patients were included in further analyses. CYP24A1 mutations were found in 16 patients (8 girls), whereas SLC34A1 was found in 4 patients (2 females). The above-mentioned 16 patients developed IH after a high dose of prophylactic vitamin D3 in the 1980s and the above-mentioned 4 subjects (Patients 9, 10, 14 and 16; Table 1) developed IH after application of vitamin D3 at a daily dose of 800–1600 IU. Among 20 patients, in whom the molecular mechanism of IH was determined, the assessment of a long-term outcome was possible in 18 patients at an average age of 23.8 years (age range 2–34) (Figure 2). Laboratory data, results of molecular analysis and ultrasonographic findings of these patients are presented in Table 1.
Table 1.
Clinical characteristics and molecular findings in 18 survivors of IIH
| Proband no. | Sex | Age at diagnosis | GFR at first presentation of IIH (eGFR; mL/min/ 1.73 m2) | Gene | Molecular result (variant 1)/(variant 2)a |
Zygosity status | Age at last observation (years) | Renal function at last observation (eGFR; mL/min/1.73 m2) | Serum calcium (mmol/L) | 25 (OH)D3 (ng/mL) | 1,25(OH)2D3 (pg/mL) | PTH (pg/mL) | Calcium:creatinine (mg/mg) | Comment | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide level | Amino acid level | ||||||||||||||
| 1 | f | 8/12 | 46 | CYP24A1 | (1186C>T)/ (1186C>T) | (R396W)/(R396W) | Hom | 34 | 58 | 2.49 | 25.2 | 56.7 | 22.4 | 0.04 | Nephrocalcinosis |
| 2 | f | 4/12 | 78 | SLC34A1 | (1425_1426del)/(?) | (C476Sfs*128)/(?) | Het | 28 | 70 | 2.41 | 28.2 | 36.4 | 21.7 | 0.12 | Nephrocalcinosis/Nephrolithiasis |
| 3 | f | 5/12 | 29 | CYP24A1 | (428_430del)/ (1186C>T) | (E143del)/(R396W) | Comp het | 30 | 98 | 2.49 | 36.4 | 93.6 | 20.4 | 0.14 | Nephrocalcinosis |
| 4 | m | 11/12 | 40 | CYP24A1 | (1186C>T)/(1226T>C) | (R396W)/(L409S) | Comp het | 25 | 78 | 2.4 | 21.8 | 63.2 | 8.7 | 0.19 | Nephrocalcinosis |
| 5 | m | 6/12 | 49 | SLC34A1 | (272_292del)/(464T>C) | (V91_A97del)/ (L155P) | Comp het | 28 | 84 | 2.4 | 31.5 | 42.3 | 12.6 | 0.25 | Nephrocalcinosis |
| 6 | m | 5/12 | 57 | CYP24A1 | (107delC)/(443T>C) | (P36Lfs*11)/(L148P) | Comp het | 27 | 85 | 2.5 | 32.1 | 51.8 | 7.9 | 0.16 | Nephrocalcinosis |
| 7 | f | 5/12 | 33 | CYP24A1 | (1186C>T)/ (1157 + 1G>A) | (R396W)/(?) | Comp het | 23 | 54 | 2.8 | 47.6 | 54.8 | 2.8 | 0.14 | Nephrocalcinosis |
| 8 | f | 10/12 | 40 | CYP24A1 | (1186C>T)/ (1186C>T) | (R396W)/(R396W) | Hom | 23 | 110 | 2.3 | 23.7 | 37.8 | 2.86 | 0.13 | Nephrocalcinosis |
| 9 | f | 8/12 | 49 | CYP24A1 | (428_430del)/ (1157 + 1G>A) | (E143del)/(?) | Comp het | 17 | 69 | 2.7 | 35.8 | 35.8 | 5.2 | 0.08 | Nephrocalcinosis |
| 10 | m | 8/12 | 100 | CYP24A1 | (1186C>T)/(?) | (R396W)/(?) | Het | 10 | 105 | 2.36 | 32.3 | 57.5 | 23.4 | 0.03 | Normal ultrasonography |
| 11 | m | 5/12 | 45 | CYP24A1 | (1186C>T)/(1186C>T) | (R396W)/(R396W) | Hom | 21 | 82 | 2.5 | 22.5 | 33.5 | 5.23 | 0.1 | Nephrocalcinosis |
| 12 | m | 14/12 | 24 | CYP24A1 | (964G>A)/(1186C>T) | (E322K)/(R396W) | Comp het | 30 | 50 (Rtx) | 2.49 | 34 | 119.1 | 18.2 | 0.13 | Increased echogenicity of renal pyramids since initial presentation. Disturbed cortico-medullary differentiation with increased echogenicity since puberty. ESRD at 27 years. Nephrocalcinosis. Renal transplantation |
| 13 | f | 6/12 | 56 | CYP24A1 | (475C>T)/(1226T>C) | (R159W)/(L409S) | Comp het | 32 | 77 | 2.54 | 39.5 | 74.2 | 2.86 | 0.12 | Nephrocalcinosis |
| 14 | m | 3/12 | 44 | CYP24A1 | (667A>T)/(1186C>T) | (R223*)/(R396W) | Comp het | 17 | 55 | 2.7 | 36.9 | 74.4 | 2.8 | 0.07 | Hyperchogenic pyramids at initial diagnosis. Normal renal ultrasonography at age of 17. |
| 15 | m | 3/12 | 23 | CYP24A1 | (443T>C)/(1186C>T) | (L148P)/(R396W) | Comp het | 34 | 75 | 2.54 | 27.1 | 65.1 | 2.22 | 0.1 | Nephrocalcinosis |
| 16 | f | 2/12 | 39 | SLC34A1 | (437C>T); (?) | (P146L)/(?) | Het | 2 | 90 | 2.55 | 30.5 | 83.2 | 8.15 | 0.15 | Nephrocalcinosis |
| 17 | m | 3/12 | 26 | CYP24A1 | (1186C>T)/(1226T>C) | (R396W)/(L409S) | Comp het | 38 | 55 (Rtx) | 2.59 | 37 | 67.2 | 39 | 0.08 | Increased echogenicity of renal pyramids since initial presentation. Disturbed cortico-medullary differentiation with increased echogenicity since puberty. ESRD at 34 years. Renal transplantation. |
| 18 | m | 6/12 | 46 | CYP24A1 | (443T>C)/(1186C>T) | (L148P)/(R396W) | Comp het | 26 | 80 | 2.55 | 37.6 | 73.3 | 2.49 | 0.07 | Nephrocalcinosis |
Variant numbering was based on the GenBank cDNA reference sequence of human CYP24A1 (NM_000782.4; NP_000773.2) and SLC34A1 (NM_003052.4; NP_003043.3), according to the guidelines of Human Genome Variation Society (HGVS; www.hgvs.org/mutnomen); single-letter abbreviations of amino acids have been used to describe molecular results at the amino acid level.
f, female; m, male; comp het, compound heterozygote; het, heterozygote, other pathogenic molecular variants not identified; hom, homozygote; Rtx, renal transplantation.
As shown in Table 1, pathogenic variants were identified in 33 alleles of CYP24A1 (n = 29) and SLC34A1 (n = 4) in our 18 IIH patients. We found 3 homozygotes (only for CYP24A1), 12 compound heterozygotes (including 1 for SLC34A1) and 3 monoallelic gene variants (heterozygotes) (Patients 2, 10 and 16), without any pathogenic variant in the second allele, however, only coding regions in one of the two mentioned genes were analysed in these cases. Three patients were included in this study since their symptoms and biochemical results were consistent with IIH diagnosis and the assessment of a long-term outcome was possible.
In total, 13 different abnormal or likely abnormal changes were identified, including missense (n = 7), frameshift (n = 2), in-frame (n = 2), splice site (n = 1) and nonsense (n = 1) mutations. Twelve of them revealed variants that were known or we already described them [2, 3]. Only one novel CYP24A1 variant, p.R159W, was identified, which affects highly conserved amino acid, found in the cytochrome P450 domain. Its frequency in various population databases is very low. In silico prediction classified this change as potentially pathogenic. All variants met the frequency criteria for recessive disorders (MAF <0.01), except for one in-frame deletion p.V91_A97del in SLC34A1, which was included in the genome aggregation database with MAF ∼0.025. This variant has already been described and, based on functional analyses, considered by them to be likely pathogenic [2]. We identified this variant in a compound heterozygous state together with p.L155P change in a boy (Patient 5) with a history of IIH as well as persistent hypercalciuria and nephrocalcinosis.
The most common variant observed in the study group was p.R396W in CYP24A1 (n = 12 patients: 3 homozygotes, 1 heterozygote and 8 compound heterozygotes; 15/29 mutated alleles). Its pathogenicity and frequency (52%) in the Polish population has been discussed in detail [3].
The average eGFR was 72 mL/min/1.73 m2 (range 15–105). However, two patients with a CYP24A1 mutation (Patients 12 and 17; Table 1) developed ESRD and underwent renal transplantation. Their eGFR at the last visit was 15 mL/min/1.73 m2. Overall, eGFR <90 mL/min/1.73 m2 was found in 14 subjects (77%), including 2 patients after renal transplantation; in 5 of them (28%), including 1 who had renal transplantation, GFR was <60 mL/min/1.73 m2 at the last follow-up visit (Figure 3). The subjects who had GFR <90 mL/min/1.73 m2 at the last follow-up visit did not differ from those who had GFR >90 mL/min/1.73 m2 at the last follow-up [median 47.5 (range 29–60) versus 43 (39.5–73) mL/min/1.73 m2, respectively] in terms of GFR at diagnosis. Two IH survivors with progressive development of ESRD had an eGFR of 24 and 26 mL/min/1.73 m2 at diagnosis and had lowered GFR (45 and 55 mL/min/1.73 m2, respectively) at the ages of 18 and 15 years, respectively (Patients 12 and 17). They developed ESRD in the third and fourth decade of life, respectively, and were then transplanted. In both cases, renal ultrasonography revealed hyperechogenic renal pyramids at the initial presentation and then, during the puberty period, increased echogenicity with disrupted corticomedullary differentiation (Table 1). Correlation between eGFR at discharge after an IH episode and at the last observation (in two patients who developed ESRD eGFR at the last observation was set as 15 mL/min/1.73 m2) did not attain statistical significance (r = 0.419, P = 0.09). Nephrocalcinosis, including both its mild variant (involving only rim) and more advanced stage (reaching the centre of pyramids), was found in 16 of 18 patients in whom ultrasonography was performed at the last follow-up visit (88.8%%), including two patients after renal transplantation in whom nephrocalcinosis was the cause of ESRD. With regard to Patient 14, rim nephrocalcinosis was found at the initial diagnosis and was still present in early childhood, but at the last follow-up visit—at the age of 17 years—ultrasonography was normal despite lower GFR and hypercalcaemia. Serum calcium levels >2.6 mmol/L were found in3 patients (16.5%) and PTH serum levels <10 pg/mL were found in 10 (55%) patients. Serum levels of 25(OH)D3 and 1,25(OH)2D3 were in the normal range in all patients. During the laboratory tests, serum calcium levels did not correlate with PTH concentrations and eGFR (Table 1) values. In one patient, mild hypercalciuria (calcium:creatinine ratio >0.2 mg/mg) was observed. In both patients (Patients 12 and 17), who developed ESRD, ultrasonography performed after an IH episode revealed increased echogenicity, suggesting mild nephrocalcinosis. In one patient (Patient 17), who underwent renal transplantation, a native renal biopsy was performed that revealed diffuse tubulointerstitial renal fibrosis with calcifications of intra- and peritubular capillaries. In two patients with SLC34A1 (Patients 2 and 16) and in one with CYP24A1 (Patient 10) mutations, variants were heterozygous and their clinical course was milder with a relatively high eGFR value both at diagnosis and at the last follow-up visit. Of three patients who had SLC34A1 variants, all had nephrocalcinosis and one patient with a heterozygous SLC34A1 variant developed nephrolithiasis.
FIGURE 3.
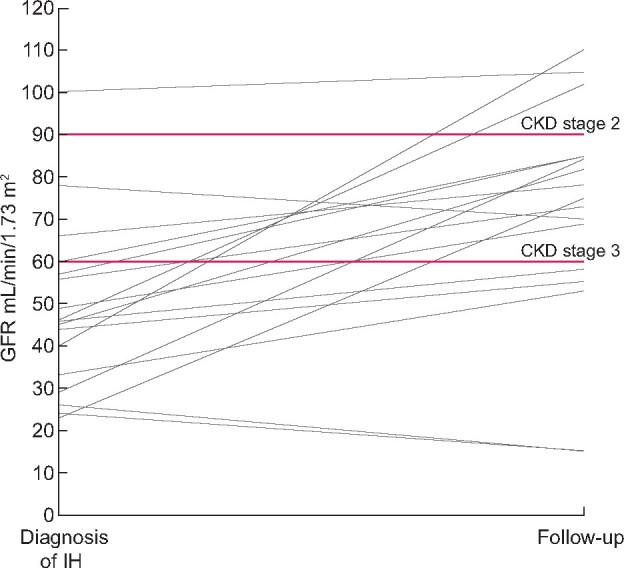
Changes of eGFR from the first observation at diagnosis of IH to the last observation at follow-up. In two patients who developed ESRD and were then transplanted the last eGFR was set as 15 mL/min/1.73 m2. Two horizontal lines were set at values of 90 and 60 mL/min/1.73 m2 to discriminate CKD Stages 2 and 3.
DISCUSSION
IH is directly linked to hypersensitivity to vitamin D. There were two episodes of endemic occurrence of this disorder as a result of prevention of rickets—the first time in the early 1950s in the UK [9, 10], as an effect of food fortification with vitamin D, and then in the 1980s in Poland and former East Germany, associated with the use of high-dose vitamin D prophylaxis [4]. Clinical symptoms were directly related to vitamin D intake. Pronicka et al.[11] postulated that the molecular mechanism of disease is based on decreased catabolism of active metabolites of vitamin D or increased synthesis. Both hypotheses were confirmed in the last decade, when two mechanisms of IH pathogenesis were proved [1, 2]. In 2011, Schlingmann et al. [1] found recessive mutations of CYP24A1 as a molecular basis for IH type 1, with autosomal recessive inheritance. CYP24A1 was found to encode 25-hydroxyvitamin D-24-hydroxylase, the key enzyme of 25(OH)D3 and 1,25(OH)D3 degradation (Figure 1). Decreased catabolism of an active form of vitamin D results in hypervitaminosis with all of its consequences. The estimated mutation frequency of CYP24A1 is 420/100 000 people [12]. This mutation is estimated to be the cause of renal calculi in 4–20% of patients with nephrolithiasis, suggesting the crucial role of vitamin D metabolism in the aetiopathogenesis of this disorder [12]. Further studies have revealed that IH may also be an effect of recessive mutation of the SLC34A1 gene [2], encoding sodium phosphate cotransporter NaPi-IIa. This form of IH is currently termed type 2. Available data demonstrate that primary renal phosphate wasting caused by abnormal cotransporter induces an excessive production of 1,25(OH)D3 with subsequent hypercalcaemia, which decreases rapidly along with phosphate supplementation. In a previous report we described CYP24A1 and SLC34A1 mutations in 11 survivors of IH (in 9 and 2 patients, respectively) [3].
In this article we report the long-term outcome in 18 subjects who suffered from IH, in whom a molecular analysis was performed and mostly biallelic variants of CYP24A1 and SLC34A1 genes were found. In three patients, only one mutant allele of CYP24A1 or SLC34A1 was found, and in these cases we cannot exclude the possibility of a second pathogenic variant not detected by available sequence analysis. Nevertheless, their IH diagnosis was additionally verified by two other parameters: clinical and biochemical pictures. Patients with heterozygous mutations and possibly a milder phenotype have been reported. It has also been noted that heterozygous carriers appear to represent a predisposition for the development of nephrolithiasis [1, 2]. As far as we know, it is the largest described group of survivors of IH with a confirmed molecular defect. The analysis of this larger group of IH survivors revealed that 14 of 18 (77%) subjects had GFR <90 mL/min/1.73 m2. In three of them their GFR was <60 mL/min/1.73 m2 at the last follow-up visit and two developed ESRD. Second, we found that despite avoidance of sun exposure, vitamin D and calcium supplementation, nephrocalcinosis and CKD developed in a large group of subjects. Third, during the laboratory tests, serum calcium levels were elevated in two subjects and lower PTH levels were found in 10 (55%) patients, which indicates ongoing exposure to higher calcium and/or active vitamin D metabolite levels. However, hypercalciuria was observed in only one subject. Altogether, these findings may explain nephrocalcinosis and tubulointerstitial injury resulting in CKD and suggest that hypercalciuria is not the main abnormality.
Data on clinical manifestations and long-term consequences of IH are fragmentary and concern only single case reports or small groups of patients. The relationship among hypercalcaemia, nephrocalcinosis and genetic variants of CYP24A1 and SLC34A1 indicates that disorders of catabolism and overproduction of vitamin D are essential in the pathogenesis of chronic kidney injury in this condition. It has been reported [5, 13–16] that patients with a history of IH (even in the normocalcaemic phase), with significant persistent hypercalciuria and elevated (in relation to estimated oral intake of vitamin D) serum levels of 25(OH)D3, especially during summer months, when serum calcium levels also increased to the upper limit of normal or above the norm.
Nephrocalcinosis and/or nephrolithiasis is a typical consequence of IH as well as CYP24A1 and SLC34A1 mutations, and it is detected in almost all affected individuals [1, 2, 12–30]. Furthermore, nephrocalcinosis and nephrolithiasis have been observed even in patients with SLC34A1 heterozygous mutations [31]. The same observations were found in our study. Nephrocalcinosis is usually permanent and asymptomatic or results in progressive CKD [17]. Figueres et al. [16] described seven individuals with a history of IH in six cases; two of them developed CKD as well as suffered from extrarenal manifestations, including calcific deposits in the cornea and osteoporosis. Patients with CKD and CYP24A1 or SLC34A1 mutations, as well as persistent nephrocalcinosis, were also reported by Dinour et al., although they did not report any case of ESRD [32, 33]. One patient was diagnosed with nephrocalcinosis antenatally and at the age of 18 years, despite proper treatment, suffered from CKD with a GFR value of 60 mL/min/1.73 m2. Meusberger et al. [13] also reported a case of the patient with CYP24A1 mutation and medullary nephrocalcinosis, CKD Stage 2, microalbuminuria, mild hypertension, nephrogenic diabetes insipidus, mild hypercalcaemia and moderate hypercalciuria who had a personal history of hypercalcaemia in the early childhood . In contrast, Murphy et al. [30] reported a 2-year-old patient with a history of symptomatic IH and nephrocalcinosis in infancy who, at the age of 2 years, had reduced GFR (74 mL/min/1.73 m2), but in whom nephrocalcinosis disappeared. It should be noted that genetic variants of CYP24A1 or SLC34A1 were found in adults who were diagnosed due to recurrent nephrolithiasis, with or without nephrocalcinosis [12, 20, 32, 34–42]. The hypothesis of CYP24A1 mutations as a cause of nephrocalcinosis or nephrolithiasis is also supported by the report of Molin et al. [43], who found biallelic mutations in 20 individuals of a cohort of 72 hypercalcaemic patients with low PTH. Nephrocalcinosis was found in 14 patients, nephrolithiasis in 4 and 1 suffered from hypercalciuria.
Our analysis has shown that although nephrocalcinosis was common, there was no direct relationship between the severity of nephrocalcinosis and a decrease of GFR. Both patients who developed ESRD and then underwent transplantation had only increased kidney echogenicity and hyperechogenic pyramids in ultrasonography. In contrast, other subjects, in whom ultrasonography revealed diffuse nephrocalcinosis, did not develop ESRD and one subject (Patient 14), who had a lowered GFR value at the age of 16 years, did not have ultrasonographic signs of nephrocalcinosis at the last follow-up visit despite nephrocalcinosis at initial presentation. These findings suggest that the determinant of progression to ESRD is not only mineral deposits with nephrocalcinosis, but rather tubulointerstitial inflammation and fibrosis. The data on histopathological changes in the kidneys of the subjects who survived IH are very limited. Gigante et al. [29] reported a case of one patient with a CYP24A1 mutation who was diagnosed with hypercalcaemia, nephrogenic diabetes insipidus and nephrocalcinosis and in whom, due to worsening renal function (CKD Stage 2), renal biopsy was performed. The histopathological examination showed chronic tubulointerstitial nephritis . Such changes were found in the renal biopsy of one of our patients, in whom diffuse tubulointerstitial fibrosis and microlithiasis foci were found. It is not known how tubulointerstitial inflammation develops in some subjects with nephrocalcinosis and CYP24A1 or SLC34A1 mutations. One may speculate that deposit composition is significant—e.g. phosphate or oxalate deposits may cause a weaker or stronger inflammatory reaction. However, as is shown in Figure 3, subjects who developed ESRD and those who had GFR <60 mL/min/1.73 m2 had the lowest GFR at discharge after an acute episode of IH. Thus the severity of the initial kidney injury rather than nephrocalcinosis seems to play a significant role as a trigger of progressive CKD with interstitial fibrosis. Although the correlation between eGFR at discharge after an IH episode and the last eGFR did not attain statistical significance (r = 0.419, P = 0.09). It suggests that analysis of a larger number of subjects could give more reliable results. Second, it should be considered that there were subtle abnormalities of calcium metabolism, because 3 patients still had elevated serum calcium concentrations and 10 had suppressed PTH levels. Ongoing exposure to subclinical disturbances of vitamin D may promote microlithiasis/nephrocalcinosis with secondary tubulointerstitial inflammation. Thus, although subjects from our cohort were characterized by 25(OH)D3 and 1,25(OH)2D3 levels in the normal range, the 25(OH)D3:1,25(OH)2D3 ratio might indicate greater exposure to active vitamin D3 over time [44]. However, we did not estimate this parameter. Third, ongoing exposure to other factors associated with nephrocalcinosis and tubulointerstitial inflammation, such as oxalate load, non-steroidal anti-inflammatory drugs and/or purines may play a role.
Fourteen of 18 subjects from our cohort were exposed to high-dose prophylaxis, which was used in the years 1970–80. This may explain a relatively high percentage of CKD in our cohort. However, four other subjects (Patients 9, 10, 14 and 16; Table 1) were exposed to relatively low doses of vitamin D3 prophylaxis (<2000 IU/day) and two of them developed CKD in adolescence (Patients 9 and 14). Therefore it suggests that even low doses of vitamin D3 may be associated with IH as well as permanent and progressive kidney injury. Concluding, the pathogenesis of progressive CKD in subjects who suffered from IH is not clear and probably multifactorial and includes both the severity of the initial injury and subtle but persistent disturbances of vitamin D metabolism.
Lowered GFR was observed not only in IH patients with CYP24A1 mutations, but also in one subject with a heterozygous SLC34A1 mutation. It suggests that exposure to greater levels of 1,25(OH)2D3, regardless of the cause, may result in kidney injury. However, the patients who were characterized by SLC34A1 and CYP24A1 heterozygous variants presented a milder clinical course with relatively well-preserved GFR at follow-up visits. One may speculate that other factors might play a role in further progression to CKD and in inducing hypercalcaemia in the neonatal period.
There are some strengths and limitations of our study. The former includes a large number of described IH cases with molecular diagnosis (observed for almost three decades). Second, we did the same clinical and laboratory tests in all subjects. Third, all subjects received the same recommendations regarding vitamin D and calcium supplementation as well as sun exposure. Nevertheless, the weak point of the study is that we measured calcium, PTH and metabolites of vitamin D at only one point in time and we did not analyse time-averaged exposure to serum calcium and vitamin D metabolites in the long-term, including periods of sun exposure.
To sum up, we found that the long-term renal prognosis of survivors of IIH is poor, with a high prevalence of CKD, nephrocalcinosis and ongoing subclinical metabolic abnormalities caused by disrupted vitamin D metabolism, despite routine recommendations for avoiding vitamin D, calcium supplementation and sun exposure. Although the majority of our patients from a historic cohort study were exposed to very high doses of vitamin D3, CKD also developed in adolescents who were exposed to relatively low pharmacological doses of vitamin D3. The tendency towards hypercalcaemia and signs of suppression of PTH secretion were present in almost all individuals, despite standard serum levels of 25(OH)D3 and 1,25(OH)2D3. Although the pathogenesis of kidney failure is not obvious, both our findings and other reports suggest that it is exposure to metabolic abnormalities, tubulointerstitial injury and probably the severity of the initial renal injury that leads to progressive CKD. Nevertheless, our results suggest that all survivors of IIH should be under constant renal care. Second, routine recommendations, on the basis of avoidance of sun and vitamin D supplements, are not sufficient, thus other preventive measures—such as early implementation of inhibitors of 25(OH)D3 and 1,25(OH)2D3 synthesis or inductor degradation—should be considered in all survivors of IIH [42, 45]. However, despite promising data, the long-term use and safety of both imidazole derivative and rifampicin in patients with CYP24A1 mutations have not been studied. This should be the subject of future research. Third, more sensitive markers of disrupted metabolism of vitamin D3, such as 1,25(OH)2D3 and the 25(OH)D3:1,25(OH)2D3 ratio, may be useful in monitoring disease activity especially in case of a CYP24A1 variant.
ACKNOWLEDGEMENTS
We express our gratitude to all the patients and their families for participating in this study.
FUNDING
The study was supported by project NSC 2014/15/B/NZ5/03541.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1. Schlingmann KP, Kaufmann M, Weber S. et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med 2011; 365: 410–421 [DOI] [PubMed] [Google Scholar]
- 2. Schlingmann KP, Ruminska J, Kaufmann M. et al. Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol 2016; 27: 604–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pronicka E, Ciara E, Halat P. et al. Biallelic mutations in CYP24A1 or SLC34A1 as a cause of infantile idiopathic hypercalcemia (IIH) with vitamin D hypersensitivity: molecular study of 11 historical IIH cases. J Appl Genetics 2017; 58: 349–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pronicka E, Kulczycka H, Rowińska E. et al. Idiopathic hypercalcemia as a syndrome of hypersensitivity to vitamin D3 in 19 infants. Pediatr Pol 1985; 60: 288–294 [PubMed] [Google Scholar]
- 5. Pronicka E, Rowińska E, Kulczycka H. et al. Persistent hypercalciuria and elevated 25-hydroxyvitamin D3 in children with infantile hypercalcaemia. Pediatr Nephrol 1997; 11: 2–6 [DOI] [PubMed] [Google Scholar]
- 6. Ciara E, Rokicki D, Lazniewski M. et al. Clinical and molecular characteristics of newly reported mitochondrial disease entity caused by biallelic PARS2 mutations. J Hum Genet 2018; 63: 473–485 [DOI] [PubMed] [Google Scholar]
- 7. Robinson JT, Thorvaldsdóttir H, Winckler W. et al. Integrative genomics viewer. Nat Biotechnol 2011; 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zemojtel T, Köhler S, Mackenroth L. et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Transl Med 2014; 6: 252ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lightwood R. Idiopathic hypercalcaemia with failure to thrive: nephrocalcinosis. Proc R Soc Med 1952; 45: 40 [Google Scholar]
- 10. Martin ND, Snodgrass GJ, Cohen RD.. Idiopathic infantile hypercalcaemia – a continuing enigma. Arch Dis Child 1984; 59: 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pronicka E, Kulczycka H, Lorenc R. et al. Increased serum level of 1,25-dihydroxyvitamin D3 after parathyroid hormone in the normocalcemic phase of idiopathic hypercalcemia. J Pediatr 1988; 112: 930–933 [DOI] [PubMed] [Google Scholar]
- 12. Nesterova G, Malicdan MC, Yasuda K. et al. 1,25-(OH)2D-24-hydroxylase (CYP24A1) deficiency as a cause of nephrolithiasis. Clin J Am Soc Nephrol 2013; 8: 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meusburger E, Mündlein A, Zitt E. et al. Medullary nephrocalcinosis in an adult patient with idiopathic infantile hypercalcaemia and a novel CYP24A1 mutation. Clin Kidney J 2013; 6: 211–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tebben PJ, Milliner DS, Horst RL. et al. Kumar R: Hypercalcemia, hypercalciuria and elevated calcitriol concentrations with autosomal dominant transmission due to CYP24A1 mutations: effects of ketoconazole therapy. J Clin Endocrinol Metab 2012; 97: 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang J, Coman D, McTaggart SJ. et al. Long-term follow-up of patients with idiopathic infantile hypercalcaemia. Pediatr Nephrol 2006; 21: 1676–1680 [DOI] [PubMed] [Google Scholar]
- 16. Figueres ML, Linglart A, Bienaime F. et al. Kidney function and influence of sunlight exposure in patients with impaired 24-hydroxylation of vitamin D due to CYP24A1 mutations. Am J Kidney Dis 2015; 65: 122–126 [DOI] [PubMed] [Google Scholar]
- 17. Ertl D, Raimann A, Csaicsich D. et al. A pediatric patient with a CYP24A1 mutation: four years of clinical, biochemical, and imaging follow-up. Horm Res Paediatr 2017; 87: 196–204 [DOI] [PubMed] [Google Scholar]
- 18. Kurnaz E, Savaş Erdeve Ş, Çetinkaya S. et al. Rare cause of infantile hypercalcemia: a novel mutation in the SLC34A1 gene. Horm Res Paediatr 2019; 91: 278–284 [DOI] [PubMed] [Google Scholar]
- 19. Sun Y, Shen J, Hu X. et al. CYP24A1 variants in two Chinese patients with idiopathic infantile hypercalcemia. Fetal Pediatr Pathol 2019; 38: 44–56 [DOI] [PubMed] [Google Scholar]
- 20. Fearn A, Allison B, Rice SJ. et al. Clinical, biochemical, and pathophysiological analysis of SLC34A1 mutations. Physiol Rep 2018; 6: e13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dinour D, Davidovits M, Aviner S. et al. Maternal and infantile hypercalcemia caused by vitamin-D-hydroxylase mutations and vitamin D intake. Pediatr Nephrol 2015; 30: 145–152 [DOI] [PubMed] [Google Scholar]
- 22. Castanet M, Mallet E, Kottler ML.. Lightwood syndrome revisited with a novel mutation in CYP24 and vitamin D supplement recommendations. J Pediatr 2013; 163: 1208–1210 [DOI] [PubMed] [Google Scholar]
- 23. Fencl F, Bláhová K, Schlingmann KP. et al. Severe hypercalcemic crisis in an infant with idiopathic infantile hypercalcemia caused by mutation in CYP24A1 gene. Eur J Pediatr 2013; 172: 45–49 [DOI] [PubMed] [Google Scholar]
- 24. Rajagopal A, Braslavsky D, Lu JT. et al. Exome sequencing identifies a novel homozygous mutation in the phosphate transporter SLC34A1 in hypophosphatemia and nephrocalcinosis. J Clin Endocrinol Metab 2014; 99: 2451–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Skalova S, Cerna L, Bayer M. et al. Intravenous pamidronate in the treatment of severe idiopathic infantile hypercalcemia. Iran J Kidney Dis 2013; 7: 160–164 [PubMed] [Google Scholar]
- 26. Madsen J, Sauer S, Beck S. et al. CYP24A1 mutation in a girl infant with idiopathic infantile hypercalcemia. J Clin Res Pediatr Endocrinol 2018; 10: 83–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dauber A, Nguyen TT, Sochett E. et al. Genetic defect in CYP24A1, the vitamin D 24-hydroxylase gene, in a patient with severe infantile hypercalcemia. J Clin Endocrinol Metab 2012; 97: E268–E274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schlingmann KP, Cassar W, Konrad M.. Juvenile onset IIH and CYP24A1 mutations. Bone Rep 2018; 9: 42–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gigante M, Santangelo L, Diella S. et al. Mutational spectrum of CYP24A1 gene in a cohort of Italian patients with idiopathic infantile hypercalcemia. Nephron 2016; 133: 193–204 [DOI] [PubMed] [Google Scholar]
- 30. Murphy J, Joseph M, Larsen CP.. Infantile nephrocalcinosis resulting from a pathogenic CYP24A1 mutation. Kidney Int Rep 2019; 4: 893–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cools M, Goemaere S, Baetens D. et al. Calcium and bone homeostasis in heterozygous carriers of CYP24A1 mutations: a cross-sectional study. Bone 2015; 81: 89–96 [DOI] [PubMed] [Google Scholar]
- 32. Dinour D, Beckerman P, Ganon L. et al. Loss-of-function mutations of CYP24A1, the vitamin D 24-hydroxylasegene, cause long-standing hypercalciuric nephrolithiasis and nephrocalcinosis. J Urol 2013; 190: 552–557 [DOI] [PubMed] [Google Scholar]
- 33. Dinour D, Davidovits M, Ganon L. et al. Loss of function of NaPiIIa causes nephrocalcinosis and possibly kidney insufficiency. Pediatr Nephrol 2016; 31: 2289–2297 [DOI] [PubMed] [Google Scholar]
- 34. Peiris E, Wusirika R.. A case report of compound heterozygous CYP24A1 mutations leading to nephrolithiasis successfully treated with ketoconazole. Case Rep Nephrol Dial 2017; 7: 167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiráčková J, Hyšpler R, Alkanderi S. et al. Novel CYP24A1 mutation in a young male patient with nephrolithiasis: case report. Kidney Blood Press Res 2019; 44: 870–877 [DOI] [PubMed] [Google Scholar]
- 36. Ferraro PM, Minucci A, Primiano A. et al. A novel CYP24A1 genotype associated to a clinical picture of hypercalcemia, nephrolithiasis and low bone mass. Urolithiasis 2017; 45: 291–294 [DOI] [PubMed] [Google Scholar]
- 37. O’Keeffe DT, Tebben PJ, Kumar R. et al. Clinical and biochemical phenotypes of adults with monoallelic and biallelicCYP24A1 mutations: evidence of gene dose effect. Osteoporos Int 2016; 27: 3121–3125 [DOI] [PubMed] [Google Scholar]
- 38. Dowen FE, Sayers JA, Hynes AM. et al. CYP24A1 mutation leading to nephrocalcinosis. Kidney Int 2014; 85: 1475. [DOI] [PubMed] [Google Scholar]
- 39. Jacobs TP, Kaufman M, Jones G. et al. A lifetime of hypercalcemia and hypercalciuria, finally explained. J Clin Endocrinol Metab 2014; 99: 708–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Colussi G, Ganon L, Penco S. et al. Chronic hypercalcaemia from inactivating mutations of vitamin D 24-hydroxylase (CYP24A1): implications for mineral metabolism changes in chronic renal failure. Nephrol Dial Transplant 2014; 29: 636–664 [DOI] [PubMed] [Google Scholar]
- 41. Wolf P, Müller-Sacherer T, Baumgartner-Parzer S. et al. A case of late oneset idiopathic infantile hypercalcemia secondary to mutations in the CYP24A1 gene. Endocr Pract 2014; 20: 91–95 [DOI] [PubMed] [Google Scholar]
- 42. Sayers J, Hynes AM, Srivastava S. et al. Successful treatment of hypercalcaemia associated with a CYP24A1 mutation with fluconazole. Clin Kidney J 2015; 8: 453–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang JCY, Nicholls H, Piec I. et al. Reference intervals for serum 24,25-dihydroxyvitamin D and the ratio with 25-hydroxyvitamin D established using a newly developed LC-MS/MS method. J Nutr Biochem 2017; 46: 21–29 [DOI] [PubMed] [Google Scholar]
- 45. Hawkes CP, Li D, Hakonarson H. et al. CYP3A4 induction by rifampin: an alternative for vitamin D inactivation in patients with CYP24A1 mutations. J Clin Endocrinol Metabol 2017; 102: 1440–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]