

SUMMARY
In pathogenic mycobacteria, transcriptional responses to antibiotics result in induced antibiotic-resistance. WhiB7 belongs to the Actinobacteria-specific family of Fe-S containing transcription factors and plays a crucial role in inducible antibiotic-resistance in mycobacteria. Here we present cryo-electron microscopy structures of Mycobacterium tuberculosis transcriptional regulatory complexes comprising RNA polymerase σA- holoenzyme, global regulators CarD and RbpA, and WhiB7, bound to a WhiB7-regulated promoter. The structures reveal how WhiB7 interacts with σA-holoenzyme while simultaneously interacting with an AT-rich sequence element via its AT-hook. Evidently, AT-hooks, rare elements in bacteria yet prevalent in eukaryotes, bind to target AT-rich DNA sequences similarly to the nuclear chromosome binding proteins. Unexpectedly, a subset of particles contained a WhiB7-stabilized closed promoter complex, revealing this intermediate’s structure, and we apply kinetic modeling and biochemical assays to rationalize how WhiB7 activates transcription. Altogether, our work presents a comprehensive view of how WhiB7 serves to activate gene expression leading to antibiotic resistance.
Keywords: Transcription, transcription factor, WhiB7, RNA polymerase, cryo-EM, transcription initiation, antibiotic resistance, iron cluster
eTOC Blurb
Mycobacteria cause diseases, including tuberculosis. The transcription factor WhiB7 induces resistance to certain antibiotics used for treatment. Lilic et al. provide the structural basis of WhiB7 activity. They show that WhiB7 interacts with DNA similarly to several eukaryotic chromosomal-binding proteins. They also present a structure of a WhiB7-stabilized transcriptional intermediate.
Graphical Abstract
INTRODUCTION
Mycobacterium tuberculosis (Mtb) is the causative agent of the disease tuberculosis (TB), which kills 1.5 million people annually (World Health Organization, 2018). Treatment of TB is complicated due to the intrinsic antibiotic resistance of this pathogen (Fonseca et al., 2015). In mycobacteria, exposure to antibiotics results in induced antibiotic resistance (Burian et al., 2012; McKinney, 2000; Morris et al., 2005). WhiB7, a member of the Actinobacteria-specific WhiB family of transcription factors (TFs), induces antibiotic-resistance in Mtb and Streptomyces against an array of antimicrobials, including streptomycin and kanamycin, two second-line treatments for TB (Geiman et al., 2006; Morris et al., 2005). WhiB7 is also the first gene to be upregulated upon exposure to low concentrations of antibiotics, suggesting it is a primary regulator in defense to antibiotics (Morris et al., 2005). Mtb has evolved multiple strategies to deal with stresses presented during infection (Shastri et al., 2018). Most of these stress responses rely on gene expression changes at the transcriptional level mediated by TFs (Manganelli et al., 2004). Therefore, it is proposed that dampening the pathogen’s transcriptional response to host immunity and chemotherapy would potentiate the current antibiotic regimen (McKinney, 2000; Shastri et al., 2018).
In bacteria, transcription initiation is performed by the ~400 kDa RNAP catalytic core enzyme (E, subunit composition α2ββ’ω) and an additional subunit, σA, creating the holoenzyme (EσA), which is capable of promoter-specific initiation (Burgess et al., 1969). EσA performs several steps before transitioning to an elongating complex (Boyaci et al., 2019a; Chen et al., 2020). In the first step, EσA forms a complex with the duplex promoter DNA through sequence-specific recognition of upstream promoter elements. This initial EσA-promoter DNA complex is referred to as the closed complex (RPc). RPc isomerizes through a series of intermediates to form the transcriptionally-competent open promoter complex (RPo) (Boyaci et al., 2019a; Buc and McClure, 1985; Chen et al., 2020; Hubin et al., 2017a; Ruff et al., 2015; Saecker et al., 2011) in which 12 to 14 base pairs of the duplex DNA are unwound to form the transcription bubble. Mtb depends on two essential non-canonical TFs, CarD and RbpA, neither found in the well-studied model bacterium Escherichia coli (Eco), to regulate these steps (Boyaci et al., 2019b).
Like CarD and RbpA, the WhiB family of TFs are absent in Eco but critical for the Mtb lifestyle. WhiBs were initially discovered in antibiotic-producing Streptomyces (Davis and Chater, 1992). Although proposed to have evolved to regulate innate resistance against antibiotics (Morris et al., 2005), the WhiB TFs are involved in multiple functions, including cell division and virulence in pathogenic Mycobacteria and sporulation and antibiotic production in Streptomyces (Bush, 2018). Consistent with this proposal, the WhiB family is unique to Actinobacteria, including the Mycobacterium and Streptomyces genera. The WhiB-like proteins are all small (~100 amino acids), with four conserved cysteines that coordinate a [4Fe-4S] cluster and contain a conserved glycine-rich sequence element. Each WhiB family-member also includes what are thought to be DNA binding determinants, but these vary between the family members (Bush, 2018). Although discovered more than 25 years ago, the biochemical roles of the WhiB proteins remain controversial (Bush, 2018). However, in vitro studies of Mycobacterium smegmatis (Msm) WhiB7 demonstrated that the purified factor interacts with the housekeeping σ-factor (σA) and activates transcription of its own promoter via an AT-rich sequence (Burian et al., 2013). More recently, ChIP-Seq studies performed in Streptomyces confirmed that WhiB (the Mtb WhiB2 homolog) binds to promoter DNA (Bush et al., 2016). The DNA binding motif of each WhiB remains undefined except for WhiB7, which contains a canonical DNA binding AT-hook determinant found in eukaryotic nuclear proteins (Morris et al., 2005). Almost all the WhiBs have been shown to interact with domain 4 of the housekeeping transcription initiation factor σA (σAD4) (Burian et al., 2013; Chen et al., 2016; Kudhair et al., 2017; Steyn et al., 2002). In Mtb, WhiB7 is critical for the antibiotic-induced expression of 12 genes, including efflux pumps and ribosomal methyltransferases (Morris et al., 2005). Therefore, characterizing and disrupting these transcriptional responses are essential for improving the efficacy of the existing repertoire of antibiotics.
Structure/function information on the WhiB family of proteins remains sparse. NMR was used to determine the [4Fe-4S] bound form of Mtb WhiB1 (Kudhair et al., 2017). More recently, a crystal structure of Mtb WhiB1 complexed with σAD4 was determined, detailing the structural basis of the interaction (Wan et al., 2020). However, an understanding of how the WhiB factors interact with both RNAP and DNA to activate transcription remained elusive. Here we present structures of Mtb transcriptional regulatory complexes comprising EσA, global regulators CarD and RbpA, and WhiB7, all bound to the WhiB7-regulated whiB7 promoter (pwhiB7). We also solved the structure of the same assembly in the absence of WhiB7. The structures reveal how WhiB7 interacts with EσA via an interface with σA while simultaneously interacting with an AT-rich sequence upstream of the promoter −35 element via its AT-hook. This structure shows that AT-hooks in bacteria bind to target AT-rich DNA sequences, similar to how nuclear chromosomal proteins bind to AT-rich DNA. Importantly, we find that a subset of particles is in the RPc form, which is stabilized by WhiB7, revealing the structural basis of RPc in Mtb. We use biochemical assays and kinetic modeling to explain this finding and to provide the basis of WhiB7 activation mechanism.
RESULTS
Cryo-EM analysis of Mtb RNAP initiation complexes on the whiB7 promoter reveal distinct transcription intermediates
EσA-CarD-RbpA, with and without WhiB7, were incubated with the WhiB7-regulated whiB7 promoter (Figure 1A) to capture de novo initiation complexes for cryo-EM analysis. Data were curated and classified (Figure S1). Analysis of the cryo-EM data from the complex without WhiB7 yielded a single class at a nominal resolution of 2.7 Å (Figures 1B, S1C, S2G–I and Table 1) comprising the initiation complex engaged with the promoter DNA, with the downstream duplex DNA and a completed transcription bubble enclosed within the RNAP active site cleft, characteristics matching previously determined RPo structures (Boyaci et al., 2019a; Hubin et al., 2017b). In the presence of WhiB7, classification yielded two structural classes (Figures 1C, S1D, S2A–F and Table 1). One class corresponds to RPo (W-RPo) derived from 73% of the particles; nominal resolution of 2.9 Å. In the other class, the initiation complex is engaged with entirely duplex promoter DNA, reminiscent of a previously determined RPc with Eco RNAP (Chen et al., 2020); we call this structure W-RPc (27% of the particles, nominal resolution of 3.4 Å).
Figure 1. Cryo-EM analysis of Mtb RNAP initiation complexes on the whiB7 promoter reveal distinct transcription intermediates.

A. Duplex Mtb whiB7 promoter fragment used for cryo-EM. The promoter elements (AT-rich WhiB7 regulatory motif; yellow, −35 element, blue; −10 element, red) and the transcription start site (+1; arrow) are denoted. The top (non-template) strand and bottom (template) strand are colored light and dark green, respectively.
B. Structural class derived from the cryo-EM data without WhiB7 on the whiB7 promoter. Cryo-EM composite maps are low-pass filtered according to the local resolution (Figure S2I) (Cardone et al., 2013) and shown as white transparent molecular surfaces. The DNA is shown as a solid surface and colored as in (A). The active site Mg2+ is indicated for reference. The number of particles and nominal resolution of each structure are indicated above the structures.
C. Structural classes derived from the cryo-EM data and with WhiB7 on the whiB7 promoter. Cryo-EM composite maps are low-pass filtered according to the local resolution (Figures S2C, F) (Cardone et al., 2013) and shown as white transparent molecular surfaces. The DNA is shown as a solid surface and colored as in (A). WhiB7 is colored in pink and the active site Mg2+ is indicated for reference. The number of particles and nominal resolution of each structure are indicated above the structures. Upper left, Coomassie stained SDS-PAGE analysis of purified σA/WhiB7 complex.
Table 1.
Cryo-EM data collection, refinement and validation statistics
| Mtb WhiB7-RPo (W-RPo) PDB ID 7KIF | Mtb WhiB7-RPc (W-RPc) PDB ID 7KIM | Mtb RPo PDB ID 7KIN | |
|---|---|---|---|
| Data collection and processing | |||
| Magnification | 105,000 | 105,000 | |
| Voltage (kV) | 300 | 300 | |
| Electron exposure (e−/Å2) | 66 | 64 | |
| Defocus range (μm) | 1–2.1 | 0.8–2.2 | |
| Pixel size (Å) | 1.1 | 1.1 | |
| Symmetry imposed | C1 | C1 | |
| Initial particle images (no.) | 4,349,833 | 2,362,200 | |
| Final particle images (no.) | 87,743 | 32,172 | 296,097 |
| Map resolution (Å) FSC threshold 0.143 | 2.94 | 3.38 | 2.74 |
| Map resolution range (Å) | 2.8–7 | 3.3–7.5 | 2.8–6.6 |
| Refinement | |||
| Initial model used (PDB code) | 6EDT | 6EDT | 6EDT |
| Model resolution range (Å) | 2.8–7 | 3.3–7.5 | 2.8–6.6 |
| Map sharpening B factor (A2) | 70.5 | 52.8 | 89.9 |
| Model composition | |||
| Non-hydrogen atoms | 30,322 | 29,573 | 29,379 |
| Protein residues | 3592 | 3572 | 3509 |
| Nucleic acid residues | 118 | 90 | 103 |
| Ligands | 4 (SF4, 1 Mg2+, 2 Zn2+) | 4 (SF4, 1 Mg2+, 2 Zn2+) | 3 (Mg2+, 2 Zn2+) |
| B factors (A2) | |||
| Protein | 104.46 | 167.51 | 100.97 |
| Nucleic acid | 221.97 | 265.08 | 191.21 |
| Ligands | 120.37 | 170.20 | 92.75 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.009 | 0.008 | 0.016 |
| Bond angles (°) | 0.865 | 0.873 | 1.047 |
| Validation | |||
| MolProbity score | 2.3 | 2.53 | 2.2 |
| Clashscore | 4.33 | 7.46 | 3.35 |
| Poor rotamers (%) | 6.85 | 7.44 | 7.82 |
| Ramachandran plota | |||
| Most favored (%) | 84.2 | 82.8 | 87.1 |
| Additional allowed (%) | 15.4 | 17 | 12.5 |
| Generously allowed (%) | 0.3 | 0.2 | 0.3 |
| Disallowed (%) | 0 | 0 | 0 |
Ramachandran plot parameters from PROCHECK
Structures of Mtb CarD-RbpA-EσA-DNA complexes with and without WhiB7
In both W-RPc and W-RPo, WhiB7 engages with the initiation complex through protein-protein interactions with σAD4 in a manner similar to the WhiB1-σAD4 crystal structure (Wan et al.,2020). The interface between WhiB7-σAD4 represents the only interactions observed between WhiB7 and the EσA in the W-RPo structure. In W-RPc (but not in W-RPo), we also observed contacts between the N-terminal tail of WhiB7 and residues of the β’-dock domain (specifically β’ residues 463–466 (Figure 2A, middle panel, inset). The dock domain forms part of the RNA exit channel during elongation. There are only two other factors we are aware of that interact with the dock. The first is σA (via the beginning of σAD4) in the holoenzyme complex. The second is the λ phage antitermination protein N (Krupp et al., 2019), which interacts with elongation complexes and is proposed to affect the RNA exit channel conformation through its interaction with the dock during elongation. We currently do not have an explanation for the significance of the interaction with WhiB7. WhiB7 is an activator but could negatively affect the transition to elongation by stabilizing σAD4 contacts with the DNA and RNAP. However, this remains to be tested.
Figure 2. Structures of Mtb CarD-RbpA-EσA-DNA complexes with and without WhiB7.

A. Overall structures of W-RPo, W-RPc, and RPo on the whiB7 promoter. Core RNAP and RbpA are shown as transparent surfaces. CarD, WhiB7 and σA are rendered in cartoon and the DNAs are shown as molecular surfaces. Each feature is colored as labeled in the key on the bottom right. Middle inset, W-RPo (blue) aligned to W-RPc (colored by key) by WhiB7 to show that the disposition of WhiB7 to the β’ subunit is the same between structures.
B. Structure of RPo on the AP3 promoter (Boyaci et al., 2019a). The upstream DNA corresponding the AT-rich motif of the whiB7 promoter is colored yellow to emphasize the upstream DNA trajectory.
C. Alignment of σAD4 from structures in (A) and (B) shows the trajectories of the upstream DNA in the different structures. Left: The presence of WhiB7 stabilizes the DNA at least 14 bps upstream of the −35 element (pink and grey versus purple). Right: The upstream trajectory of the whiB7 promoter, in the presence of WhiB7, differs from that of RPo with the AP3 promoter (pale green); WhiB7 would clash with the upstream DNA of RPo with the AP3 promoter.
In both structures with WhiB7, the C-terminal AT-hook of WhiB7 binds an AT-rich region upstream of the −35 promoter element (Figures 1 and 2A). The AT-rich region is critical for WhiB7 to activate transcription from this promoter (Burian et al., 2013). Comparing W-RPo to the previously determined Mtb RPo on the rrnAP3 promoter (Boyaci et al., 2019a) shows that the WhiB7-DNA interaction alters the trajectory of the upstream duplex DNA (Figure 2B and C, left). In the presence of WhiB7, the cryo-EM density allows modeling of the upstream duplex DNA to at least −51, while in the absence of WhiB7, the upstream duplex DNA is visible only to - 43 (Figures 2A and C). Most of the WhiB7 induced conformational changes to the promoter DNA occur at and upstream of the −35 element. While the refined models indicate that WhiB7 does not significantly alter the −35 element-σAD4 interaction (Figure 3A), the DNA density, in the absence of WhiB7, starting at the −35 element, becomes increasingly poor in the upstream direction (Figure 3B), requiring that the manual modeling of the −35 element of RPo be completed with a low-resolution (low-pass filtered) map (Cardone et al., 2013) (Figure 3B, top). The density maps’ differences between W-RPo and RPo suggest that the interactions between the −35 element and σAD4 are less stable in the absence of WhiB7.
Figure 3. WhiB7 stabilizes σAD4 contacts with the −35 promoter element.

A. The W-RPo and RPo structures were superimposed by aligning α-carbon positions of σA, the CarD-CTD, and the RbpA-linker (root-mean-square deviation of 0.535 Å over 417 α-carbon atoms). The resulting DNA structures closely superimposed. WhiB7 is shown in faded pink to increase the clarity of σAD4. The disordered and unmodeled regions of the transcription bubble are indicated by spheres.
B. The sharpened (blue mesh) and local-resolution filtered (gray transparent surface) cryo-EM maps are show for RPo (top) and W-RPo (bottom), with the structures superimposed. The view shows a zoom-in of the boxed area in (A), reoriented as shown. Cryo-EM density for σAD4 contacts with the −35 promoter region in RPo is weak in the sharpened map, and density in the local-resolution filtered map disappears about six base-pairs upstream of the −35 motif. Cryo-EM density for the σAD4 contacts with the −35 promoter region in W-RPo is well-resolved and better connected in both the sharpened (blue mesh) and local-resolution filtered (light grey surface) maps, and density extends well upstream of the AT-rich DNA bound by the WhiB7 AT-hook. Maps were normalized in PyMOL and contoured at 4σ.
The weak interaction between the −35 element and σAD4 of RPo in the absence of WhiB7 can be attributed to the non-optimal −35 motif. The consensus −35 element is TTGACA on the non-template strand and AACTGT on the template strand (Shultzaberger et al., 2007). The crystal structure of Thermus aquaticus σAD4 with the −35 element revealed that the guanosine base in the template strand formed two hydrogen bonds with an Arg that corresponds to R500 of Mtb σAD4 (Campbell et al., 2002). We note that the −35 element of the whiB7 promoter has a cytosine in the template strand at this position and could not form favorable polar interactions with the Arg side chain. The loss of these two hydrogen bonds would negatively affect the −35 element-σAD4 contacts, explaining the fragmented density in the absence of WhiB7 (Figure 3B, top). WhiB7 binding to the upstream AT-rich region compensates for the poor whiB7 promoter −35 element (Figure 3B, bottom).
In W-RPo and RPo, the DNA conformation and contacts with the main DNA-interacting subunits, σA, CarD, and RbpA, are essentially identical. This similarity is most apparent with the downstream DNA contacts with the holoenzyme, where the cryo-EM maps are better resolved. The only difference was that the template strand −11 and −10 nucleotides of W-RPo were not modeled because the cryo-EM density was absent. This observation suggests that W-RPo may be less stable. However, this could also result from the slightly lower resolution of the W-RPo (2.9 Å) versus the RPo (2.7Å) structure. On this basis, we conclude that the WhiB7-upstream DNA interaction stabilizes binding between the holoenzyme via σAD4 and the promoter DNA at the −35 element but does not significantly affect downstream promoter interactions.
WhiB7 interactions with σAD4
Like the interactions between WhiB1 and σAD4 observed in the crystal structure (Wan et al., 2020), most of the interactions between WhiB7 and σAD4 are nonpolar (Figures 4A and B). However, in contrast to WhiB1-σAD4, two new salt bridges are observed: Between WhiB7-D26 with σA-R519, and WhiB7-E61 with σA-R515. Docking analyses predicted the latter interaction, and subsequent mutational analyses showed that each of these residues was required for Msm to grow in the presence of antibiotics (Burian et al., 2013). A sequence alignment of 13 WhiB7 homologs (Figure S3A) reveals that E61 is invariable, pointing to its importance for the WhiB7-σAD4 interaction. WhiB7-E61 is part of an EPW motif (absolutely conserved in WhiB7 sequences) just upstream of the glycine-rich motif, also conserved in all WhiB-family members (Bush, 2018). The EPW motif appears to be key for the WhiB7-σAD4 interface, making multiple interactions with σAD4. We note that E61 of the EPW motif is present as an ERF motif in the WhiB2 (known as WblE or WhiB in Streptomyces) class of factors, suggesting it may also play a role in interacting with σAD4. A sequence alignment of the 13 Mtb σ-factors shows that most of the residues involved in WhiB1 and WhiB7 interactions are not conserved, suggesting that these two factors only regulate σA (Figure S3B).
Figure 4. WhiB7 interactions with σAD4 and DNA.

A. Structure-based sequence alignment of Mtb WhiB7 and WhiB1. Residues of WhiB1 that interact with σAD4 (Wan et al., 2020) are highlighted by the nature of interactions and compared to those of WhiB7 (this study). Residues of σAD4 participating in ionic interactions are indicated above or below the sequences. The DNA binding AT-hook motif of WhiB7 is boxed. The interactions between WhiB7 and σAD4 were determined using a 4.5 Å distance cutoff (CCP4) (Winn et al., 2011) between carbon atoms for nonpolar interactions, a 4.5 Å cutoff between basic and acidic side chains for ionic interactions, and a 3.5 Å cutoff for hydrogen bonds between donors and acceptors. Identical residues are bolded orange, homologous residues colored orange, and the conserved cysteines that chelate the 4Fe-4S cluster are in bolded purple. The secondary structure of each protein is shown in red: H indicates helices.
B. Residues of WhiB7 and σAD4 that interact with each other are shown in stick, and each protein’s backbone is shown in cartoon tube. Dotted lines connect residues of σAD4 and WhiB7 that participate in ionic or hydrogen bond interactions. The 4Fe-4S cluster is drawn as spheres.
C. Top left: WhiB7 AT-hook interactions with the AT-rich DNA are indicated. Both the AT-hook and the DNA are drawn as cartoon sticks and colored, as indicated. Bottom left: A top view showing the WhiB7 AT-hook fits intimately into the minor groove of the AT-rich motif. The DNA is rendered in surface, and backbone oxygens are colored red to highlight the ionic and hydrogen bonds between WhiB7 and the DNA. Top Right: an alignment of the AT-rich DNA of W-RPo to that of the crystal structure of the human HMGA1 AT-hook with DNA (Fonfría-Subirós et al., 2012) shows that the AT-hooks superimpose well. Bottom Right: Image in the top inset shows the W-RPo structure for reference with the region of interest boxed.
The interfaces of WhiB1 and WhiB7 on σAD4 almost completely overlap (Figure S4A), indicating that the factors compete with each other (and likely the other five WhiBs) for binding to σA. Therefore, the expression and availability of each WhiB factor, the binding affinity for σAD4, and additional binding partners (Bush et al., 2016) are expected to determine RNAP occupancy. Comparing the structures of WhiB1 and WhiB7 with and without σAD4 reveals that the 4Fe-4S cluster of WhiB7 is more exposed to solvent (Figure S4B) due to the WhiB1 N-terminus (not conserved with WhiB7) helping to bury the 4Fe-4S cluster. Therefore, the state of oxidative stress in the cell could also determine each factor’s availability as WhiB7 would be expected to be more sensitive to oxidative stress.
WhiB7 AT-hook/AT-rich DNA interactions are similar to the eukaryotic nuclear chromosomal proteins/DNA interactions
WhiB7 is the only representative of the WhiBs to have a canonical DNA binding motif, a conserved AT-hook near its C-terminus (Morris et al., 2005). AT-hooks are prevalent in eukaryotic chromosomal binding proteins but have been reported in Myxococcus xanthus CarD (not related to the Mtb CarD) (Aravind, 1998; Nicolas et al., 1996). The core AT-hook sequence motif (RGRP) is often flanked by basic residues and prolines (Figure 4A), tethering proteins to the DNA (Aravind, 1998). Structures of AT-hook-containing eukaryotic nuclear proteins bound to AT-rich elements revealed that the RGR motif forms extensive hydrophobic and polar contacts exclusively in the DNA minor groove (Fonfría-Subirós et al., 2012; Huth et al., 1997). The local resolution of the cryo-EM maps around the WhiB7 AT-hook (~4.5 Å; Figures S2 and S4C) did not allow de novo model building. However, since the AT-hook sequences of WhiB7 and the HMGA protein are identical in sequence, we modelled the WhiB7 AT-hook by superimposing the AT-rich DNA from the HMGA crystal structure (PDB ID: 3UXW) onto the AT-rich region of the whiB7 promoter. The resulting position of the HMGA AT-hook fit the density well (Figure S4C), and subsequent modeling and restrained refinement resulted in only small adjustments (Figure 4C). The WhiB7 AT-hook forms a network of van der Waal’s interactions, hydrogen bonds and salt bridges with the DNA. The glycine and prolines appear to dictate the hook’s backbone conformation, while the basic residues form polar and ionic interactions with the DNA backbone (Figure 4C). The Args also slither into the minor groove, forming extensive nonpolar interactions with the DNA ribose carbons. Lastly, R85 is positioned to make a base-specific hydrogen bond with O2 from a thymine in the middle of the AT-rich sequence.
An Mtb closed promoter complex (RPc) structure
In the dataset with WhiB7, 27% of particles are found in W-RPc. Previous work revealed the structure of an Eco RPc (Chen et al., 2020). Here, we compare the conformational changes observed between RPc and RPo in Mtb, and between the RPcs of Eco and Mtb. We focus on CarD, RbpA, the N-terminus of σAD2, and three mobile modules that form the RNAP pincers and secure the DNA in a large cleft upon promoter melting (Boyaci et al., 2019a; Chakraborty et al., 2012; Chen et al., 2020). The first module, which makes up the bottom pincer, is the clamp (colored pink, Figure 5), which includes σAD2, critical for recognizing and binding the promoter - 10 element (Boyaci et al., 2018; Feklistov et al., 2017). The top pincer is made up of two domains: The first is the β-lobe, which secures the downstream duplex DNA in RPo (colored blue, Figure 5). The second domain is the β-protrusion, also involved in securing the promoter DNA (colored cyan, Figure 5). In Mtb RNAP, the essential transcription factor CarD is required for efficient promoter melting (Srivastava et al., 2013; Stallings et al., 2009). CarD binds the β- protrusion at the upstream edge of the transcription bubble and prevents the bubble from collapsing, stabilizing RPo (Bae et al., 2015a; Boyaci et al., 2019a; Hubin et al., 2017a; Rammohan et al., 2015) (colored green, Figure 5).
Figure 5. Comparisons of structural differences between W-RPc, W-RPo and Eco RPc.

A. Structural alignment of the core module (Boyaci et al., 2018) of W-RPc to W-RPo shows that in WRPc, σA N-terminal domain helix (W-RPc σ-NTDh; orange) occupies the space in the channel where the downstream-DNA (colored yellow in this model) eventually rests in W-RPo. Upon W-RPo formation, the helix is moved (W-RPo σ-NTDh; brick red) to interact with the β- lobe so that the DNA can be placed in its RPo position. The structure shown is W-RPo with the superimposed W-RPc σ-NTD. Core, β lobe, β protrusion, clamp and CarD are colored as listed in key on the right.
B. Structural alignment of the β-protrusion between W-RPo and W-RPc reveals that CarD from W-RPc would clash with the double-stranded −10 element DNA. In W-RPc, the loops on the CarD-CTD (colored purple) are disordered, presumably due to the clash with the double-stranded DNA. In W-RPo, RbpA-R79 is positioned to form an ionic bond (2.0 Å distance) with OP1 of the non-template strand DNA −14 position, an interaction shown previously to be critical for RbpA transcription activation function (Hubin et al., 2017). However, RpbA-R79 in W-RPc is 19 Å away from the non-template DNA −14 phosphate oxygens, and the closest nucleotide is the non-template strand −16 position, located 8.7 Å away. Thus, RbpA-DNA backbone contacts are not established in RPc. Dashed lines indicate distances, with the distance of 8.7 Å noted for reference.
C. Structural alignment of the core module of Eco RPc (Boyaci et al., 2018) and W-RPc reveals that the downstream DNA trajectory changes due to CarD-CTD (green). The structure shown is W-RPc, except for the Eco RPc DNA (slate). Core, β lobe, β protrusion, clamp and CarD are colored as listed in key in (A).
D. Distances between the clamp and the β-lobe (α-carbons of σA-F233 and β-G284), the clamp and β-protrusion (α-carbons of σA-R309 and b-E402), and the clamp and CarD-CTD (a-carbons of σA-T345 and CarD-R87) were measured for W-RPc (left) and W-RPo (right) using PyMOL (The PyMOL Molecular Graphics System, Version 2.3.5 Schrodinger, LLC). These distances are illustrated by white double-edged arrows and noted on the “top” pincer domains. Core, β lobe, β protrusion, clamp, CarD, σ-NTDh and DNA are colored as listed in key in (A).
N-terminal to the conserved structural domain σAD2, Actinobacterial Group 1’s have a conserved linker helix extending from the N-terminus of σAD2 (σ-NTDh). The σ-NTDh is preceded by a large N-terminal extension (206 residues in Mtb σA) that appears to be an intrinsically disordered region [IDR (Hubin et al., 2017b)]. The IDR has not been observed in any structural context, but in previously observed mycobacterial RPos (Boyaci et al., 2019a; Hubin et al., 2017a; Lin et al., 2017) as well as W-RPo, the σ-NTDh crosses the RNAP cleft at a more or less right angle and interacts with the β-lobe. In W-RPc, where the RNAP cleft is much more open, the σ-NTDh also crosses the cleft to interact with the β-lobe but at a much shallower angle (Figure 5A). The position of the σ-NTDh clashes with the ultimate position of the downstream duplex DNA in RPo; the rotation to the position in RPo allows the downstream duplex DNA to be accommodated (Figure 5A). In W-RPc, the downstream promoter DNA is located outside of the cleft in front of the σ-NTDh, while in RPo, the transcription bubble and downstream duplex DNA are inside the cleft behind the σ-NTDh. Thus, an intermediate along the RPo formation pathway must exist (between RPc and RPo) where the σ-NTDh (and associated IDR) swing open and create a gap between the clamp and the β-lobe to allow the DNA to enter the cleft (Boyaci et al., 2019a).
As observed in previously determined RPo structures containing CarD (Bae et al., 2015b; Boyaci et al., 2019a), the CarD C-terminal domain (CarD-CTD) in the W-RPo and RPo structures determined here interacts with the upstream edge of the transcription bubble, positioning a universally conserved ‘wedge’-Trp in the widened DNA major groove. Previous modeling indicated that the expected path of the duplex DNA in RPc would clash with the CarD-CTD. We proposed that the clash had to be relieved either by RNAP pincer opening, by a conformational change of the CarD-CTD (moving it out of the way of the DNA), or both (Bae et al., 2015b; Srivastava et al., 2013). The W-RPc structure shows that the CarD-CTD in W-RPc becomes very flexible (poor cryo-EM density) and density for the DNA-proximal end of the CarD-CTD (where the wedge-Trp resides) is completely absent (Figure 5B), suggesting that CarD moves out the way of the DNA in the RPc state. In addition, the RNAP pincers in RPc is open compared with W-RPo (Figures 5D). Thus, both pincer opening and CarD-CTD flexibility allow space for the duplex −10 element DNA in W-RPc. In the Eco RPc (Chen et al., 2020), the DNA trajectory differs from that observed for Mtb. The Eco RPc downstream DNA is tilted towards where CarD is in the W-RPc: This suggests that CarD alters the DNA trajectory in RPc, offering a structural explanation for how CarD affects promoter melting by possibly pushing the DNA towards the clamp and consequently increasing interactions with the DNA-melting residues of σA in clamp and DNA.
RbpA, a σ-tether (Chen et al., 2021), has been shown to contact the non-template promoter strand via a conserved Arg (RbpA-R79), an interaction critical for its role as a transcriptional activator (Hubin et al., 2015). RbpA interacts with σA and core RNAP similarly in all three structures reported here (Figure 2A), as well as to previously determined RPo structures (Boyaci et al., 2019a; Hubin et al., 2017b, 2017a). RbpA-R79 is positioned to form an ionic bond with the phosphate backbone of the −14 non-template base in both W-RPo and RPo (Figure 5B illustrates the interaction for W-RPo). However, in W-RPc, because of the trajectory of the DNA, RbpA-R79 is 19 Å away from the −14 non-template base phosphate backbone and 8.7 Å away from the closest atom of the DNA; too far to form favorable molecular interactions (Figure 5B). This finding explains the results of our previous kinetic analysis showing that RbpA has a negligible effect on RPc formation at two promoters (pvapB and prrnAP3) (Hubin et al., 2017a).
To assess the width of the RNAP active site cleft, we measured distances between an α-carbon on the clamp (the bottom pincer) and an α-carbon on three positions of the top pincer, CarD, the β-protrusion, and the β-lobe (Figure 5D). Each of these distances increased 10 Å in W-RPc compared with W-RPo (Figure 5D). We ranked a series of RNAP structures according to the measured pincer opening, from most open to most closed (Fig. S4D):
Mtb Fidaxomicin-RNAP > Mtb W-RPc > Mtb free RNAP > Eco RPc > Mtb RP2 > Mtb Rpo
This analysis reveals the distances between pincers necessary to accommodate the double- stranded −10 element, and that Mtb free RNAP is in the “ready” conformation to form RPc.
Kinetic modeling of WhiB7’s effect on the RPo formation pathway
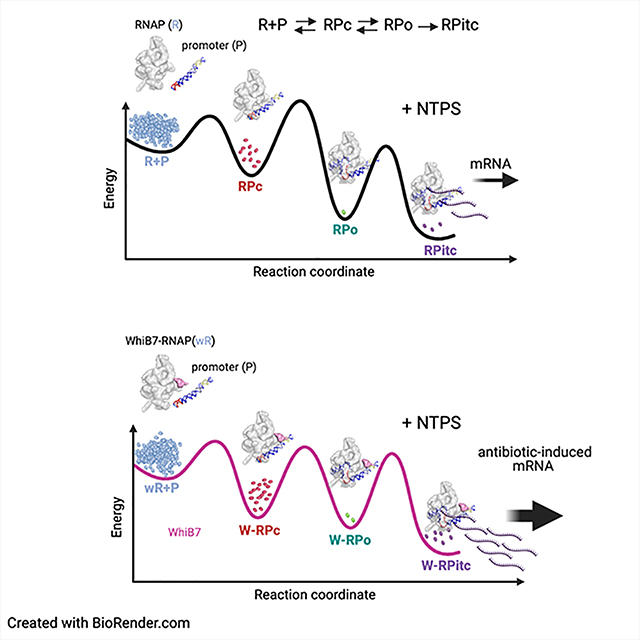
Our in vitro transcription assays show that WhiB7 activates transcription from the whiB7 promoter approximately 2-fold (Figure 6A). For cryo-EM sample preparation, the EσA and associated factors were equilibrated with whiB7 promoter DNA without nucleotide substrates. In the absence of WhiB7, only RPo was observed (Figure 1B), while in the presence of the activator WhiB7, 73% of the particles formed RPo and 27% of the particles comprised an RPc intermediate (Figure 1C). This finding is consistent with electrophoresis mobility shift assays (EMSAs), demonstrating that WhiB7 increases the shift by RNAP of pwhiB7 DNA by increasing the population of RPc (Figure 6B).
Figure 6. Kinetic modeling of WhiB7’s effect on the RPo formation pathway.

A. Representative transcription assays showing abortive transcripts synthesized from Mtb RNAP and Mtb RNAP/WhiB7 holoenzymes. The assay was performed in triplicate and the results were quantified and shown in the histogram. The activity of Mtb RNAP was normalized to 1 and the values are the average of triplicate experiments. The error bars denote the mean standard errors.
B. Native EMSA showing that WhiB7 increases Mtb holo-RbpA/CarD shifting of the pwhiB7 DNA. In the presence of WhiB7, two shifted bands are observed; one is resistant to heparin (RPo), the other is heparin-sensitive (RPc).
C. The transcript flux calculator (Galburt, 2018) was used to illustrate that an activator like WhiB7 could alter the kinetic parameters in a simple four-state kinetic model (Walter et al., 1967): R + P ⇄ RPc ⇄ RPo → RPITC to increase the steady state flux into RPITC by a factor of approximately 2. (top) Kinetic model (Galburt, 2018) and the definition of the kinetic parameters (see text). The table on the right lists the values of the rate constants for basal and activated transcription, yielding the energy profiles shown on the left (black line, basal; green line, activated) (Galburt, 2018). The calculated transcript flux for the basal conditions was normalized to 1. The activated conditions increase the transcript flux by a factor of 2.2 (Galburt, 2018). (bottom) Under the equilibrium conditions of cryo-EM grid preparation (no nucleotide substrates, so no transition of RPo into RPitc): R + P ⇄ RPc ⇄ RPo. The basal kinetic parameters (no WhiB7) yield only RPo at equilibrium, while the same changes in the kinetic parameters that activate transcription ~2-fold (A) result in the appearance of RPc in equilibrium with RPo at a ratio of about 1:3, similar to the experimental observation (Figure 1C). The concentration profiles were calculated from the kinetic parameters using Kintek Explorer (Johnson et al., 2009). To achieve this result, the rate of dissociation of RPc back to R+P was decreased, while the rate of dissociation of RPo back to RPc was increased (top). Examination of the energy landscapes shows that these changes effectively stabilize RPc with respect to RPo (top).
D. A schematic model for how WhiB7 could affect the free energy profile for a two-step mechanism of RPo formation on the whiB7 promoter (R+P O RPc O RPo) is shown. Superimposed are blob renditions of stable states (RPc and RPo), as observed in our structures (Figure 1B, C). The black line indicates the reaction in the absence of WhiB7 and the green indicates how WhiB7 could change the profile to both activate transcription and stabilize RPc with respect to RPo, as observed in (B).
Heparin has long been used as a DNA competitor that can distinguish various types of RNAP-DNA complexes (Walter et al., 1967); heparin blocks free RNAP from binding promoter DNA, RPc is rapidly disrupted, while RPo and elongation complexes are generally resistant (Straney and Crothers, 1985). Our EMSA results (Figure 6B) show that in the absence of WhiB7, the free DNA is shifted into a single, heparin-resistant band (RPo). In the presence of WhiB7, two shifted bands appear; one heparin-resistant band corresponding to RPo, the other heparin-sensitive and assigned as RPc. The ratio of RPc to RPo observed in the EMSA is much higher than observed by cryo-EM, but the caging effect of the polyacrylamide gel in the EMSA experiment could effectively stabilize relatively weakly-bound complexes (such as RPc).
To qualitatively reconcile this seemingly counterintuitive result, we used the transcript flux calculator (Galburt, 2018) to illustrate that an activator like WhiB7 could alter the kinetic parameters in a simple four-state kinetic model (Walter et al., 1967): R + P ⇄ RPc ⇄ RPo → RPITC to increase the steady state flux into RPITC by approximately 2-fold (Figure 6C top). The definition of the kinetic parameters are as follows: R and P, representing free RNAP and free promoter DNA respectively, bind to form the intermediate RPc with forward and reverse rate constants k1 and k−1. RPc isomerizes to RPo with rate constants k2 and k−2. In the presence of nucleotide substrates, RPo irreversibly transitions into an initial transcription complex (RPitc) with rate constant k3. The values of the rate constants for basal and activated transcription are tabulated in Figure 6C (top), with the resulting energy profiles shown on the right (black line, basal; green line, activated) (Galburt, 2018).
The cryo-EM samples for grid preparation were prepared under equilibrium conditions with no nucleotide substrates and therefore would not transition from RPo into RPitc: Rather, the equilibrium equation can be represented by R + P ⇄ RPc ⇄ RPo [Figure 6C (bottom)]. Using these conditions, the basal kinetic parameters (no WhiB7) yield only RPo at equilibrium, while the same changes in the kinetic parameters that activate transcription ~2-fold [Figure 6C (top)] result in the appearance of RPc in equilibrium with RPo at a ratio of about 1:3 [Figure 6C (bottom)], similar to our experimental observation (Figure 1C). To achieve this result, the rate of dissociation of RPc back to R+P was decreased, while the rate of dissociation of RPo back to RPc was increased. Examination of the energy landscapes shows that these changes effectively stabilize RPc with respect to RPo [Figure 6C (top)]. We note that these changes to the kinetic parameters that qualitatively mimic our experimental results are not unique, other combinations of changes to the kinetic parameters may yield similar results. Thus, this represents a possible mechanistic basis for WhiB7 function, but other possibilities exist. A schematic model for how WhiB7 could affect the free energy profile for a two-step mechanism of RPo formation on the whiB7 promoter (R+P ⇔ RPc ⇔ RPo) is illustrated in Figure 6D. This model provides an explanation for how WhiB7 could change the profile to both activate transcription and stabilize RPc with respect to RPo, as observed (Figure 6D).
DISCUSSION
Mtb does not contain homologs of the well-characterized Eco iron-binding TFs FNR and SoxR, but instead has seven WhiB proteins implicated in controlling responses to numerous infection-relevant stresses: hypoxia, antibiotics, ROS, NO, and invasion of macrophages (Flentie et al., 2016). Here we present structures of WhiB7 in complex with Mtb RNAP holoenzyme on a WhiB7-regulated promoter. The structures reveal the nature of interactions between WhiB7 and the housekeeping σA factor and with DNA, showing the structural basis of WhiB7 activity. WhiB7 contains an AT-hook, a DNA binding motif common in eukaryotic nuclear chromosomal binding proteins but unusual in bacteria. The structural similarity between the binding of the AT-hook and AT-rich DNA revealed in this work is striking. It is unclear if there is an evolutionary relationship between AT-hooks in WhiB7s and the eukaryotic chromatin proteins. It is worth noting the bacterial chromosomal histone-like nucleoid family of proteins have been purported to encode AT-like hooks (XRG motifs) (Qin et al.). However, the sequence conservation to the eukaryotic/WhiB7 AT-hooks is weak and the mode of DNA binding does not appear to be conserved (Cordeiro et al., 2011), suggesting they are not canonical AT-hooks.
The structures also offer perspectives into how promoter melting initiates in mycobacteria by revealing the WhiB7-stabilized RPc structure. While the complex in the absence of WhiB7 yielded a single population comprising RPo, in the presence of WhiB7 about 73% of the particles formed RPo while 27% of the particles formed RPc. RPc was previously observed with Eco RNAP (Chen et al., 2020) but is a newly observed promoter melting intermediate for Mtb RNAP. Previous structures of RPo containing CarD (Bae et al., 2015b; Boyaci et al., 2019a) and the RPo structures determined here (Figures 1B, C) establish that the CarD C-terminal domain (CTD), including an absolutely conserved Trp residue (W85 of Mtb CarD), interacts intimately with the promoter DNA, inserting into the widened minor groove at the upstream edge of the transcription bubble. Modeling of the expected DNA path in RPc (Hubin et al., 2017a), subsequently supported by the Eco RNAP RPc structure (Chen et al., 2020), indicated a significant steric clash of the duplex −10 element in RPc with the CarD-CTD [also see (Rammohan et al., 2015) and (Srivastava et al., 2013)]. The Mtb RPc structure here shows that CarD remains bound to the complex and that the path of the duplex DNA also diverges from that seen Eco RPc. The promoter-proximal part of the CarD-CTD is very flexible and becomes disordered in the presence of the RPc DNA, indicating that the CarD-CTD essentially moves out of the way of the DNA and also appears to redirect the path of the DNA towards the clamp (Figure 5B and C).
The finding that we observe RPc only in the presence of WhiB7 indicates that WhiB7 stabilizes RPc with respect to W-RPo. Because of the incubation time required to prepare the cryo-EM samples and freeze the grids, we believe the cryo-EM samples are at equilibrium. Thus, the equilibrium population of W-RPc increases while the population of W-RPo decreases compared to the absence of WhiB7 (Figure 1B, C). This finding is reflected in the EMSA (Figure 6B), where RPc is observed, but only in the presence of WhiB7. This may seem counterintuitive to the finding that WhiB7 stimulates transcription (Figure 6A), but transcription activity does not necessarily reflect the equilibrium population of RPo but instead reflects the steady-state flux through the system (Figure 6C). We use kinetic modeling tools to show that in the steady-state transcription assay conditions, WhiB7 could stimulate transcription but also stabilize the RPc intermediate at equilibrium conditions (Figure 6C, D).
Recent work has demonstrated that CarD and RbpA regulate initiation on the ribosomal RNA operon promoter (Boyaci et al., 2019b; Hubin et al., 2017a; Jensen et al., 2019; Rammohan et al., 2015, 2016). Specifically, CarD and RbpA were shown to stabilize a partially melted intermediate called RP2 (Boyaci et al., 2019a; Hubin et al., 2017a). WhiB7, on the other hand, stabilizes an earlier intermediate, RPc, suggesting that promoter recruitment is the rate-limiting step on WhiB7 regulated promoters. We note that promoters known to be regulated by WhiB7 have suboptimal −35 elements (Burian et al., 2012), which are known to affect the formation of RPc. Therefore, WhiB7 appears to serve as an aid to strengthen interactions with upstream promoter elements and to compensate for weak −35 promoter interactions with σAD4. Most promoters in Mtb lack strong −35 elements, especially when compared to those in Eco (Cortes et al., 2013; Hubin et al., 2017b; Shell et al., 2015). It is therefore conceivable that the seven WhiB factors serve to compensate for the weak −35 element, offering additional layers of regulation to fine-tune gene expression. Thus, it appears that Actinobacteria have evolved a unique set of transcription factors to optimize gene expression of intrinsically weak promoters, expanding possibilities for regulation.
Limitations of the Study
The proposal that WhiB7 increases the rate of RPc formation while simultaneously decreasing the stability of RPo is based on a transcriptional flux model. While we show using EMSAs that WhiB7 increases the population of RPc, kinetic parameters need to be verified experimentally. The kinetic parameters used in Figure 6 are reasonable but hypothetical - they are intended to illustrate that the behavior of the system can qualitatively capture the experimentally observed behavior of the WhiB7 activation mechanism. The kinetic parameters used here are not intended to represent the actual kinetic parameters for the whiB7 promoter, which are unknown. We acknowledge that changing rate parameters of the transcriptional flux calculator would provide alternate models to the one we provide but that this model fitted our transcriptional and EMSA data best.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elizabeth A. Campbell (campbee@rockefeller.edu)
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-22886 (W-RPo), EMD-22887 (W-RPc), EMD-22888 (RPo). The atomic coordinates have been deposited in the Protein Data Bank under accession codes 7KIF (W-RPo), 7KIM (W-RPc), 7KIN (RPo).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For protein expression, BL21 (DE3) pLysS [ F– ompT hsdSB (rB–, mB–) gal dcm (DE3) pLysS(CamR), BL21 (DE3) [F– ompT gal dcm lon hsdSB(rB – mB – ) λ(DE3 [lacl lacUV5-T7p07 ind1 sam7 nin5]) [malB+ ]K-12(λS )], and Rosetta 2 (DE3) [F- ompT hsdSB(rB- mB-) gal dcm pRARE2 (CamR)] were used for heterologous protein expression. Mtb RNAP subunits, CarD, RbpA, σA and WhiB7 are proteins found in M. tuberculosis.
METHOD DETAILS
Structural biology software was accessed through the SBGrid consortium(Morin et al., 2013).
Protein Expression and Purification.
Mtb σA/WhiB7
A co-expression plasmid of Mtb σA and WhiB7 was made in the pCDFDuet-1 backbone (Novagen) to express 6His-WhiB7 with Strep-tag II-σA. Competent Eco BL21 (DE3) (Novagen) cells were transformed with the pCDFDuet-1–6His-WhiB7-Strep-tag II-σA, and cells were grown to an Optical Density (O.D.) of 0.5 at λ600 nM and induced with 1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) for 16 hours (h) at 18 °C. The cells were resuspended in lysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 2 mM DTT) and lysed by continuous flow through a French press (Avestin). WhiB7 and σA proteins were first co-purified by Ni2+-affinity chromatography. The sample was then subjected to StrepTrap HP chromatography, and the WhiB7-σA protein complex eluted with lysis buffer plus 2.5 mM desthiobiotin. A Coomassie stained SDS-PAGE analysis of purified σA/WhiB7 complex is shown in Fig. 1C. The 4Fe-4S cluster presence was monitored by measuring absorbance at λ420 nm.
Mtb CarD
Mtb CarD was overexpressed as previously described (Davis et al., 2015; Srivastava et al., 2013). In brief, Mtb CarD in the overexpression vector pET SUMO (Invitrogen) was transformed into BL21(DE3) (Novagen), and expression was then induced with 1 mM IPTG for 4 h at 28 °C. The overexpressed protein was purified by Ni2+-affinity chromatography, and eluted protein was cleaved with ULP-1 protease (Invitrogen). The cleaved protein complex was loaded onto a Ni2+- affinity; the flow-through was collected and further purified by size exclusion chromatography (Superdex 200, Cytiva).
Mtb RNAP
Mtb RNAP was expressed and purified as previously described(Boyaci et al., 2018, 2019a; Davis et al., 2015). Briefly, plasmid pMP61 was used to express the WT Mtb RNAP (Boyaci et al., 2019a). The Mtb core RNAP subunits were co-overexpressed in E. coli Rosetta 2 (DE3) cells overnight at room temperature for ~16 hours after induction with 0.1 mM IPTG. Cells were lysed by continuous flow through a French press (Avestin) and the lysate was precipitated by the addition of 0.6% polyethyleneimine (PEI). PEI pellets were washed three times in lysis buffer with 0.3M NaCl, and RNAP was eluted with lysis buffer and 1M NaCl. Further purification was done by Ni2+-affinity chromatography and size exclusion chromatography.
Mtb RbpA
Mtb RbpA was expressed and purified as described (Hubin et al., 2015, 2017a). Briefly, Mtb rbpA cloned in pET20b, was transformed into Eco BL21(DE3) pLys. Transformed cells were grown to an O.D. of 0.4 at λ 600 nM. Cells were then placed on ice for 15-min and induced with 1 mM IPTG for 3 h. Protein was purified by ion-exchange chromatography (HiTrap Q Sepharose Fast Flow and Mono Q 5/50 GL; Cytiva) and size-exclusion chromatography (HiLoad 16/60 Superdex 200; Cytiva).
Mtb σA/RbpA
Mtb σA and RbpA were co-expressed and purified as described (Hubin et al., 2017a).
DNA for Cryo-EM samples
The full sequence of the whiB7 promoter (−70 to +30) fragment used for cryoEM is listed below. Oligos were ordered gel purified from Integrated DNA Technologies, Coralville, IA:
WhiB7
Top Strand:
5’CTGTACCGGCAAACGCGCAGGTCAGAAAATCGGTTGTGGTCAGCTGCTGCCACCGGTTAACCTCCAGGTCGCATTCTGCTGCCAGCCTGGAGATGGCATT3’
Bottom Strand
3’GACATGGCCGTTTGCGCGTCCAGTCTTTTAGCCAACACCAGTCGACGACGGTGGCCAATTGGAGGTCCAGCGTAAGACGACGGTCGGACCTCTACCGTAA5’
In vitro Transcription Assay
Transcription assays were performed with 50 nM of Mtb RNAP holoenzymes with or without WhiB7 in transcription buffer [10 mM Tris HCl, pH 7.9, 80 mM K-acetate, 10 mM, MgCl2, 1 mM DTT, 5 μg/ml bovine serum albumin (BSA) and 0.1 mM EDTA]. Both holoenzymes were prepared as described in protein preparation for cryo-EM grids. After purification on Superose 6 Increase 10/300 GL column (Cytiva), holoenzymes were mixed with 250 nM CarD while the RNAP/WhiB7 holoenzyme was mixed with 250 nM RbpA. The whiB7 promoter (−70 to +30) was then added (10 nM) to the holoenzymes, and the samples were incubated for 15 min at 37 °C to allow the formation of RNAP open complex. Transcription was initiated by adding a nucleotide mixture consisting of 250 μM CpG dinucleotide (Trilink Biotechnologies, San Diego, CA), 50 μM CTP, and 1.25 μCi (15 nM)- [α-P32 ] CTP. Each reaction was allowed to proceed for 10 min at 37 °C, and reactions were quenched by the addition of 2x stop buffer (0.5X TBE, pH 8.3, 8 M urea, 30 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol). Reactions were heated to 95°C for 10 min and loaded onto a polyacrylamide gel [23% Acrylamide/Bis acrylamide (19:1), 6M urea, and 1X TBE, pH8.3]. Transcription products were visualized by using Typhoon 9400 Variable Imager (Amersham Biosciences) and quantified using Image J(Schneider et al., 2012). Quantified values were plotted in a histogram and the mean standard errors were calculated from three independent data sets.
Preparation of Mtb RNAP/WhiB7 and Mtb RNAP/no WhiB7 for cryo-EM
Mtb holo RNAP with WhiB7 was formed by mixing RNAP core with 5-fold molar excess of σA- WhiB7. Mtb holo RNAP without WhiB7 was formed by mixing RNAP core with 5-fold molar excess of σA-RbpA. The formed holoenzymes were purified over a Superose 6 Increase 10/300 GL column (Cytiva) in gel filtration buffer (20 mM Tris-HCl pH 8.0, 150 mM K-glutamate, 5 mM MgCl2, 2.5 mM DTT). The eluted RNAP holoenzymes were concentrated to 5 mg/mL (12 μM) by centrifugal filtration (Amicon Ultra). RNAP holoenzymes were mixed with a 5-fold molar excess of CarD and incubated for 10 minutes at 37oC. The 5-fold molar excess of RbpA was added to RNAP holo with WhiB7 protein and incubated for 5 minutes at 37oC. The whiB7 promoter fragment was added last (final concentration of 12 μM) to the samples and incubated for 15 minutes at 37oC, before cryo-EM grid preparation.
Native electrophoretic mobility shift assays
Mtb holo RNAP with and without WhiB7 were assembled as described in the preparation of holoenzymes for cryo-EM, except the concentrations of Mtb holo RNAP were 1 μM, CarD 5 μM, RbpA 5 μM, and the DNA 100 nM. Reactions were run on a 4.5% polyacrylamide native gel (37.5:1 acrylamide:bis-acrylamide) in 1x TBE (89 mM Tris, 89 mM boric acid, 1 mM EDTA) at 4 °C. The gel was stained with Gel-Red (Biotium).
Cryo-EM grid preparation
C-flat holey carbon grids (CF-1.2/1.3–4Au, Protochips, Morrisville, NC) were glow-discharged for 20 sec before the application of 3.5 μL of the samples. OG, (n-Octyl-β-D-Glucoside, Anatrace, Maumee, OH), was added to the samples to a final 0.1% (w/v). Using a Vitrobot Mark IV (Thermo Fisher Scientific Electron Microscopy, Hillsboro, OR), grids were blotted and plunge- froze into liquid ethane with 100% chamber humidity at 22°C.
Cryo-EM data acquisition and processing
Mtb RNAP/WhiB7.
Grids were imaged using a 300 keV Titan Krios (Thermo Fisher Scientific Electron Microscopy) equipped with a K2 Summit direct electron detector (Gatan, Pleasanton, CA). Dose-fractionated movies were recorded in counting mode using Leginon (Nicholson et al., 2010) at a nominal pixel size of 1.1 Åover a defocus range of −1 μm to −2.1 μm. Movies were recorded with a dose rate of 8 electrons/physical pixel/s over a total exposure of 10 s (50 subframes of 0.2 s). A total of 4,653 movies were collected for the first dataset. A total of 3,841 movies were collected for the second dataset. Dose-fractionated movies were gain-normalized, drift-corrected, summed, and dose-weighted using MotionCor2. The contrast transfer function was estimated for each summed image using Patch CTF estimation in cryoSPARC2 (Punjani et al., 2017). Blob picker in cryoSPARC2 was used to pick particles (no template was supplied). A total of 2,587,166 particles were picked from the first dataset, and 1,762,667 particles were picked from the second dataset. Particles were extracted in cryoSPARC2 using a box size of 300 pixels. Ab-initio model was obtained and used as a 3D template. Four rounds of cryoSPARC2 adaptation of “random-phase 3Dclassification” (Gong et al., 2016) heterogeneous refinements (X=4) with 8 heterogeneous classes (N=8) were performed in cryoSPARC2 to curate and classify particles. Curated particles from each dataset were refined in the Local CTF refinement (Punjani et al., 2017) and polished in Relion (Scheres, 2012; Zivanov et al., 2018). After polishing, particles from two datasets were combined and they were classified (N=6) in RELION using signal subtraction(Bai et al.), with masked region around the WhiB7, σAD4, and upstream DNA. Classified particles were reverted to the non-subtracted particles and refined using cryoSPARC2 Hetero Refinement followed by the Non-uniform refinement. Particles with WhiB7 density were combined and classified in cryoSPARC2 hetero refinement. After classification, two classes were observed with DNA configurations corresponding to previously described Mtb RNAP RPo (Boyaci et al., 2019a) and one corresponding to the RPc complex. The particles from the RPo and RPc classes were further refined using cryoSPARC2 non-uniform refinement. Particles were refined by local (BETA) refinement in cryoSPARC2 with masking the region around WhiB7, upstream DNA, and σAD4. A composite map was generated in Phenix (Adams et al., 2010) from the local refinement RPo (or RPc maps) and non-uniform RPo (or RPc maps). The RPo composite map contained 87,743 particles with a nominal resolution of 2.94 Å determined by using the gold-standard FSC 0.143 cutoff (Afonine et al., 2018). The RPc composite map contained 32,712 particles with a nominal resolution of 3.38 Å determined by using the gold-standard FSC 0.143 cutoff (Afonine et al., 2018). Local resolution calculations were generated using blocres and blocfilt commands from the Bsoft package (Cardone et al., 2013) by using composite half maps.
Mtb RNAP/no WhiB7.
Grids were imaged using a 300 keV Titan Krios (Thermo Fisher Scientific Electron Microscopy) equipped with a K2 Summit direct electron detector (Gatan, Pleasanton, CA). Dose-fractionated movies were recorded in counting mode using Leginon (Nicholson et al., 2010) at a nominal pixel size of 1.1 Å over a defocus range of −0.8 μm to −2.2 μm. Movies were recorded with a dose rate of 8 electrons/physical pixel/s over a total exposure of 10 s (50 subframes of 0.2 s). A total of 5,080 movies were collected. Dose-fractionated movies were gain-normalized, drift-corrected, summed, and dose-weighted using MotionCor2 (Zheng et al., 2017). The contrast transfer function was estimated for each summed image using Patch CTF estimation in cryoSPARC2. Blob picker in cryoSPARC2 was used to pick particles (no template was supplied). A total of 2,362,200 particles were picked and extracted in cryoSPARC2 using a box size of 300 pixels. Ab-initio model was obtained and used as a 3D template. Four rounds of cryoSPARC2 adaptation of “random-phase 3Dclassification” (Gong et al., 2016) heterogeneous refinements (X=4) with 8 heterogeneous classes (N=8) were performed in cryoSPARC2 to curate and classify particles. Curated particles were polished in Relion (Scheres, 2012; Zivanov et al., 2018). Polished particles were classified in cryoSPARC2 hetero refinement but only one DNA configuration was observed corresponding to previously described Mtb RNAP RPo (Boyaci et al., 2019a). The particles from the RPo class were further refined using cryoSPARC2 Local CTF refinement and non-uniform refinement (Punjani et al., 2017). The RPo class contained 296,097 particles with a nominal resolution of 2.74 Å determined by using the gold-standard FSC 0.143 cutoff (Afonine et al., 2018). Local resolution calculations were generated using blocres and blocfilt commands from the Bsoft package (Cardone et al., 2013).
Model building and refinement
For initial models of the complexes, the Mtb RNAP cryo-EM structure (PDB ID 6EDT for the RPo classes (Boyaci et al., 2019a)), Eco RNAP cryo-EM structure (PDB ID 6PSQ for the RPc class (Chen et al., 2020)), HMGA-1 crystals structure (PDB ID 3UXW for the AT-hook (Fonfría-Subirós et al., 2012)) were manually fit into the cryo-EM density maps using UCSF Chimera (Pettersen et al., 2004) and real-space refined using PHENIX (Adams et al., 2010). For real-space refinement, rigid body refinement was followed by all-atom refinements with Ramachandran and secondary structure restraints. Refined models were inspected and modified in Coot (Emsley and Cowtan, 2004). The refined models were ‘shaken’ by introducing random shifts to the atomic coordinates with RMSD of 0.15 Å to the final models of Mtb RNAP/WhiB7 RPo, 0.16 Å to the final models of Mtb RNAP/WhiB7 RPc, and 0.14 Å to the final model of Mtb RNAP/no WhiB7 (Adams et al., 2010). Shaken models were refined against the first half map and these refined models were used to calculate the FSC against the same first half maps (FSChalf1 or work), the second half maps (FSChalf2 or free) that were not used for the refinement, and combined (full) maps by using phenix.mtriage (Afonine et al., 2018). Masked log file for the combined (full) maps was plotted in PRISM and the FSC 0.143 was marked on the graph.
QUANTIFICATION AND STATISTICAL ANALYSIS
For calculations of Fourier shell correlations (FSC) in Figures S2B, S2E, and S2H, the FSC cutoff criterion of 0.143 (Afonine et al., 2018) was used. Image J (Schneider et al., 2012) was used to visualize and quantify gels. To quantify the transcription assays (Figure 6A), mean values and the standard error of the mean from at least three independent measurements were calculated. Structural biology software was accessed through the SBGrid consortium (Morin et al., 2013).The local resolution of the cryo-EM maps (Figures S2C, S2F, and S2I) was estimated using blocres (Cardone et al., 2013) with the following parameters: box size 15, verbose 7, sampling 1.1, and cutoff 0.5. The quantification and statistical analyses for model refinement and validation were generated using PHENIX (Adams et al., 2010).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| E. coli BL21(DE3) | Novagen | N/A |
| E. coli BL21(DE3) pLysS | Novagen | N/A |
| Rosetta 2 (DE3) | Novagen | N/A |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| n-Octyl-b-D-Glucopyranoside | Anatrace | Cat# O311 |
| Mtb RNAP (cryo-EM samples) | Boyaci et al., 2019a | N/A |
| Mtb CarD | Srivastava et al., 2013 | N/A |
| Mtb σA/WhiB7 | This paper | N/A |
| Mtb RbpA | Hubin et al., 2015 | N/A |
| Mtb σA/RbpA | Boyaci et al., 2018 | N/A |
| Mtb σA | Davis et al., 2015 | N/A |
| Polyethyleneimine | Fisher Scientific | Cat# AC178572500 |
| Desthiobiotin | Sigma Aldrich | Cat# D1411 |
| ULP-1 protease | Invitrogen | Cat# 12588018 |
| Critical commercial assays | ||
| Deposited data | ||
| Coordinates of Mtb W-RPo | This paper | PDB: 7KIF |
| Coordinates of Mtb W-RPc | This paper | PDB: 7KIM |
| Coordinates of Mtb RPo | This paper | PDB: 7KIN |
| Cryo-EM map of Mtb W-RPo | This paper | EMD-22886 |
| Cryo-EM map Mtb W-RPc | This paper | EMD-22887 |
| Cryo-EM map of Mtb RPo | This paper | EMD-22888 |
| Abortive initiation transcription gel | This paper; Mendeley Data | https://data.mendeley.com/datasets/jph6vjvhbs/draft?a=394ea63c-e6a8-4415-acda-1725bd491dd6 |
| EMSA gel shifts | This paper; Mendeley Data | https://data.mendeley.com/datasets/fh9639t5sc/draft?a=48f930a7-2899-494e-a494-eb7f3ae0ecac |
| Experimental models: cell lines | ||
| Experimental models: organisms/strains | ||
| Mycobacterium tuberculosis | N/A | N/A |
| Oligonucleotides | ||
| WhiB7 top strand: CTGTACCGGCAAACGCGCAGG TCAGAAAATCGGTTGTGGTCA GCTGCTGCCACCGGTTAACC TCCAGGTCGCATTCTGCTGC CAGCCTGGAGATGGCATT |
This paper, IDT | N/A |
| WhiB7 bottom strand: AATGCCATCTCCAGGCTGGCA GCAGAATGCGACCTGGAGGTT AACCGGTGGCAGCAGCTGACC ACAACCGATTTTCTGACCTGCG CGTTTGCCGGTACAG |
This paper, IDT | N/A |
| Recombinant DNA | ||
| Mtb RNAP, pMP61 | Boyaci et al., 2019a | N/A |
| pCDFDuet-1 | Novagen | N/A |
| pCDFDuet-1/ σA/WhiB7 | This paper | N/A |
| pET SUMO/CarD | Srivastava et al., 2013 | N/A |
| pET20b/RbpA | Hubin et al., 2015 | N/A |
| pAC27/ σA | Davis et al., 2015 | N/A |
| Software and algorithms | ||
| Bayesian Polishing | Zivanov et al., 2018 | https://github.com/3dem/relion |
| blocfilt | Cardone et al., 2013 | https://lsbr.niams.nih.gov/bsoft/programs/blocres.html |
| blocres | Cardone et al., 2013 | https://lsbr.niams.nih.gov/bsoft/programs/blocres.html |
| Coot | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| cryoSPARC | Punjani et al., 2017 | https://cryosparc.com/ |
| GraphPad Prism | GraphPad | http://www.graphpad.com/scientific-software/prism/ |
| ImageQuant 5.2 | GE Healthcare, Pittsburgh PA | N/A |
| MotionCor2 | Zheng et al., 2017 | N/A |
| MTRIAGE | Afonine et al. 2018 | https://www.phenix-online.org/documentation/reference/mtriage.html |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org/documentation/index.html |
| RELION | Scheres, 2012 | https://github.com/3dem/relion |
| SBGrid | Morin et al., 2013 | https://sbgrid.org/ |
| The PyMOL Molecular Graphics System | Schrïdinger, LLC | https://pymol.org/2/ |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera |
| MegAlign Pro | DNASTAR | https://www.dnastar.com/?utm_source=software&utm_medium=MegAlignPro |
| Other | ||
| C-flat CF-1.2/1.3 400 mesh gold grids | Electron Microscopy Sciences | Cat# CF413-100-Au |
| HiLoad 16/600 Superdex 200 pg | Cytiva | Cat# 28989335 |
| HiTrap IMAC HP | Cytiva | Cat# 17092003 |
| StrepTrap HP | Cytiva | Cat# 28907547 |
| Isotope [α32P] CTP | Perkin Elmer | Cat # BLU013H250UCI |
| Superose 6 INCREASE 10/300 GL | Cytiva | Cat# 29091596 |
| Mono Q 5/50 GL | Cytiva | Cat# 17516601 |
Highlights.
Structure of an M. tuberculosis transcription initiation complex with regulator WhiB7
WhiB7 interacts with initiation complex via protein-protein contacts with σ-factor
WhiB7-AT-hook interacts with promoter DNA via upstream AT-rich sequence
WhiB7 activates transcription by stabilizing a closed promoter complex
Acknowledgments
We thank M. Ebrahim, J. Sotiris, and Honkit Ng at The Rockefeller University Evelyn Gruss Lipper Cryo-electron Microscopy Resource Center. Some of this work was performed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247), NYSTAR, and the NIH National Institute of General Medical Sciences (GM103310) with additional support from the Agouron Institute (F00316) and NIH (OD019994). The authors are grateful for support from NIH grant R01 GM114450 to E.A.C. We thank R. Froom, L. Zhang, and J. Rock for discussion of the manuscript. We thank James Chen, Ruby Froom and Hande Boyaci for advice on data processing.
Footnotes
The authors declare no competing interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D: Biological Crystallography 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Klaholz BP, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, Adams PD, and Urzhumtsev A (2018). New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallographica Section D l 74, 814–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L (1998). AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Research 26, 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Feklistov A, Lass-Napiorkowska A, Landick R, and Darst SA (2015a). Structure of a bacterial RNA polymerase holoenzyme open promoter complex. ELife 4, e08504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Chen J, Davis E, Leon K, Darst SA, and Campbell EA (2015b). CarD uses a minor groove wedge mechanism to stabilize the RNA polymerase open promoter complex. ELife 4, e08505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Rajendra E, Yang G, Shi Y, and Scheres SH Sampling the conformational space of the catalytic subunit of human γ-secretase. ELife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyaci H, Chen J, Lilic M, Palka M, Mooney RA, Landick R, Darst SA, and Campbell EA (2018). Fidaxomicin jams Mycobacterium tuberculosis RNA polymerase motions needed for initiation via RbpA contacts. ELife 7, e34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyaci H, Chen J, Jansen R, Darst SA, and Campbell EA (2019a). Structures of an RNA polymerase promoter melting intermediate elucidate DNA unwinding. Nature 565, 382–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyaci H, Saecker RM, and Campbell EA (2019b). Transcription initiation in mycobacteria: a biophysical perspective. Transcription 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buc H, and McClure WR (1985). Kinetics of open complex formation between Escherichia coli RNA polymerase and the lac UV5 promoter. Evidence for a sequential mechanism involving three steps. Biochemistry 24, 2712–2723. [DOI] [PubMed] [Google Scholar]
- Burgess RR, Travers AA, Dunn JJ, and Bautz EKF (1969). Factor Stimulating Transcription by RNA Polymerase. Nature 221, 43–46. [DOI] [PubMed] [Google Scholar]
- Burian J, Ramón-García S, Sweet G, Gómez-Velasco A, Av-Gay Y, and Thompson CJ (2012). The Mycobacterial Transcriptional Regulator whiB7 Gene Links Redox Homeostasis and Intrinsic Antibiotic Resistance. Journal of Biological Chemistry 287, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burian J, Yim G, Hsing M, Axerio-Cilies P, Cherkasov A, Spiegelman GB, and Thompson CJ (2013). The mycobacterial antibiotic resistance determinant WhiB7 acts as a transcriptional activator by binding the primary sigma factor SigA (RpoV). Nucleic Acids Research 41, 10062–10076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush MJ (2018). The actinobacterial WhiB-like (Wbl) family of transcription factors: The Actinobacterial WhiB-like (Wbl) Family of Transcription Factors. Molecular Microbiology 110, 663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush MJ, Chandra G, Bibb MJ, Findlay KC, and Buttner MJ (2016). Genome-Wide Chromatin Immunoprecipitation Sequencing Analysis Shows that WhiB Is a Transcription Factor That Cocontrols Its Regulon with WhiA To Initiate Developmental Cell Division in Streptomyces. MBio 7, e00523–16, /mbio/7/2/e00523–16.atom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EA, Muzzin O, Chlenov M, Sun JL, Olson CA, Weinman O, Trester-Zedlitz ML, and Darst SA (2002). Structure of the bacterial RNA polymerase promoter specificity sigma subunit. Molecular Cell 9, 527–539. [DOI] [PubMed] [Google Scholar]
- Cardone G, Heymann JB, and Steven AC (2013). One number does not fit all: mapping local variations in resolution in cryo-EM reconstructions. Journal of Structural Biology 184, 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Wang D, Ebright YW, Korlann Y, Kortkhonjia E, Kim T, Chowdhury S, Wigneshweraraj S, Irschik H, Jansen R, et al. (2012). Opening and Closing of the Bacterial RNA Polymerase Clamp. Science 337, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chiu C, Gopalkrishnan S, Chen AY, Olinares PDB, Saecker RM, Winkelman JT, Maloney MF, Chait BT, Ross W, et al. (2020). Stepwise Promoter Melting by Bacterial RNA Polymerase. Molecular Cell 78, 275–288.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Boyaci H, and Campbell EA (2021). Diverse and unified mechanisms of transcription initiation in bacteria. Nature Reviews Microbiology 19, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Hu Y, Wang Z, Chen S, and Feng L (2016). Genome-wide characterization of monomeric transcriptional regulators in Mycobacterium tuberculosis. Microbiology 162, 889–897. [DOI] [PubMed] [Google Scholar]
- Cordeiro TN, Schmidt H, Madrid C, Juárez A, Bernadó P, Griesinger C, García J, and Pons M (2011). Indirect DNA readout by an H-NS related protein: structure of the DNA complex of the C-terminal domain of Ler. PLoS Pathogens 7, e1002380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes T, Schubert OT, Rose G, Arnvig KB, Comas I, Aebersold R, and Young DB (2013). Genome-wide Mapping of Transcriptional Start Sites Defines an Extensive Leaderless Transcriptome in Mycobacterium tuberculosis. Cell Reports 5, 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis NK, and Chater KF (1992). The Streptomyces coelicolor whiB gene encodes a small transcription factor-like protein dispensable for growth but essential for sporulation. Molecular and General Genetics 232, 351–358. [DOI] [PubMed] [Google Scholar]
- Davis E, Chen J, Leon K, Darst SA, and Campbell EA (2015). Mycobacterial RNA polymerase forms unstable open promoter complexes that are stabilized by CarD. Nucleic Acids Research 43, 433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: Model-building tools for molecular graphics. Acta Crystallographica Section D: Biological Crystallography 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Feklistov A, Bae B, Hauver J, Lass-Napiorkowska A, Kalesse M, Glaus F, Altmann K-H, Heyduk T, Landick R, and Darst SA (2017). RNA polymerase motions during promoter melting. Science 356, 863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flentie K, Garner AL, and Stallings CL (2016). Mycobacterium tuberculosis Transcription Machinery: Ready To Respond to Host Attacks. Journal of Bacteriology 198, 1360–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfría-Subirós E, Acosta-Reyes F, Saperas N, Pous J, Subirana JA, and Campos JL (2012). Crystal Structure of a Complex of DNA with One AT-Hook of HMGA1. PLoS ONE 7, e37120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca JD, Knight GM, and McHugh TD (2015). The complex evolution of antibiotic resistance in Mycobacterium tuberculosis. International Journal of infectious Diseases 32, 94–100. [DOI] [PubMed] [Google Scholar]
- Galburt EA (2018). The calculation of transcript flux ratios reveals single regulatory mechanisms capable of activation and repression. Proceedings of the National Academy of Sciences USA 115, E11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiman DE, Raghunand TR, Agarwal N, and Bishai WR (2006). Differential Gene Expression in Response to Exposure to Antimycobacterial Agents and Other Stress Conditions among Seven Mycobacterium tuberculosis whiB-Like Genes. Antimicrobial Agents and Chemotherapy 50, 2836–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Qian H, Zhou X, Wu J, Wan T, Cao P, Huang W, Zhao X, Wang X, Wang P, et al. (2016). Structural Insights into the Niemann-Pick C1 (NPC1)-Mediated Cholesterol Transfer and Ebola Infection. Cell 165, 1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubin EA, Tabib-Salazar A, Humphrey LJ, Flack JE, Olinares PDB, Darst SA, Campbell EA, and Paget MS (2015). Structural, functional, and genetic analyses of the actinobacterial transcription factor RbpA. Proceedings of the National Academy of Sciences USA 112, 7171–7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubin EA, Fay A, Xu C, Bean JM, Saecker RM, Glickman MS, Darst SA, and Campbell EA (2017a). Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. ELife 6, e22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubin EA, Lilic M, Darst SA, and Campbell EA (2017b). Structural insights into the mycobacteria transcription initiation complex from analysis of X-ray crystal structures. Nature Communications 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huth JR, Bewley CA, Nissen MS, Evans JN, Reeves R, Gronenborn AM, and Clore GM (1997). The solution structure of an HMG-I(Y)-DNA complex defines a new architectural minor groove binding motif. Nature Structural Biology 4, 657–665. [DOI] [PubMed] [Google Scholar]
- Jensen D, Manzano AR, Rammohan J, Stallings CL, and Galburt EA (2019). CarD and RbpA modify the kinetics of initial transcription and slow promoter escape of the Mycobacterium tuberculosis RNA polymerase. Nucleic Acids Research 47, 6685–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Simpson ZB, and Blom T (2009). Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal Biochem 387, 20–29. [DOI] [PubMed] [Google Scholar]
- Krupp F, Said N, Huang Y-H, Loll B, Bürger J, Mielke T, Spahn CMT, and Wahl MC (2019). Structural Basis for the Action of an All-Purpose Transcription Anti-termination Factor. Molecular Cell 74, 143–157.e5. [DOI] [PubMed] [Google Scholar]
- Kudhair BK, Hounslow AM, Rolfe MD, Crack JC, Hunt DM, Buxton RS, Smith LJ, Le Brun NE, Williamson MP, and Green J (2017). Structure of a Wbl protein and implications for NO sensing by M. tuberculosis. Nature Communications 8, 2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Mandal S, Degen D, Liu Y, Ebright YW, Li S, Feng Y, Zhang Y, Mandal S, Jiang Y, et al. (2017). Structural Basis of Mycobacterium tuberculosis Transcription and Transcription Inhibition. Molecular Cell 66, 169–179.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli R, Proveddi R, Rodrigue S, Beaucher J, Gaudreau L, and Smith I (2004). σ Factors and Global Gene Regulation in Mycobacterium tuberculosis. Journal of Bacteriology 186, 895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney JD (2000). In vivo veritas: The search for TB drug targets goes live. Nature Medicine 6, 1330–1333. [DOI] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, and Sliz P (2013). Collaboration gets the most out of software. ELife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RP, Nguyen L, Gatfield J, Visconti K, Nguyen K, Schnappinger D, Ehrt S, Liu Y, Heifets L, Pieters J, et al. (2005). Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences USA 102, 12200–12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson WV, White H, and Trinick J (2010). An approach to automated acquisition of cryoEM images from lacey carbon grids. Journal of Structural Biology 172, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas FJ, Cayuela ML, Martinez-Argudo IM, Ruiz-Vazquez RM, and Murillo FJ (1996). High mobility group I(Y)-like DNA-binding domains on a bacterial transcription factor. Proceedings of the National Academy of Sciences USA 93, 6881–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Qin L, Erkelens AM, Ben Bdira F, and Dame RT The architects of bacterial DNA bridges: a structurally and functionally conserved family of proteins. Open Biology 9, 190223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammohan J, Ruiz Manzano A, Garner AL, Stallings CL, and Galburt EA (2015). CarD stabilizes mycobacterial open complexes via a two-tiered kinetic mechanism. Nucleic Acids Research 43, 3272–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammohan J, Ruiz Manzano A, Garner AL, Prusa J, Stallings CL, and Galburt EA (2016). Cooperative stabilization of Mycobacterium tuberculosis rrnA P3 promoter open complexes by RbpA and CarD. Nucleic Acids Research 44, 7304–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff EF, Record MT, and Artsimovitch I (2015). Initial events in bacterial transcription initiation. Biomolecules 5, 1035–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saecker RM, Record MT, and deHaseth PL (2011). Mechanism of Bacterial Transcription Initiation: RNA Polymerase - Promoter Binding, Isomerization to Initiation-Competent Open Complexes, and Initiation of RNA Synthesis. Journal of Molecular Biology 412, 754–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW (2012). RELION: Implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastri MD, Shukla SD, Chong WC, Dua K, Peterson GM, Patel RP, Hansbro PM, Eri R, and O’Toole RF (2018). Role of Oxidative Stress in the Pathology and Management of Human Tuberculosis. Oxidative Medicine and Cellular Longevity 2018, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell SS, Wang J, Lapierre P, Mir M, Chase MR, Pyle MM, Gawande R, Ahmad R, Sarracino DA, Ioerger TR, et al. (2015). Leaderless Transcripts and Small Proteins Are Common Features of the Mycobacterial Translational Landscape. PLoS Genetics 11, e1005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultzaberger RK, Chen Z, Lewis KA, and Schneider TD (2007). Anatomy of Escherichia coli σ 70 promoters. Nucleic Acids Research 35, 771–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava DB, Leon K, Osmundson J, Garner AL, Weiss LA, Westblade LF, Glickman MS, Landick R, Darst SA, Stallings CL, et al. (2013). Structure and function of CarD, an essential mycobacterial transcription factor. Proceedings of the National Academy of Sciences USA 110, 12619–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings CL, Stephanou NC, Chu L, Hochschild A, Nickels BE, and Glickman MS (2009). CarD Is an Essential Regulator of rRNA Transcription Required for Mycobacterium tuberculosis Persistence. Cell 138, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyn AJC, Collins DM, Hondalus MK, Jacobs WR, Kawakami RP, and Bloom BR (2002). Mycobacterium tuberculosis WhiB3 interacts with RpoV to affect host survival but is dispensable for in vivo growth. Proceedings of the National Academy of Sciences USA 99, 3147–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straney DC, and Crothers DM (1985). Intermediates in transcription initiation from the E. coli lac UV5 promoter. Cell 43, 449–459. [DOI] [PubMed] [Google Scholar]
- Walter G, Zillig W, Palm P, and Fuchs E (1967). Initiation of DNA-Dependent RNA Synthesis and the Effect of Heparin on RNA Polymerase. European Journal of Biochemistry 3, 194–201. [DOI] [PubMed] [Google Scholar]
- Wan T, Li S, Beltran DG, Schacht A, Zhang L, Becker DF, and Zhang L (2020). Structural basis of non-canonical transcriptional regulation by the σA-bound iron-sulfur protein WhiB1 in M. tuberculosis. Nucleic Acids Research 48, 501–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011). OverView of the it CCP4 suite and current developments. Acta Crystallographica Section D 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2018). Global tuberculosis report 2018.
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. ELife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-22886 (W-RPo), EMD-22887 (W-RPc), EMD-22888 (RPo). The atomic coordinates have been deposited in the Protein Data Bank under accession codes 7KIF (W-RPo), 7KIM (W-RPc), 7KIN (RPo).