

Abstract
PURPOSE
The anti-CD19 chimeric antigen receptor T-cell therapy tisagenlecleucel (CTL019) has an 81% response rate in children with relapsed or chemotherapy refractory (r/r) B-cell acute lymphoblastic leukemia (ALL). Cytokine release syndrome (CRS) is a life-threatening treatment-related toxicity that limits the full therapeutic potential in adults. We report outcomes for adults with r/r ALL treated with an optimized CTL019 dosing and CRS management strategy.
METHODS
Adults with r/r B-cell ALL received CTL019 in 1 of 2 trials. Patients received lymphodepletion followed by CTL019 as either a one-time infusion or fractionated infusions split over 3 days (day 1, 10%; day 2, 30%; day 3, 60%), which allowed for day 2 and day 3 doses to be held for early CRS. Total planned CTL019 dose varied with adaptive protocol modifications in response to efficacy and CRS toxicity.
RESULTS
Thirty-five adults with r/r ALL received CTL019 in 1 of 3 dosing cohorts. The low-dose cohort (n = 9) received single or fractionated dosing and had manageable toxicity with a 33% complete remission (CR) rate. In the high-dose single infusion cohort, 3 of 6 patients with refractory CRS concurrent with culture-positive sepsis died, and 3 achieved CR. The 20 patients in the high-dose fractionated (HDF) cohort had a 90% CR rate and manageable CRS. The HDF cohort had the highest survival, with a 2-year overall survival of 73% (95% CI, 46% to 88%) and event-free survival of 49.5% (95% CI, 21% to 73%).
CONCLUSION
Fractionated dosing of CTL019 with intrapatient dose modification optimizes safety without compromising efficacy in adults with r/r ALL.
INTRODUCTION
T cells engineered to express a chimeric antigen receptor (CAR) that targets CD19 have generated high remission rates in pediatric and adult patients with relapsed or chemotherapy refractory (r/r) acute lymphoblastic leukemia (ALL).1-6 The immune activation on which this efficacy depends is also responsible for the life-threatening toxicity of cytokine release syndrome (CRS). CRS correlates with the activation and expansion of anti-CD19 CAR T cells and clinically manifests with fevers that can progress to life-threatening vasodilatory shock and capillary leak with hypoxic respiratory failure.7 With efficacy clearly established, a better understanding of treatment-related toxicities and evaluation of CRS mitigation strategies are critical.
On August 30, 2017, the US Food and Drug Administration approved the first CD19-directed CAR T-cell therapy tisagenlecleucel (formerly CTL019) for the treatment of pediatric and young adult patients up to age 25 years with B-cell precursor ALL that is refractory or in second or greater relapse. We now report outcomes using CTL019 in 35 older adult patients with r/r ALL. Compared with the pediatric cohort, we have observed significant toxicity in adults, with 3 CRS-related deaths. We then modified the protocol to ask whether mitigation strategies, including a fractionated dosing scheme, could maintain high response rates with acceptable tolerability in adult patients with ALL.
METHODS
Study Design
Thirty-five adult patients with r/r ALL were treated at the University of Pennsylvania in 2 single-arm, open-label studies. The studies were designed to determine the safety and efficacy of autologous T cells transduced through a lentiviral vector with a CAR composed of a murine anti-CD19 single-chain variable (scFv) region and T-cell receptor CD3ζ/4-1BB costimulatory domains (CTL019). Our pilot first-in-human study (ClinicalTrials.gov identifier: NCT01029366) was open to all patients with CD19+ malignancies, of whom 5 had r/r ALL and are reported here. The second study (ClinicalTrials.gov identifier: NCT02030847) was a follow-up specific to patients with r/r ALL. Eligible patients in both studies were age ≥ 18 years with r/r CD19+ B-cell ALL. Patients with relapse after blinatumomab or allogeneic hematopoietic stem-cell transplantation (HSCT) were eligible provided that they did not have active graft-versus-host disease. Patients with active disease in their CNS were excluded. The study protocols and their amendments were approved by the institutional review board of the Hospital of the University of Pennsylvania and conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonization of Good Clinical Practice guidelines, including written informed consent. Research that involved recombinant DNA was conducted under biosafety level 2 containment and was approved by the institutional biosafety committee at the University of Pennsylvania in accordance with the National Institutes of Health Guidelines for Research Involving Recombinant DNA Molecules.
Peripheral blood T cells for manufacturing were collected from patients by leukapheresis and stimulated with beads coated with antibodies directed against CD3 and CD28. Cells were transduced with a lentiviral vector that encoded anti-CD19 scFv linked to 4-1BB and CD3ζ signaling domains, as described previously.8,9 Lymphodepleting chemotherapy before CTL019 infusion was recommended but not mandated for patients with WBC ≤ 1,000/μL. In both studies, the choice of lymphodepletion was left to the discretion of the treating physician.
The dosing and timing of CTL019 varied with protocol amendments in response to toxicity and efficacy. Depending on timing of enrollment, patients were treated in 1 of 3 dosing cohorts (Table 1). Patients either received a planned high dose of CTL019 cells (5 × 108) or a low dose of cells (5 × 107). The cells were then administered as a single infusion, or the total planned dose was split into fractions, with 10% of the dose delivered on day 1 (D1), 30% on D2, and the remaining 60% on D3. In the cohorts treated with a fractionated dosing approach, D2 and/or D3 doses were held if the patient experienced early signs of CRS, including fever (Table 1).
TABLE 1.
Dosing Cohorts (N=35)

Patients treated in the initial pilot trial were treated with the high-dose fractionated (HDF) approach according to the design of our first-in-human study. In the follow-up study, the first patients were treated with a high-dose single infusion (HDS) of 5 × 108 CAR T cells. Because of CRS-related deaths, the protocol was modified, and patients were treated with a lower dose of 5 × 107 cells with either single infusion or fractionated dosing (low-dose [LD]) cohort. Because of low efficacy in the LD cohort, the protocol was again modified, and patients were treated with the higher dose but with the fractionated dosing scheme (HDF), which was safe and effective in the pilot study (Fig 1).
FIG 1.

Flow diagram for adult patients with acute lymphoblastic leukemia treated with CTL019. CR, complete remission; CRS, cytokine release syndrome; NR, no response; HDF, high-dose fractionated; LD, low dose; HDS, high-dose single infusion.
Toxicity Assessment
CRS was graded according to the Penn grading scale10,11 (Appendix Table A1, online only). Other adverse events were graded according to the National Cancer Institute CTCAE (version 4.03).
Response Assessment
Disease response assessments were done at protocol-defined time points. The primary efficacy end point was overall complete remission (CR) rate at D28. CR was defined as < 5% bone marrow blasts; CRi was defined as CR without full hematopoietic recovery. Patients were determined to be minimal residual disease (MRD) negative if bone marrow blasts were < 0.01% by multiparameter flow cytometry. Secondary efficacy end points were time-to-event analysis of overall survival (OS) and event-free survival (EFS).
Statistical Analysis
Means, medians, and ranges were calculated for groups. OS and EFS were estimated by Kaplan-Meier curves, and dosing cohort differences were tested using the log-rank test. OS and EFS by subsequent HSCT were compared using a landmark analysis, which compares only those patients in both groups that were free of an event at 5.2 months postinfusion, the maximum time of HSCT.12 This approach addresses the immortality time bias in survival curves caused by HSCT-eligible patients having to remain event free until the time of HCST.13 D28 response rates and frequency of severe (grade 4/5) CRS by cohort were tested using Fisher’s exact test. The association between the number of doses received and baseline disease burden was assessed using Spearman’s rank correlation coefficient. Statistics were calculated using SAS 9.4 (SAS Institute, Cary, NC) and R version 3.4 (R Foundation for Statistical Computing, Vienna, Austria) software.
RESULTS
Patient Characteristics
A total of 49 patients were enrolled (7 in the first-in-human study and 42 in the follow-up study). Fourteen patients did not receive study treatment for the following reasons (Fig 1): manufacturing failure (n = 1), patients sought alternative treatment (n = 3), died (n = 5), or were medically unfit to receive treatment because of rapid disease progression or comorbid illness (n = 5). Thirty-five patients with r/r ALL (median age, 34 years; range, 21-70 years) received CTL019 through 3 dosing cohorts and were included in the study analysis (Table 2). Thirteen patients (37%) had experienced relapse after prior HSCT, and 11 (31%) had received prior blinatumomab. The median number of prior therapies was 3 (range, 1-7 therapies), and 11 patients (31%) had primary refractory disease. For patients with bone marrow biopsies available between lymphodepleting chemotherapy and CTL019 infusion (n = 29), the majority (93%) had > 5% bone marrow involvement.
TABLE 2.
Patient Demographics and Baseline Characteristics

Lymphodepleting chemotherapy was administered to 33 of 35 patients 1-2 weeks before CTL019 infusion. The majority of patients (n = 25) received single-agent cyclophosphamide (300 mg/m2 every 12 hours × 6), and 5 patients received cyclophosphamide (500 mg/m2 × 2 days) with fludarabine (30 mg/m2 × 4 days). One patient received clofarabine and one patient received methotrexate, and cytarabine and cyclophosphamide, vincristine, and doxorubicin. Two patients did not receive lymphodepletion at physician discretion because of leukopenia (WBC ≤ 60/μL [below the level of quantification] and WBC = 200/μL).
Response Rates and Survival
The overall CR rate by D28 for patients in all treatment cohorts was 69% (95% CI, 51% to 83%). Response rates, treatment-related toxicity, and survival varied significantly by dosing cohort (Table 3). In the HDS cohort (n = 6), 3 patients died before disease response assessment as a result of complications of CRS and infections; the surviving 3 patients achieved CR. In the LD cohort (n = 9), patients had manageable CRS, but only 33% achieved CR. In the HDF cohort (n = 20), toxicity was manageable, and the CR rate was 90%. All patients who achieved CR and had concurrent bone marrow assessment for MRD by flow cytometry (n = 19) were MRD negative.
TABLE 3.
Outcomes by Cohort

Long-Term Survival
At a median follow-up of 13 months (range, 0.2-52.7 months), the median OS for the entire cohort was 19.1 months (95% CI, 6.2 months to not estimable), and the median EFS was 5.6 months. OS and EFS were significantly improved in the HDF cohort (n = 20) compared with the HDS and LD cohorts. In the HDF cohort, median OS was not reached, with a 2-year survival rate of 73% (95% CI, 46% to 88%); median EFS was 19.4 months, and the 2-year EFS rate was 49.5%. For HDS, the 2-year OS and EFS rates were both 17% (95% CI, 0.8% to 52%). For LDS, the 2-year OS and EFS rates were 22% (95% CI, 3% to 51%) and 0% (95% CI, 0% to 33%), respectively (Fig 2; Table 3).
FIG 2.
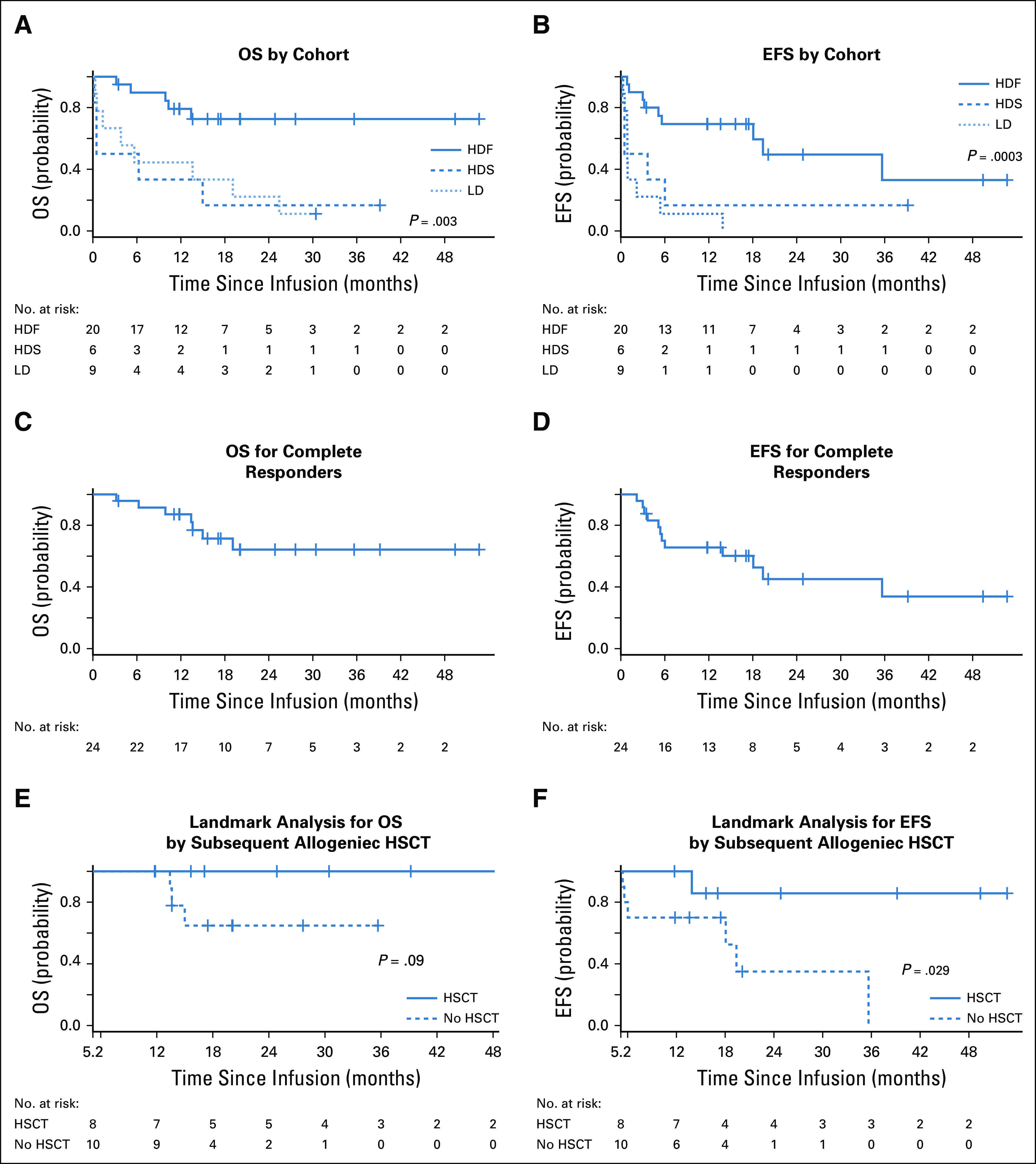
Kaplan-Meier graphs of overall survival (OS) and event-free survival (EFS). (A) OS by cohort. (B) EFS by cohort. (C) OS for complete responders. (D) EFS for complete responders. (E) Landmark analysis for OS by subsequent allogeneic hematopoietic stem-cell transplantation (HSCT). (F) Landmark analysis for EFS by subsequent allogeneic HSCT. HDF, high-dose fractionated; HDS, high-dose single infusion; LD, low dose.
We evaluated the effect of age on survival. OS and EFS curves were similar between patients age ≥ 35 years (n = 17) versus younger patients (n = 18; log-rank P = .56 and 1.00, respectively). Cox regression was used to examine the effect of age as a continuous covariate, and no significant effect of age was found; a 10-year increase in age was associated with 1.03-fold increase in the hazard of death (95% CI, 0.76 to 1.41) and a 1.15-fold increase in the hazard of an event (95% CI, 0.86 to 1.53).
For patients who achieved CR (n = 24), the median OS was not reached, with a 2-year survival rate of 64% (95% CI, 38% to 82%), and the median EFS was 19.4 months (95% CI, 5.6 months to not estimable; Figs 2C and 2D). Nine of the 24 patients who achieved CR received a consolidative allogeneic HSCT in CR, and 15 received no therapy after CTL019 unless relapse occurred. The median time to subsequent HSCT was 2.6 months (range, 1.7-5.2 months). For patients bridged and not bridged to HSCT, median ages were 38.7 years (range, 24.4-50.5 years) and 35.5 years (range, 27.2-63.0 years), respectively. To estimate the impact of HSCT in responding patients, we did a landmark analysis for OS and EFS by subsequent HSCT and found a nonsignificant improvement in OS and a significant improvement in EFS in patients who received HSCT (Figs 2E and 2F).
Toxicity
Treatment-related neurocognitive toxicity of any grade affected 14 patients (40%). Grade 1-2 toxicity occurred in 13 patients (37%) and grade 3 in 2 (6%), and no patient had grade 4 neurotoxicity. One patient in the LD cohort died as a result of intracranial hemorrhage D17 after CART infusion in the setting of persistent disease and thrombocytopenia. The most prevalent treatment-related adverse event was CRS, which was graded with the Penn grading scale11 (Appendix Table A1). CRS of any grade occurred in 33 patients (94%). Grade 1-2 CRS occurred in 8 patients (23%), grade 3 in 19 (54%), grade 4 in 3 (9%), and grade 5 in 3 (9%). The frequency of grade 4/5 CRS varied by treatment cohort (Fisher’s exact P = .017; Table 3). The 3 patients with grade 5 CRS were all in the HDS cohort, and all had concurrent infections. All 3 patients (ages 32, 56, and 63 years) died as a result of refractory hypotension, having developed pulmonary edema that required mechanical ventilation. All 3 patients received at least 2 doses of tocilizumab and systemic corticosteroids, and all had concern for concurrent sepsis at the time of death (1 with influenza B, 1 with Stenotrophomonas pneumonia, and 1 with Pseudomonas pneumonia). Only 1 grade 4 CRS and no grade 5 CRS were observed in the 20 patients treated in the HDF cohort.
Fractionated Dosing
The 20 patients in the HDF cohort had planned infusions of CTL019 over 3 days, with 10% delivered on D1, 30% on D2, and 60% on D3. D2 and D3 were withheld for early signs of clinical CRS, such as fever, that occurred after the first or second dose. Nine patients received 1 dose, 4 received 2 doses, and 7 received all 3 doses. Only 2 of the 20 patients in the HDF cohort did not achieve CR; both received 2 doses of cells.
Given that baseline disease burden in ALL is a predictor for severe CRS, we analyzed whether baseline disease burden (available for 15 of 20 patients in the HDF cohort) correlated with the number of fractions given. Both evaluable patients with < 5% bone marrow blasts received all 3 doses of CTL019. Of note, of the 9 patients in the HDF cohort with high disease burden (> 50%), 3 received D1 only, 3 received D1 and D2 only, and 3 received all 3 doses of cells. The Spearman’s rank correlation coefficient was 0.12 (95% CI, −0.42 to 0.6; P = .66).
DISCUSSION
Tisagenlecleucel (formerly CTL019) has been found to be effective with manageable toxicity in pediatric, adolescent, and young adult patients with r/r ALL. Similar to other therapies for ALL, older adults may have a differential ability to tolerate treatment-related toxicity. Because CAR T cells are living drugs capable of marked in vivo expansion, the identification of an ideal infusion dose with a traditional phase I design is challenged by the fact that the infusional dose is only one of many factors that influence peak CAR T-cell levels, which correlates with toxicity and efficacy. We hypothesized that a fractionated dosing scheme, where D2 and D3 doses are held in response to early clinical CRS, allows individualized dose modifications for a better balance of efficacy and safety compared with a single dose for all patients. Our initial findings preliminarily support this hypothesis. For example, only 3 of the 9 patients in the LD cohort (planned dose of 5 × 107 CAR T cells) achieved a CR, which implies that for unselected patients, this dose may be insufficient. The 20 patients treated in the HDF cohort had the potential to receive 5 × 108 CAR T cells but in fractions of 10%, 30%, and 60%. Of note, 9 of the 20 patients developed CRS after the first 10% dose (which is 5 × 107 cells, as in the LD cohort), and no additional cells were given. All 9 achieved CR compared with only 33% in the LD cohort who received the same dose of cells. The fractionated dosing scheme allows for adaptive dosing where only those patients most likely to respond to the 5 × 107 dose (early CRS) will receive this lower dose; if these patients went on to receive the full dose despite early CRS, it is possible that they would have more severe toxicity, and our strategy potentially mitigates this. Those patients who did not have early CRS received subsequent doses but did not experience excessive CRS-related toxicity. Of the 20 patients total treated in the HDF cohort, 90% achieved CR and only 1 had severe CRS (grade 4 per Penn grading scale) because of a requirement for high-dose vasopressor support, and no patients required mechanical ventilation. In contrast, when we treated 6 patients in the HDS cohort (5 × 108 flat dose), there were 3 CRS-related deaths. Our analysis is limited by the small sample size, which prevents a multivariable analysis but supports further exploration of a fractionated dosing scheme in future trials of high-risk patients.
Because high baseline disease burden in ALL is associated with severe CRS, some investigators have adopted a risk adaptive strategy to infuse a lower dose of anti-CD19 CAR T cells for patients with higher disease burdens.3-6 Of note, in our HDF cohort, high disease burden did not always predict the number of fractions patients received, which suggests that additional modifiers contribute to CRS risk. These findings suggest that a fractionated dosing scheme may be a personalized approach to improve the therapeutic index of CAR T cells beyond the capability of a strategy of dose modification on the basis of disease burden.
In our study, patients who achieved an initial CR had a median OS that was not reached and median EFS of 19.4 months. A key question is whether to consolidate a fit patient in MRD-negative remission from anti-CD19 CAR T cell therapy with allogeneic HSCT. Treatment-related morbidity and mortality as a result of HSCT needs to be balanced against relapse risk. In addition, some CAR T-cell products with the 4-1BB costimulatory domain such as CTL019 can have functional persistence in vivo that offers the potential for ongoing tumor surveillance, which could be lost if a patient proceeds to allogeneic HSCT. Single-center studies have evaluated different patient populations with different cellular products that contain different costimulatory domains with conflicting results. In a pediatric single-center study with a product using a CD28 costimulatory domain, improved EFS and OS for patients in MRD-negative CR were seen in those bridged to HSCT.14 Another single-center study in adults that used a different anti-CD19 CAR T-cell product that also contained a CD28 costimulatory domain found no difference in outcomes for patients bridged to HSCT versus observation.5 Most pediatric patients treated with tisagenlecleucel, which has a 4-1BB costimulatory domain, were not bridged to transplantation, and a subset has had durable remissions that correlate with in vivo functional persistence, although outcomes by initial bridge to HSCT versus observation are unknown.3,4 In our study that uses this product for adults, we performed a landmark analysis and found improved EFS and OS in recipients of HSCT, although of importance, durable remissions with observation only were also seen. Interpretation of these data, including our own, must be done with caution because the study was not designed to answer this question and there is likely significant bias that influences HSCT decisions. The decision to proceed with HSCT will be individualized by patient age, preferences, performance status, prior therapies, and cellular product used. The finding of CAR T-cell persistence or ongoing B-cell aplasia (which implies functional persistence of CAR T cells) may further influence this decision.
The promise of CAR T cells will be fully realized when toxicity management and efficacy are optimized. After testing different dose levels and schedules, we believe that a fractionated dosing scheme using CTL019 for adult patients with r/r ALL allows for adaptive dosing and maximizes safety while preserving efficacy. These strategies now warrant definitive testing in future trials.
Appendix
TABLE A1.
Penn Grading System for CRS

SUPPORT
Supported by Novartis.
AUTHOR CONTRIBUTIONS
Conception and design: Noelle V. Frey, Edward Pequignot, Saar Gill, Shannon L. Maude, Stephan A. Grupp, Bruce L. Levine, Carl H. June, David L. Porter
Administrative support: Joan Gilmore
Provision of study material or patients: Saar Gill, Selina M. Luger, Alison W. Loren, Alexander E. Perl, Joan Gilmore, Bruce L. Levine
Collection and assembly of data: Noelle V. Frey, Elizabeth O. Hexner, Edward Pequignot, Selina M. Luger, Alison W. Loren, Alexander E. Perl, Joan Gilmore, Simon F. Lacey, Jos J. Melenhorst, Bruce L. Levine
Data analysis and interpretation: Noelle V. Frey, Pamela A. Shaw, Elizabeth O. Hexner, Edward Pequignot, Saar Gill, James K. Mangan, Alison W. Loren, Alexander E. Perl, Stephan A. Grupp, Nirav N. Shah, Simon F. Lacey, Jos J. Melenhorst, Bruce L. Levine, David L. Porter
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Optimizing Chimeric Antigen Receptor T-Cell Therapy for Adults With Acute Lymphoblastic Leukemia
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/journal/jco/site/ifc.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Noelle V. Frey
Consulting or Advisory Role: Novartis, Kite Pharma, Gilead
Research Funding: Novartis
Pamela A. Shaw
Patents, Royalties, Other Intellectual Property: Patent owned by the University of Pennsylvania currently licensed to Novartis for an algorithm that predicts severe cytokine release syndrome for chimeric antigen receptor T-cell 19 therapy; receive 10% of the licensing fees
Elizabeth O. Hexner
Research Funding: Blueprint Medicines (Inst), Tmunity Therapeutics (Inst)
Edward Pequignot
Patents, Royalties, Other Intellectual Property: As part of the University of Pennsylvania’s role in the US Food and Drug Administration approval of chimeric antigen receptor T-cell (CAR-T) therapy and for my part as an employee of the University of Pennsylvania involved in its research in CAR-T; have received royalties of approximately $300 over the past 2 years
Saar Gill
Stock and Other Ownership Interests: Carisma Therapeutics
Honoraria: Fate Therapeutics, Sensei Biotherapeutics, Aro Biotherapeutics
Research Funding: Novartis (Inst)
Patents, Royalties, Other Intellectual Property: Patents for chimeric antigen receptor T cells for acute myeloid leukemia
Selina M. Luger
Honoraria: Pfizer, Daiichi Sankyo, Agios
Research Funding: Cyclacel (Inst), ARIAD (Inst), Onconova Therapeutics (Inst), Biosight (Inst), Roche (Inst), Genentech (Inst), Kura (Inst), Celgene (Inst)
Travel, Accommodations, Expenses: Hemedicus, PER
James K. Mangan
Consulting or Advisory Role: Agios
Alexander E. Perl
Honoraria: Agios, Actinium Pharmaceuticals, Astellas Pharma, Daiichi Sankyo, Jazz Pharmaceuticals, Novartis, Newlink Genetics, Takeda Pharmaceuticals, AbbVie
Consulting or Advisory Role: Arog, Astellas Pharma, Actinium Pharmaceuticals, Daiichi Sankyo, AbbVie
Research Funding: Actinium Pharmaceuticals (Inst), Astellas Pharma (Inst), Bayer AG (Inst), BioMed Valley Discoveries (Inst), Daiichi Sankyo (Inst), Fujifilm (Inst), Novartis (Inst)
Travel, Accommodations, Expenses: Agios, AbbVie, Astellas Pharma, Daiichi Sankyo, Jazz Pharmaceuticals, Novartis, Newlink Genetics, Takeda Pharmaceuticals, Arog
Shannon L. Maude
Consulting or Advisory Role: Novartis, Kite Pharma
Travel, Accommodations, Expenses: Novartis, Kite Pharma
Stephan A. Grupp
Consulting or Advisory Role: Novartis, Jazz Pharmaceuticals, Janssen Pharmaceuticals, Cellular Biomedicine Group, TCR2 Therapeutics, Humanigen, Roche, Vertex, Adaptimmune
Research Funding: Novartis, Kite Pharma, Gilead, SERVIER
Nirav N. Shah
Stock and Other Ownership Interests: Exelixis, Oncosec, Geron, Cidara Therapeutics, Cell Vault
Consulting or Advisory Role: Jazz Pharmaceuticals, Kite Pharma, Incyte, Juno Therapeutics, Celgene
Research Funding: Miltenyi Biotec
Travel, Accommodations, Expenses: Miltenyi Biotec
Simon F. Lacey
Research Funding: Novartis (Inst), Tmunity (Inst)
Patents, Royalties, Other Intellectual Property: Patents and intellectual property related to chimeric antigen receptor T-cell technology licensed to Novartis (Inst)
Jos J. Melenhorst
Consulting or Advisory Role: Shanghai Unicar Therapy, Simcere of America, IASO Biotherapeutics
Research Funding: Novartis (Inst), Incyte (Inst)
Patents, Royalties, Other Intellectual Property: Dr. Melenhorst is a patent inventor for CTL019
Bruce L. Levine
Stock and Other Ownership Interests: Tmunity Therapeutics
Honoraria: Novartis, Terumo, Sysmex, Draper, AstraZeneca
Consulting or Advisory Role: Brammer Bio, Avectas, CRC Oncology, Cure Genetics, Ori Biotech, Vycellix
Research Funding: Novartis, Tmunity Therapeutics
Patents, Royalties, Other Intellectual Property: Intellectual property and patents in the field of cell and gene therapy
Travel, Accommodations, Expenses: GE Healthcare, BrammerBio, Avectas, CRC Oncology
Carl H. June
Stock and Other Ownership Interests: Celldex, Tmunity Therapeutics, Cabaletta, Carisma Therapeutics, Cytosen
Honoraria: Novartis, Pfizer, Johnson & Johnson
Consulting or Advisory Role: Celldex, Viracta Therapeutics, Cabaletta, Carisma Therapeutics, Kiadis Pharma, WIRB-Copernicus Group, Janssen Oncology
Research Funding: Novartis, Tmunity Therapeutics
Patents, Royalties, Other Intellectual Property: Intellectual property licensed to Novartis, royalties paid to the University of Pennsylvania; Office of Naval Research, intellectual property and patent royalties; intellectual property licensed to Tmunity Therapeutics (Inst)
David L. Porter
Employment: Genentech (I), Roche (I)
Stock and Other Ownership Interests: Genentech (I), Roche (I)
Consulting or Advisory Role: Novartis, Kite Pharma, Incyte, Gerson Lehrman Group, Bellicum Pharmaceuticals, Glenmark
Research Funding: Novartis
Patents, Royalties, Other Intellectual Property: Dr. Porter is a patent inventor for CTL019
Travel, Accommodations, Expenses: Kite Pharma, Novartis
Other Relationship: National Marrow Donor Program, American Board of Internal Medicine
No other potential conflicts of interest were reported.
REFERENCES
- 1.Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults Blood 1293322–33312017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial Lancet 385517–5282015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia N Engl J Med 3711507–15172014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia N Engl J Med 378439–4482018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JH, Rivière I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia N Engl J Med 378449–4592018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients J Clin Invest 1262123–21382016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frey NV, Porter DL.Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia Hematology (Am Soc Hematol Educ Program) 2016567–5722016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo Mol Ther 171453–14642009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Porter D, Frey N, Wood PA, et al: Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol 11:35, 2018 [Erratum: J Hematol Oncol 11:81, 2018] [Google Scholar]
- 12.Anderson JR, Cain KC, Gelber RD.Analysis of survival by tumor response J Clin Oncol 1710–7191983 [DOI] [PubMed] [Google Scholar]
- 13.Mi X, Hammill BG, Curtis LH, et al. Use of the landmark method to address immortal person-time bias in comparative effectiveness research: A simulation study Stat Med 354824–48362016 [DOI] [PubMed] [Google Scholar]
- 14. Shalabi H, Delbrook C, Stetler-Stevenson M, et al: Chimeric antigen receptor T-cell therapy can render patients with ALL into PCR-negative remission and can be an effective bridge to transplant. Biol Blood Marrow Transplant 24:S25-S26, 2018. [Google Scholar]
- 15. Van Belle G. Statistical Rules of Thumb. Hoboken, NJ, Wiley, 2011. [Google Scholar]