

Graphical abstract
Keywords: SARS‐CoV‐2, Spike glycoprotein, Coronavirus variant, Residue mutation, Binding affinity
Highlights
-
•
Theoretical investigation of the binding affinity of the SARS-CoV-2 variants.
-
•
The 501Y.V1 variant yielded the highest enhancements in binding affinity.
-
•
The binding affinity mainly enhanced by forming hydrogen bonds and salt bridges.
Abstract
During the rapid worldwide spread of SARS-CoV-2, the viral genome has been undergoing numerous mutations, especially in the spike (S) glycoprotein gene that encode a type-I fusion protein, which plays an important role in the infectivity and transmissibility of the virus into the host cell. In this work, we studied the effect of S glycoprotein residue mutations on the binding affinity and mechanisms of SARS-CoV-2 using molecular dynamics simulations and sequence analysis. We quantitatively determined the degrees of binding affinity caused by different S glycoprotein mutations, and the result indicated that the 501Y.V1 variant yielded the highest enhancements in binding affinity (increased by 36.8%), followed by the N439K variant (increased by 29.5%) and the 501Y.V2 variant (increased by 19.6%). We further studied the structures, chemical bonds, binding free energies (enthalpy and entropy), and residue contribution decompositions of these variants to provide physical explanations for the changes in SARS-CoV-2 binding affinity caused by these residue mutations. This research identified the binding affinity differences of the SARS-CoV-2 variants and provides a basis for further surveillance, diagnosis, and evaluation of mutated viruses.
1. Introduction
The novel coronavirus disease (COVID-19), caused by severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2) [1], has emerged as a severe epidemic and a serious risk to the global public health and economy since its first outbreak in December 2019 [2], [3], [4]. Many studies have indicated that human angiotensin converting enzyme 2 (hACE2) is the entry receptor of SARS-CoV-2 [5], [6], [7], [8], which is similar to that of SARS-CoV [9], [10]. SARS-CoV-2 has been reported to use the homotrimeric spike (S) glycoprotein on the virion surface to implement receptor recognition and membrane fusion [2]. The S protein includes two main subunits (S1 and S2) that form the receptor-binding domain and the fusion peptide domain, in which S1 identifies and binds to the host cell and S2 implements membrane fusion. It has been reported that the receptor binding domain (RBD) within S1 subunit is a key functional component for binding to hACE2, and this binding is also a critical initial step in viral infection [1], [6], [8], [11]. Moreover, neutralizing monoclonal antibodies of the immune system have been reported to carry out their neutralization activity by binding to the RBD within the S protein and disrupting virus binding [12], [13], [14], [15], [16]. Therefore, an in-depth investigation of RBD binding to hACE2 is essential for drug design and antibody development.
However, residue mutations in S glycoprotein can significantly affect virus infectivity and transmission efficiency, thus influencing the efficacy of neutralizing antibodies. During the global spread of SARS-CoV-2, a large number of reports have indicated that S glycoprotein mutations can enhance the infection of the virus, but the mechanism has not been explained [17], [18], [19], [20]. For example, the N439K variant, in which the ASN439 residue in the RBD is mutated to LYS439, first emerged in Scotland and spread widely in many European countries. N439K was reported to significantly enhance the binding affinity of S glycoprotein to the hACE2 receptor and influence the activity of neutralizing antibodies [21], [22]. Another widespread SARS-CoV-2 variant is 501Y.V1, which is considered to be the most severe mutation based on experimental evaluation of the structures and functions of the virus [23], [24], [25], with increased binding affinity to the hACE2 receptor. The third SARS-CoV-2 variant found to have severe effects was reported at the end of 2020, when the 501Y.V2 variant was first discovered in South Africa and then found to be widely spread to a series of countries, including the UK, Switzerland, Finland, Japan and Australia, which has been observed to have an increased receptor-binding ability [25], [26]. The 501Y.V2 variant has three residue mutations in the S glycoprotein (K417N, E484K and N501Y mutations), which allow the virus to more easily bind to human cells [25], [27]. K417N refers to the residue mutation from LYS417 to ASN417, E484K represents the residue mutation from GLU484 to LYS484, and N501Y denotes the residue mutation from ASN501 to TYR501 [28]. It is very rare for three mutations to appear in one variant at the same time, especially during large outbreaks. Therefore, it is very important to study the binding affinity of receptors to the 501Y.V2 variant, which is difficult to observe in the laboratory and can serve as an identification indicator of virus infection after mutation.
In this work, we studied the structures, chemical bond changes, binding free energies (enthalpy and entropy), and residue contribution decompositions of the three most widespread and important S glycoprotein variants (501Y.V1, 501Y.V2, N439K) of SARS-CoV-2. As shown in Fig. 1(a), the 501Y.V1 variant contains N501Y mutation in the RBD, the 501Y.V2 variant contains the mutations of K417N, E484K, and N501Y in the RBD, and the N439K variant contains the mutation of N439K in the RBD. Based on structural analysis and molecular dynamics simulations, we studied the effects of the three variants on hACE2 binding affinity and mechanisms found the following discoveries. (1) Structural and sequence analysis showed that the molecular weight and stability changed significantly, but the aliphatic index and half-life did not change after the residue mutations, as shown in Table S1 of the supplementary information (SI). The 501Y.V1 and 501Y.V2 variants exhibited fewer hydrophobic properties and more lyotropic properties than wild-type SARS-CoV-2. (2) The root-mean-square deviations (RMSDs) and root-mean-square fluctuations (RMSFs) of the 501Y.V1, 501Y.V2, and N439K variants were all greater than those of wild-type SARS-CoV-2, indicating the increased flexibility of the SARS-CoV-2 residues after mutations and the enhanced binding affinities between the three variants and the hACE2 receptor. (3) The binding free energy calculations indicated that the 501Y.V1, 501Y.V2 and N439K variants all had significant increases in binding affinity, with enhancements of 36.8%, 19.6% and 29.5%, respectively, indicating the enhanced binding affinity of the SARS-CoV-2 variant. (4) The mutated residues enhanced the binding affinity mainly by forming hydrogen bonds and salt bridges, thus changing the electrostatic and van der Waals energies and enhancing the hydrophilic interactions. For example, the 501Y.V1 and N439K variants were both capable of creating new hydrogen bonds, resulting in the enhancement of their binding affinities to hACE2. (5) The N501Y mutation waw present not only in the 501Y.V1 variant but also in the 501Y.V2 variant. However, the 501Y.V1 variant had a stronger binding affinity to hACE2 than the 501Y.V2 variant, suggesting that the K417N and E484K mutations in the 501Y.V2 variant weaken the binding affinity between the SARS-CoV-2 RBD and hACE2 compared to the 501Y.V1 variant by breaking hydrogen bonds.
Fig. 1.

Illustration of the SARS-CoV-2 glycoprotein. (a) Schematic domains of the wild-type SARS-CoV-2 spike monomer and the three variants (501Y.V1, 501Y.V2 and N439K), with the N501Y mutation appearing in both the 501Y.V1 and 501Y.V2 variants. (b) Sequence and secondary structure of the wild-type SARS-CoV-2 RBD. (c) Crystal structure of SARS-CoV-2 S glycoprotein with the RBD highlighted in red. Thr333-Gly526 refers to the sequence of the RBD region. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2. Results and discussion
2.1. Structural and sequence-based analysis
The SARS-CoV-2 S glycoprotein, a large homotrimeric protein, plays a major role in viral when entry into host cells. Recent studies on the S glycoprotein and its interaction with the cell receptor (hACE2) have shown that their binding is a critical initial step for SARS-CoV-2 to enter host cells, in which the binding of the RBD domain induces the dissociation of the S1 subunit from hACE2 and promotes the transformation of the S2 subunit to a stable post-fusion state for membrane fusion [6], [7], [8], [11], [29].
The S protein is comprised of a S1 subunit and a S2 subunit. As shown in Fig. 1 (a), the S1 subunit of the S protein is composed of an N-terminal domain (NTD), receptor binding domain (RBD), subdomain 1 (SD1) and subdomain 2 (SD2), while the S2 subunit is comprised of a fusion peptide (FP), heptad repeat 1 (HR1), heptad repeat 2 (HR2), transmembrane region (TM) and intracellular domain (IC). The mutations in the RBD of the three studied variants (501Y.V1, 501Y.V2 and N439K) are also shown in Fig. 1 (a). Both the 501Y.V1 and N439K variants have single mutated residues in the RBD, while the 501Y.V2 variant is comprised of three mutated residues (K417N, E484K, and N501Y). The secondary structure, along with the sequence of the RBD, is shown in Fig. 1 (b). The SARS-CoV-2 RBD possesses seven sheets and five helices and loops, including twisted five-stranded antiparallel sheets designated , , 3, and , three short connecting helices and loops (, and ), and an extended insertion between the and sheets designated the receptor binding motif (RBM) (containing the , and sheets, and the and helices and loops). Previous cryo-electron microscopy research [6] has also determined the crystal structure of the SARS-CoV-2 S glycoprotein with one SB open as shown in Fig. 1 (c), where the RBD is highlighted in red.
The sequences of the wild-type SARS-CoV-2 RBD and the three mutated variants were analyzed by the ProtParam tool [30]. Table S1 in SI shows the physical and chemical parameters of the wild-type SARS-CoV-2 RBD and the three variants, including formula, molecular weight, theoretical pI, instability index, aliphatic index, grand average of hydropathicity (GRAVY), and estimated half-life (mammalian reticulocytes, in vitro). As can be observed in Table S1, the molecular weights of the mutated structures changed obviously, but the aliphatic indexes and half-lives did not. The instability indexes of the four structures in Table S1 were all below 40, indicating that the structures were stable with or without the studied mutations. Furthermore, the 501Y.V2 and 501Y.V2 variants exhibited the same GRAVY (-0.205), which was less negative than that of the wild-type RBD (-0.216), indicating that they were less hydrophobic and more lyotropic.
2.2. Binding affinity
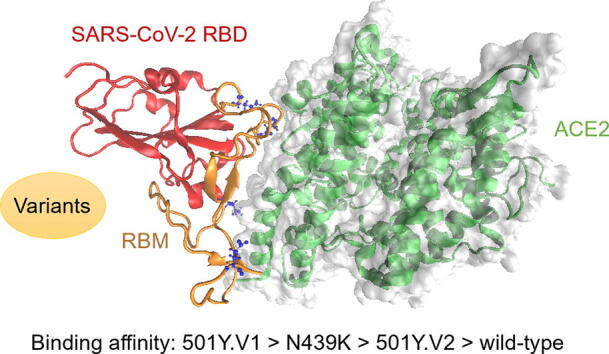
In this study, we investigated the binding affinity of the SARS-CoV-2 RBD and hACE2 complex (PDB ID: 6M0J) [29]. The crystal structure of the RBD-hACE2 complex is shown in Fig. 2 (a) with a solid surface model, where the receptor-binding motif (RBM) region is highlighted in orange and the mutated residues of the three variants are colored in blue with a ball-and-stick model. Previous cryo-electron microscopy results have shown that the RBM is essential for RBD contact with the hACE2 receptor [5], [31]. To evaluate the impact of the mutations in the SARS-CoV-2 RBD on binding affinity, the three variants (501Y.V1, 501Y.V2 and N439K) with different mutations in the RBD were selected for further molecular dynamics (MD) simulations. After energy minimization, heating and density equilibration, the RBD-hACE2 complex solution was equilibrated with a 1.2 ns MD simulation in an NPT ensemble with a temperature of 300 K and a pressure of 1 atm. Then, a 40 ns production run was performed while keeping the system stable with the convergence of temperature, density and total energy.
Fig. 2.

The structure, root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) of the wild-type SARS-CoV-2 RBD and hACE2 complex over 40 ns MD simulations. (a) Crystal structure of the SARS-CoV-2 RBD (wild-type) bound to hACE2, where the RBM region is highlighted in orange, the RBD region in red, and the mutated residues of the three variants in blue with the ball-and-stick model. (b) RMSD of all the non-hydrogen atoms of the RBD-hACE2 complex. (c) RMSF of all non-hydrogen atoms of the RBD-hACE2 complex. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The RMSD was analyzed to verify the convergence of the RBD-hACE2 complex during MD simulation. Fig. 2(b) shows the RMSDs of all non-hydrogen atoms for the wild-type RBD-hACE2 complex over the entire production simulation. The RBD-hACE2 complex remained stable from 10 ns to 40 ns with an RMSD of 1.9–2.0 Å. The RMSDs of the three mutated RBD-hACE2 complexes are shown in Fig. S1(a)-S3(a) of the SI. The results show that the RMSDs of variants 501Y.V1, 501Y.V2 and N439K were approximately 2.0–2.1 Å, which were slightly greater than that of the wild-type RBD-hACE2 complex. In addition, to detect the fluctuation and flexibility of residues on the RBD-hACE2 complexes, we calculated the RMSFs during the MD simulations. The RMSFs of the three variants compared with wild-type complex are shown in Fig. S1(b)-S3(b) of the SI. The three variants and the wild-type RBD-hACE2 complexes presented similar flexibility patterns, but the fluctuations of the RBD domain for the three variants were higher than those of the wild-type RBD, indicating a significant flexibility increase in the RBD residues after the mutations.
Table S2 shows the calculated solvent accessible surface area (SASA) and the radius of gyration (Rg), which can evaluate the solvent exposure degree and the structural compactness of the RBD-hACE2 complex. The results show that there was no obvious divergence in Rg between the four RBD-hACE2 complexes. The SASA value of wild-type RBD-hACE2 was 34996.9 Å2, which was slightly smaller than those of the 501Y.V1 (35165.5 Å2), 501Y.V2 (35496.9 Å2) and N439K (35783.6 Å2) variants.
Since the RMSDs of the four complexes reached convergence at 10 ns, the MD trajectory of 10–40 ns was used for the binding energy calculation, residue contribution decomposition and interaction analysis. Table 1 shows the calculated binding free energies of the four RBD-hACE2 complexes (one wild-type and three mutated RBDs), including the total enthalpy () and entropy contribution (). The total enthalpies were calculated by the Molecular Mechanics-Generalized Born Surface Area (MM/GBSA) method with 150 frames within the 30 ns MD trajectories, including the contributions of the van der Waals energy, the electrostatic energy, and the polar and nonpolar solvation free energies but without entropy, which plays an essential role in the binding process. The entropy contributions were obtained from all frames during the 30 ns trajectories using the interaction entropy (IE) method, which has been developed and successfully applied to many large protein systems [32], [33].
Table 1.
The calculated enthalpies (), entropy contributions () and binding free energies () of the four RBD-hACE2 complexes. denotes the binding free energy deviation between the variant complexes and the wild-type SARS-CoV-2 complex. All values of energy are shown in kcal/mol.
| Complexes | ||||
|---|---|---|---|---|
| RBD (wild)-hACE2 | −46.633 | 33.668 | −12.965 | – |
| RBD (501Y.V1)-hACE2 | −53.073 | 35.332 | −17.741 | −4.775 |
| RBD (501Y.V2)-hACE2 | −49.714 | 34.213 | −15.501 | −2.536 |
| RBD (N439K)-hACE2 | −47.632 | 30.838 | −16.794 | −3.829 |
From the calculated total enthalpy () in Table 1, the SARS-CoV-2 RBD-hACE2 complex of the 501Y.V1 variant exhibited the strongest (the lowest) binding energy of −53.073 kcal/mol, while the wild-type RBD-hACE2 complex showed the weakest (the highest) binding energy of −46.633 kcal/mol. However, the total enthalpy did not take into account the entropy contribution, which depends on the alternation of motional freedom induced by RBD binding to hACE2 and contributes significantly to the total binding free energy. After considering the entropy contribution, the total binding free energies () in Table 1 indicated that the three variants all showed stronger binding free energies than the wild-type RBD-hACE2 complex (−12.965 kcal/mol), which is consistent with previous experimental investigations [23], [24], [25], [27], [34], [35], [36]. For the three variants, the 501Y.V1 variant exhibited the strongest binding energy of the RBD-hACE2 complex at −17.741 kcal/mol, followed by the N439K variant (-16.794 kcal/mol) and the 501Y.V2 variant (−15.501 kcal/mol).
However, compared with the single N501Y mutation, the additional K417N and E484K mutations resulted in a decreased affinity between 501Y.V2 and hACE2 (as shown in Table 1). In addition, the N439K variant showed only slightly lower binding free energy than the wild-type SARS-CoV-2. From Table 1, the 501Y.V1, 501Y.V2 and N439K variants had a binding free energy increased by 36.8%, 19.6% and 29.5%, with binding energy changes () of −4.775, −2.536, and −3.829 kcal/mol, respectively. The N501Y mutation significantly increased the binding affinity of the variant, while N439K showed little effect on binding affinity. We also presented the experimental relative equilibrium dissociation constant (KD) of the four RBD-hACE2 complexes (wild-type, 501Y.V1, 501Y.V2, N439K) compared with the wild-type SARS-CoV-2 complex [22], [23], [29], [31], [37], [38], [39] and the comparison of binding free energies changes () of the three variants between theoretical calculations and experimental results in Supporting Information (Table S3 and S4). As Tables S3-S4 shown, the 501Y.V1 variant is commonly considered to significantly increase the binding affinity of RBD to ACE2, while 501Y.V2 and N439K show small increasement in binding affinity. Overall, our results show that the mutations (N501Y, K417N, E484K, and N439K) in the three variants were all capable of enhancing the binding affinity and thus probably increasing the infectivity of the virus, and this result agrees well with previously published experimental results [40].
2.3. Residue contribution
To understand the biophysical preference of the SARS-CoV-2 RBD for binding to hACE2, we further analyzed the residue contributions, hydrogen bonds and salt bridges of the SARS-CoV-2 RBD. Table 2 shows the top five residues with the dominant contributions of the four SARS-CoV-2 RBDs binding with hACE2, where the ASN501 residue in the wild-type SARS-CoV-2 RBD was mutated to the TYR501 residue in the 501Y.V1 and 501Y.V2 variants. From Table 2, TYR 505, GLN493 and PHE486 were the top residues with dominant contributions to the binding of wild-type RBD-hACE2, which is consistent with previous experimental results [5], [29], [40], [41]. The binding interfaces of the four RBD-hACE2 complexes, including hydrogen bonds, salt bridges and residues with dominant binding contributions in the ball-and-stick model, are shown in Fig. 3. From Fig. 3 (a)-(d), GLN493 in the SARS-CoV-2 RBD broke the salt bridge between LYS31 and GLU35 in hACE2 and formed hydrogen bonds with these two residues. During the binding process, PHE486 in the SARS-CoV-2 RBD enhanceed the hACE2 binding affinity by creating a hydrophobic pocket involving MET82 and TYR83 in hACE2 (Fig. 3 (a)-(d)). For wild-type SARS-CoV-2, the TYR505 residue contributed the most to the binding of RBD with hACE2, with a large electrostatic energy and van der Waals energy. However, the mutated TYR501 became the residue with the greatest contribution for binding to hACE2 in the 501Y.V1 and 501Y.V2 variants (-5.11 kcal/mol for 501Y.V1 and −6.102 kcal/mol for 501Y.V2, both of which are highlighted in orange in Table 2), compared with ASN501 (-2.785 kcal/mol), which was the residue with the fourth greatest contribution for the wild-type SARS-CoV-2 RBD (as shown in Table 2).
Table 2.
Top five residues with dominant contributions of the four SARS-CoV-2 RBDs binding with hACE2. The TYR501 residue in the 501Y.V1 and 501Y.V2 variants is mutated from the ASN501 residue of the wild-type SARS-CoV-2 RBD.
| RBDs | Top contributed residues |
||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Wild-type | TYR 505 | GLN 493 | PHE 486 | ASN 501 | THR 500 |
| 501Y.V1 | TYR 501 | PHE 486 | TYR 449 | GLN 493 | TYR 505 |
| 501Y.V2 | TYR 501 | TYR 505 | GLN 493 | PHE 486 | THR 500 |
| N439K | TYR 505 | GLN 498 | GLN 493 | TYR 449 | PHE 486 |
Fig. 3.

The SARS-CoV-2 RBD-hACE2 interfaces and interactions. Binding interfaces between (a) the wild-type, (b) the 501Y.V1 variant, (c) the 501Y.V2 variant, and (d) the N439K variant SARS-CoV-2 RBD and hACE2, including hydrogen bonds, salt bridges and residues with the dominant binding contributions. The residues of hACE2 are colored green, residues with dominant binding contributions are colored red, and the mutated residues are colored orange. The dashed circles in (b), (c), and (d) denote the newly formed hydrogen bonds after mutation. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
These studied mutations mainly enhanced the binding affinity of the SARS-CoV-2 RBD by forming hydrogen bonds and salt bridges, thus changing the electrostatic and van der Waals energies and promoting hydrophilic interactions. Fig. 3 (b)-(d) show the interfaces and residues between the SARS-CoV-2 RBD and hACE2 for the wild-type 501Y.V1 variant, 501Y.V2 variant and N439K variant, respectively. In the 501Y.V1 and 501Y.V2 variants, the mutation from ASN501 to TYR501 formed strong hydrogen bonds and salt bridges between residues TYR501-TYR41 and THR500-ASP355, as shown in Fig. 3 (b) and (c). In addition to the newly formed hydrogen bond between TYR501 and TYR41, a weaker hydrogen bond between residues GLU484 in RBD and LYS31 in hACE2 was also generated in the 501Y.V1 variant. However, such weaker hydrogen bonds were destroyed in the 501Y.V2 variant. In addition to breaking the weak hydrogen bond between GLU484 and LYS31, the hydrogen bond between LYS417 and ASP30 in wild-type SARS-CoV-2 was also broken in the 501Y.V2 variant. Therefore, compared with the 501Y.V1 variant, the 501Y.V2 variant had a weaker binding affinity for the cellular target in the host. Fig. 3 (d) shows the composite structure of the N439K variant, which formed new but weak hydrogen bonds between residues LYS439 and GLU329, thus slightly enhancing the binding affinity of the SARS-CoV-2 RBD (Fig. 3 (d)). From the perspective of chemical bond generation and breaking, Fig. 3 explains why 501Y.V1 had the highest binding affinity to humans among the three variants, followed by N439K and 501Y.V2.
Fig. 4 shows the contributions of the binding free energy components for the top 5 residues in the SARS-CoV-2 RBD, which were decomposed into van der Waals energy (vdW), electrostatic energy (Ele), polar solvation energy (Polar) and non-polar solvation energy (Non-polar). As shown in Fig. 4 and Tables S5-S8, the enhancement of binding affinity for variants was mainly attributed to the energetic changes in electrostatic and van der Waals interactions, which is consistent with the formation of salt bridges and hydrogen bonds (Fig. 3). In summary, the mutations in the variants mainly enhanced the binding affinity by forming hydrogen bonds and salt bridges, thus changing the electrostatic, van der Waals and polar energies.
Fig. 4.

Decomposing the binding free energy of the top 5 residues into van der Waals interactions (vdW), electrostatic interactions (Ele), polar solvation energy (Polar) and non-polar energy (Non-polar). (a) Wild-type, (b) 501Y.V1, (c) 501Y.V2 and (d) N439K variants of SARS-CoV-2 RBD.
3. Conclusions
In conclusion, we investigated the binding affinity and mechanisms of the three most widespread SARS-CoV-2 variants (501Y.V1, 501Y.V2, N439K) based on molecular dynamics simulations and sequence analysis. Compared with wild-type SARS-CoV-2, we demonstrated that the 501Y.V1 and 501Y.V2 variants showed less hydrophobicity and more solubility behaviors and that the molecular weight and stability of the three variants were significantly changed. According to the calculations of the binding free energies, the binding affinities of 501Y.V1, N439K, and 501Y.V2 variants were significantly increased by 36.8%, 29.5%, and 19.6%, respectively. We further analyzed the interaction and residue contribution and demonstrated that the mutated residues enhance the binding affinity mainly by forming hydrogen bonds and salt bridges, which changed the electrostatic and van der Waals energies and enhance the hydrophilic effect. For example, both the 501Y.V1 and N439K variants were able to form new hydrogen bonds, thus enhancing their binding affinity with hACE2. The method and results presented in this paper will provide a theoretical basis for further surveillance, diagnosis, and evaluation of the binding affinity of variant viruses.
4. Methods
4.1. 3D structures
In this study, the crystal structure of the wild-type SARS-CoV-2 spike receptor-binding domain (RBD) bound to the human cell receptor (hACE2) was taken from the RCSB Protein Data Bank (PDB ID: 6M0J) [42], which was determined by Lan et al. [29], at a resolution of 2.45 Å. SWISS-MODEL [43], [44], a protein structure homology-modeling server, was used to prepare 3D structures of the three mutated viruses (501Y.V1, 501Y.V2, N439K) with the crystal structure of the complex of wild-type RBD-hACE2 (6M0J) as the target template.
4.2. Sequence-based structural analysis
In this study, the sequences and structures of the wild-type SARS-CoV-2 RBD and three mutated variants were analyzed based on previous experimental studies [29], [40]. In addition, the physical and chemical parameters for the wild SARS-CoV-2 RBD and the three variants, including the formula, molecular weight, theoretical pI, instability index, aliphatic index, grand average of hydropathicity (GRAVY), and estimated half-life (mammalian reticulocytes, in vitro), were obtained by ProtParam [30], a tool for protein sequence analysis.
4.3. Molecular dynamic simulation
In this study, parallel molecular dynamics (MD) simulations and binding free energy calculations of the RBD-hACE2 complexes were performed using Amber16 [45] software. The ff14SB force field [46] was used to generate the parameters of the RBD-hACE2 complexes. An explicit solvent model was used during the MD simulations by solvating the RBD-hACE2 complexes in a rectangular TIP3P [47] water box with a 12 Å buffer. Counterions (Na+ and Cl-) were added to neutralize the whole system. A 30,000-step energy minimization was first performed on the system. Then the solution system was gradually heated from 0 to 300 K with an NVT ensemble (N, V and T denote the number of particles, volume, and temperature, respectively) during a 100 ps simulation, followed by a 100 ps simulation for density equilibration with weak restraints (10 kcal mol−1 Å−2) on the RBD-hACE2 complex. Subsequently, the system was equilibrated for 1 ns in an NPT ensemble with a temperature of 300 K and pressure of 1 atm. Finally, a 40 ns production run without any restriction was performed with the NPT ensemble for each solution system, with all hydrogen atoms constrained using the SHAKE algorithm. The cutoff distance for long-range nonbonded interactions was set to 8 Å and the time step was set to 2 fs. The energy minimization and MD simulation were all performed with the sander program in Amber16. The last 30 ns trajectory of the production run was used to calculate the binding free energy. The RMSD, RMSF, SASA and Rg were analyzed with the cpptraj program in Amber16.
4.4. Binding free energy calculation
The binding free energy () of the RBD-hACE2 complex system were obtained by the following equation:
| (1) |
where , , and were the free energies of the complex, RBD and hACE2, respectively.
For each subsystem, the free energy was calculated by the following equation:
| (2) |
where and were the total enthalpy from the generalized Born (GB) model, including the internal and solvation energies, and the contribution of entropy, respectively.
The total enthalpy was obtained by the Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) [48], [49] method using MMPBSA.py script in Amber16. Here, for each system, 150 frames were extracted from the 30 ns trajectory for calculation. The enthalpy was calculated by the following equation:
| (3) |
where and were the gas phase energy and the solvation energy, respectively, and , and were the van der Waals energy, the electrostatic energy, and the polar and nonpolar solvation free energies, respectively.
The contribution of entropy was calculated by the interaction entropy (IE) [32], [33] method with the following equations:
| (4) |
where was , and was the fluctuation of the RBD-hACE2 interaction energy () around the average interaction energy (). was calculated by the following equation:
| (5) |
where and were calculated by the following equations:
| (6) |
| (7) |
5. Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding
Research reported in this publication was supported by the SJTU Global Strategic Partnership Fund (2020 SJTU-HUJI), the National Natural Science Foundation of China (Nos. 21901157 and 81901069) and the National Key R&D Program of China (2021YFC2100100).
CRediT authorship contribution statement
Yanqiang Han: Conceptualization, Methodology, Formal analysis, Investigation, Writing – original draft, Visualization. Zhilong Wang: Data curation, Software, Visualization, Validation. Zhiyun Wei: Conceptualization, Methodology, Supervision, Resources, Writing - review & editing. Igor Schapiro: Software, Data curation, Validation. Jinjin Li: Conceptualization, Formal analysis, Funding acquisition, Project administration, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.07.026.
Contributor Information
Zhiyun Wei, Email: zhiyun_wei@163.com.
Jinjin Li, Email: lijinjin@sjtu.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Lu R., Zhao X., Li J., Niu P., Yang B.o., Wu H. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu F., Zhao S.u., Yu B., Chen Y.-M., Wang W., Song Z.-G. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu N. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan R., Zhang Y., Li Y., Xia L.u., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Letko M., Marzi A., Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simmons G., Zmora P., Gierer S., Heurich A., Pöhlmann S. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res. 2013;100:605–614. doi: 10.1016/j.antiviral.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belouzard S., Chu V.C., Whittaker G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci. 2009;106:5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tian X. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect. 2020;9:382–385. doi: 10.1080/22221751.2020.1729069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ju B., Zhang Q.i., Ge J., Wang R., Sun J., Ge X. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584(7819):115–119. doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- 13.Liu L., Wang P., Nair M.S., Yu J., Rapp M., Wang Q. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584(7821):450–456. doi: 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- 14.Cao Y., Su B., Guo X., Sun W., Deng Y., Bao L. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell. 2020;182(1):73–84.e16. doi: 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi R., Shan C., Duan X., Chen Z., Liu P., Song J. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020;584(7819):120–124. doi: 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- 16.Hansen J., Baum A., Pascal K.E., Russo V., Giordano S., Wloga E. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369(6506):1010–1014. doi: 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W. Tracking Changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182(4):812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saha P, Banerjee AK, Tripathi PP, Srivastava AK, Ray, U. A virus that has gone viral: amino acid mutation in S protein of Indian isolate of Coronavirus COVID-19 might impact receptor binding, and thus, infectivity. Biosci. Rep. 40, BSR20201312 (2020). [DOI] [PMC free article] [PubMed]
- 19.Sheikh J.A. Emerging genetic diversity among clinical isolates of SARS-CoV-2: lessons for today. Infect Genet Evol. 2020;84 doi: 10.1016/j.meegid.2020.104330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Dorp L. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect Genet Evol. 2020;83 doi: 10.1016/j.meegid.2020.104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.COG-UK update on SARS-CoV-2 Spike mutations of special interest. 2020. https://www.cogconsortium.uk/wp-content/uploads/2021/02/Report-2_COG-UK_SARS-CoV-2-Mutations.pdf.
- 22.Thomson E.C. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell. 2021;184:1171–1187.e20. doi: 10.1016/j.cell.2021.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Starr T.N., Greaney A.J., Hilton S.K., Ellis D., Crawford K.H.D., Dingens A.S. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182(5):1295–1310.e20. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu H., Chen Q.i., Yang G., He L., Fan H., Deng Y.-Q. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science. 2020;369(6511):1603–1607. doi: 10.1126/science.abc4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu J. Emerging SARS-CoV-2 variants reduce neutralization sensitivity to convalescent sera and monoclonal antibodies. Cell Mol Immunol. 2021;18:1061–1063. doi: 10.1038/s41423-021-00648-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature. 2021;592(7854):438–443. doi: 10.1038/s41586-021-03402-9. [DOI] [PubMed] [Google Scholar]
- 27.Tegally H. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat Med. 2021;27:440–446. doi: 10.1038/s41591-021-01255-3. [DOI] [PubMed] [Google Scholar]
- 28.GISAID - Novel variant combination in spike receptor binding site. https://www.gisaid.org/references/gisaid-in-the-news/novel-variant-combination-in-spike-receptor-binding-site/.
- 29.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 30.Gasteiger E. et al. Protein Identification and Analysis Tools on the ExPASy Server. in The Proteomics Protocols Handbook (ed. Walker, J. M.) 571–607 (Humana Press, 2005). doi:10.1385/1-59259-890-0:571.
- 31.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan L., Liu X., Zhang J.Z.H. Interaction entropy: a new paradigm for highly efficient and reliable computation of protein-ligand binding free energy. J. Am. Chem. Soc. 2016;138:5722–5728. doi: 10.1021/jacs.6b02682. [DOI] [PubMed] [Google Scholar]
- 33.Huang K., Luo S., Cong Y., Zhong S., Zhang J.Z.H., Duan L. An accurate free energy estimator: based on MM/PBSA combined with interaction entropy for protein–ligand binding affinity. Nanoscale. 2020;12(19):10737–10750. doi: 10.1039/c9nr10638c. [DOI] [PubMed] [Google Scholar]
- 34.Leung K., Shum M.H., Leung G.M., Lam T.T., Wu J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance. 2021;26:2002106. doi: 10.2807/1560-7917.ES.2020.26.1.2002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davies N.G., Abbott S., Barnard R.C., Jarvis C.I., Kucharski A.J., Munday J.D. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372(6538):eabg3055. doi: 10.1126/science.abg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eurosurveillance editorial Team. Updated rapid risk assessment from ECDC on the risk related to the spread of new SARS-CoV-2 variants of concern in the EU/EEA – first update. Eurosurveillance 26, 2101211 (2021). [DOI] [PMC free article] [PubMed]
- 37.Liu H. The basis of a more contagious 501Y.V1 variant of SARS-CoV-2. Cell Res. 2021;31:720–722. doi: 10.1038/s41422-021-00496-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian F. et al. Mutation N501Y in RBD of Spike Protein Strengthens the Interaction between COVID-19 and its Receptor ACE2. bioRxiv 2021.02.14.431117 (2021) doi:10.1101/2021.02.14.431117.
- 39.Laffeber C., de Koning K., Kanaar R., Lebbink J.H.G. Experimental evidence for enhanced receptor binding by rapidly spreading SARS-CoV-2 variants. J Mol Biol. 2021;433 doi: 10.1016/j.jmb.2021.167058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yi C. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell Mol Immunol. 2020;17:621–630. doi: 10.1038/s41423-020-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shang J., Ye G., Shi K.e., Wan Y., Luo C., Aihara H. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581(7807):221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berman H.M. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waterhouse A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed]
- 44.Studer G. et al. QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 36, 1765–1771 (2020). [DOI] [PMC free article] [PubMed]
- 45.Case D.A. AMBER 2016. Univ. Calif. San Franc. 2016;810 [Google Scholar]
- 46.Maier J.A. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price D.J., Brooks C.L. A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys. 2004;121:10096–10103. doi: 10.1063/1.1808117. [DOI] [PubMed] [Google Scholar]
- 48.Miller BR et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 8, 3314–3321 (2012). [DOI] [PubMed]
- 49.Feig M. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem. 2004;25:265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.