

Abstract
Neuroendocrine (NE) cells are epithelial cells that possess many of the characteristics of neurons, including the presence of secretory vesicles and the ability to sense environmental stimuli. The normal physiologic functions of solitary airway NE cells remain a mystery. We show that mouse and human airway basal stem cells sense hypoxia. Hypoxia triggers the direct differentiation of these stem cells into solitary NE cells. Ablation of these solitary NE cells during hypoxia results in increased epithelial injury, whereas the administration of the NE cell peptide CGRP rescues this excess damage. Thus, we identify stem cells that directly sense hypoxia and respond by differentiating into solitary NE cells that secrete a protective peptide that mitigates hypoxic injury.
Airway neuroendocrine (NE) cells were first identified as epithelial cells that store and secrete amines and peptides from membrane-bound vesicles, mirroring the process of neurotransmitter release from neurons. Indeed, both neurons and airway NE cells secrete serotonin and calcitonin gene–related peptide (CGRP) (1, 2). NE cell hyperplasia occurs in diverse lung diseases, including neuroendocrine hyperplasia of infancy (NEHI), sudden infant death syndrome (SIDS), asthma, congenital pneumonia, pulmonary hypertension, cystic fibrosis, congenital diaphragmatic hernia, and chronic obstructive pulmonary disease (COPD) (3, 4). However, both the cause and relevance of NE cell hyperplasia remain unknown.
NE cells can be found as solitary cells or clustered as neuroepithelial bodies (NEBs). In mice, solitary pulmonary NE cells are found in the trachea and primary bronchi, but in humans, they are scattered throughout the airway tree. By contrast, the neuroepithelial bodies (NEBs) of mice are clustered groups of NE cells found specifically at airway branch points, whereas their presumed human NEB counterparts are less stereotypically distributed throughout the airway epithelium (5, 6). NEBs are reported to perform various physiologic functions, including oxygen sensing (7), mechanotransduction (8, 9), modulating pulmonary blood flow (9), chemosensation (9), and regulating inflammation (10). Additionally, murine NEBs are thought to serve as niches for adjacent progenitor cells (11). However, the physiologic functions of solitary NE cells are largely unknown. One study of human solitary NE cells in vitro suggests that they function as airway chemosensory cells in vivo (12). Human solitary NE cells secrete CGRP in culture, and it is hypothesized that this neuropeptide links epithelial stimulus detection to the modulation of stem cell behavior (12).
Murine NEBs contain a subpopulation of NE stem cells that rarely produce new NE cells under steady-state conditions. However, following injury, NE stem cells generate more NE cells, leading to larger NEBs. The NE stem cells of NEBs are also plastic and contribute to the lineage of non-NE epithelial cells in their immediate vicinity (13). Despite this plasticity, genetic ablation of NEBs fails to elicit a gross effect on airway epithelial regeneration (14). By contrast, solitary NE cells display rapid turnover and are regularly replaced by basal stem cells (15, 16). The divergent lineage and kinetics of solitary NE cells and NEB-localized NE cells suggests that their functions may be distinct.
After birth, as the airway epithelium is first exposed to ambient oxygen, NE cell numbers decline (17–19). Hypoxia is known to delay the normal postnatal disappearance of NE cells in rabbit airway epithelia, but the mechanism is not understood (17). Elimination of oxygen-sensing prolyl hydroxylases (PHDs) from NEB-resident NE cells results in NEB growth (20). Because solitary NE cells are derived from basal stem cells rather than preexisting NE cells, we postulated that basal stem cells might possess an oxygen-sensing mechanism that triggers hypoxia-stimulated stem cells to undergo solitary NE cell differentiation. We further speculated that these hypoxia-induced solitary NE cells would have a protective physiologic function in the setting of hypoxia. The coincident occurrence of solitary NE cells and basal cells is characteristic of the majority of the human airway tree, so we used the murine trachea as a model system to study solitary NE cell biology because it is the only part of the murine respiratory tree that contains both solitary NE cells and basal stem cells (21).
Hypoxia induces solitary NE cell differentiation
When mice were exposed to hypoxic conditions (FiO2 8%) (fig. S1), we observed a significant time-dependent increase in solitary NE cells (Fig. 1, A and B). To exclude the possibility of injury-induced nonspecific expression of NE cell markers by non-NE cells, we assessed the expression of the NE cell fate–determining transcription factor Ascl1. Ascl1-CreERT2::Rosa26-tdTomato (hereafter referred to as Ascl1-tdTomato) mice were exposed to 20 days of hypoxia (fig. S2A), and tamoxifen was administered to label Ascl1+ NE cells. We observed a significant increase in Ascl1-tdTomato+ cells (fig. S2, B and C), confirming NE cell differentiation. Finally, quantifying the fraction of cells possessing NE cell–specific vesicles by electron microscopy confirmed the potent induction of bona fide solitary NE cells (Fig. 1, C and D).
Fig. 1. Hypoxia leads to an increase in solitary neuroendocrine (NE) cell numbers in the adult mouse trachea.
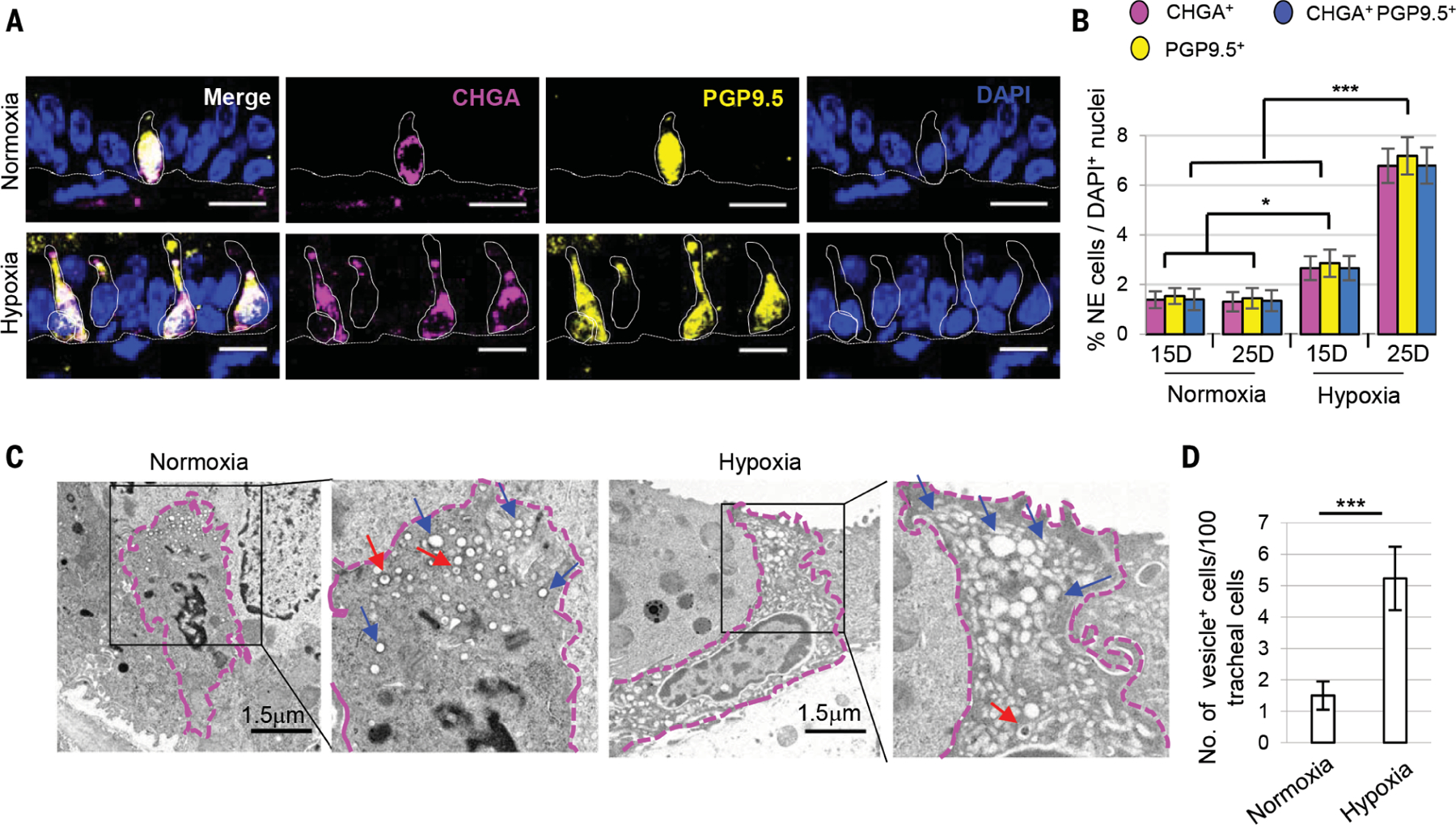
(A) Immunostaining for NE cell markers PGP9.5 (yellow) and CHGA (magenta). (B) Quantification of the percentage of CHGA+, PGP9.5+ and double-positive solitary NE cells in tracheal sections (n = 8). Dotted lines indicate basement membrane and cell borders. (C) Transmission electron microscopy (TEM) images of solitary NE cells and (D) quantification of cells with NE vesicles after 25 days of normoxia and hypoxia (n = 2) (total 300 nuclei from each condition). Blue arrows indicate empty core vesicles, and red arrows point to empty degranulating vesicles. Dotted magenta lines demarcate NE cell borders. n= biological replicates/condition repeated three times (three independent experiments excluding TEM experiments). ***p < 0.001, **p < 0.01, *p < 0.05; error bars, means ± SD. Scale bars, 20 μm (unless indicated otherwise).
We next sought to determine the cellular origin of the hypoxia-induced solitary NE cells. Because the deletion of PHDs, which mimics hypoxia, results in NEB growth (20), we assessed whether hypoxia could similarly cause solitary NE cells to self-renew. Preexisting solitary NE cells in Ascl1-tdTomato mice were lineage labeled, and then animals were subjected to hypoxia. The incorporation of 5-bromodeoxyuridine (BrdU) over the entire period of hypoxia was measured as an index of replication (fig. S3A). Although solitary NE cell numbers increased significantly after hypoxia (fig. S3, B and C), we did not observe any lineage-labeled cells or chromogranin A–positive (CHGA+) solitary NE cells positive for Ki67 (fig. S3, B and D), and very few arose from NE cell replication (4.41% of lineage-labeled NE cells were BrdU+) (fig. S3, E and F). Thus, NE cell replication does not account for the accumulation of solitary NE cells.
Basal stem cells are the source of hypoxia-induced solitary NE cells
To assess whether basal stem cells are the source of hypoxia-induced solitary NE cells, we lineage labeled basal stem cells using p63-CreER::R26R-tdTomato mice (hereafter referred to as p63-tdTomato, 97% labeling efficiency) and subjected the mice to hypoxia (fig. S4). This revealed that the basal stem cells are indeed the source of hypoxia-induced solitary NE cells (Fig. 2A). Furthermore, electron microscopy revealed the presence of basal stem cells that expressed abundant vesicles. After hypoxia, 4 ± 0.49% of basal cells exhibited NE-specific vesicles (Fig. 2B), which were normally absent. These data point to a direct differentiation of basal cells into solitary NE cells.
Fig. 2. Hypoxia leads to increased solitary NE cell numbers by accelerating the differentiation of basal stem cells into NE cells.
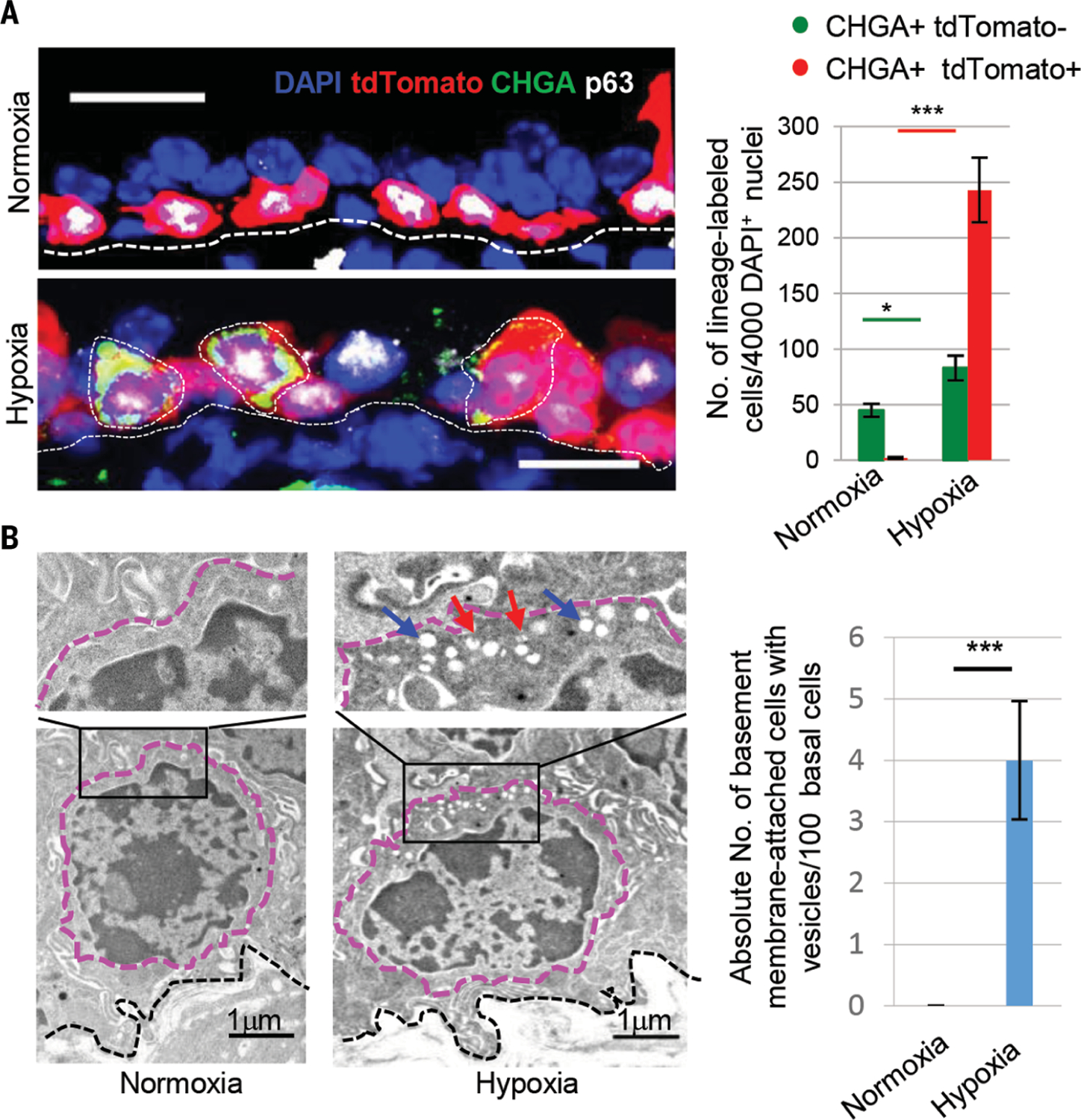
(A) Immunostaining for CHGA (green) and p63 (white) and quantification of the absolute number of indicated cells from p63-tdTomato lineage–labeled mice exposed to hypoxia. (B) Transmission electron microscopy images of tracheal sections after 25 days of normoxia or hypoxia. After hypoxia, cells with characteristic features of basal cells contain apically located secretory vesicles. Basal cells with and without secretory vesicles are quantified (200 nuclei from each condition). Dotted black lines indicate basement membrane, magenta lines demarcate cell boundaries. Blue arrows point to empty core vesicles, and red arrows point to empty degranulated vesicles. n.s., not significant; ***p < 0.001, *p < 0.05; error bars, means ± SD. Scale bars, 20 μm (unless indicated otherwise).
Although basal cells clearly contribute to the pool of hypoxia-induced solitary NE cells, a significant increase in the number of unlabeled CHGA+ tdTomato− cells after basal cell lineage tracing was observed, implying contributions from other cells (Fig. 2A). Because airway secretory cells have been shown to be plastic (22), we lineage labeled secretory cells using Scgb1a1-CreERT2::Rosa26-tdTomato mice (labeling efficiency 62 ± 5.02%) and then subjected the mice to hypoxia (fig. S5A). A small increase in the fraction of lineage-labeled CHGA+ solitary NE cells occurred (fig. S5B), revealing that secretory cells can undergo limited NE cell differentiation. Of the non-neuroendocrine cells contributing to the pool of hypoxia-induced solitary NE cells, basal cells are the major contributors (76%), followed by secretory cells (4 to 8%) (fig. S5C).
Basal stem cells sense hypoxia
We next sought to determine the molecular basis of NE cell differentiation. Oxygen-requiring PHDs act as molecular oxygen sensors (23). During normoxia, PHDs hydroxylate hypoxia-inducible factor alpha (HIFα), which in turn leads to HIFα ubiquitination and degradation. This fails to occur during hypoxia, and stabilized HIFα activates numerous genes associated with the hypoxia response. To address whether PHDs play a similar role in the airway epithelium, we used small molecules that stabilize HIFs in a hypoxia-mimetic state, including the PHD inhibitor FG-4592 and a HIF-a-pVHL interaction inhibitor, CoCl2 (24, 25). Both inhibitors stabilized HIF-1a and HIF-2a in cultured epithelium (fig. S6, A to C). Ascl1-tdTomato mice were treated with varying doses of FG-4592 over 20 days, and then tamoxifen was administered to label Ascl1+ solitary NE cells (fig. S7A). A significant dose-dependent increase in tdTomato+ cells occurred (fig. S7, B and C). Analogous results were obtained with an independent NE cell lineage driver, Cgrp-CreERT2::Rosa26-tdTomato (hereafter Cgrp-tdTomato) (fig. S7, D and E). A significant increase in solitary NE cells was also observed after CoCl2 administration (fig. S7, F to H).
To assess whether molecular oxygen sensing specifically occurs within the stem cell compartment per se, we genetically activated HIF signaling in vivo by conditionally deleting all three PHDs exclusively in basal stem cells using mice harboring p63-CreERT2 and Rosa26-tdTomato::Phd1fl/fl::Phd2fl/fl::Phd3fl/fl alleles (hereafter referred as p63-Phds) (fig. S8). This stabilized HIF-1a and HIF-2a in a two-dimensional culture system (fig. S9, A to C). After tamoxifen-induced deletion, CHGA+ NE cell numbers increased (Fig. 3A). This demonstrates that stem cells harbor a molecular oxygen-sensing mechanism and that stem cell–specific HIF activation triggers those same stem cells to directly differentiate into NE cells.
Fig. 3. Stem cells sense hypoxia using prolyl hydroxylases and then differentiate into solitary NE cells.
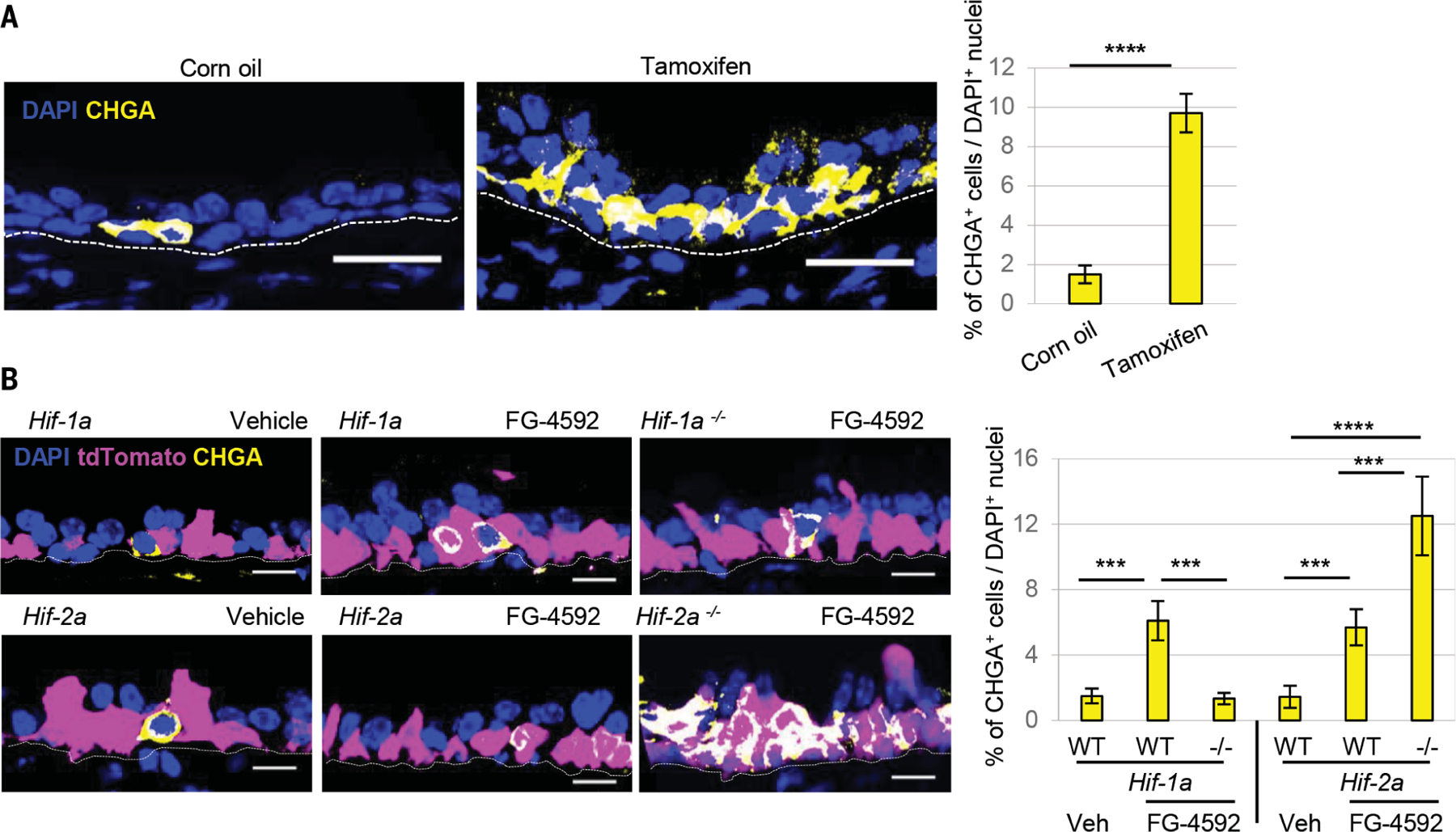
(A) Immunostaining for CHGA (yellow) and quantification of NE cells after deletion using p63-CreER::PhDs mice. (B) Immunostaining for CHGA (yellow) and quantification of CHGA+ cells from p63-Hif1α or p63-Hif2α mice treated with FG-4592 (n = 5). In the merged image, the overlap of yellow (CHGA), blue (DAPI, 4´,6-diamidino-2-phenylindole), and tdTomato appears white. ****p < 0.0001, ***p < 0.001; error bars, means ± standard deviation. Scale bars, 20 μm.
To identify the specific HIF isoform involved in solitary NE cell differentiation, we specifically activated either Hif1a or Hif2a by conditionally deleting one Hif at a time in basal stem cells (p63-CreERT2::Rosa26-tdTomato::Hif-1afl/fl or Hif-2afl/fl) and stabilizing the other through FG-4592 administration (fig. S10). Enforced HIF1a stabilization and Hif-2a deletion augmented FG-4592–induced CHGA+ NE cell induction, whereas HIF2a-stabilization and Hif-1a deletion completely blocked it (Fig. 3B). Thus, the stem cell NE differentiation program is Hif1a dependent, and Hif2a is a negative regulator of this process.
Hypoxia-induced solitary NE cells mediate a protective tissue response
The epithelial damage induced by hypoxia is characterized by epithelial cell apoptosis and a compensatory stem cell hyperplasia (Fig. 4, A and B, columns 1 and 3, and Fig. 5, A and B). We hypothesized that hypoxia-induced solitary NE cells could mediate a protective tissue response in this setting. To assess the functional consequences of solitary NE cell loss, we used compound mice carrying the NE cell–specific CGRP-CreER driver allele and a floxed DTA allele. Intranasal administration of a low dose of 4-hydroxytamoxifen was used to achieve airway-specific NE cell ablation, as evidenced by a reduction in CHGA staining (efficiency ~88 ± 2%) (Fig. 5C). NE cell ablation during normoxia did not significantly alter epithelial apoptosis or cell proliferation (Fig. 4B, columns 1 and 2, and Fig. 5, A and B), whereas in the setting of hypoxia, it resulted in a marked increase in apoptosis (Fig. 4B columns 3 and 4, and Fig. 5A) and a drop in epithelial cell proliferation (Fig. 4B, column 3 and 4, and Fig. 5B). Ciliated cells were most prone to apoptosis, whereas basal stem cells were the most hypoxia resistant (fig. S11, A and B). Basal cells were also the dominant cell population undergoing hypoxia-induced proliferation, although secretory cells also divided (fig. S12, A and B). NE cell ablation has no effect during normoxia, but in the setting of hypoxia, the loss of NE cells results in a significant decrease in basal stem cell and secretory cell progenitor cell proliferation (fig. S12, A and B).
Fig. 4. Solitary NE cells are required for the repair of hypoxia-induced injury.
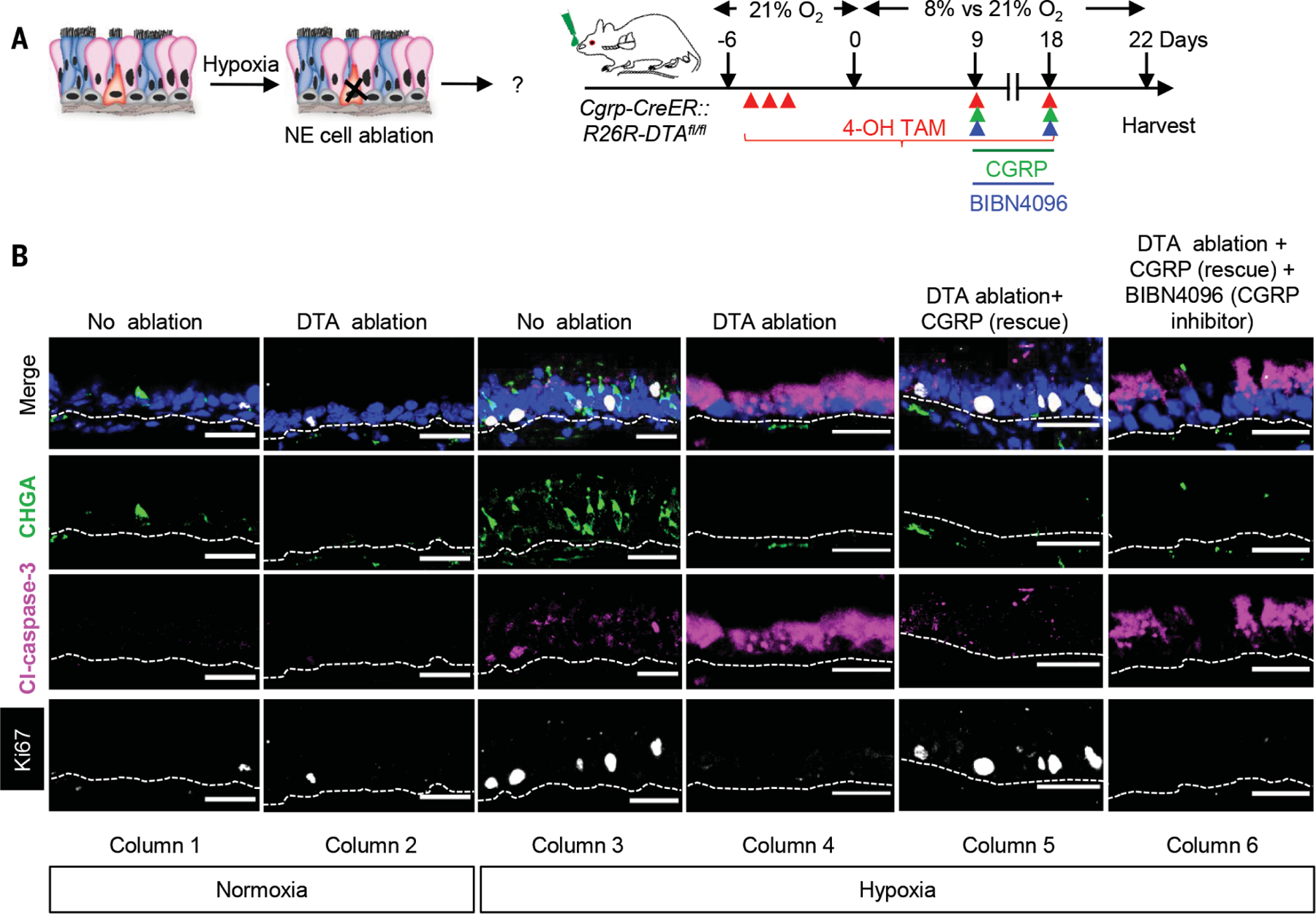
(A) Schematic representation of the protocol for assessing the functional consequences of NE cell ablation in the setting of hypoxia-induced injury. (B) Immunostaining for CHGA (green), Cleaved caspase-3 (magenta) and Ki67 (white) on normoxic and hypoxic tracheal epithelium with and without NE cell ablation and in combination with CGRP rescue and CGRP receptor inhibitor administration. (n = 5). ***p < 0.001, **p < 0.01, *p < 0.05; error bars, means ± standard deviation. Scale bars 30 μm.
Fig. 5. CGRP stimulates progenitor replication and prevents epithelial apoptosis.
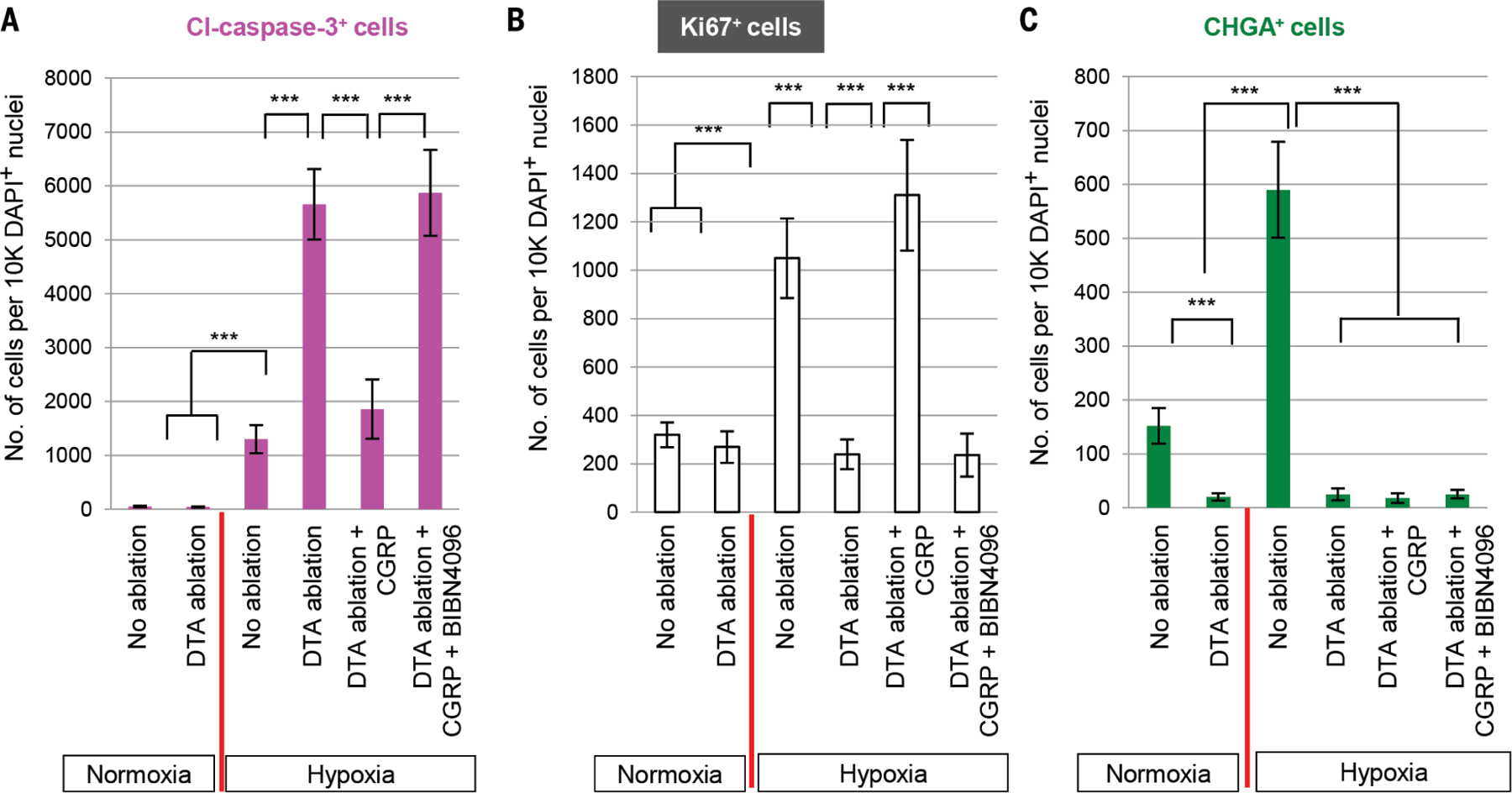
Quantification of (A) Cleaved caspase-3+ cells, (B) Ki67+, and (C) CHGA+ cells under normoxic or hypoxic conditions with and without NE cell ablation coupled to CGRP administration or blockade. Tissue samples from Fig. 4. ***p < 0.001; error bars, means ± SD.
Next, we tried to identify an NE cell–associated factor that promotes stem and progenitor cell hyperplasia and/or prevents apoptosis in the setting of hypoxia. We noted that CGRP levels increase after hypoxia (26) and that CGRP serves as a mitogen in rat lung alveolar epithelial cells (27) and murine submucosal gland progenitor cells (28). We then confirmed that epithelial CGRP expression in the trachea is found exclusively in Ascl1-positive solitary NE cells (fig. S13, A and B). At baseline, 54% of Ascl1-positive NE cells are CGRP immunoreactive, which sharply increases to 92% after hypoxia (fig. S13C). Nasal administration of CGRP to normoxic wild-type mice resulted in increased epithelial cell proliferation (fig. S13D). Costaining with cell type–specific markers and Ki67 identified replicating basal stem cells (CK5+Ki67+) and a smaller increase in replicating secretory cells (SCGB1A1+Ki67+) (fig. S13,D to F). Furthermore, we were able to block CGRP-stimulated proliferation using the CGRP-receptor inhibitor BIBN-4096 (29) (fig. S13, D to F).
Next, we intranasally administered CGRP peptide after NE cell ablation to assess whether the presence of CGRP could compensate for the loss of whole NE cells. CGRP restored epithelial proliferation to amounts normally observed after hypoxia and also prevented epithelial apoptosis (Fig. 4B, columns 3 to 5, and Fig. 5,A and B). Furthermore, proliferation was restored in both basal stem cells and secretory cells (fig. S14, A and B). Ciliated cells were the predominant cell type protected from apoptosis, although basal stem cells were also protected (fig. S11B). The CGRP-receptor inhibitor BIBN-4096 appropriately blocked the CGRP-mediated effects on cell type–specific apoptosis and proliferation (Fig. 4B, columns 5 and 6, and figs. S11A, columns 5 and 6, and S14). TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining confirmed these findings (fig. S15, A and B).
To determine whether CGRP sourced from cells other than solitary NE cells contributes to epithelial protection, we assessed which other cell types were ablated in our Cgrp-CreERT2:: Rosa26-DTA model. We noted that tracheal innervation was lost, so we used an Ascl1-CreERT2:: Rosa26-DTA model to eliminate solitary NE cells and spare CGRP-expressing neuronal populations (fig. S16). The results closely mirrored our findings with the Cgrp-CreERT2::Rosa26-DTA model (fig. S17). Additionally, the administration of CGRP inhibitor to hypoxic animals with intact solitary NE cells resulted in a significant increase in apoptosis and a drop in proliferation, consistent with a protective role for CGRP (fig. S17, B, C, columns 5 and 7, D, and E). Moreover, hypoxia led to increased epithelial expression of the CGRP receptors RAPM1 and CALCRL (fig. S18, A and B), suggesting increased CGRP signal reception.
We next sought to assess whether CGRP secreted by NEBs was acting as a long-range protective factor, originating in the intrapulmonary airway epithelium where NEBs occur, and acting directly or indirectly on the remote tracheal epithelium. Using Ascl1-tdTomato mice to lineage label preexisting NEBs, we confirmed that hypoxia triggers NEB hyperplasia, consistent with the previously reported finding that NEBs lacking PHDs become larger (20). Although there was no significant increase in the total number of NEBs (fig. S19, A to D), scattered NEBs did contain rare Ki67 cells (4 to 10 NEBs per left lobe of the lung) (fig. S19C). However, despite the efficient ablation of tracheal solitary NE cells in our model system, NEBs were unaffected (fig. S20). This suggests that solitary NE cells are the source of protective CGRP in our model system and that this peptide acts locally to protect adjacent epithelial cells from hypoxia-induced damage. However, these findings do not exclude a physiologic role for pools of CGRP produced elsewhere.
Finally, we assessed the effects of hypoxia on human airway epithelia grown from airway basal stem cells. Stabilizing HIFs by adding FG-4592 significantly increased the number of solitary NE cells (fig. S21). This system should prove useful in defining the role of solitary NE cells in diseases characterized by NE cell excess, paralleling the use of such models in dissecting the root mechanisms of cystic fibrosis.
Discussion
We have shown that hypoxia-induced murine solitary NE cells are necessary for repairing hypoxia-induced epithelial damage. The mechanism invokes the secretion of a protective paracrine signal. We speculate that the diffuse distribution of solitary NE cells throughout the human airway, in contrast to the discrete location of murine NEBs at airway bifurcations, forms the basis of a distributed tissue-wide protection system in which effective epithelial repair can be fostered throughout the length of the airway tree.
Oxygen therapy is used in the setting of respiratory failure in many diseases associated with NE cell excess, ranging from severe asthma to cystic fibrosis to COPD, but oxygen has also been associated with multiple forms of toxicity (30). If hypoxia-induced solitary NE cells are indeed protective in disease states, supportive oxygen therapy might result in unintended consequences by reducing the physiologic stimulus for generating protective NE cells. By contrast, in neuroendocrine hyperplasia of infancy (NEHI), where NE cell excess appears to be a primary pathology, supplemental oxygen might act as a primary therapy by triggering a reduction in pathologic NE cell numbers. Indeed, some patients with NEHI have a sudden improvement with oxygen administration that is uncharacteristic of related respiratory disorders of childhood, which display a slow and graded response to oxygen therapy (31).
With regards to normal physiology, our findings may help explain why NE cell numbers decrease after birth (17–19, 32) because oxygen levels increase after air breathing, as well as why high altitude is associated with increased numbers of NE cells (33). It may also explain why Notch loss-of-function experiments produce increased numbers of NE cells in the embryonic epithelium, whereas they do not in the air-breathing adult. Finally, it is of interest to note that sudden infant death syndrome (SIDS) has been associated with hypoxia, NE cell excess, and elevated serotonin levels (34, 35).
In this study, we have identified a conserved tissue-level response to a fundamental form of stress, oxygen deprivation. In this instance, sentinel stem cells detect hypoxia and produce a specific protective cell type that is needed to mitigate the effects of the hypoxia. Although we have identified CGRP as a protective airway solitary NE cell factor, NE cells can secrete a host of other neuropeptides and amines. Thus, we speculate that different forms of pulmonary injury might engender varied protective NE cell responses. It will be of great interest to assess whether other organ-specific stem cells more generally execute their own specific protective behaviors when triggered by hypoxia. It will also be important to determine whether the stem cells of other organs respond to local hypoxia by generating their own distinctive populations of protective NE cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Rajagopal lab and R. Chivukula for constructive criticism. We thank P. Chuang for providing Cgrp-CreER. We thank New England Donor Services (NEDS) for providing human airway samples. Finally, we thank E. Cutz for his constructive criticism and for reminding us of the history of NE cell biology.
Funding:
J.R. is supported by the New York Stem Cell Foundation, the National Institutes of Health–National Heart, Lung, and Blood Institute (RO1HL118185, and 1R01HL148351–01A1), and the Ludwig Cancer Institute at Harvard. J.X. is supported by R01CA193455. Electron microscopy was performed in the Microscopy Core of the MGH Program in Membrane Biology, which is partially supported by an Inflammatory Bowel Disease Grant (DK043351) and a Boston Area Diabetes and Endocrinology Research Center (BADERC) Award (DK057521).
Footnotes
Competing interests: V.K.M. is a paid adviser to Janssen Pharmaceutics and 5am Ventures and is a founder of and owns equity in Raze Therapeutics. V.K.M., I.H.J., and W.M.Z. are listed as inventors on patent application (WO2017027810A2) submitted by Massachusetts General Hospital on therapeutic uses of hypoxia for mitochondrial disease.
Data and materials availability: p63-creER mice are available from J. Xu under a materials transfer agreement with the Baylor College of Medicine. All data are available in the manuscript or the supplementary materials.
REFERENCES AND NOTES
- 1.Feyrter F, Virchows Arch. Pathol. Anat. Physiol 320, 551–563 (1951). [DOI] [PubMed] [Google Scholar]
- 2.Feyrter F, Virchows Arch. Pathol. Anat. Physiol. Med 325, 723–732 (1954). [DOI] [PubMed] [Google Scholar]
- 3.Cutz E, Semin. Diagn. Pathol 32, 420–437 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Cutz E, Yeger H, Pan J, Ito T, Curr. Respir. Med. Rev 4, 174–186 (2008). [Google Scholar]
- 5.Noguchi M, Sumiyama K, Morimoto M, Cell Rep 13, 2679–2686 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Kuo CS, Krasnow MA, Cell 163, 394–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cutz E, Jackson A, Respir. Physiol 115, 201–214 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Linnoila RI, Lab. Invest 86, 425–444 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Cutz E, Pan J, Yeger H, Domnik NJ, Fisher JT, Semin. Cell Dev. Biol 24, 40–50 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Branchfield K et al. , Science 351, 707–710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds SD et al. , Am. J. Physiol. Lung Cell. Mol. Physiol 278, L1256–L1263 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Gu X et al. , Am. J. Respir. Cell Mol. Biol 50, 637–646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouadah Y et al. , Cell 179, 403–416.e23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song H et al. , Proc. Natl. Acad. Sci. U.S.A 109, 17531–17536 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montoro DT et al. , Nature 560, 319–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson JK et al. , Cell Rep 12, 90–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keith IM, Will JA, Thorax 36, 767–773 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Track NS, Cutz E, Life Sci 30, 1553–1556 (1982). [DOI] [PubMed] [Google Scholar]
- 19.Moosavi H, Smith P, Heath D, Thorax 28, 729–741 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan J, Bishop T, Ratcliffe PJ, Yeger H, Cutz E, Hypoxia (Auckl.) 4, 69–80 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rock JR, Randell SH, Hogan BLM, Dis. Model. Mech 3, 545–556 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tata PR et al. , Nature 503, 218–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willam C, Nicholls LG, Ratcliffe PJ, Pugh CW, Maxwell PH, Adv. Enzyme Regul 44, 75–92 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Wu K et al. , Brain Res 1632, 19–26 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Dai Z-J et al. , J. Exp. Clin. Cancer Res 31, 28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Springall DR et al. , J. Pathol 155, 259–267 (1988). [DOI] [PubMed] [Google Scholar]
- 27.Kawanami Y et al. , Respir. Res 10, 8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie W et al. , J. Clin. Invest 121, 3144–3158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doods H et al. , Br. J. Pharmacol 129, 420–423 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tinits P, Ann. Emerg. Med 12, 321–328 (1983). [DOI] [PubMed] [Google Scholar]
- 31.Caimmi S et al. , Ital. J. Pediatr 42, 84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spindel ER et al. , J. Clin. Invest 80, 1172–1179 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gosney JR, Anat. Rec 236, 105–107, discussion 108–112 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Aita K et al. , Leg. Med 2, 134–142 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Siren PMA, Ups. J. Med. Sci 121, 199–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.