

Abstract
Considerable effort has been devoted to the development of first-row transition-metal catalysts containing redox-active imino-pyridine ligands that are capable of storing multiple reducing equivalents. This property allows abundant and inexpensive first-row transition metals, which favor sequential one-electron redox processes, to function as competent catalysts in the concerted two-electron reduction of substrates. Herein we report the syntheses and characterization of a series of iron complexes that contain both π-donating thiolate and π-accepting (α-imino)-N-heterocycle redox-active ligands, with progressively larger N-heterocycle rings (imidazole, pyridine, and quinoline). A cooperative interaction between these complementary redox-active ligands is shown to dictate the properties of these complexes. Unusually intense charge-transfer (CT) bands, and intraligand metrical parameters, reminiscent of a reduced (α-imino)-N-heterocycle ligand (L•−), initially suggested that the electron-donating thiolate had reduced the N-heterocycle. Sulfur K-edge X-ray absorption spectroscopic (XAS) data, however, provides evidence for direct communication, via backbonding, between the thiolate sulfur and the formally orthogonal (α-imino)-N-heterocycle ligand π*-orbitals. DFT calculations provide evidence for extensive delocalization of bonds over the sulfur, iron, and (α-imino)-N-heterocycle, and TD-DFT shows that the intense optical CT bands involve transitions between a mixed Fe/S donor, and (α-imino)-N-heterocycle π*-acceptor orbital. The energies and intensities of the optical and S K-edge pre-edge XAS transitions are shown to correlate with N-heterocycle ring size, as do the redox potentials. When the thiolate is replaced with a thioether, or when the low-spin S = 0 Fe(II) is replaced with a high-spin S = 3/2 Co(II), the N-heterocycle ligand metrical parameters and electronic structure do not change relative to the neutral L0 ligand. With respect to the development of future catalysts containing redox-active ligands, the energy cost of storing reducing equivalents is shown to be lowest when a quinoline, as opposed to imidazole or pyridine, is incorporated into the ligand backbone of the corresponding Fe complex.
Graphical Abstract
INTRODUCTION
Base-metal catalysis is an important area of research aimed at the design of abundant and inexpensive first-row transition-metal catalysts that are capable of operating under mild conditions.1–7 One limitation of first-row transition metals, however, is that they favor sequential one-electron, as opposed to two-electron (2e−), redox changes. Many important chemical transformations involve concerted 2e− chemistry, and Nature has developed methods of circumventing this limitation, by using redox-active ligands,1,8–13 such as porphyrins or thiolates, or cooperative interactions between metal ions incorporated into clusters or bimetallic systems.14–18 This is illustrated by the numerous examples of first-row transition metal-containing enzymes that promote complex, multielectron processes.14,15,17–22 For example, soluble methane monooxygenase (sMMO) and cytochrome P450 (Cmpd I) both generate high-valent iron oxidants through a concerted 2e− O—O bond-cleaving step, involving O2 as the terminal oxidant.17,23 With P450, proton-induced heterolytic cleavage of the Fe(III)-OOH (Cmpd 0) O—O bond,24 generates a highly reactive Fe(IV)═O (P450-I).25–28 The two electrons required for this step originate from two different sources: one from the Fe and the other from the conjugated porphyrin and axial cysteinate.29 Density functional theory (DFT) calculations indicate that a significant amount of unpaired spin density resides on the cysteinate sulfur,29 indicating that P450-I is best described as a (por/cysS)•+Fe(IV)═O moiety.11,30 The role of the axial cysteinate is not limited to its redox capabilities, however, as the sulfur has also been shown to increase the basicity of the oxo moiety.23 In addition, the cysteinate is proposed to facilitate protonation of the peroxo, and heterolytic, as opposed to homolytic, cleavage of the O—O bond, via what is referred to as the “push-effect” (Figure 1).24,31–33 The low-energy vO—O stretch (at 799 cm−1)34,35 of Cmpd 0 has been attributed to the influence of the axial cysteinate and low S = 1/2 spin-state of the Fe(III). The thiolate also influences the redox-active frontier orbitals via a highly covalent interaction with the appropriate symmetry metal d-orbitals. This cooperativity between cysteinate, Fe, and porphyrin π-orbitals is essential both for the 2e− O—O bond-cleaving step, and for the organic redox reservoir to delocalize the radical cation of Cmpd I.13,23,36
Figure 1.
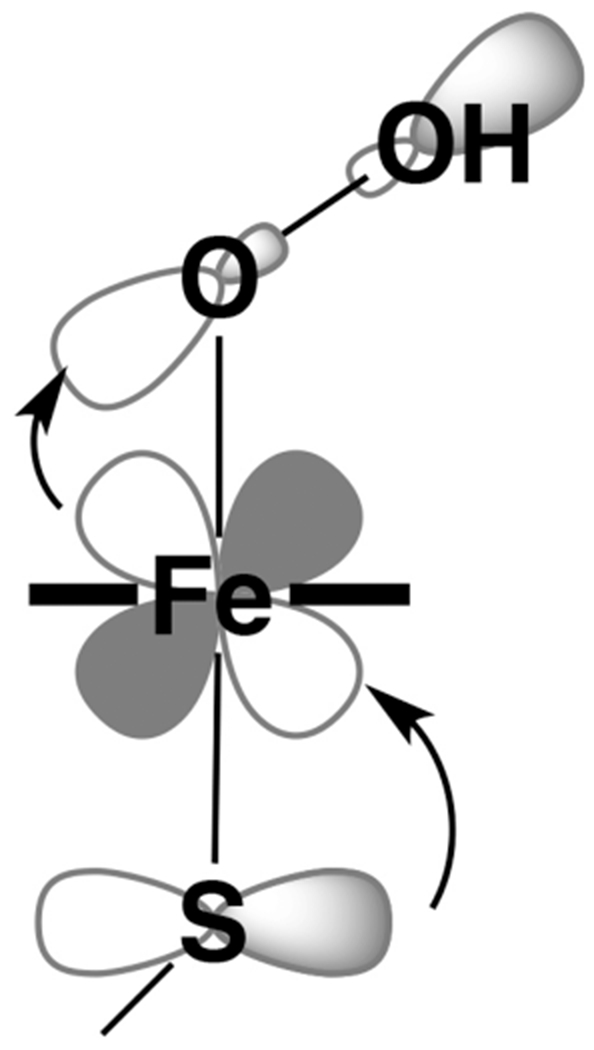
Thiolate-induced push mechanism of P450 O–O bond cleavage.
The involvement of a redox-active organic cofactor in promoting P450 chemistry has inspired considerable efforts toward the development of first-row transition-metal catalysts containing redox-active ligands.1,5,6,10,37–39 One such ligand moiety is the iminopyridine, which is capable of storing multiple reducing equivalents,1,5,6,8–10,37–40 thereby allowing complexes of first-row transition metals to function as competent catalysts in reactions involving two electrons. For example, using iminopyridine ligands, Chirik and Ritter have shown that base-metal catalyzed transformations involving alkene hydrogenation, C—C bond activation, and diene hydrosilylation proceed through concerted 2e− oxidative-addition, and reductive-elimination steps,4,41–43 reactivity that is typically associated with more costly second- and third-row transition metal catalysts.1 Distributing charge over one or more ligands has been shown to alleviate the electronic demands at the metal, and avoid high- and low-valent oxidation states, thus increasing catalytic turnover numbers, by minimizing catalyst degradation.43–46
Appreciable overlap between metal and ligand orbitals establishes the communication that is necessary for cooperative redox events.5,36,47,48 Determining how one can optimize orbital overlap in synthetic systems is imperative to achieving catalytic function attainable by Nature. To that end, we describe herein the synthesis and detailed spectroscopic, crystallographic, and electrochemical characterization of a family of novel (α-imino)-N-heterocycle complexes containing a thiolate in the coordination sphere. A synergistic interaction between the π-donating thiolate, the metal ion, and the π-accepting (α-imino)-N-heterocycle is shown to govern the properties of these complexes, and activate the (α-imino)-N-heterocycle ligand π*-framework via a cis-type push-effect, reminiscent of P450.31–33
RESULTS
In order to determine how to optimize orbital overlap in synthetic transition-metal complexes containing synergistic π-donor and π-acceptor redox-active ligands, and favor cooperative redox events, we have synthesized, and spectroscopically, crystallographically, and electrochemically characterized, a family of novel (α-imino)-N-heterocycle-containing transition-metal complexes (Figure 2) containing a thiolate in the coordination sphere, [FeII(tame-N2S(py)2)]22+ (1), [FeII(tame-N2S(quino)2)]22+ (2), [FeII(tame-N2S(MeIm)2)]22+ (3), [CoII(tame-N2S(MeIm)2)]22+ (4), as well as [FeII(tame-N2SBz(MeIm)2(MeCN))]2+ (5), which contains a thioether in the coordination sphere.
Figure 2.
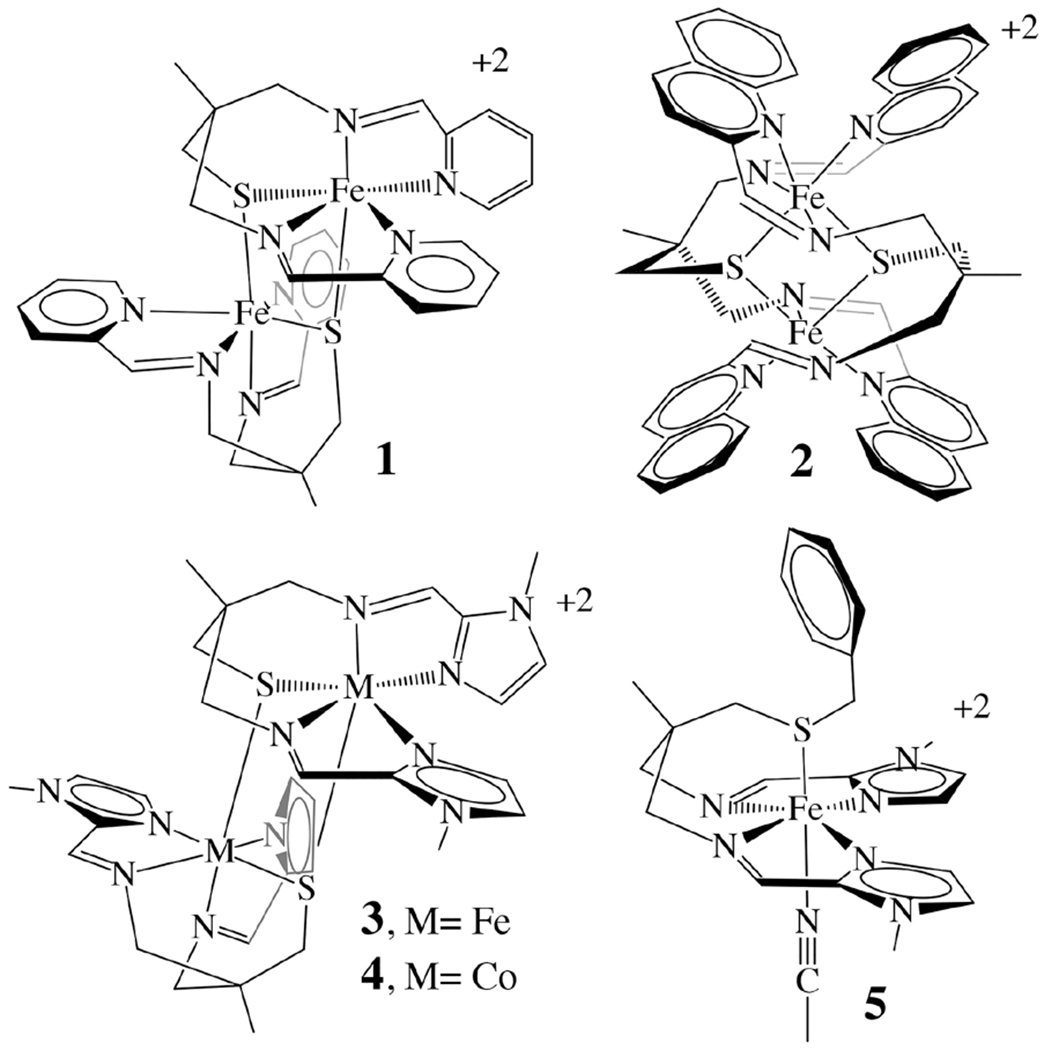
Complexes studied herein.
Synthesis and Characterization of [FeII(tame-N2S-(py)2)]22+ (1).
The synthesis of a ligand framework that incorporates both a thiolate and an (α-imino)-pyridine moiety was carried out according to the method outlined in Scheme 1. The tripodal thiolate/amine ligand precursor, tame-N2S, was synthesized via a previously reported method,49 with slight modifications involving the use of mesylate, as opposed to tosylate, leaving groups, and Boc-protection of the amines prior to deprotection of the benzyl-protected thiolate (Scheme S1). A Schiff-base condensation between the tripodal thiolate/amine (tame-N2S) and 2 equiv of 2-pyridine carboxaldehyde in the presence of FeCl2 was found to afford [FeII(tame-N2S-(py)2)]22+ (1) containing intact (α-imino)-pyridine moieties (Scheme 1). As shown in the ORTEP diagram of Figure 3, the expected five-coordinate thiolate [FeII(tame-N2S(py)2)]+ complex was found to form a dimeric structure, with overall C2 symmetry, and an Fe(μ-SR)2Fe core. The thiolates are symmetrically bridging, and derived from the chelating pentadentate ligand, as well as from that of the adjacent chelated metal ion. Two imine and two pyridine nitrogens complete the pseudo-octahedral coordination sphere. The planar bidentate (α-imino)-pyridine moieties of 1 are roughly orthogonal to one another, with a dihedral angle of 70.0° between N(2)C(12)C(13)N(3) and N(1)C(6)C(5)N(4) planes. The Fe—N bond lengths (Table 1) are in the range typically observed with low-spin (S = 0) ferrous complexes,50,51 and the Fe—S bond lengths are significantly shorter than that of a high-spin (S = 2) Fe(II) ion,52,53 but comparable to that of a low-spin (S = 1/2) Fe(III) ion.54–57
Scheme 1.
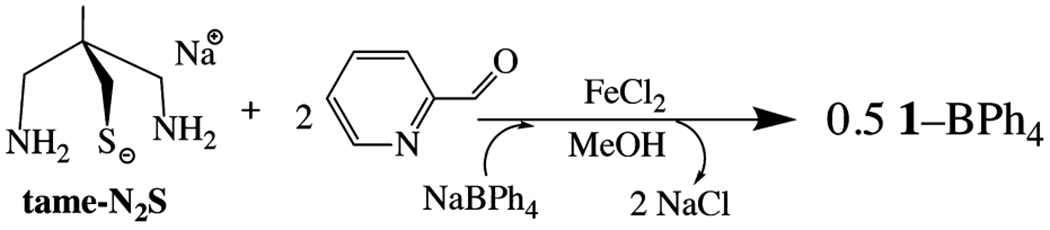
Figure 3.
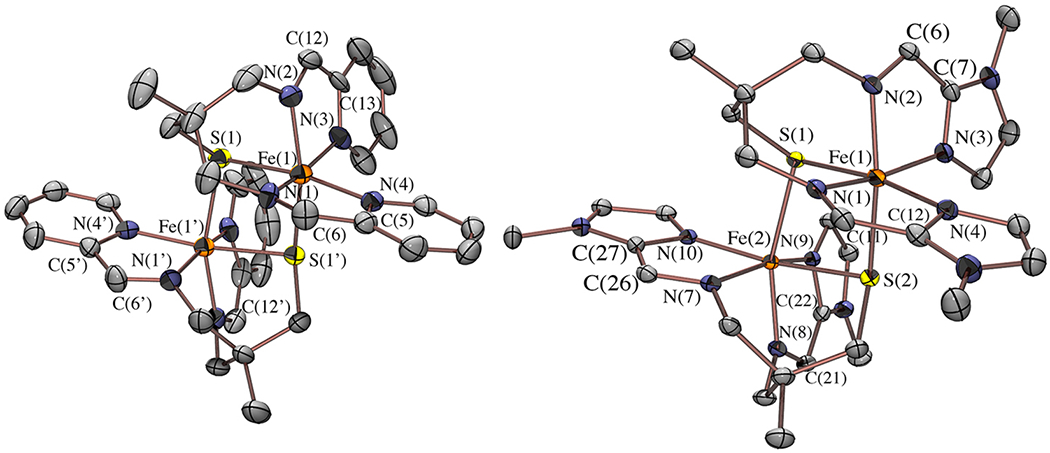
ORTEP of dicationic [FeII(tame-N2S(py)2)]22+ (1) and [FeII(tame-N2S(MeIm)2)]22+ (3) with counterions, solvents of crystallization, and hydrogen atoms omitted for clarity.
Table 1.
Selected M—L Bond Distances (Å) for 1–5
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| M(1)–S(1) | 2.284(1) | 2.2734(3) | 2.2883(8) | 2.3930(5) | 2.271(1) |
| M(1)–S(2) | 2.290(1)a | 2.2789(4)a | 2.3109(8) | 2.4653(5) | N/A |
| M(2)–S(1) | N/A | N/A | 2.2965(8) | 2.4616(5) | N/A |
| M(2)–S(2) | N/A | N/A | 2.2825(8) | 2.3912(5) | N/A |
| M(1)–N(1)imine | 1.908(4) | 1.949(1) | 1.917(2) | 2.162(2) | 1.948(4) |
| M(1)–N(2)imine | 1.916(4) | 1.949(1) | 1.923(2) | 2.145(2) | 1.941(4) |
| M(1)–N(3)NHet | 1.997(4) | 1.996(1) | 1.955(2) | 2.141(2) | 1.996(4) |
| M(1)–N(4)NHet | 1.984(4) | 1.987(1) | 1.966(2 | 2.120(2) | 1.981(4) |
| M(2)–N(7)imine | N/A | N/A | 1.933(2) | 2.123(2) | N/A |
| M(2)–N(8)imine | N/A | N/A | 1.925(2) | 2.2078(16) | N/A |
| M(2)–N(9)NHet | N/A | N/A | 1.970(2) | 2.121(2) | N/A |
| M(2)–N(10)NHet | N/A | N/A | 1.973(2) | 2.156(2) | N/A |
For this structure, S(2) and Fe(2) are related to S(1) and Fe(1) via a crystallographic inversion center, and are thus labeled S(1)′ or Fe(1′) in the supplemental crystallographic tables (Supporting Information), where S(1) and S(1)′ are derived from two different pentadentate ligands.
It is well-established that redox-active (α-imino)-pyridine ligands have characteristic intraligand bond lengths depending on their formal oxidation state.10 Three redox states are accessible, L0, L•−, and L2− (Scheme 2). Comparison of the intraligand bond lengths of 1 with those previously established for the neutral and reduced ligands indicates that the two (α-imino)-pyridine arms of 1 (Py(1) and Py(2) of Scheme 2) have Cim—Cipso distances close to that of a monoreduced open-shell L•− oxidation state (Table 2), and are thus decidedly “activated”.9,10 Strong reducing agents are typically required to access the L•− oxidation state.6,9,10,45,58
Scheme 2.
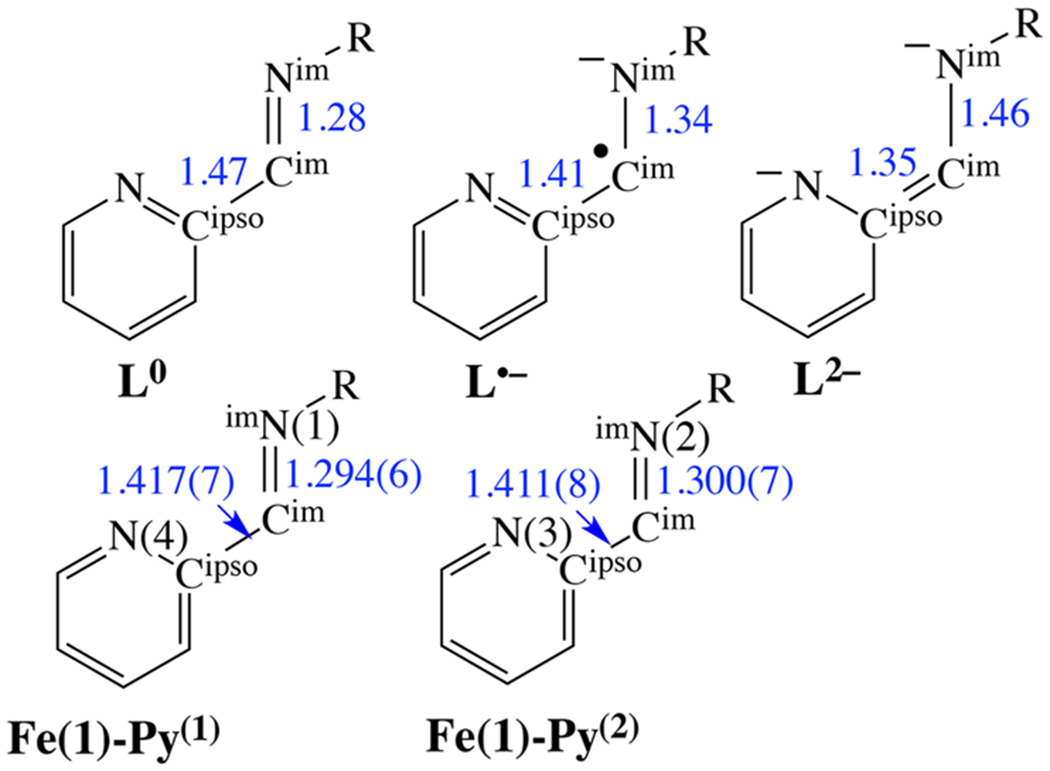
Comparison of the Metrical Parameters within the Structurally Distinct (α-Imino)-pyridine Fragments of 1, with Those Reported for the Three Different Redox States of This Ligand Fragment8
Table 2.
Selected Intraligand Bond Distances (Å) for 1–5 versus Typical Neutral and Monoreduced Radical Anion (α-Imino-N-heterocycle) Ligands10
| 1 | 2 | 3 | 4 | 5 | typical L0 | typical L•− | |
|---|---|---|---|---|---|---|---|
| Nim(1)–Cima | 1.294(6) | 1.297(4) | 1.281(5) | 1.275(2) | 1.281(5) | 1.28 | 1.34 |
| Nim(2)–Cimc | 1.300(7) | 1.297(3) | 1.287(5) | 1.274(2) | 1.287(5) | 1.28 | 1.34 |
| Cim(1)–Cipsoa | 1.417(7) | 1.427(4) | 1.432(6) | 1.447(3) | 1.436(6) | 1.47 | 1.41 |
| Cim(2)–Cipsoc | 1.411(8) | 1.427(4) | 1.436(6) | 1.449(3) | 1.432(6) | 1.47 | 1.41 |
| Nim(1)–Cimb | 1.294(6) | 1.301(3) | N/A | 1.283(2) | N/A | 1.28 | 1.34 |
| Nim(2)–Cimd | 1.300(7) | 1.294(3) | N/A | 1.272(2) | N/A | 1.28 | 1.34 |
| Cim(1)–Cipsob | 1.417(7) | 1.430(4) | N/A | 1.442(3) | N/A | 1.47 | 1.41 |
| Cim(2)–Cipsod | 1.411(8) | 1.423(4) | N/A | 1.451(3) | N/A | 1.47 | 1.41 |
Imine(1) bound to Fe(1).
Imine(1) bound to Fe(2).
Imine(2) bound to Fe(1).
Imine(2) bound to Fe(2).
However, π-backbonding has also been shown to induce, albeit only slight (vide infra), activation of the (α-imino)-pyridine moiety in low-spin Fe(II) complexes, previous examples of which have bond lengths much closer to that of a closed-shell L0.6,7 In the absence of an exogenous reductant, ligand activation of 1 (Table 2) could be due to an unusually large degree of π-backbonding. Given the redox-active nature of (α-imino)-pyridines, it is also possible that electronic rearrangement to an Fe(III)-(L)(L•−) configuration has occurred in 1 as a consequence of having a thiolate in the coordination sphere. Both the metal—ligand53,54,57,59–63 and intraligand metrical parameters would be consistent with this. In this scenario, the radical could be either localized, or delocalized, across both (α-imino)-pyridine moieties as previously described by Lu and Wieghardt.10 The 1H NMR spectrum of 1 (Figure 4) shows sharp peaks in the diamagnetic region, appearing to rule out the presence of a paramagnetic species in solution, and favor a closed-shell low-spin Fe(II)L0 electronic description. Ligand radicals can, however, be strongly coupled (J = −330 cm−1 to −1370 cm−1) antiferromagnetically to a paramagnetic metal ion,9,10 meaning that the observed diamagnetic properties of 1 do not necessarily preclude an Fe(III)—(L)(L•−) electronic description.
Figure 4.
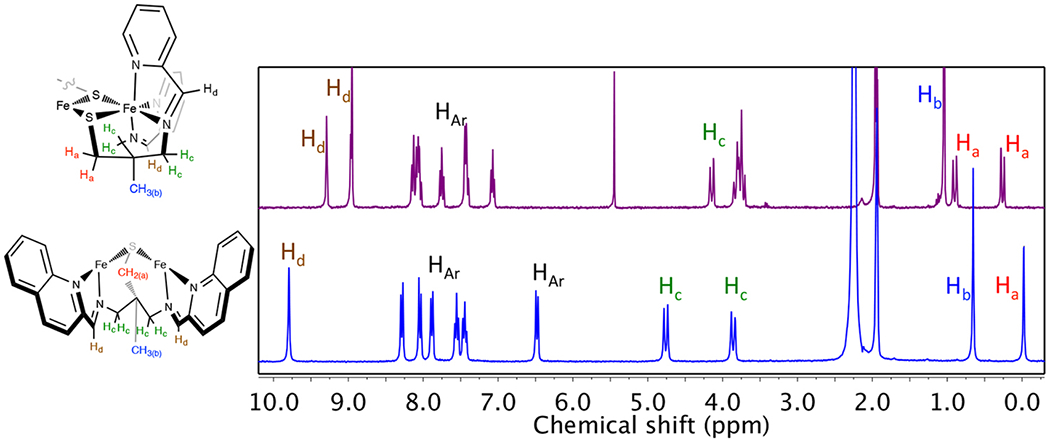
1H NMR of [FeII(tame-N2S(py)2)]22+ (1, top) versus [FeII(tame-N2S(quino)2)]22+ (2, bottom) in MeCN-d3 at 298 K. Only one-half of each dimer is shown. The other half is related by symmetry.
Synthesis and Characterization of [FeII(tame-N2S-(quino)2)]22+ (2) and [FeII(tame-N2S(MeIm)2)]22+ (3).
In order to determine how the relative energies of the redox-active ligand π*- and Fe d-orbitals influence the properties of the π-donor/π-acceptor complexes, we systematically varied the N-heterocycle ring size, from a pyridine ring in 1, to a quinoline in 2, and an imidazole in 3 (Figure 2). The corresponding iron complexes, [FeII(tame-N2S(quino)2)]22+ (2) and [FeII(tame-N2S(MeIm)2)]22+ (3), were synthesized in the same manner as the pyridine derivative (Scheme 1), using 2-quinolinecarboxaldehyde and 1-methyl-2-imidazolecarboxaldehyde in place of 2-pyridinecarboxaldehyde, respectively. These two derivatives form bimetallic iron complexes in the solid state containing two bridging thiolates, analogous to the pyridine derivative (Figures 3 and 5). The Fe—L bond lengths of 1–3 are similar (Table 1), with some minor differences, suggesting that all three complexes contain Fe in the same oxidation state and spin state. The bridging Fe—S distances are more inequivalent in the imidazole structure 3, relative to 1, suggesting that the bridge would more readily cleave (vide infra). In addition, all three complexes have similar intraligand imine C═N and Cimine—Cipso metrical parameters (Table 2 and Figures S1–S2), which are indicative of partially populated redox-active orbitals, as discussed previously. The tripodal tame-N2S backbone of C2h symmetric 2 has a unique bridging mode, where it spans two metal centers, as opposed to binding to a single metal ion as seen in structures 1 and 3. Reflecting these differences, the Fe—S bonds of 2 are noticeably shorter than those in the pyridine and imidazole complexes 1 and 3. The distinct bridging motif of the quinoline complex 2 persists in solution as indicated by the 1H NMR spectrum of Figure 4. While C2h symmetric [FeII(tame-N2S(quino)2)]22+ (2) displays a singlet corresponding to equivalent CH2 protons (Ha) adjacent to the sulfur, and a singlet corresponding to the imine proton (Hd), the less symmetric C2 [FeII(tame-N2S(py)2)]22+ (1) displays two Ha doublets, and two Hd singlets indicating that the imine and methylene protons adjacent to the sulfur are inequivalent, the latter of which are diastereotopic.
Figure 5.
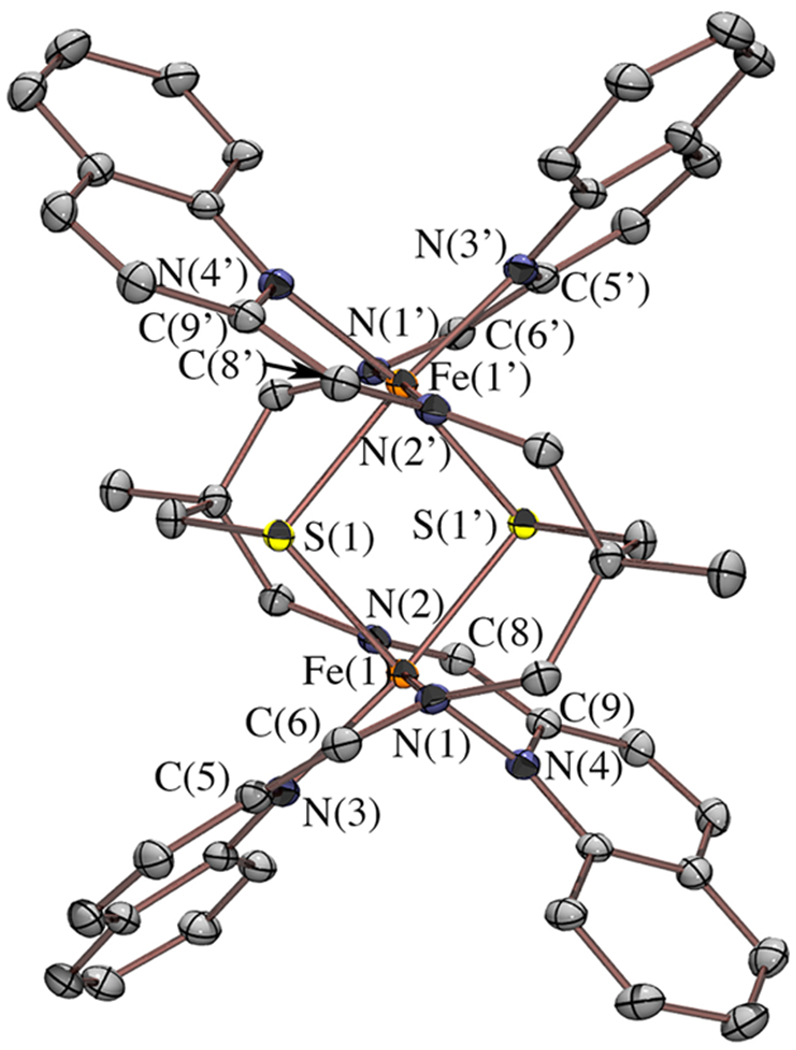
ORTEP of dicationic [FeII(tame-N2S(quino)2)]22+ (2) with counterions, solvents of crystallization, and hydrogen atoms omitted for clarity.
Dissociation of Dicationic 3 into Monocationic Monomers in Solution.
Despite their analogous bridging modes, in solution, the pyridine and imidazole complexes 1 and 3 display subtle spectroscopic differences. For example, in contrast to the diamagnetic 1H NMR spectrum of 1, that of 3 consists of mainly paramagnetic signals (Figure S3), suggesting that more than one species is present in solution. Given the more asymmetric Fe2S2 core (Table 1), and longer, weaker Fe(1)—S(2) bond in 3, it is likely that the thiolate bridge is readily cleaved in solution, leading to an equilibrium mixture of monomeric and dimeric species. Such a change in speciation could induce a change in spin-state, leading to the observed paramagnetic features in the 1H NMR. In order to distinguish between a paramagnetic monomer ⇋ diamagnetic dimer equilibrium, and the possibility that that 3 possesses a thermally accessible higher-spin-state, we collected high-resolution LTQ ion trap mass spectral data for 3 in MeCN solution. This data shows isotopomers differing by single, as opposed to half, mass units (Figure S4), consistent with a monocationic monomer. In contrast, using identical mass spectrometer settings, the LTQ ion trap mass spectroscopy data of 2 only contains isotopomers differing by half-mass units (Figure S5), consistent with a dicationic dimer. Complex 1 displays a mixture of single and half mass unit isotopomers (Figures S6 and S7), consistent with a mixture of monocationic and dicationic species. In order to determine the spin-state of the monocationic species, we measured the solution-state μeff for 3 over the temperature range 238–335 K in MeCN-d3 using the Evans method (Figure S8). The magnetic moment was shown to increase as a function of temperature (from μeff = 2.9 μB at 298 K, to μeff = 3.7 μB at 330 K). A magnetic moment of 3.7 μB suggests that the monomer is high-spin S = 2, as opposed to intermediate-spin S = 1, since the latter would have a maximum spin-only μeff of 2.83 μB, and 3.7 μB exceeds that number. Since a monomeric species would be entropically favored at higher temperatures, this T-dependent magnetic data supports the possibility that, in solution, a paramagnetic monomeric species exists in equilibrium with the crystallographically characterized diamagnetic dimer 3. Athough one cannot rule out the possibility that dimeric 3 possesses a thermally accessible higher-spin-state, the observation of both a dimer and monomer in high-resolution mass spectral data provides evidence for an equilibrium. Since a monomeric species would be entropically favored at higher temperatures, the increase in magnetic moment as the temperature is increased from 298 to 330 K would be consistent with a paramagnetic monomer. One can calculate the fraction of paramagnetic monomer species at 298 K under the conditions used for the Evans method measurements (5.02 μmol of Fe in 550 μL of MeCN-d3) using the ratio μeff2(expt)/μeff2(theoretical). If 100% of the solution consisted of an S = 2 monomer, then μeff(theoretical) would be 4.9 μB. A magnetic moment of 2.9 μB at 298 K would thus indicate that 35% of the iron is a paramagnetic monomer (1.76 μmol) in solution, and 65% a diamagnetic dimer (3.26 μmol of Fe, or 1.63 μmol of dimer). The effective magnetic moment squared (μeff2) was used for this calculation because the measurable macroscopic quantity χp is dependent on μeff2. One can then use this information to calculate the dimer/monomer equilibrium constant at 298 K, Keq298 K = (3.20 × 10−3)2/(2.96 × 10−3) = 3.46 × 10−3 M, as described in the Supporting Information. Assuming that both dimeric 3 and its monomeric derivative contribute to the absorbance at λmax = 619 nm, we can use Beer’s law (path length l = 1 cm) and the equilibrium concentrations of dimer and monomer at 298 K to determine the extinction coefficients for these two species. Using electronic absorption spectroscopy, equilibrium monomer and dimer concentrations, [M]eq and [D]eq, were determined at 298 K at two different initial [Fe]init concentrations, using two simultaneous equations as described in the Supporting Information. The absorbance response to concentration changes was nonlinear, providing evidence that more than one species contributes to the absorbance, the relative ratios of which are concentration-dependent, at the wavelength (λmax = 619 nm) monitored. The nonlinear Beer’s law behavior would be consistent with a dimer/monomer equilibrium, and is inconsistent with population of a higher-spin-state being responsible for the increase in magnetic moment (Figure S8). Using this method, we determined that solutions of 3 ([3]init = 0.120 and 0.240 mM) consist mainly of monomers at 298 K ([M]eq = 0.113 and 0.213 mM, respectively).
Finally, in order to probe the possibility of an open coordination site in the monomeric form of 3, preliminary reactivity studies were conducted with small molecules. In contrast to 1 and 2, which show no signs of reaction with Bu4NN3, the paramagnetic peaks shift in the 1H NMR spectrum of 3 upon the addition of Bu4NN3 (Figure S3). Complexes 1 and 2 take hours, or months, respectively, to react with O2, whereas 3 reacts with O2 within ~2 min (kobs = 4.9 × 10−3 s−1), as determined using electronic absorption spectroscopy (Figure S9) and 1H NMR. In addition, preliminary results indicate that 3 reacts with CO. The increased reactivity of 3 would be consistent with the presence of a coordinatively unsaturated, or solvent-bound, monomeric Fe(II) species in solution. A detailed investigation of the reactivity of 3 with a variety of small molecules will be the subject of a subsequent paper.
Electronic Absorption Spectroscopy.
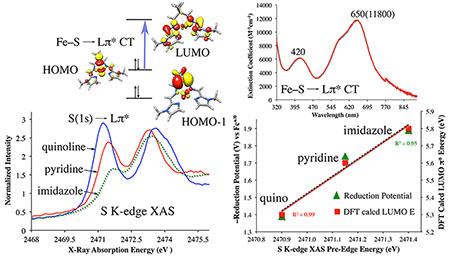
The electronic absorption spectra of 1–3 are richly featured, with multiple transitions in the visible region. Solutions of 1 and 2 are intensely turquoise in color, while 3 is an intense royal blue. Complexes 1 and 2 have three charge-transfer (CT) bands in the range 400–720 nm (Figure 6), whereas 3 has only a single resolved absorption band at λmax (ϵ, M−1 cm−1) = 619 (11 050) nm (Figure 7). The fact that solutions of 3 consist mainly of a paramagnetic (S = 2) monomeric species, whereas those of 1 and 2 consist mainly, or solely, of the crystallographically characterized dimer, explains why the solution spectrum of 3 is distinctly different from that of 1 or 2. The energy of the lowest-energy band correlates with the N-heterocycle ring size, shifting to lower energies as the ring size increases. This implies that transitions associated with these bands involve N-heterocycle π*-acceptor orbitals (vide infra). Intense (ε ≥ 104 M−1 cm−1), low-energy π-to-π* CT transitions (λmax ≤ 700 nm) are a hallmark of iron complexes bearing reduced, L•−, (α-imino)-pyridine ligands.10,40,64,65,10,40,64,65 In contrast, closed-shell L0 (α-imino)-pyridine Fe complexes have Fe → π* MLCT bands with extinction coefficients in the range 700–6000 M−1 cm−1 and λmax-values in the range 408–600 nm.6,66,67 This suggests that the N-heterocycle ligands of 1–3 are reduced. Thiolate ligands have been shown to significantly influence the intensity of M → π* MLCT bands,68,69 however, making it possible that the rather large extinction coefficients of 1–3 (Figure 6) reflect enhancement of the putative Fe → π* MLCT transitions via thiolate π-donation (vide infra).
Figure 6.
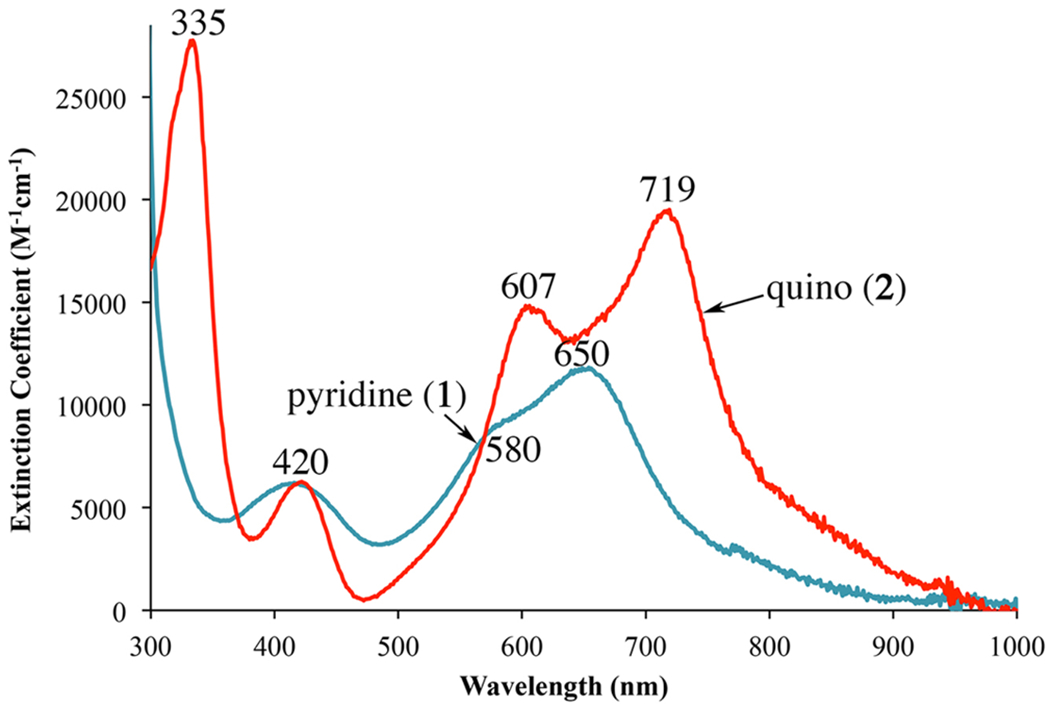
Electronic absorption spectra of 1 and 2 in MeCN at 298 K.
Figure 7.
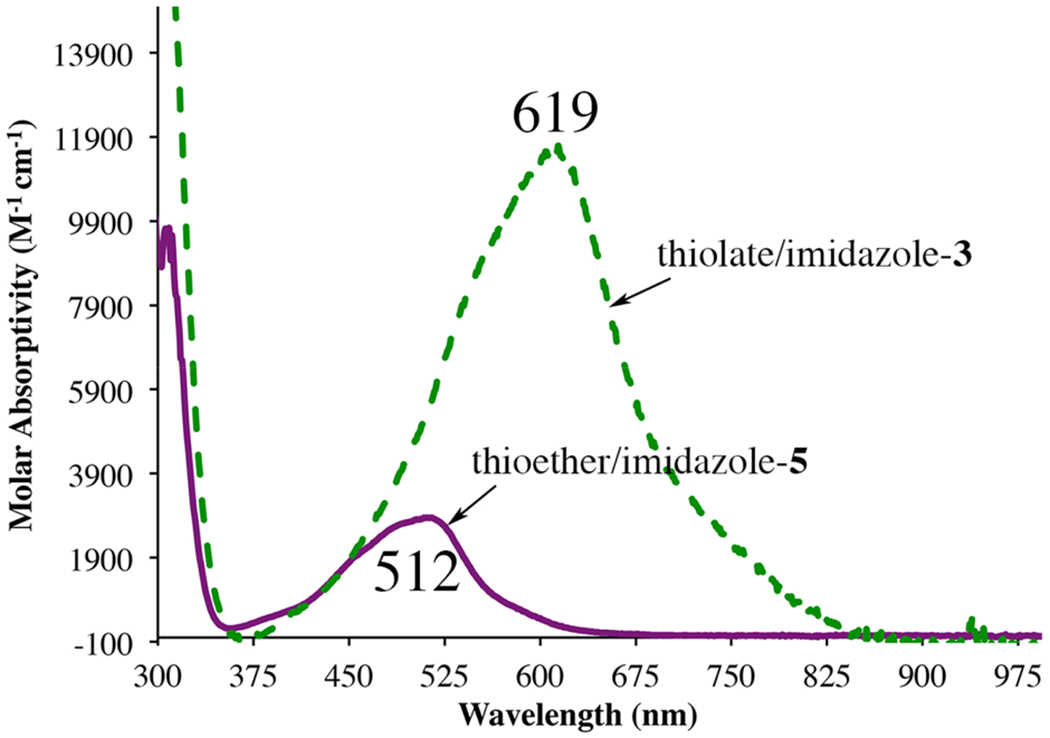
Comparison of the electronic absorption spectra of 3, which contains a thiolate in the coordination sphere, versus 5, which contains a thioether in place of the thiolate, illustrating the influence of the π-donating thiolate on spectral intensity.
Electrochemistry.
All three of the complexes described herein (1, 2, and 3) display multiple redox events within the MeCN solvent window. This includes at least two oxidation waves (Figure 8 and Figure S10), the lowest of which is reversible (or quasireversible) (Table 3). The potentials of these oxidation events are in the range typically observed for metal-centered oxidation of RS—Fe(II) complexes,55,70,71 but also directly correlate with N-heterocycle ring size (E1/2(2) > E1/2(1) > E1/2(3)). The latter reflects the metal orbital stabilization that occurs as a result of overlap with π-acceptor N-heterocycle orbitals, the extent of which increases as the energy match optimizes with increasing N-heterocycle ring size. The observation of two oxidation waves (tentatively assigned as Fe(II)Fe(II) → Fe(II)Fe(III) → Fe(III)Fe(III)) with a more than 500 mV separation would be consistent with the close proximity of two metal ions in these bimetallic structures, and the expectation that electrochemical events would not be independent of one another. The irreversibility of the more anodic wave suggests that the bimetallic species dissociate upon oxidation. Reduction waves (Figure S11) are also observed for all three complexes in the range −1.39 V to −1.89 V (vs Fc0/+, Table 3), with potentials that correlate with N-heterocycle ring size (Epc(2) < Epc(1) < Epc(3)). Although these potentials are ~700 mV more negative than most Fe(II) complexes bearing redox-active iminopyridine ligands, these redox events can be attributed to N-heterocycle ligand-centered reduction (vide infra),10 and likely reflect the influence of the electron-donating thiolate. The relative energies of these reduction potentials would be consistent with the expected drop in energy of the lowest unoccupied molecular orbital (LUMO) as the extent of conjugation within the π-system increases. Consistent with the 1H NMR, magnetic, and electronic absorption data, the imidazole complex 3 reproducibly displays an additional oxidation wave (Epa(2)) at +152 mV versus Fc0/+ (Table 3 and Figure S10), attributable to the monomer that is proposed to exist in equilibrium with the crystallized dimer.
Figure 8.
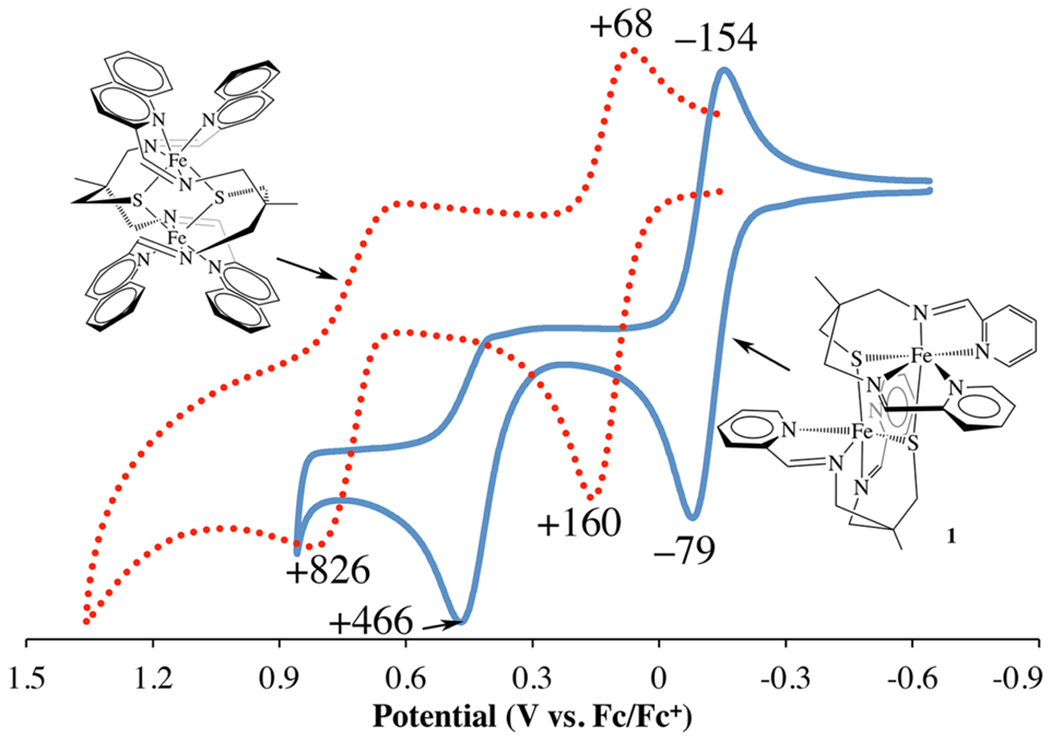
Cyclic voltamagram of 1 and 2 in MeCN at 298 K showing reversible and irreversible oxidation waves. Potentials shown are in mV vs Fc0/+.
Table 3.
Redox Potentials for 1–3 in MeCN versus Fc/Fc+
| 1 | 2 | 3 | |
|---|---|---|---|
| E1/2(1) | −0.116 V | +0.112 V | −0.489 V |
| Epa(2) | N/A | N/A | +0.152 V |
| Epa | +0.466 V | +0.826 V | +0.722 V |
| Epc | −1.74 V | −1.39 V | −1.89 V |
Sulfur K-Edge X-ray Absorption Spectroscopy.
Sulfur K-edge X-ray absorption spectroscopy (XAS) is well-established as a sensitive measure of metal—sulfur covalency, as well as ligand radical character.72–78 Excitation of a sulfur 1s electron to low-lying unoccupied orbitals probes both the energy of the acceptor orbital, and the extent of mixing between this orbital and the thiolate sulfur, as reflected in peak intensity. The acceptor orbital may be either a metal 3d (in the case of first-row transition metals), or an unoccupied ligand-based orbital with S 3p character. The S K-edge XAS spectra of solid-state samples of 1–3 are shown in Figure 9 (top), and Figure S12 (full range). These spectra exhibit two intense pre-edge peaks: a prominent lower-energy pre-edge feature at ~2471 eV, and a higher-energy feature at ~2473 eV (Table S1). The lowest-energy peak systematically shifts in energy and intensity in a manner that correlates with the N-heterocycle ring size (vide infra), analogous to the experimentally-determined reduction potentials (Table 3).
Figure 9.
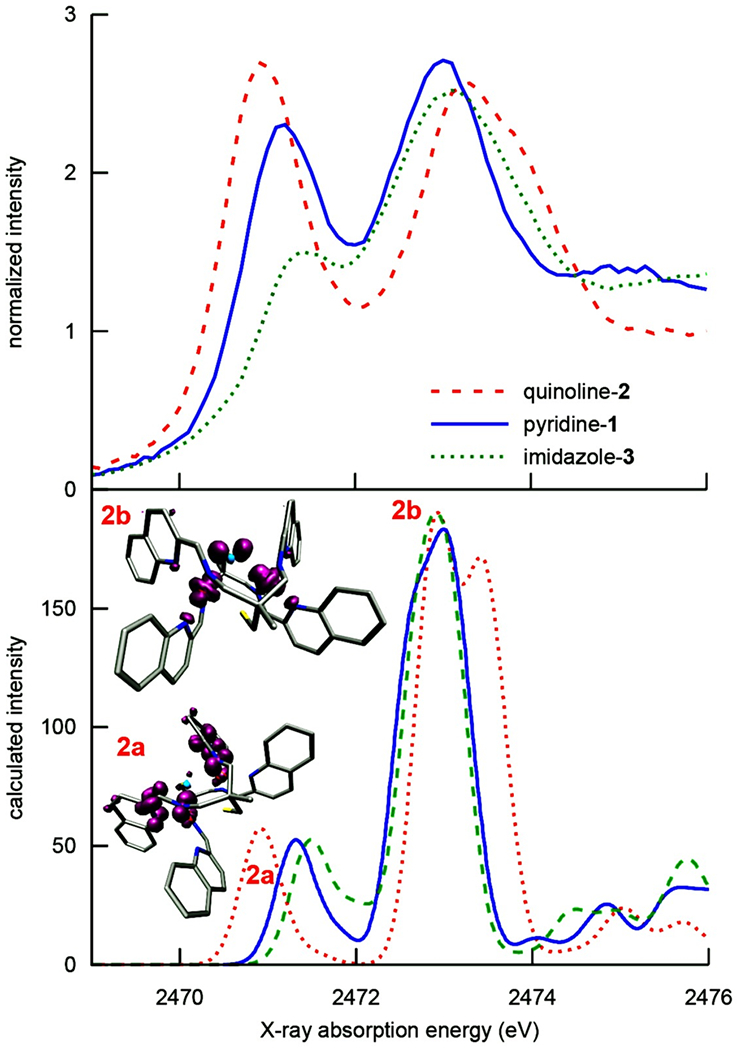
Experimental (top) and TD-DFT-calculated (bottom) S K-edge XAS pre-edge features associated with 1–3. Representative transition difference densities for quinoline compound peaks (denoted 2a and 2b) are shown at an isovalue of 0.0015a03, with regions of increased electron density (acceptors) shown in purple. The calculated spectrum was shifted by 40.2 eV to align theory with experiment.
The relative energies of the higher-energy pre-edge feature (Figure 9, top right) do not display the same trends seen with the lower-energy pre-edge peak. For example, the peak for the quinoline complex 2 is highest, as opposed to lowest, in energy (Table S1). With a low-spin d6 Fe(II) complex, the metal-centered t2g d-orbitals are completely filled, and thus, transitions would have to involve the unoccupied Fe eg* d-orbitals as the acceptor orbitals. The highest-energy pre-edge feature of 2 (Table S1) reflects the shorter Fe—S bond lengths relative to 1 and 3 (Table 1), which should serve to increase both the covalency of this bond, and the ionization potential of the S 1s electrons. We note that there should also be S(1s) → C—S σ* and S(1s) → S 4p transitions to higher energies, possibly near, or mixed in with, the higher-energy pre-edge feature of Figure 9. These interpretations are explored in more detail using the density functional theory (DFT) calculations described below.
Density Functional Theory Calculations.
Density functional theory (DFT) calculations were performed in order to generate a molecular orbital picture of complexes 1–3, and provide a clear understanding of the electronic structure of each. Geometry optimizations were initiated from crystallographic coordinates, using both closed-shell and broken-symmetry singlet configurations. In all cases the lowest-energy closed-shell solution, with neutral ligands and low-spin S = 0 Fe(II) ions, provided the closest reproduction of metrical parameters. In particular, the unusual intraligand distances associated with the coordinated (α-imino)-N-heterocycles are calculated to within 0.005 Å (Table S2), suggesting that the electronic effects responsible for these structural features are successfully captured by the calculations. A qualitative MO diagram for the crystallized dimeric form of 3 is shown in Figure 10, and for 1 in Figure S16; a quantitative comparison is included in the Discussion section. Consistent with the diamagnetic 1H NMR of 1 (Figure 4), all of the t2g-type orbitals in the DFT-optimized structures are doubly occupied (Figure 10 and Figure S16). The sulfur character of both the dxy, and dxz (or dyz) orbitals (Table S3) and d-orbital character in the bonding S p-orbital (Figure 10) indicate that the Fe d-orbitals have substantial covalent mixing with the bridging thiolate sulfur orbitals, as is typical for Fe-thiolate complexes.54 Consistent with the conjugated portion of the ligand scaffold being π-acidic in nature, the LUMO orbitals are found to be the (α-imino)-N-heterocycle π*, with the Fe eg* orbitals at higher energy (Figure 10). Analogous electronic structures were determined for 1 (Figure S16) and 2 (Table S3). Inspection of the calculated orbital compositions in Table S3 reveals that the quinoline complex 2 has the most substantial covalent mixing between the ligand π* and thiolate orbitals, and the most Fe character in the ligand π*-orbitals. The extent of covalent S(3p)/Lπ* mixing, and % Fe character in Lπ*, is less with 1, relative to 2, and smallest with 3 (Table S3). Interestingly, while the sulfur contribution to the t2g orbitals is largest for 2, sulfur character in the eg* orbitals is largest for 3. This highlights the π-donating nature of the thiolate ligands, which dominates when M/π-acceptor interactions are attenuated with the smaller ring system.
Figure 10.
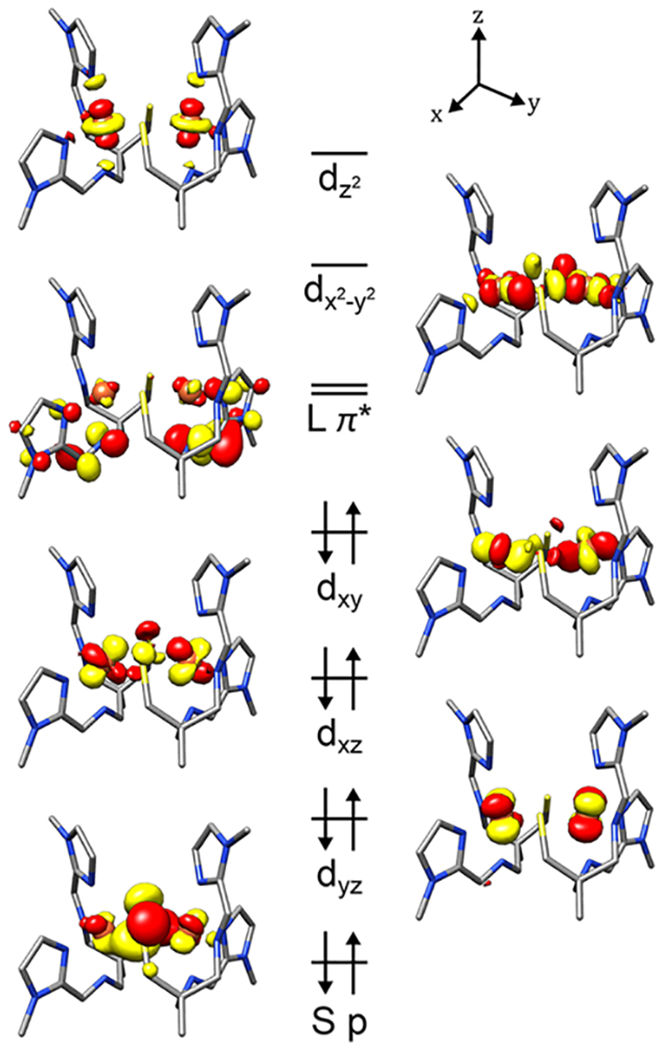
Qualitative molecular orbital diagram for imidazole-3. The the t2g orbitals are the highest energy doubly-occupied orbitals, the ligand π*-orbitals are the LUMOs, and the eg* orbitals lie to higher energy. Molecular orbitals are shown at an isovalue of 0.05 a0−3/2.
While the relative energies of the molecular orbitals [M(t2g) < L(π*) < M(eg*)] are not surprising in light of the abundant literature precedent,3,4,6,41,51 the apparent mixing of the thiolate sulfur orbitals into the redox-active (α-imino)-N-heterocycle ligand orbitals (Table S3) is highly unusual. The intensities of the optical (Figures 6 and 7) and S K-edge pre-edge XAS (Figure 9) transitions further suggest that there is substantial communication between the thiolate sulfur 3p and (α-imino)-N-heterocycle ligand π*-orbitals. On the basis of the orbital compositions in Table S3, there is extensive delocalization of the dx2–y2 over both the sulfur (17.8–21.1%) and Lπ* (12.4–22.7%) orbitals, and this appears to be the most involved of the Fe 3d orbitals in facilitating the orbital mixing that engenders this spectral intensity. The nature of the underlying electronic transitions involved in both types of absorption spectroscopy was investigated using time-dependent DFT (TD-DFT) calculations.
The putative charge-transfer nature of the transitions involved in the electronic absorption spectra prompted the use of the range-separated hybrid density functional CAM-B3LYP for these calculations. As shown in Figure 11, the TD-DFT-calculated electronic absorption spectra for 1 and 2 both have intense transitions in the visible region. The relative energies and intensities are in good agreement, although the absolute transition energies are overestimated by approximately 200 nm when compared to the experimental data (Figure 6). This is a well-known shortcoming of TD-DFT.79 The nature of these transitions can be examined with reasonable fidelity, however, via the transition difference densities (Figure 11). For both complexes, the donors and acceptors associated with the intense visible region CT transitions are shown to be mixed Fe—S, and (α-imino)-N-heterocycle π* in nature, respectively (Figure 11). The iron 3d-orbitals thus appear to mediate charge transfer from the thiolate 3p to the formally orthogonal (α-imino)-N-heterocycle π* LUMO.
Figure 11.
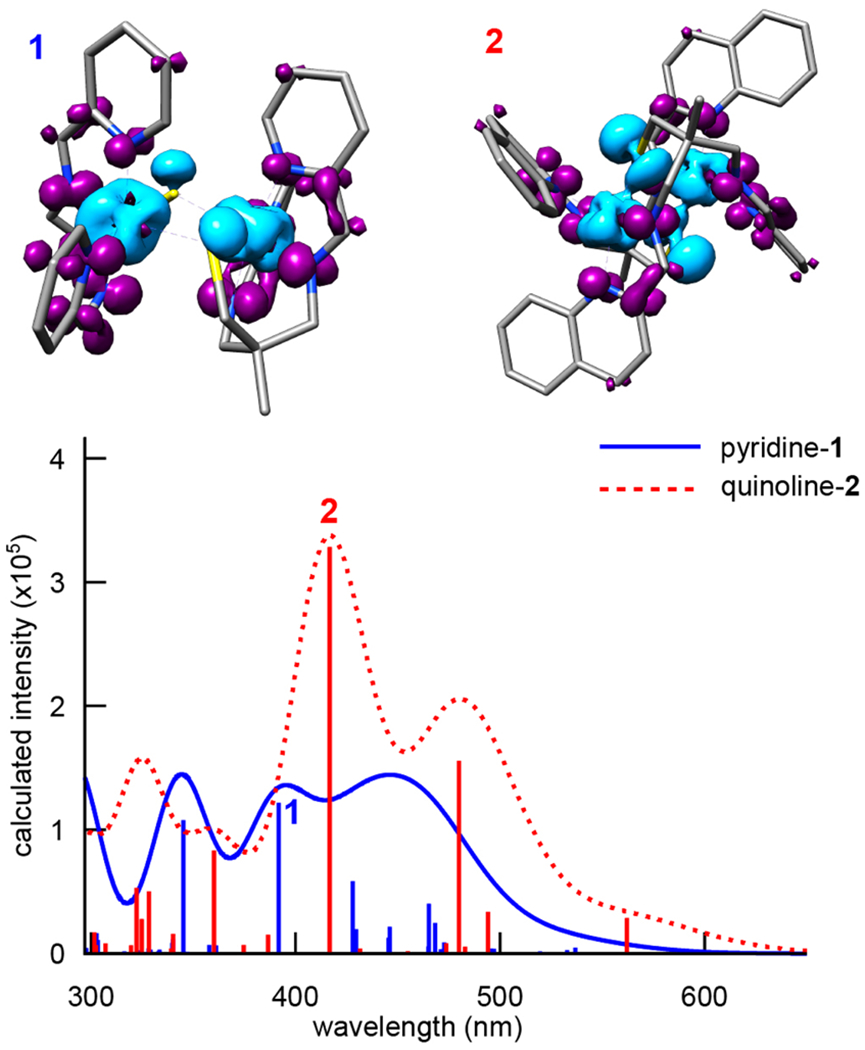
TD-DFT-calculated electronic absorption spectra of 1 and 2. Transition difference densities for the most intense transitions are shown at an isovalue of 0.0015 a0−3, with areas of decreased electron density (donors) shown in teal and increased electron density (acceptors) shown in purple.
As discussed previously, S K-edge XAS provides13,73 a direct probe of the thiolate bonding in these complexes. The TD-DFT-calculated pre-edge features of the S K-edge XAS spectra of 1–3 are shown at the bottom of Figure 9, and employed the CAM-B3LYP functional. Comparison with the experimental S K-edge pre-edge XAS spectra (Figure 9, top) indicates that the energetic trends observed for both features are accurately reproduced in the calculated spectra. Examination of the transition difference densities (Figures S13–S15) establishes that the lower-energy S K-edge pre-edge XAS feature indeed corresponds to a sulfur 1s → (α-imino)-N-heterocycle π* LUMO transition, as proposed above. Inspection of the transition difference densities for the transitions underlying the higher-energy pre-edge feature, however, reveals a combination of Fe eg*-type and C—S σ*-acceptor orbitals, with significant sulfur np character (Figure 9, bottom, Figures S11–S13). The experimental intensities of the first pre-edge feature (Figure 9, top) provide evidence for an increase in sulfur 3p/ligand π* LUMO mixing as the N-heterocycle ring size increases: imidazole (3) < pyridine (1) < quinoline (2). Thiolate π-donation into the (α-imino)-N-heterocycle π* LUMO is likely mediated via backbonding from the filled Fe t2g-type orbitals. It is noted that the TD-DFT-calculated intensities of the S K-edge XAS pre-edge feature do not capture the experimental trend, although this shortcoming can be rationalized by the difficulty associated with calculating both CT transitions and the onset of a continuum, using TD-DFT.79 Both of these factors could alter the relative intensities of the lower- and higher-energy pre-edge features. The agreement between the TD-DFT-calculated electronic absorption intensities and the experimental intensities, as well as the trend in energies as a function of ring size, however, suggests that the electronic effects influencing the CT intensity may in fact be successfully captured by the calculations, and that the discrepancies in the S K-edge XAS spectra could simply be due to problems modeling the onset of the continuum.
57Fe Mössbauer Spectroscopy.
The electronic environment of the iron center of 1–3 was also investigated using 57Fe Mössbauer spectroscopy. The zero-field 57Fe Mössbauer spectra of 2 and 3 are shown in Figure 12, and that of 1 is shown in Figure S15. The compounds exhibit well-resolved quadrupole doublets with isomer shift parameters, δ, in the range 0.36–0.41 mm/s, and relatively small quadrupole splitting parameters, ΔEQ, in the range 0.38–0.87 mm/s (Table 4). The isomer shift parameter, δ, depends on the charge density at the Mössbauer nucleus, reflects the oxidation state and spin-state, and is well below the range established for five- and six-coordinate high-spin Fe(II) sites (>1 mm/s).80 Unfortunately, these values are consistent not only with low-spin Fe(II), but also with Fe(III), low-spin or high-spin. Fortunately, the latter cases, i.e., the presence of oxidized Fe(III), can be ruled out for the following reasons: (i) The compounds exhibit a diamagnetic ground state which could not be explained by any kind of antiferromagnetic spin coupling between high-spin S = 5/2 Fe(III) and a ligand radical with S′ = 1/2. (ii) Low-spin Fe(III) can be excluded because the corresponding 3d t2g5 configuration would exhibit substantial charge asymmetry in the 3d shell, which would give rise to substantial valence contribution to the electric field gradient (efg), and a corresponding large quadrupole splitting parameter, |ΔEQ| > 1–3 mm/s,80 which is not experimentally observed for compounds 1–3. Hence, the Mössbauer spectra corroborate the presence of low-spin Fe(II) for compounds 1–3. This is also supported by the reasonable match between experimental and calculated Mössbauer parameters obtained from the orbital analysis of the DFT results given above.
Figure 12.
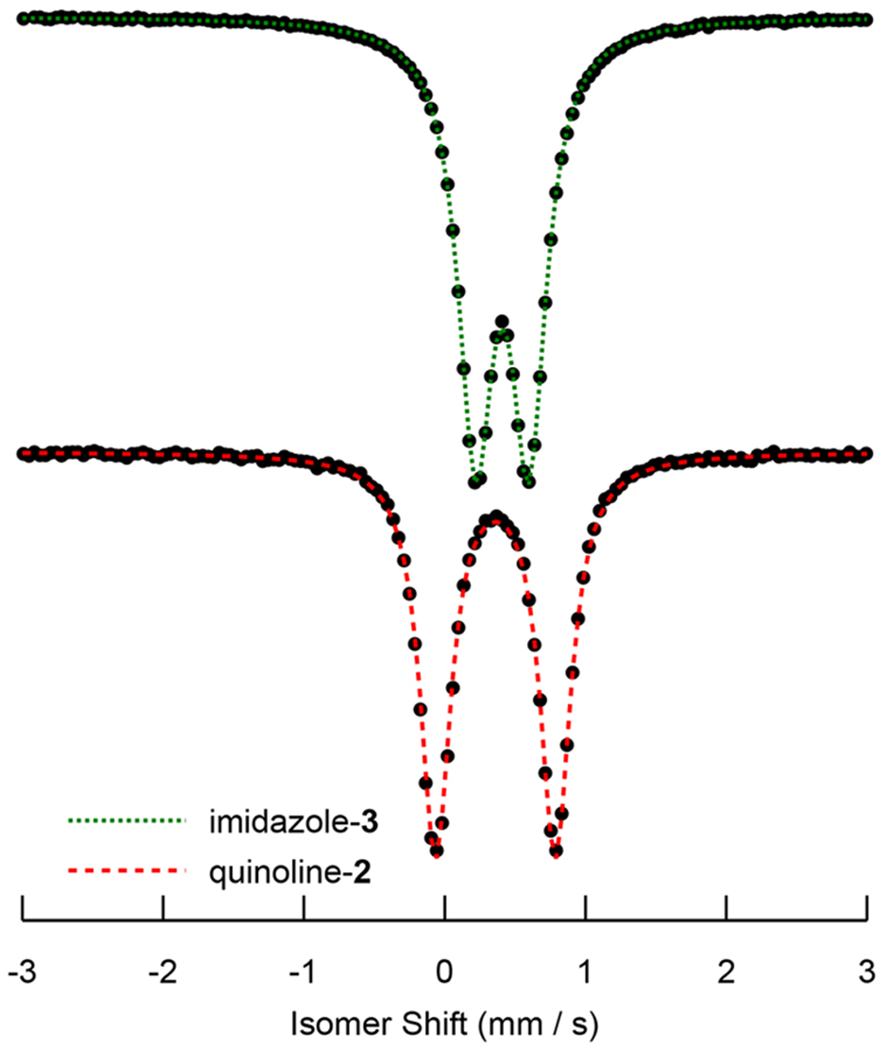
Zero-field 57Fe Mossbauer spectra of 2 and 3.
Table 4.
Comparison of the Experimental and Calculated Mossbauer Parameters for 1–3
| N-heterocycle | δ (mm/s) | |ΔEQ| (mm/s) | BP86 calcd δ (mm/s) | BP86 calcd ΔEQ (mm/s) | |
|---|---|---|---|---|---|
| 1 | pyridine | 0.36 | 0.87 | 0.22 | 1.07 |
| 2 | quinoline | 0.36 | 0.85 | 0.22 | −1.09 |
| 3 | imidazole | 0.41 | 0.38 | 0.39 | 0.82 |
Isomer shifts theoretically correlate with the average Fe—L bond length.81 Iron—ligand (Fe—L) bond lengths in 1–3 are modulated by the π-acceptor (α-imino)-N-heterocycle moieties, such that a decrease in Fe → L π-back-donation should lead to a more positive δ parameter. The isomer shift δ parameters of Table 4 therefore indicate that Fe → L π-back-donation is smallest with the imidazole complex 3 (δ = 0.41 mm/s), and comparable in the pyridine and quinoline complexes 1 and 2 (both δ = 0.36 mm/s). While the former conclusion is in good agreement with the S K-edge XAS spectra and TD-DFT calculations (Figure 9, Table S3), one might have anticipated that the Mössbauer parameters of 2 would be indicative of more π-backbonding than 1. However, just as π-accepting ligands can depopulate 3d orbitals, π-donating ligands such as thiolates can increase the effective 3d orbital population. Comparison of the metrical parameters of 1 and 2 indeed reveals that the Fe—S bonds in 2 are shorter by 0.01 Å, indicative of increased S → Fe π-donation in 2. Thus, the π-donating thiolate ligands compensate for the increased π-backbonding with the π-acceptor imino-N-heterocycle π*-orbitals in 2 by forming more covalent Fe—SR bonds with more generous S → Fe π-donation, in agreement with the DFT-calculated dyz-, dxz-, and L π*-orbital compositions (Table S3). This coincidently results in identical isomer shift parameters for 2 and 1. This phenomenon of offsetting perturbations to δ has been previously investigated theoretically in some detail for other complexes bearing covalent π-bonding ligands.82,83
While δ parameters are sensitive to electron density at the 57Fe nucleus, the quadrupole splitting parameter, ΔEQ, provides a measure of the asymmetry of the efg at the nucleus. In the limit of crystal field theory, an Oh S = 0 d6 Fe(II) ion has a vanishing valence contribution to the efg that should result in the coalescence of the two quadrupole doublet peaks. However, the lowered symmetry, π-donating thiolate, and π-accepting (α-imino)-N-heterocycle of 1–3 should contribute finite covalence and a lattice contribution to the efg, in agreement with the experimental data. As with the isomer shift parameter, the quadrupole splitting parameter, ΔEQ, of 3 is smaller than that of 1 and 2 (Table 4). This suggests that there is less asymmetry in the covalent bonding of 3, and, being isostructural to 1, implies that the bonding in 3 may be less covalent overall. Finally, the DFT-calculated Mössbauer parameters shown in Table 4 are in excellent agreement with the experimentally observed trends, reinforcing the accuracy of the calculated electronic structures. In summary, the 57Fe Mössbauer data and accompanying DFT-calculated Mössbauer parameters suggest that the extent of π-back-donation into the π-accepting ligands correlates with the size of the N-heterocycle, as proposed above. These data also provide evidence for a compensatory contraction of the Fe—S bonds in 2 to enable stronger covalent π-bonding to the (α-imino)quinoline ligand.
Examining the Influence of the Thiolate Ligand and Metal Ion.
The electronic structures of 1–3 developed thus far highlight the importance of the thiolate and Fe valence orbitals in tuning back-donation to the (α-imino)-N-heterocycle ligand π* LUMO and activating the ligand. To systematically explore these effects, derivatives of imidazole 3 were synthesized in which either the metal ion or the thiolate was varied. The cobalt imidazole derivative, [CoII(tame-N2S(MeIm)2)]22+ (4), was synthesized using a method analogous to that used to synthesize 3. As shown in Figure 13, 4 has a similar C2 symmetric structure in the solid state, with Co—L bond lengths consistent with high-spin Co(II) (Table 1). The solution-state magnetic moment of 4 (μeff = 3.98 μB) is consistent with an S = 3/2 spin-state.
Figure 13.
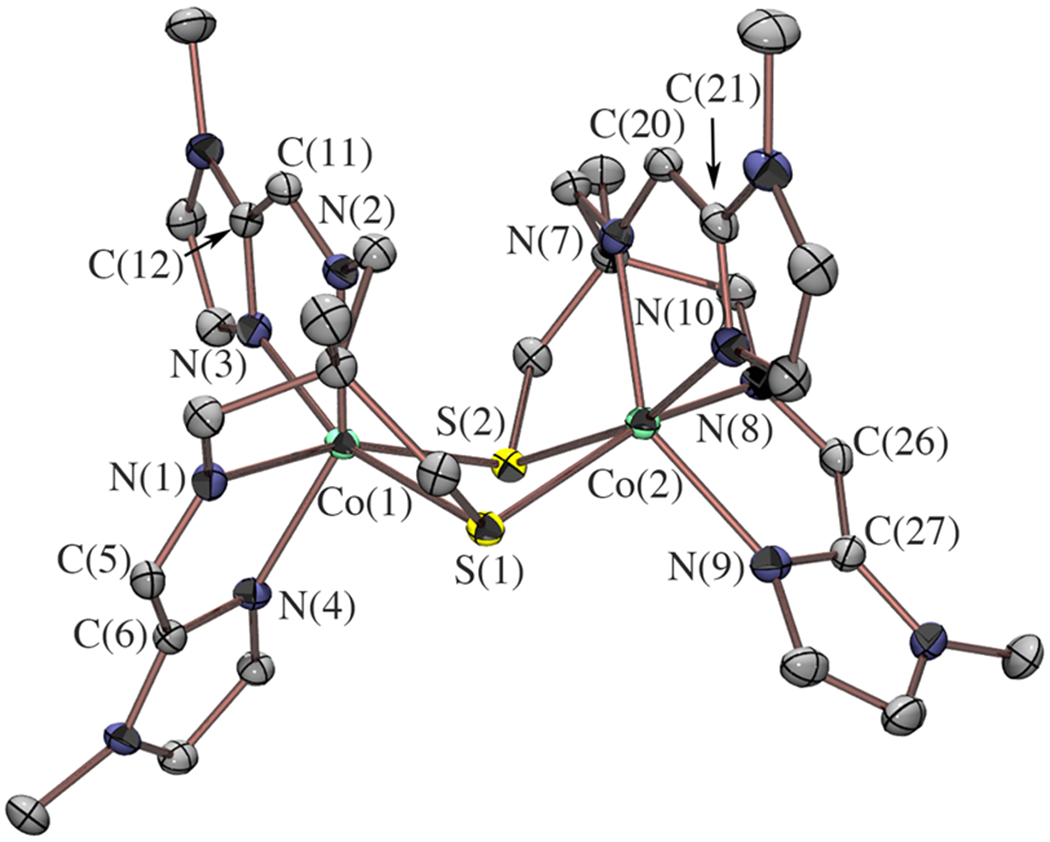
ORTEP of dicationic [CoII(tame-N2S(MeIm)2)]22+ (4) with counterions, solvents of crystallization, and hydrogen atoms omitted for clarity.
A notable difference between structures 4 and 3 is that the Co2S2 core of 4 is significantly more puckered (Figure 13), with a separation between the M—M and S—S centroids of 0.72 Å, compared to 0.34 Å for the Fe2S2 core of 3. This is likely a result of the change in d-electron configuration (d7 Co(II) vs d6 Fe(II)). In addition, the M—S bond lengths in 4 are significantly elongated compared to those of 3 (Table 1), by an average of 0.13 Å. This likely reflects the high- versus low-spin-states of 4 and 3, respectively. The more puckered core and longer M—S bonds of 4 decrease RS → M(t2g) π-donation, which should decrease the extent of π-back-donation into the π-acidic (α-imino)-N-heterocycle π* LUMO. Distinct differences in intraligand metrical parameters for cobalt-4 versus iron-3 (Table 2) provide evidence to support this. While the intra-ligand bond lengths of the Fe complex, 3, resemble a reduced L•− ligand (Cim—Nim = 1.297(3) Å, Cim—Cipso = 1.427(3) Å), intra-ligand bond lengths in the Co complex 4 (Cim—Nim = 1.276(5) Å, Cim—Cipso = 1.447(4) Å) more closely resemble those of a closed-shell L0 ligand (Table 2). Consistent with this picture, involving less effective RS/M/(α-imino)-imidazole LUMO π-orbital overlap, the electronic absorption spectrum of 4 (Figure S18) notably lacks the intense low-energy CT transitions observed in those of 1–3 (Figures 6 and 7).
The influence of the thiolate ligand in activating the π-acidic (α-imino-N-heterocycle) ligand was also investigated, by synthesizing a thioether derivative of 3. In contrast to thiolate structures 1 – 4, thioether [FeII(tame-N2SBz(MeIm)2(MeCN))]2+ (5) is monomeric in the solid state (Figure 14), with a pseudo-octahedral coordination sphere that is completed by an axial MeCN trans to the thioether. Although the short Fe—N and Fe—S bond lengths are most consistent with a low-spin S = 0 ferrous ion at 100 K (the temperature at which X-ray crystallographic data was collected), the 1H NMR spectrum of 5 at 298 K displays paramagnetically shifted peaks (Figure S19), and the ambient temperature solution magnetic moment of 5, μeff = 5.04 μB, is consistent with an S = 2 spin-state, indicating that there is a thermally accessible higher-spin-state. Although the Fe—N bond lengths of the thiolate complex 3 and thioether complex 5 are comparable, the intraligand Cim—Nim and Cim—Cipso metrical parameters in 5 are closer to that of a closed-shell (α-imino)-N-heterocycle (Table 2). Furthermore, although 5 retains an Fe → (α-imino)-pyridine π* CT transition in the visible region of the electronic absorption spectrum (Figure 7), it is significantly lower in intensity, and to higher energy (λmax (ϵ, M−1 cm−1) = 512 (2850) nm), relative to that of the thiolate complex 3 (λmax (ϵ, M−1 cm−1) = 619 (11 500) nm). These differences in intraligand metrical parameters, and electronic absorption spectral properties, suggest that the π-donating thiolate ligand plays a key role in activating the π-accepting (α-imino)-N-heterocycle ligand.
Figure 14.
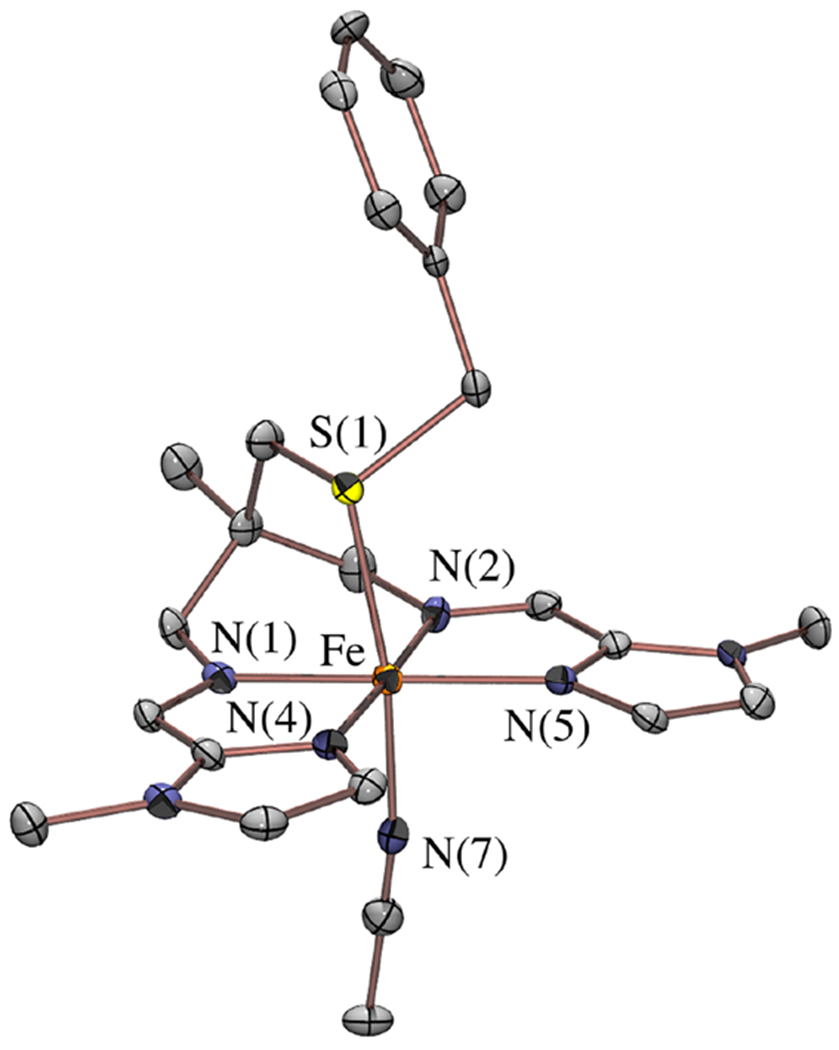
ORTEP of dicationic 5 with counterions, solvents of crystallization, and hydrogen atoms omitted for clarity.
DISCUSSION
The interplay between metal and ligand redox orbitals has been clearly established as an integral component in the function of both metalloenzymes and synthetic transition-metal catalysts containing redox-active ligands.6,11,23,25,26,84,85 As detailed herein, the synthesis and characterization of complexes 1–5 provide insight into the cooperativity between the frontier orbitals of a π-donating thiolate, Fe, and π-accepting (α-imino)-N-heterocycle ligands with extended π-systems. Cytochrome P450 is the classic biological example that implements cooperativity between redox-active orbitals in order to efficiently promote catalysis.11,23,25,26,84,85 While many synthetic catalysts featuring redox-active ligands1,6,41,86,87 derive inspiration from P450,31–33 only a limited number incorporate thiolate ligands.38,39 Thiolate ligands have well-established functional roles in biology, which include tuning redox potentials,55,57,88 stabilizing low-spin-states,54,56,57,63 or using a compensatory effect to maintain a constant metal ion Lewis acidity.59 They can also influence the properties of other ligands in the coordination sphere, by labilizing trans ligands,89–91 or altering basicity and O—H bond strengths.11,84,92–96 With P450, the cysteine thiolate directly participates in the redox chemistry, functioning as part of the reservoir of oxidizing equivalents necessary to form Cmpd I.11,84,97,98 Despite this diverse biological utility, an in-depth understanding of the impact that thiolate ligands have on the electronic structure of synthetic complexes containing redox-active ligands has yet to be fully elucidated.
The series of complexes, 1–5, described herein illustrates the ability of π-donating thiolate ligands to tune intraligand metrical parameters and the valence electronic structure of π-acceptor ligands. For thiolate complexes 1–3, unusual activation of the (α-imino)-N-heterocycle ligand is observed (Table 2), wherein imine Cim═N bond lengths are elongated, and Cim—Cipso bonds are shortened, relative to that predicted for a closed-shell ligand, L0, indicating that electron density has shifted into the LUMO orbital of the (α-imino)-N-heterocycle ligand framework.10 Cobalt(II)-containing 4 and the thioether complex 5 do not exhibit the same ligand activation. As discussed above, the origin of ligand activation in 1–3 could either be attributed to a formal reduction of the ligand to L•− (with strong antiferromagnetic coupling to the resultant low-spin S = 1/2 Fe(III) ion), or to extraordinarily large amounts of π-backbonding from low-spin Fe(II) to the π-acidic ligand,6 both of which would give rise to the observed diamagnetic 1H NMR spectra of 1 and 2. The Mössbauer parameters of 1–3 and the metal—ligand bond lengths of 1–3 and 5 are most consistent with low-spin Fe(II), however. The comparable Fe—N bond lengths in thioether-containing 5, and thiolate-containing 3, as well as the absence of perturbed intraligand metrical parameters in 5, support the latter description as well, in agreement with spectroscopic data and DFT calculations (vide supra).
The extent of metal-to-ligand π-backbonding largely depends on the population and energy of the metal π-symmetry orbitals, which can be modulated by metal oxidation state and spin-state, as well as the nature of supporting ligands. Low-spin ferrous ions, such as those in 1–3 and 5, have a (t2g)6 configuration and are optimally suited for π-back-donation to a π-acceptor ligand. In contrast, the high-spin Co(II) ions in 4 have a (t2g)5(eg*)2 configuration, with one fewer electron in the π-symmetry d-orbitals. Additionally, population of the eg*-orbitals serves to elongate the metal—ligand bonds, which would weaken π-backbonding to Co(II) (Table 1). Given that thiolates are known to be effective π-donors, two of the three π-symmetry t2g-orbitals of 1–3 should be relatively electron-rich. The metal-centered π-symmetry t2g-orbitals are also of the appropriate symmetry to back-bond into the (α-imino)-N-heterocycle π*-orbitals. However, effective communication between all of these these orbitals requires efficient π-overlap, which, of course, is dependent on metal—ligand bond lengths and bond angles. This is evident in the metrical parameters of 4 compared to 3, where longer metal—ligand bonds and a more puckered diamond core in the former result in negligible (α-imino)-N-heterocycle ligand activation. The comparable Fe—N bond lengths, but contrasting intraligand distances in thioether 5 versus thiolate 3, illustrate the influence of the thiolate on π-backbonding to the (α-imino)imidazole ligand, in the solid state. In the solution state, this is supported by the significantly less intense CT bands for Co(II)-4 (ε = 654 M−1 cm−1, Figure S18) and the thioether complex 5 (ε = 2850 M−1 cm−1, Figure 7) versus the unusually intense Fe → (α-imino)pyridine π* CT bands (ε > 10 000 M−1 cm−1) for the thiolate complexes 1–3 (Figures 6 and 7). The intensities of the latter are indicative of optimal orbital overlap.54 This is supported by the TD-DFT-calculated transition difference densities (Figure 11), which indicate that the charge-donating thiolate contributes to the intense transitions. Additionally, the Fe → (α-imino)pyridine π* CT band associated with 5 is shifted to higher energy relative to 1–3. The latter reflects the higher energy of the d-orbitals in 1–3 caused by π-donation from the thiolate, and the well-established ability of thiolate ligands to tune the energies of redox-active metal orbitals.
Evidence for the direct involvement of the π-donating thiolate ligands in activating the (α-imino)-N-heterocycle ligand π*-orbitals is provided by the S K-edge XAS spectra, which show electronic transitions from the thiolate sulfur 1s-orbital into the (α-imino)-N-heterocycle LUMO π* (Figure 9). Assignment of this S K-edge XAS pre-edge feature is supported by the strong correlation between its transition energy and intensity, and the N-heterocycle ring size (Figure 15). The intensity of the S K-edge XAS pre-edge transitions increases as the thiolate character of the LUMO π*-orbitals increases (Table S3). A quantitative comparison of reduction potentials (Table 3), S K-edge XAS pre-edge energies (Table S1), and the DFT-calculated LUMO (α-imino)-N-heterocycle π*-orbital energies for 1–3 shows (Figure 15) that these properties are highly correlated.
Figure 15.
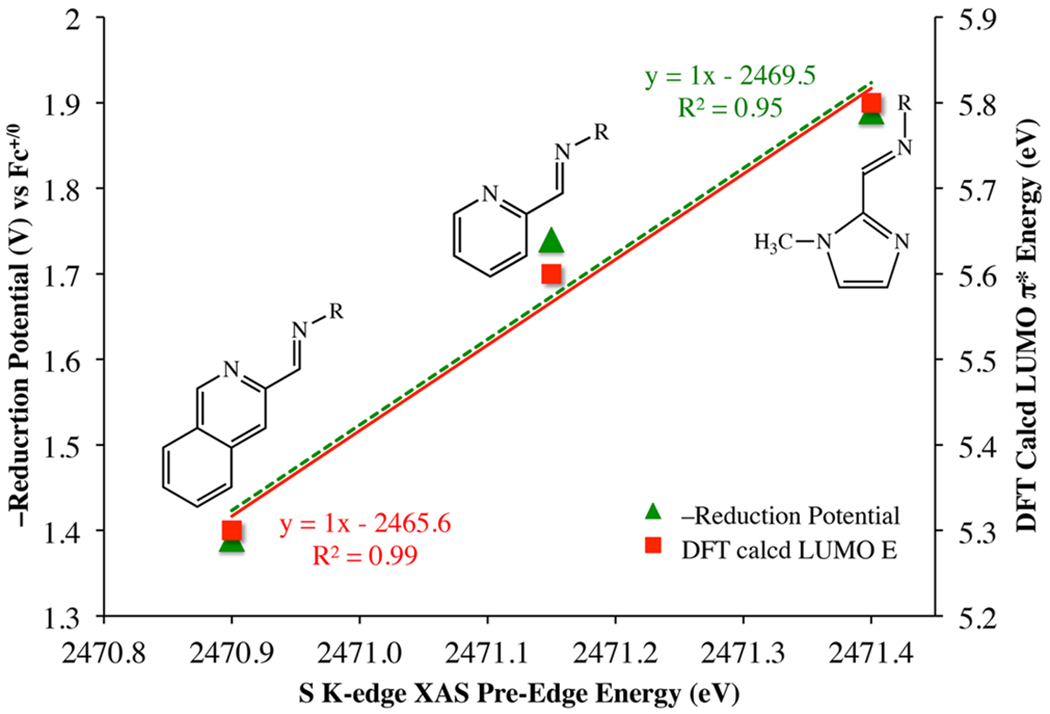
Correlation between reduction potentials, S K-edge XAS pre-edge energies, and DFT-calculated LUMO energies (referenced to their respective HOMO energies), illustrating the effect of variations in N-heterocycle ring size.
CONCLUSIONS
Herein we have examined the geometric and electronic structure of a family of metal complexes containing both a π-donating thiolate ligand, and π-acceptor (α-imino)-N-heterocycle ligands. A combined analysis of X-ray crystal structures and spectroscopic parameters, supported by theoretical calculations, indicates that the ground state electronic structure of 1–3 is heavily delocalized over the thiolate ligand, the metal center, and the (α-imino)-N-heterocycle. Each of these complexes is shown to contain low-lying LUMOs that are α-imino-N-heterocycle ligand π* in character, and HOMOs which consist of a highly covalent mix of sulfur 3p and metal t2g-type 3d-orbitals. This electronic structure results in unusually intense RS—Fe → (α-imino)-N-heterocycle π* CT bands in the electronic absorption spectra, the intensities of which are reminiscent of complexes containing reduced L•− ligands. Sulfur K-edge X-ray absorption spectroscopy (XAS) and TD-DFT provide evidence for an interaction between the π-donating thiolate and π-accepting (α-imino)-N-heterocycle ligand π*-orbitals, via the metal ion d-orbitals. The energies and intensities of the lowest-energy S K-edge XAS pre-edge feature are shown to correlate with reduction potentials, DFT-calculated (α-imino)-N-heterocycle LUMO energies, and N-heterocycle ring size (Figure 15). Communication between the π-donor and π-acceptor orbitals is shown to generate an activated α-imino-N-heterocycle ligand with intraligand metrical parameters close to those of a monoreduced L•−. This is unprecedented, and distinctly different from most complexes containing (α-imino)pyridine-ligands, where ligand activation is only observed upon the addition of a strong reducing agent. Four components of the geometric and electronic structures of 1–3 were shown to be responsible for the observed ligand activation: (1) an approximate energy match between the metal ion and (α-imino)-N-heterocycle ligand π*-orbitals, (2) effective orbital spatial overlap, (3) a low-spin-state, and most importantly, (4) the inclusion of a π-donating thiolate in the coordination sphere. The Fe 3d-orbitals were shown to mediate charge-transfer from the thiolate sulfur 3p-orbital to the formally orthogonal (α-imino)-N-heterocycle π* LUMO. We liken this interaction to the proposed “push-effect” of the P450 ferric hydroperoxo Cmpd 0, wherein electron donation from the cysteinate ligand into the Fe 3d-orbitals results in the population of the peroxo O—O σ*-orbital.31–33
The study reported herein also highlights key properties likely to be beneficial for the development of redox-active (α-imino)-N-heterocycle base-metal catalysts capable of promoting concerted 2e− reactions.1–3 By incorporating redox-active ligands that are capable of both donating and accepting electron density to and from the metal ion, charge distribution could be modulated in a facile manner, thereby potentially improving catalyst performance. We demonstrated that we can modulate the extent of metal—ligand redox-active orbital coupling by altering the size of the ligand π-system. As the energies of the ligand π*-orbitals decrease in response to increasing ring size, the extent of orbital overlap with the highly covalent Fe—SR moiety was shown, by the intensities of S K-edge XAS pre-edge transitions, to systematically increase. In addition, the electron-donating thiolate ligands were shown to raise the energy of the redox-active metal ion t2g-orbitals closer to the (α-imino)-N-heterocycle ligand π*-orbitals, thereby optimizing the facile transfer of charge. The energy cost of storing reducing equivalents was shown to be lowest when a quinoline is incorporated into the ligand framework.
Supplementary Material
ACKNOWLEDGMENTS
J.A.K. acknowledges the NIH (GM45881) for funding, and J.K.K., E.B., and S.D. acknowledge the Max Planck Society for funding. J.A.R. was funded, in part, by a graduate study scholarship from the German Academic Exchange Service (DAAD). Aleksandr Forov is thanked for assistance during beam time, the ESRF is acknowledged for providing beamtime, and Pieter Glatzel is thanked for technical assistance with data collection at beamline ID26.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.7b02748.
Experimental details regarding the synthesis and characterization of the tame-N2SH ligand, and metal complexes derived therefrom; 1H NMR spectrum of 3 and 5; magnetic moment vs T plot of 3; cyclic voltammograms for 1–3; DFT generated MO diagram and Mössbauer spectrum of 1; electronic absorption spectrum of 4; crystallographic tables and metrical parameters for DFT optimized structures of 1–5; and experimental details for the S K-edge XAS (PDF)
Accession Codes
CCDC 1584846–1584850 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).Chirik PJ; Wieghardt K Radical Ligands Confer Nobility on Base-Metal Catalysts. Science 2010, 327, 794–795. [DOI] [PubMed] [Google Scholar]
- (2).Chirik P; Morris R Getting Down to Earth: The Renaissance of Catalysis with Abundant Metals. Acc. Chem. Res 2015, 48, 2495–2495. [DOI] [PubMed] [Google Scholar]
- (3).Tondreau AM; Stieber CE; Milsmann C; Lobkovsky E; Weyhermuller T; Semproni SP; Chirik PJ Oxidation and Reduction of Bis(imino)pyridine Iron Dinitrogen Complexes: Evidence for Formation of a Chelate Trianion. Inorg. Chem 2013, 52, 635–646. [DOI] [PubMed] [Google Scholar]
- (4).Darmon JM; Stieber CE; Sylvester KT; Fernandez I; Lobkovsky E; Semproni SP; Bill E; Wieghardt K; DeBeer S; Chirik PJ Oxidative Addition of Carbon—Carbon Bonds with a Redox-Active Bis(imino)pyridine Iron Complex. J. Am. Chem. Soc 2012, 134, 17125–17137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sazama GT; Betley TA Multiple, Disparate Redox Pathways Exhibited by a Tris(pyrrolido)ethane Iron Complex. Inorg. Chem 2014, 53, 269–281. [DOI] [PubMed] [Google Scholar]
- (6).Dugan TR; Bill E; MacLeod KC; Christian G. j.; Cowley RE; Brennessel WW; Ye S; Neese F; Holland PL Reversible C—C Bond Formation between Redox-Active Pyridine Ligands in Iron Complexes. J. Am. Chem. Soc 2012, 134, 20352–20364. [DOI] [PubMed] [Google Scholar]
- (7).Butschke B; Fillman KL; Bendikov T; Shimon LJW; Diskin-Posner Y; Leitus G; Gorelsky SI; Neidig ML; Milstein D Inorg. Chem 2015, 54, 4909–4926. [DOI] [PubMed] [Google Scholar]
- (8).Ray K; Bill E; Weyhermuller T; Wieghardt K Redox-Noninnocence of the S,S-Coordinated Ligands in Bis(benzene-1,2-dithiolato)iron Complexes. J. Am. Chem. Soc 2005, 127, 5641–5654. [DOI] [PubMed] [Google Scholar]
- (9).Bart SC; Chlopek K; Bill E; Bouwkamp MW; Lobkovsky E; Neese F; Wieghardt K; Chirik PJ Electronic Structure of Bis(imino)pyridine Iron Dichloride, Monochloride, and Neutral Ligand Complexes: A Combined Structural, Spectroscopic, and Computational Study. J. Am. Chem. Soc 2006, 128, 13901–13912. [DOI] [PubMed] [Google Scholar]
- (10).Lu CC; Bill E; Weyhermuller T; Bothe E; Wieghardt K Neutral Bis(r-iminopyridine)metal Complexes of the First-Row Transition Ions (Cr, Mn, Fe, Co, Ni, Zn) and Their Monocationic Analogues: Mixed Valency Involving a Redox Noninnocent Ligand System. J. Am. Chem. Soc 2008, 130, 3181–3197. [DOI] [PubMed] [Google Scholar]
- (11).Green MT C—H bond activation in heme proteins: the role of thiolate ligation in cytochrome P450. Curr. Opin. Chem. Biol 2009, 13, 84–88. [DOI] [PubMed] [Google Scholar]
- (12).Meunier B; de Visser SP; Shaik S Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- (13).Dey A; Jiang Y; Ortiz de Montellano P; Hodgson KO; Hedman B; Solomon EI S K-edge XAS and DFT Calculations on Cytochrome P450: Covalent and Ionic Contributions to the Cysteine-Fe Bond and Their Contribution to Reactivity. J. Am. Chem. Soc 2009, 131, 7869–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Askerka M; Brudvig GW; Batista VS The O2-Evolving Complex of Photosystem II: Recent Insights from Quantum Mechanics/Molecular Mechanics (QM/MM), Extended X-ray Absorption Fine Structure (EXAFS), and Femtosecond X-ray Crystallography Data. Acc. Chem. Res 2017, 50, 41–48. [DOI] [PubMed] [Google Scholar]
- (15).Hoffman BM; Lukoyanov D; Dean DR; Seefeldt LC Nitrogenase: A Draft Mechanism. Acc. Chem. Res 2013, 46, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Citek C; Lyons CT; Wasinger EC; Stack TD Self-assembly of the oxy-tyrosinase core and the fundamental components of phenolic hydroxylation. Nat. Chem 2012, 4, 317–322. [DOI] [PubMed] [Google Scholar]
- (17).Tinberg CE; Lippard SJ Dioxygen Activation in Soluble Methane Monooxygenase. Acc. Chem. Res 2011, 44, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).McEvoy JP; Brudvig GW Water-Splitting Chemistry of Photosystem II. Chem. Rev 2006, 106, 4455–4483. [DOI] [PubMed] [Google Scholar]
- (19).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hegg EL Unraveling the Structure and Mechanism of Acetyl-Coenzyme A Synthase. Acc. Chem. Res 2004, 37, 775–783. [DOI] [PubMed] [Google Scholar]
- (21).Que L; Tolman WB Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [DOI] [PubMed] [Google Scholar]
- (22).Kovaleva EG; Neibergall MB; Chakrabarty S; Lipscomb JD Finding Intermediates in the O2 Activation Pathways of Non-Heme Iron Oxygenases. Acc. Chem. Res 2007, 40, 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rittle J; Green MT Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- (24).Groves JT Using push to get pull. Nat. Chem 2014, 6, 89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ortiz de Montellano PR Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Denisov IG; Makris TM; Sligar SG; Schlichting I Structure and Chemistry of Cytochrome P450. Chem. Rev 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- (27).Groves JT; Haushalter RC; Nakamura M; Nemo TE; Evans BJ High-Valent Iron-Porphyrin Complexes Related to Peroxidase and Cytochrome P-450. J. Am. Chem. Soc 1981, 103, 2884–2886. [Google Scholar]
- (28).Groves JT; Watanabe Y Reactive Iron Porphyrin Derivatives Related to the Catalytic Cycles of Cytochrome P-450 and Peroxidase. Studies of the Mechanism of Oxygen Activation. J. Am. Chem. Soc 1988, 110, 8443–8452. [Google Scholar]
- (29).Green MT Evidence for Sulfur-Based Radicals in Thiolate Compound I Intermediates. J. Am. Chem. Soc 1999, 121, 7939–7940. [Google Scholar]
- (30).Krest CM; Onderko EL; Yosca TH; Calixto JC; Karp RF; Livada J; Rittle J; Green MT Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem 2013, 288, 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dawson JH; Holm RH; Trudell JR; Barth G; Linder RE; Bunnenberg E; Djerassi C; Tang SC Chloroperoxidase. Evidence for P-450Type Heme Environment from Magnetic Circular Dichroism Spectroscopy. J. Am. Chem. Soc 1976, 98, 3707–3709. [DOI] [PubMed] [Google Scholar]
- (32).Yamaguchi K; Watanabe Y; Morishima I Direct Observation of the Push Effect on the O-O Bond Cleavage of Acylperoxoiron(III) Porphyrin Complexes. J. Am. Chem. Soc 1993, 115, 4058–4065. [Google Scholar]
- (33).Geri JB; Shanahan JP; Szymczak NK Testing the Push—Pull Hypothesis: Lewis Acid Augmented N2 Activation at Iron. J. Am. Chem. Soc 2017, 139, 5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mak PJ; Denisov IG; Victoria D; Makris TM; Deng T; Sligar SG; Kincaid JR Resonance Raman detection of the hydroperoxo intermediate in the cytochrome P450 enzymatic cycle. J. Am. Chem. Soc 2007, 129, 6382–6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Denisov IG; Mak PJ; Makris TM; Sligar SG; Kincaid JR J. Phys. Chem. A 2008, 112, 13172–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Schoneboom JC; Lin H; Reuter N; Thiel W; Cohen S; Ogliaro F; Shaik S The Elusive Oxidant Species of Cytochrome P450 Enzymes: Characterization by Combined Quantum Mechanical/Molecular Mechanical (QM/MM) Calculations. J. Am. Chem. Soc 2002, 124, 8142–8151. [DOI] [PubMed] [Google Scholar]
- (37).Jiang Y; Widger LR; Kasper GD; Siegler MA; Goldberg DP Iron(II)-Thiolate S-Oxygenation by O2: Synthetic Models of Cysteine Dioxygenase. J. Am. Chem. Soc 2010, 132, 12214–12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Badiei YM; Siegler MA; Goldberg DP O2 Activation by Bis(imino)pyridine Iron(II)-Thiolate Complexes. J. Am. Chem. Soc 2011, 133, 1274–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Widger LR; Jiang Y; Siegler MA; Kumar D; Latifi R; de Visser SP; Jameson GNL; Goldberg DP Synthesis and Ligand Non-Innocence of Thiolate-Ligated (N4S) Iron(II) and Nickel(II) Bis(imino)pyridine Complexes. Inorg. Chem 2013, 52, 10467–10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lu CC; Weyhermüller T; Bill E; Wieghardt K Inorg. Chem 2009, 48, 6055–6064. [DOI] [PubMed] [Google Scholar]
- (41).Chirik PJ Iron- and Cobalt-Catalyzed Alkene Hydrogenation: Catalysis with Both Redox-Active and Strong Field Ligands. Acc. Chem. Res 2015, 48, 1687–1695. [DOI] [PubMed] [Google Scholar]
- (42).Wu JY; Moreau B; Ritter T J. Am. Chem. Soc 2009, 131, 12915–12917. [DOI] [PubMed] [Google Scholar]
- (43).Wu JY; Stanzl BN; Ritter T J. Am. Chem. Soc 2010, 132, 13214–13216. [DOI] [PubMed] [Google Scholar]
- (44).Yu RP; Darmon JM; Milsmann C; Margulieux GW; Stieber SCE; DeBeer S; Chirik PJ J. Am. Chem. Soc 2013, 135, 13168–13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bart SE; Lobkovsky AE; Chirik PJ Preparation and Molecular and Electronic Structures of Iron(0) Dinitrogen and Silane Complexes and Their Application to Catalytic Hydrogenation and Hydrosilation. J. Am. Chem. Soc 2004, 126, 13794–13807. [DOI] [PubMed] [Google Scholar]
- (46).Bouwkamp MW; Bowman CA; Lobkovsky EA; Chirik PJ J. Am. Chem. Soc 2006, 128, 13340. [DOI] [PubMed] [Google Scholar]
- (47).Sazama GT; Betley TA Inorg. Chem 2010, 49, 2512–2524. [DOI] [PubMed] [Google Scholar]
- (48).King ER; Betley TA J. Am. Chem. Soc 2009, 131, 14374–14380. [DOI] [PubMed] [Google Scholar]
- (49).Lugo-Mas P; Taylor W; Schweitzer D; Theisen RM; Xu L; Shearer J; Swartz RD; Gleaves MC; DiPasquale A; Kaminsky W; Kovacs JA Properties of Square-Pyramidal Alkyl—Thiolate FeIII Complexes, Including an Analogue of the Unmodified Form of Nitrile Hydratase. Inorg. Chem 2008, 47, 11228–11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Roelfes G; Vrajmasu V; chen K; Ho RYN; Rohde J-U; Zondervan C; la Crois RM; Schudde EP; Lutz M; Spek AL; Hage R; Feringa BL; Munck E; Que L Jr End-on and Side-on Peroxo Derivatives of Non-Heme Iron Complexes with Pentadentated Ligands: Models for Putative Intermedates in Biological Iron/Dioxygen Chemistry. Inorg. Chem 2003, 42, 2639–2653. [DOI] [PubMed] [Google Scholar]
- (51).Tondreau AM; Milsmann C; Patrick AD; Hoyt HM; Lobkovsky E; Wieghardt K; Chirik PJ Synthesis and Electronic Structure of Cationic, Neutral, and Anionic Bis(imino)pyridine Iron Alkyl Complexes: Evaluation of Redox Activity in Single-Component Ethylene Polymerization Catalysts. J. Am. Chem. Soc 2010, 132, 15046–15059. [DOI] [PubMed] [Google Scholar]
- (52).Shoner SC; Nienstedt A; Ellison JJ; Kung I; Barnhart D; Kovacs JA Structural Comparison of Thiolate–Ligated M(II)=Fe(II), Co(II), Ni(II), and Zn(II) Ions Wrapped in a Chiral Helical Ligand. Inorg. Chem 1998, 37, 5721–5725. [Google Scholar]
- (53).Shearer J; Nehring J; Kaminsky W; Kovacs JA; Lovell S Modeling The Reactivity of Superoxide Reducing Metalloenzymes With a Nitrogen and Sulfur Coordinated Iron Complex. Inorg. Chem 2001, 40, 5483–5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kennepohl P; Neese F; Schweitzer D; Jackson HL; Kovacs JA; Solomon EI Spectroscopy of Non-Heme Iron Thiolate Complexes: Insight into the Electronic Structure of the Low-Spin Active Site of Nitrile Hydratase. Inorg. Chem 2005, 44, 1826–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kovacs JA; Brines LM Understanding How the Thiolate Sulfur Contributes to the Function of the Non-Heme Iron Enzyme Superoxide Reductase. Acc. Chem. Res 2007, 40, 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kovacs JA Synthetic Analogues of Cysteinate-Ligated Non-Heme Iron, and Non-Corrinoid Cobalt Enzymes. Chem. Rev 2004, 104, 825–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Jackson HL; Shoner SC; Rittenberg D; Cowen JA; Lovell S; Barnhart D; Kovacs JA Probing the Influence of Local Coordination Environment on the Properties of Fe-Type Nitrile Hydratase Model Complexes. Inorg. Chem 2001, 40, 1646–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Stieber SCE; Milsmann C; Hoyt JM; Turner ZR; Finkelstein KD; Wieghardt K; DeBeer S; Chirik PJ Bis(imino)pyridine Iron Dinitrogen Compounds Revisited: Differences in Electronic Structure Between Four- and Five-Coordinate Derivatives. Inorg. Chem 2012, 51, 3770–3785. [DOI] [PubMed] [Google Scholar]
- (59).Lugo-Mas P; Dey A; Xu L; Davin SD; Benedict J; Kaminsky W; Hodgson KO; Hedman B; Solomon EI; Kovacs JA How Does Single Oxygen Atom Addition Affect the Properties of an Fe-Nitrile Hydratase Analogue? The Compensatory Role of the Unmodified Thiolate. J. Am. Chem. Soc 2006, 128, 11211–11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Shearer J; Jackson HL; Schweitzer D; Rittenberg DK; Leavy TM; Kaminsky W; Scarrow RC; Kovacs JA The first example of a nitrile hydratase model complex that reversibly binds nitriles. J. Am. Chem. Soc 2002, 124, 11417–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Schweitzer D; Shearer J; Rittenberg DK; Shoner SC; Ellison JJ; Loloee R; Lovell SC; Barnhart D; Kovacs JA Enhancing Reactivity via Ligand Constraints. Inorg. Chem 2002, 41 , 3128–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Ellison JJ; Nienstedt A; Shoner SC; Barnhart D; Cowen JA; Kovacs JA Reactivity of Five-Coordinate Models for the Thiolate-Ligated Fe Site of Nitrile Hydratase. J. Am. Chem. Soc 1998, 120, 5691–5700. [Google Scholar]
- (63).Shoner S; Barnhart D; Kovacs JA A Model for the Low-Spin, Non-Heme, Thiolate-Ligated Iron Site of Nitrile Hydratase. Inorg. Chem 1995, 34, 4517–4518. [Google Scholar]
- (64).Chłopek K; Bill E; Weyhermüller T; Wieghardt K Inorg. Chem 2005, 44, 7087–7098. [DOI] [PubMed] [Google Scholar]
- (65).Ghosh P; Bill E; Weyhermüller T; Wieghardt K J. Am. Chem. Soc 2003, 125, 3967–3979. [DOI] [PubMed] [Google Scholar]
- (66).Noveron JC; Herradora R; Olmstead MM; Mascharak PK Low-spin iron(III) complexes with N,S coordination: syntheses, structures, and properties of bis(N-2-mercaptophenyl-20-pyridylmethyleniminato)iron(III) tetraphenylborate and bis(N-2-mercapto-2-methylpropyl-2-pyridylmethyleniminato)iron(III) tetraphenylborate. Inorg. Chim. Acta 1999, 285, 269–276. [Google Scholar]
- (67).Shejwalkar P; Rath NP; Bauer EB New iron(II) a-iminopyridine complexes and their catalytic activity in the oxidation of activated methylene groups and secondary alcohols to ketones. Dalton Trans 2011, 40, 7617–7631. [DOI] [PubMed] [Google Scholar]
- (68).Kato M; Okamura T; Yamamoto H; Ueyama N Effects of the Intramolecular NHâââS Hydrogen Bond in Mononuclear Platinum(II) and Palladium(II) Complexes with 2,2-Bipyridine and Benzenethiol Derivatives. Inorg. Chem 2005, 44, 1966–1972. [DOI] [PubMed] [Google Scholar]
- (69).Broering EP; Dillon S; Gale EM; Steiner RA; Telser J; Brunold TC; Harrop TC Accessing Ni(III)-Thiolate Versus Ni(II)-Thiyl Bonding in a Family of Ni–N2S2 Synthetic Models of NiSOD. Inorg. Chem 2015, 54, 3815–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Shearer J; Fitch SB; Kaminsky W; Benedict J; Scarrow RC; Kovacs JA How does cyanide inhibit superoxide reductase? Insight from synthetic FeIIIN4S model complexes. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3671–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Brines LM; Shearer J; Fender JK; Schweitzer D; Shoner SC; Barnhart D; Kaminsky W; Lovell S; Kovacs JA Periodic Trends within a Series of Five-Coordinate Thiolate-Ligated [M(II)(S(Me2)N(4)(tren))](+) (M = Mn, Fe, Co, Ni, Cu, Zn) Complexes, Including a Rare Example of a Stable Cu(II)-Thiolate. Inorg. Chem 2007, 46, 9267–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Dey A; Jenney FE; Adams MW; Johnson MK; Hodgson KO; Hedman B; Solomon EI Sulfur K-edge X-ray absorption spectroscopy and density functional theory calculations on superoxide reductase: role of the axial thiolate in reactivity. J. Am. Chem. Soc 2007, 129, 12418–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Dey A; Chow M; Taniguchi K; Lugo-Mas P; Davin SD; Maeda M; Kovacs JA; Odaka M; Hedman B; Hodgson KO; Solomon EI S K-edge XAS and DFT Calculations on Nitrile Hydratase: Geometric and Electronic Structure of the Non-Heme Iron Active Site. J. Am. Chem. Soc 2006, 128, 533–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Glaser T; Hedman B; Hodgson KO; Solomon EI Ligand K-Edge X-ray Absorption Spectroscopy: A Direct Probe of Ligand-Metal Covalency. Acc. Chem. Res 2000, 33, 859–868. [DOI] [PubMed] [Google Scholar]
- (75).Szilagyi RK; Bryngelson PA; Maroney MJ; Hedman B; Hodgson KO; Solomon EI S K-edge X-ray absorption spectroscopic investigation of the Ni-containing superoxide dismutase active site: new structural insight into the mechanism. J. Am. Chem. Soc 2004, 126, 3018–3019. [DOI] [PubMed] [Google Scholar]
- (76).Rose K; Shadle SE; Eidsness MK; Kurtz DM; Scott RA; Hedman B; Hodgson KO; Solomon EI Investigation of Iron-Sulfur Covalency in Rubredoxins and a Model System Using Sulfur K-Edge X-ray Absorption Spectroscopy. J. Am. Chem. Soc 1998, 120, 10743–10747. [Google Scholar]
- (77).Ray K; DeBeer George S; Solomon E; Wieghardt K; Neese F Description of the Ground-State Covalencies of the Bis(dithiolato) Transition-Metal Complexes from X-ray Absorption Spectroscopy and Time-Dependent Density-Functional Calculations. Chem. - Eur. J 2007, 13, 2783–2797. [DOI] [PubMed] [Google Scholar]
- (78).Kowalska JK; Hahn AW; Albers A; Schiewer CE; Bjornsson R; Lima FA; Meyer F; DeBeer S X-ray Absorption and Emission Spectroscopic Studies of [L2Fe2S2]n Model Complexes: Implications for the Experimental Evaluation of Redox States in Iron–Sulfur Clusters. Inorg. Chem 2016, 55, 4485–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Neese F Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev 2009, 253, 526–563. [Google Scholar]
- (80).Gütlich P; Bill E; Trautwein AX Mössbauer Spectroscopy and Transition-Metal Chemistry; Springer Verlag: Berlin, 2011. [Google Scholar]
- (81).Neese F; Petrenko T In Mössbauer Spectroscopy and Transition-Metal Chemistry; Gütlich P, Bill E, Trautwein AX, Eds.; Springer Verlag: Berlin, 2011; pp 137–199. [Google Scholar]
- (82).Ye S; Bill E; Neese F Electronic Structures of the [Fe(N2)(SiPiPr3)]+1/0/−1 Electron Transfer Series: A Counter-intuitive Correlation between Isomer Shifts and Oxidation States. Inorg. Chem 2016, 55, 3468–3474. [DOI] [PubMed] [Google Scholar]
- (83).Kupper C; Rees JA; Dechert S; DeBeer S; Meyer F Complete Series of {FeNO}8, {FeNO}7, and {FeNO}6 Complexes Stabilized by a Tetracarbene Macrocycle. J. Am. Chem. Soc 2016, 138, 7888–7898. [DOI] [PubMed] [Google Scholar]
- (84).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C–H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Newcomb M; Zhang R; Chandrasena RE; Halgrimson JA; Horner JH; Makris TM; Sligar SG Cytochrome p450 compound I. J. Am. Chem. Soc 2006, 128, 4580–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Chirik PJ Preface: Forum on Redox-Active Ligands. Inorg. Chem 2011, 50, 9737–9740. [DOI] [PubMed] [Google Scholar]
- (87).Feller M; Ben-Ari E; Diskin-Posner Y; Carmieli R; Weiner L; Milstein D O2 Activation by Metal-Ligand Cooperation with IrI PNP Pincer Complexes. J. Am. Chem. Soc 2015, 137, 4634–4637. [DOI] [PubMed] [Google Scholar]
- (88).Villar-Acevedo G; Nam E; Fitch S; Benedict J; Freudenthal J; Kaminsky W; Kovacs JA Influence of Thiolate Ligands on Reductive N–O Bond Activation. Oxidative Addition of NO to a Biomimetic SOR Analogue, and its Proton-Dependent Reduction of Nitrite. J. Am. Chem. Soc 2011, 133, 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Shearer J; Kung IY; Lovell S; Kaminsky W; Kovacs JA Why Is There an “Inert” Metal Center in the Active Site of Nitrile Hydratase? Reactivity and Ligand Dissociation from a Five-Coordinate Co(III) Nitrile Hydratase Model. J. Am. Chem. Soc 2001, 123, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Kitagawa T; Dey A; Lugo-Mas P; Benedict J; Kaminsky W; Solomon E; Kovacs JA A Functional Model for the Metalloenzyme Superoxide Reductase. J. Am. Chem. Soc 2006, 128, 14448–14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Shearer J; Kung I; Lovell S; Kovacs JA A Co(III) Complex in a Mixed Sulfur/Nitrogen Ligand Environment: Modeling the Substrate– and Product–Bound Forms of the Metalloenzyme Thiocyanate Hydrolase. Inorg. Chem 2000, 39, 4998–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Green MT; Dawson JH; Gray HB Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry. Science 2004, 304, 1653–1656. [DOI] [PubMed] [Google Scholar]
- (93).Coggins MK; Brines LM; Kovacs JA Synthesis and Structural Characterization of a Series of Mn(III)-OR Complexes, Including a Water-Soluble Mn(III)-OH that Promotes Aerobic Hydrogen Atom Transfer. Inorg. Chem 2013, 52, 12383–12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Behan RK; Hoffart LM; Stone KL; Krebs C; Green MT Evidence for basic ferryls in cytochrome P450. J. Am. Chem. Soc 2006, 128, 11471–11474. [DOI] [PubMed] [Google Scholar]
- (95).Sivaramakrishnan S; Ouellet H; Matsumura H; Guan S; Moenne-Loccoz P; Burlingame AL; Ortiz de Montellano PR Proximal Ligand Electron Donation and Reactivity of the Cytochrome P450 Ferric–Peroxo Anion. J. Am. Chem. Soc 2012, 134, 6673–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Shaik S; Lai W; Chen H; Wang Y The Valence Bond Way: Reactivity Patterns of Cytochrome P450 Enzymes and Synthetic Analogs. Acc. Chem. Res 2010, 43, 1154–1165. [DOI] [PubMed] [Google Scholar]
- (97).Krest CM; Silakov A; Rittle J; Yosca TH; Onderko EL; Calixto JC; Green MT Significantly shorter Fe–S bond in cytochrome P450-I is consistent with greater reactivity relative to chloroperoxidase. Nat. Chem 2015, 7, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Yosca TH; Ledray AP; Ngo J; Green MT A new look at the role of thiolate ligation in cytochrome P450. JBIC, J. Biol. Inorg. Chem 2017, 22, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.