

Abstract
Objective
To evaluate the aryl hydrocarbon receptor (AHR)-dependent transforming growth factor alpha (TGF-α)/vascular endothelial growth factor B (VEGF-B) ratio, which regulates the effects of metabolic, dietary, and microbial factors on acute and chronic CNS inflammation, as a potential marker in multiple sclerosis (MS).
Methods
TGF-α, VEGF-B, and AHR agonistic activity were determined in serum of 252 patients with relapsing-remitting (RR) MS, primary and secondary progressive MS, as well as during active disease (clinically isolated syndrome [CIS] and RRMS relapse).
Results
The TGF-α/VEGF-B ratio and AHR agonistic activity were decreased in all MS subgroups with a stable disease course as compared to controls. During active CNS inflammation in CIS and RRMS relapse, the TGF-α/VEGF-B ratio and AHR agonistic activity were increased. Conversely, in patients with minimal clinical impairment despite long-standing disease, the TGF-α/VEGF-B ratio and AHR agonistic activity were unaltered. Finally, the TGF-α/VEGF-B ratio and AHR agonistic activity correlated with neurologic impairment and time to conversion from CIS to MS.
Conclusions
The AHR-dependent TGF-α/VEGF-B ratio is altered in a subtype, severity, and disease activity–specific manner and correlates with time to conversion from CIS to MS. It may thus represent a novel marker and serve as additive guideline for immunomodulatory strategies in MS.
Classification of Evidence
This study provides Class III evidence that serum levels of AHR, TGF-α, and VEGF-B distinguish subtypes of MS and predict the severity and disease activity of MS.
Metabolic, environmental, and microbial factors modify the disease course and outcome in multiple sclerosis (MS).1,2 The ligand-activated transcription factor aryl hydrocarbon receptor (AHR) acts as an immunometabolic interface and induces ligand- and cell-type–specific transcriptional programs in the peripheral and central immune compartment.2-8 Deficiency of AHR in microglia, the resident macrophage cell population in the CNS, exacerbates disease in an animal model of MS. This effect is mediated by an imbalance of secreted mediators inversely regulated by the AHR in microglia: although ligand-induced nuclear translocation of the AHR induces the expression of transforming growth factor alpha (TGF-α), it inhibits vascular endothelial growth factor B (VEGF-B). Both cytokines act on astrocytes to indirectly promote or inhibit recovery, respectively. Reduced levels of circulating AHR ligands have been described in patients with MS and correlate with disease duration and severity as well as progressive CNS atrophy.9 Together with observations of reduced TGF-α and increased VEGF-B in chronic MS lesions, the AHR-controlled TGF-α to VEGF-B ratio may represent a meaningful marker to evaluate the disease stage and risk of progression in individual MS patients.5,6,10-13 Therefore, the objective of this study is to investigate whether the AHR-dependent TGF-α/VEGF-B ratio is altered in patients with MS and whether it correlates with prognostic parameters.
Here, we demonstrate that the AHR-dependent TGF-α/VEGF-B ratio is altered in a subtype, severity, and disease activity–specific manner in serum of patients with MS, providing a signature specific to defined disease stages. It is important that changes in the AHR-dependent TGF-α/VEGF-B ratio correlate with time to conversion from clinically isolated syndrome (CIS) to MS and thus may help to predict the course of individual patients. The AHR-dependent TGF-α/VEGF-B ratio may thus serve as a prognostic marker, representing the interface between environmental, microbial, and metabolic factors and local immune reactions in the CNS. Future studies are warranted to test its suitability as a potential parameter to improve monitoring and treatment in MS.
Methods
Primary Research Questions
The primary research questions of this study were to analyze whether serum levels of AHR agonistic activity, TGF-α, VEGF-B, and/or the TGF-α/VEGF-B ratio differ between controls and patients with MS as well as between different MS subgroups. Furthermore, we wanted to test whether serum levels of AHR agonistic activity correlate with the TGF-α/VEGF-B ratio and to investigate whether these parameters correlate with the Expanded Disability Status Scale (EDSS) in patients with MS. Finally, we addressed the question of whether serum levels of AHR agonistic activity correlate with time to definite MS diagnosis in patients with CIS. This study provides Class III evidence to the above-mentioned primary research questions.
Samples
All serum samples were collected and stored at −80°C using a standardized protocol. Samples were collected at the Biobank of the Department of Neurology at Technical University of Munich, Germany, which is part of the Joint Biobank Munich in the framework of the German Biobank Node from 2010 to 2019. MS subgroups were classified according to the 2017 McDonald MS Diagnostic Criteria.14 Patients with autoimmune or inflammatory diseases besides MS such as infections, bowel diseases, or a reduced kidney function as well as patients receiving acute MS relapse treatment such as steroids were excluded from the study. Benign MS was defined as patients with an EDSS of 0 despite a disease course of RRMS for 5 years or longer to ensure a relevant disease duration. Patients were followed up regularly in the university hospital's MS outpatient clinic and were evaluated by neurologists. Only patients with complete clinical data sets regarding the analyzed parameters were included into the study. Cohorts of patients with admission due to primary headache or idiopathic intracranial hypertension but no other neurologic or inflammatory condition served as age- and sex-matched controls. All available biobank samples meeting the above-mentioned inclusion criteria were included into the analyses. Clinical records of 1,448 potentially eligible patients with MS were reviewed and examined for eligibility. In total, 252 patients met all inclusion criteria and were included. Of 227 potentially eligible and examined control patients, 43 were confirmed eligible as age- and sex-matched controls and included into the study.
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the standing ethical committee of the Klinikum rechts der Isar. All participants provided written informed consent within the framework of the Biobank resources at the Department of Neurology at Technical University of Munich, Germany, which is part of the national competence network MS.
Quantitative Determination of TGF-α and VEGF-B Levels in Serum by ELISA
Commercial TGF-α ELISA Kit (EHTGF-Α, Thermo Scientific) and VEGF-B ELISA Kit (CSB-E04758h, CUSABIO) were used for the quantitative determination of TGF-α and VEGF-B levels. The TGF-α to VEGF-B ratio was determined by dividing patients' serum TGF-α levels by their respective VEGF-B levels.
Determination of AHR agonistic activity in serum was performed as described before.10,11 Twenty thousand HEK293 cells per well were plated in 96-well flat bottom plates. Twenty-four hours after plating, cells were transfected with equal amounts of pGud-Luc15 and pTK-Renilla (Renilla luciferase under control of constitutively active thymidine kinase promoter, Promega) using FuGENE-HD Transfection Reagent (Promega) as suggested by the manufacturer. Twenty-four hours later, transfected cells were incubated with Dulbecco's Modified Eagle's Medium supplemented with 10% of patient serum in duplicates. Luciferase activity was analyzed 24 hours later using the Dual-Luciferase Reporter System (Promega). Firefly luciferase activity was divided by Renilla luciferase activity, which was normalized to control levels set to 100%.
Statistical analysis was performed using Prism software (GraphPad Prism 8.4, San Diego, CA) or IBM SPSS Statistics 27.0.0.0, and detailed analysis descriptions are provided in the figure legends. The data did not follow a normal distribution (D'Agostino-Pearson normality test was performed for each set of data); therefore, nonparametric tests were used. All p values were corrected for multiple testing using the Dunn multiple comparison test.
Data Availability
Anonymized data will be shared by request to any qualified investigator.
Results
Decreased TGF-α/VEGF-B Ratio and AHR Agonistic Activity in Patients With MS
To investigate the role of TGF-α and VEGF-B in different subtypes of MS, we quantified serum levels of TGF-α and VEGF-B in patients with stable relapsing-remitting MS (RRMS), primary progressive MS (PPMS), and secondary progressive MS (SPMS) as well as age- and sex-matched controls (Table 1). TGF-α was reduced in patients with RRMS (Figure 1A) and SPMS (Figure 1B), but not in PPMS (Figure 1B). Conversely, VEGF-B levels were increased in patients with SPMS, but not in RRMS and PPMS (Figure 1, C and D). To integrate these observations by accounting for these formally independent parameters, we determined the TGF-α/VEGF-B ratio, which we had shown previously to be decreased in chronic MS lesions and to represent a marker for neurodegeneration and astrogliosis.6,9 Indeed, the TGF-α/VEGF-B ratio was diminished in patients with RRMS and SPMS, but not in PPMS (Figure 1, E and F), and exhibited enhanced statistical significance levels when compared with the individual parameters, suggesting an additive value of using this ratio instead of individual parameters alone.
Table 1.
Characteristics of Patients With MS and Controls

Figure 1. TGF-α/VEGF-B Ratio and AHR Agonistic Activity Are Decreased in Patients With MS.

TGF-α (A, B) and VEGF-B (C, D) levels as well as AHR agonistic activity (G, H) in serum samples of patients with RRMS in remission (n = 98), SPMS (n = 44), PPMS (n = 36), and their respective controls (A, C, E, G: n = 43; B, D, F, H: n = 16) were assessed. TGF-α and VEGF-B levels were measured in pg/mL using a human TGF-α or VEGF-B ELISA. TGF-α/VEGF-B ratio (E, F) was determined by dividing TGF-α levels by VEGF-B levels. An AHR ligand–sensitive luciferase assay was used. Relative activity was calculated by dividing firefly luciferase activity (pGud-Luc) by Renilla luciferase activity (pTK-Renilla). Values are means of duplicate measurements. Lines represent median and interquartile range. Significance levels were derived using nonparametric tests (Mann-Whitney test and Kruskal-Wallis test with the Dunn multiple comparison test correcting for multiple comparisons). ****p < 0.0001, ***p < 0.001, **p < 0.001, *p < 0.01, and ns = nonsignificant. AHR = aryl hydrocarbon receptor; MS = multiple sclerosis; PPMS = primary progressive MS; RRMS = relapsing-remitting MS; SPMS = secondary progressive MS; TGF-α = transforming growth factor alpha; VEGF-B = vascular endothelial growth factor B.
The expression of TGF-α and VEGF-B is influenced by the extent of AHR activation in a given cell type.6 Thus, we studied the levels of AHR ligands in our MS and control cohorts using a cell-based AHR ligand activity–responsive reporter system.11,15-17 In agreement with previous studies, we detected a global decrease in AHR agonistic activity in all MS groups compared with controls (Figure 1, G and H). Of note, AHR ligand levels were not influenced by age (eFigure 1A, links.lww.com/NXI/A533), sex, or MS subtype (eTable 1, links.lww.com/NXI/A532).
Together, AHR agonistic activity was decreased in patients with MS concomitantly with a decreased TGF-α/VEGF-B ratio. To integrate these observations, the TGF-α/VEGF-B ratio was plotted against AHR agonistic activity. Indeed, a high TGF-α/VEGF-B ratio correlated with elevated AHR agonistic activity in all patient and control samples (eFigure 1C, links.lww.com/NXI/A533). Thus, the TGF-α/VEGF-B ratio and AHR agonistic activity intercorrelate and are decreased in patients with MS as compared to controls, supporting a role for the AHR-dependent TGF-α/VEGF-B ratio in MS.
TGF-α/VEGF-B Ratio and AHR Agonistic Activity Are Increased in MS Relapses
Because AHR ligands and AHR agonistic activity have been shown to be increased during acute inflammation,11,18,19 we hypothesized that this might translate into increased TGF-α/VEGF-B ratios during active CNS inflammation. We thus analyzed an additional group of patients with RRMS relapse before treatment with corticosteroids. Relapse was defined as experiencing a new neurologic symptom explained by a new CNS lesion, persisting over at least 24 hours and not accompanied by an increased temperature or other causal condition. Indeed, during active CNS inflammation in patients with RRMS relapse, TGF-α (Figure 2A) was increased, whereas VEGF-B levels were unaltered (Figure 2B). Conversely, the TGF-α/VEGF-B ratio (Figure 2C) and AHR agonistic activity (Figure 2D) were increased as compared to RRMS in remission. To further corroborate these observations, we analyzed an additional cohort of untreated patients with CIS, the first clinical episode of neurologic symptoms caused by inflammation or demyelination in the CNS, which does not fulfill the diagnostic criteria for MS but converts into MS frequently.14 Similar to our observation in RRMS during relapse, TGF-α (Figure 3A) showed a tendency toward an increase, whereas VEGF-B (Figure 3B) levels were decreased. These opposing observations were further strengthened in a significant increase in the TGF-α/VEGF-B ratio in patients with RRMS with acute relapse (Figure 3C). Finally, AHR agonistic activity was elevated in patients with CIS as compared to patients with RRMS in remission (Figure 3D). Thus, acute inflammatory attacks correlate with increases in the TGF-α/VEGF-B ratio and AHR ligand levels, which might constitute a counterregulatory mechanism elicited in acute CNS inflammation.
Figure 2. TGF-α/VEGF-B Ratio and AHR Agonistic Activity Are Upregulated in RRMS During Relapse Compared With RRMS in Remission.

TGF-α (A) and VEGF-B (B) levels as well as AHR agonistic activity (D) were assessed in serum samples of patients with RRMS in remission (n = 98), RRMS during relapse (n = 54), and controls (n = 44). TGF-α and VEGF-B levels were measured in pg/mL using a human TGF-α or VEGF-B ELISA. TGF-α/VEGF-B ratio (C) was determined by dividing TGF-α levels by VEGF-B levels. An AHR ligand–sensitive luciferase assay was used. Relative activity was calculated by dividing firefly luciferase activity (pGud-Luc) by Renilla luciferase activity (pTK-Renilla). Values are means of duplicate measurements. Lines represent median and interquartile range. Significance levels were derived using the nonparametric Kruskal-Wallis test with the Dunn multiple comparison test correcting for multiple comparisons. ****p < 0.0001, ***p < 0.001, **p < 0.001, *p < 0.01, and ns = nonsignificant. AHR = aryl hydrocarbon receptor; RRMS = relapsing-remitting multiple sclerosis; TGF-α = transforming growth factor alpha; VEGF-B = vascular endothelial growth factor B.
Figure 3. TGF-α/VEGF-B Ratio and AHR Agonistic Activity Are Upregulated in Clinically Isolated Syndrome Compared With RRMS in Remission.

TGF-α (A) and VEGF-B (B) levels as well as AHR agonistic activity (D) were assessed in serum samples of patients with RRMS in remission (n = 98), CIS (n = 20), and controls (n = 44). TGF-α and VEGF-B levels were measured in pg/mL using a human TGF-α or VEGF-B ELISA. TGF-α/VEGF-B ratio (C) was determined by dividing TGF-α levels by VEGF-B levels. An AHR ligand–sensitive luciferase assay was used. Relative activity was calculated by dividing firefly luciferase activity (pGud-Luc) by Renilla luciferase activity (pTK-Renilla). Values are means of duplicate measurements. Lines represent median and interquartile range. Significance levels were derived using the nonparametric Kruskal-Wallis test with the Dunn multiple comparison test correcting for multiple comparisons. ****p < 0.0001, ***p < 0.001, **p < 0.001, *p < 0.01, and ns = nonsignificant. AHR = aryl hydrocarbon receptor; CIS = clinically isolated syndrome; RRMS = relapsing-remitting multiple sclerosis; TGF-α = transforming growth factor alpha; VEGF-B = vascular endothelial growth factor B.
Unaffected TGF-α/VEGF-B Ratio and AHR Agonistic Activity in Patients With Benign MS
Benign MS is defined by a mild disease subtype characterized by low disability levels as reflected by the EDSS despite long-standing disease. Because elevated TGF-α/VEGF-B ratios and AHR agonistic activity promote anti-inflammatory effects in an animal model of MS,1,6,13,20 we hypothesized that patients with benign MS might have unaffected TGF-α/VEGF-B ratios and AHR agonistic activity. Therefore, we analyzed serum samples of RRMS patients with benign MS (Table 1) with an EDSS of 0 despite long-standing disease. Indeed, levels of TGF-α, VEGF-B, the TGF-α/VEGF-B ratio, and AHR agonistic activity were unaltered compared with controls (eFigure 2, links.lww.com/NXI/A534). Collectively, patients with benign MS exhibited no changes in the TGF-α/VEGF-B ratio or AHR agonistic activity as compared to controls.
TGF-α/VEGF-B Ratio and AHR Ligand Levels Correlate With Disease Severity
Because our previous studies in serum samples of patients with MS as well as in the animal model of MS had suggested a protective role of AHR ligand levels in autoimmune CNS inflammation, we sought to determine whether the TGF-α/VEGF-B ratio and AHR net activity were linked to disease severity. Indeed, both elevated TGF-α/VEGF-B ratios (Figure 4A) and increases in AHR agonistic activity (Figure 4B) correlated with lower disease severity as determined by the EDSS.
Figure 4. TGF-α/VEGF-B Ratio and AHR Agonistic Activity Correlate With the EDSS in RRMS.

TGF-α/VEGF-B ratio (A) and AHR agonistic activity (B) in serum samples of patients with RRMS in remission (n = 98) were assessed. TGF-α and VEGF-B levels were measured in pg/mL using a human TGF-α or VEGF-B ELISA. TGF-α/VEGF-B ratio was determined by dividing TGF-α levels by VEGF-B levels. An AHR ligand–sensitive luciferase assay was used. Relative activity was calculated by dividing firefly luciferase activity (pGud-Luc) by Renilla luciferase activity (pTK-Renilla). Values are presented as means of duplicate measurements. Line shows linear regression of the TGF-α/VEGF-B ratio or AHR agonistic activity and patients' EDSS. Significance levels were derived using Spearman correlation analysis (A: Spearman r = −0.2427; R2 = 0.059; B: Spearman r = −0.2433; R2 = 0.059). AHR = aryl hydrocarbon receptor; EDSS = Expanded Disability Status Scale; RRMS = relapsing-remitting multiple sclerosis; TGF-α = transforming growth factor alpha; VEGF-B = vascular endothelial growth factor B.
AHR Ligand Levels Determine Disease Severity in MS and Time to Conversion in CIS
Having correlated the AHR-dependent TGF-α/VEGF-B ratio with measures of disease severity and activity in CIS and RRMS and observed high TGF-α/VEGF-B ratios and AHR agonistic activity in patients with low EDSS scores, we aimed to investigate whether these parameters also affected the risk of conversion from CIS to definite MS, a clinically important parameter to stratify patients in need of early disease-modifying therapy. Indeed, high AHR agonistic activity during acute CIS was associated with a delayed conversion to RRMS (Figure 5). Together, our findings suggest that determining the status of the AHR-dependent TGF-α/VEGF-B ratio may represent a tool to be used in concert with MRI and additional clinical and paraclinical parameters to determine the need for more aggressive immunotherapies in patients presenting with a first inflammatory episode in CIS.
Figure 5. AHR Agonistic Activity Correlates With Time to Conversion From CIS to MS.
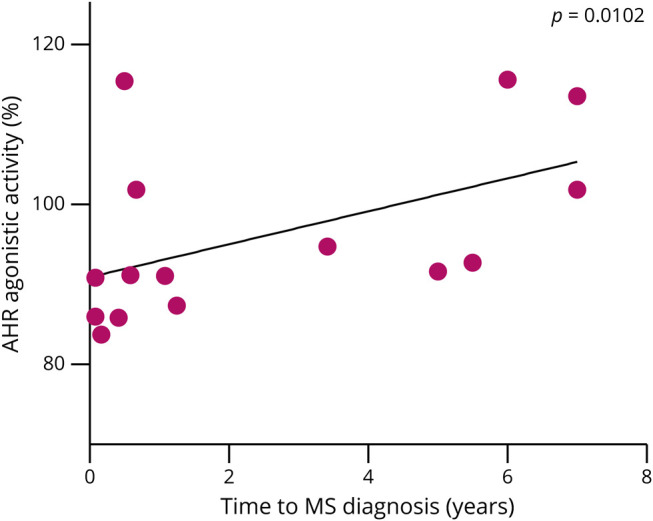
AHR agonistic activity in serum samples of patients with CIS who later converted into RRMS (n = 15) was assessed. An AHR ligand–sensitive luciferase assay was used. Relative activity was calculated by dividing firefly luciferase activity (pGud-Luc) by Renilla luciferase activity (pTK-Renilla). Values are means of duplicate measurements. Line shows linear regression of AHR agonistic activity and patients' time to RRMS diagnosis. Significance levels were derived using Spearman correlation analysis (Spearman r = 0.6512; R2 = 0.424). AHR = aryl hydrocarbon receptor; CIS = clinically isolated syndrome; MS = multiple sclerosis; RRMS = relapsing-remitting multiple sclerosis.
TGF-α/VEGF-B Ratio and AHR Agonistic Activity as Potential Disease Markers
To quantify the association of the performed assays with diagnostic status, we performed multivariable logistical regression analyses in our patient cohorts.
First, we used logistic regression (eTable 2, links.lww.com/NXI/A532) to analyze the association of the AHR agonistic activity and the TGF-α/VEGF-B ratio with MS case/control status. All 3 MS subtypes (RRMS, SPMS, and PPMS) were combined into the case group. Here, serum levels of TGF-α, VEGF-B, and AHR agonistic activity were used as predictors and age and sex as covariates. The model correctly classified 81% of the cases, was statistically significant, χ2 = 50.15, p < 0.001, and explained 32.4% of the variance (Nagelkerke's pseudo-R2). All 3 factors (TGF-α, VEGF-B, and AHR agonistic activity) contributed significantly to the model. The overall sensitivity of this model was 84.0%, with a specificity of 52.3%. Age was statistically significant (p = 0.032) because the SPMS and PPMS subgroups were older than the RRMS and control groups.
Second, we used multinomial logistic regression (eTable 3, links.lww.com/NXI/A532) to analyze the association of the 3 assays with the 3 MS subgroups (RRMS, SPMS, and PPMS). As before, serum levels of TGF-α, VEGF-B, and AHR agonistic activity were used as predictors and age and sex as covariates. Because patients with SPMS and PPMS were older than patients with RRMS, the association of age was significant. The overall multinomial logistic regression model was statistically significant, χ2 = 110.152, p < 0.0001. The model explained 53.4% (Nagelkerke's pseudo-R2) of the variance and correctly classified 68.0% of the cases into their respective subgroups. TGF-α and AHR agonistic activity contributed significantly to the model. Together, these analyses suggest that the AHR-dependent TGF-α/VEGF-B ratio may represent a meaningful combinatorial parameter to distinguish controls from patients with MS and to stratify patients with MS into their respective disease subtypes. Of note, future studies are needed to verify these correlations in larger cohorts and to test the suitability of these parameters as novel markers in MS.
Discussion
In previous studies, we have reported that lower AHR ligand levels in MS are linked to increased disease severity, progressive CNS atrophy, and an increasing lesion load.9 Furthermore, we have found that the activation of AHR promotes an increase in the TGF-α/VEGF-B ratio in microglia and induces tissue regenerative effects in an animal model of autoimmune CNS inflammation.6 However, AHR ligand agonistic activity is globally decreased in patients with MS,11 which may lead to hypoactivation of AHR and contribute to tissue degeneration in MS. In this context, the AHR-controlled TGF-α/VEGF-B ratio has not been examined so far. Here, we analyzed the TGF-α/VEGF-B ratio and AHR agonistic activity in serum of patients with MS to assess the validity of a putative AHR-dependent TGF-α–VEGF-B correlation in MS. We found a corresponding decrease in the TGF-α/VEGF-B ratio and AHR agonistic activity in patients with RRMS and progressive MS compared with controls. During acute CNS inflammation, such as in CIS or RRMS relapses, upregulation of both the TGF-α/VEGF-B ratio and AHR agonistic activity was observed. Finally, patients with a low EDSS exhibited higher TGF-α/VEGF-B ratios than those with more severe disease throughout all RRMS stages, and AHR ligand levels in patients with CIS correlated with the time to conversion to definitive MS. Thus, the AHR-controlled TGF-α/VEGF-B ratio is decreased in MS, correlates with disease severity and activity, and may thus provide additive information to existing markers in MS.
Reasons for decreased AHR agonistic activity in patients with MS, such as genetic variants possibly altering biochemical pathways involved in the uptake and metabolism of AHR ligands21-23 as well as gut microbial factors,24-26 have been outlined in detail before.11,18,27 However, little is known about the conditions that regulate the TGF-α/VEGF-B ratio in patients with MS. So far, it has been shown that microbial metabolites signaling through the microglial AHR regulate the production of TGF-α and VEGF-B in an animal model of experimental autoimmune encephalomyelitis mediated by AHR-responsive elements in the TGF-α and VEGF-B promoters.6 In our study, increased AHR agonistic activity was linked to an increased TGF-α/VEGF-B ratio in human serum samples in line with those observations, suggesting the regulation of the TGF-α/VEGF-B ratio by the AHR in humans as well.
Inflammation has been shown to promote AHR agonistic activity in patients with MS, partly because of an increased production of AHR ligands.11,28,29 Accordingly, we detected a consensual increase in the AHR agonistic activity and in the TGF-α/VEGF-B ratio in patients with acute CNS inflammation during RRMS relapse or CIS. However, the roles of TGF-α and VEGF-B in acute CNS inflammation have not been elucidated. TGF-α is a transforming growth factor expressed in multiple organs as well as in glial and neuronal cells in the CNS.30 In our previous studies, we have shown that TGF-α decreases the expression of proinflammatory chemokines (CCL2 and CSF2) and cytokines (IL-6) induced by TNF-alpha and IL-1β in murine astrocytes.6 By contrast, VEGF-B promotes the expression of proinflammatory CCL2 and neurotoxic NOS2.6 Although the AHR seems to be an important regulator of TGF-α and VEGF-B during CNS inflammation, it remains unclear to what extent other transcription factors binding to the promoters of TGF-α or VEGF-B might play a role during acute inflammation. So far, little is known about transcription factors regulating VEGF-B expression besides AHR because studies showed that growth factors, hypoxia, or hormones do not regulate VEGF-B expression.31,32 However, regulation of TGF-α expression has been reviewed before.30 Among other factors, TGF-α expression is promoted by glucocorticoids, thyroid hormone,33 and estrogens.34 Therefore, physiologic changes, inflammation, exogenous hormone administration, and patients' individual hormone levels could modulate TGF-α levels. Hence, endogenously increased glucocorticoid levels during acute inflammation could contribute to higher TGF-α/VEGF-B ratios observed in patients during RRMS relapses.
Indeed, to exclude MS relapse treatment as a confounding factor, none of our patients received steroid medication as an exogenous source when serum samples were drawn. Similarly, higher estrogen levels in women compared with men could account for higher TGF-α/VEGF-B ratios in females vs males observed in our study as an AHR-independent modifier because AHR agonistic activities did not differ between females and males. In these lines, higher TGF-α/VEGF-B ratios due to higher estrogen levels in females, especially during pregnancy, could contribute to a lower disease activity during pregnancy and a better MS prognosis in women compared with men. In addition, AHR ligands are induced during pregnancy,23 possibly further increasing the TGF-α/VEGF-B ratio. A possible neuroprotective role of the TGF-α/VEGF-B ratio in patients with MS is further suggested by our observations of higher TGF-α/VEGF-B ratios in patients with a low EDSS during both remission and RRMS relapses.
The nominal overall sensitivity of this model distinguishing controls from patients with MS was 84%, with a specificity of 52%. These analyses were performed in a limited number of patients and small group sizes, which is why our statistical analyses need to be reproduced and solidified in future studies including larger patient and control groups. However, considering other established markers, e.g., prostate-specific antigen in prostate cancer (sensitivity of 86% and specificity of 33%35) or oligoclonal bands in MS (sensitivity of 88% and specificity of 86% in patients with MS36), the analyses derived from our limited cohort suggest that the AHR agonistic activity and the TGF-α/VEGF-B ratio might potentially prove useful in MS, if additional and larger studies come to similar conclusions. If so, this model could be useful to predict MS subgroups (RRMS, SPMS, and PPMS) because it classified more than two-thirds of the patients correctly according to the patients' MS subtypes. Further studies and modifications, e.g., including clinical and imaging data and adding further parameters, could potentially corroborate the AHR agonistic activity and TGF-α/VEGF-B ratio as clinically useful tools in MS. In this context, when compared with the widely accepted biomarker, oligoclonal bands, they have a specificity of 86% vs the present model of just 52%.
In addition to these considerations, additional limitations of this study should be borne in mind. First, some of our cohorts, such as the group of patients with CIS, were limited in sample size. Although the groups were matched for age, sex, treatment, and disease duration, additional confounding factors such as variance in sample processing, storage conditions, or degradation of AHR ligands, TGF-α, or VEGF-B cannot be excluded. Also, differences in patients' diets, composition of gut microbiota, environmental factors, or hormone levels could also have affected AHR ligands, TGF-α, and VEGF-B levels. It is possible that endogenous and exogenous AHR agonists in concert with dietary, microbial, environmental, metabolic, and genetic variations influence AHR agonist levels and therefore modulate TGF-α and VEGF-B, thus favoring a high TGF-α/VEGF-B ratio with neuroprotective effects on the course of MS. Individual levels of AHR agonists might differ because of genetic variants in the production of AHR ligands by the patients' metabolism or supply by gut microbiota. Also, dietary intake of different substances, such as tryptophan, a source for the synthesis of AHR ligands, might also play a role as a modifying factor in MS.5,24,37-40 Furthermore, the extent of an individual's capacity to upregulate AHR agonistic activity and the TGF-α/VEGF-B ratio during acute inflammation might be related to the efficiency in controlling immunopathology in MS lesions. Similarly, higher levels of TGF-α in women could contribute to better disease outcomes compared with male patients with MS. Finally, limited sample sizes in our study need to be taken into account when considering biologically meaningful as opposed to statistically significant results. However, given our previous studies on AHR as a potential marker of disease course in MS, the changes determined for AHR and the TGF-α/VEGF-B ratio in this study may represent both significant and biologically meaningful parameters in MS.
To further evaluate the relevance of AHR ligands and the TGF-α/VEGF-B ratio in MS, future studies such as longitudinal patient analyses and assessment of therapeutic potential of modulation of these factors in animal models are needed. Considering available data so far, the AHR-dependent TGF-α/VEGF-B ratio may give new insights into the mechanisms by which multiple external and internal factors modulate the disease course and may represent potentially druggable targets in MS.
Glossary
- AHR
aryl hydrocarbon receptor
- CIS
clinically isolated syndrome
- EDSS
Expanded Disability Status Scale
- MS
multiple sclerosis
- PPMS
primary progressive MS
- RRMS
relapsing-remitting MS
- SPMS
secondary progressive MS
- TGF-α
transforming growth factor alpha
- VEGF-B
vascular endothelial growth factor B
Appendix. Authors
Contributor Information
Ana Cirac, Email: a.cirac@tum.de.
Thanos Tsaktanis, Email: thanos.tsaktanis@uk-erlangen.de.
Tobias Beyer, Email: t.beyer@tum.de.
Mathias Linnerbauer, Email: mathias.linnerbauer@fau.de.
Till Andlauer, Email: till.andlauer@tum.de.
Verena Grummel, Email: v.grummel@tum.de.
Lucy Nirschl, Email: lucy.nirschl@tum.de.
Lena Loesslein, Email: lena.loesslein@uk-erlangen.de.
Francisco J. Quintana, Email: fquintana@rics.bwh.harvard.edu.
Bernhard Hemmer, Email: hemmer@tum.de.
Study Funding
A.C. and T.T. were funded by the Clinical Research Council (Kommission für Klinische Forschung [KKF]), Klinikum rechts der Isar. T.T. received a SEED fellowship provided by the Disease-related Competence Network Multiple Sclerosis (Krankheitsbezogenes Kompetenznetz Multiple Sklerose [KKNMS]). V.R. was supported by a Heisenberg fellowship, supply support (Sachmittelbeihilfe), and transregional research center support provided by the German Research Foundation (DFG, RO4866-3/1, RO4866-4/1, SFB TRR274) as well as a European Research Council (ERC) Starting Grant provided by the European Union's Horizon 2020 Research and Innovation Program (851693 HICI). B.H. received funding for the study by the European Union's Horizon 2020 Research and Innovation Program (grant MultipleMS, EU RIA 733161) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy—ID 390857198). B. Hemmer and T. Andlauer are associated with DIFUTURE (Data Integration for Future Medicine, BMBF 01ZZ1804[A-I]). The Biobank of the Department of Neurology as part of the Joint Biobank Munich in the framework of the German Biobank Node supported the study.
Disclosure
The authors report no disclosures. The study is not industry sponsored. Go to Neurology.org/NN for full disclosures.
References
- 1.International Multiple Sclerosis Genetics Consortium. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476(7359):214-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rothhammer V, Quintana FJ. Environmental control of autoimmune inflammation in the central nervous system. Curr Opin Immunol. 2016;43:46-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol. 2019;19(3):184-197. [DOI] [PubMed] [Google Scholar]
- 4.Wheeler MA, Jaronen M, Covacu R, et al. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell. 2019;176(3):581-596.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothhammer V, Mascanfroni ID, Bunse L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22(6):586-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothhammer V, Borucki DM, Tjon EC, et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018;557(7707):724-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mascanfroni ID, Takenaka MC, Yeste A, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med. 2015;21(6):638-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zelante T, Iannitti RG, Cunha C, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39(2):372-385. [DOI] [PubMed] [Google Scholar]
- 9.Tsaktanis T, Beyer T, Nirschl L, et al. Aryl hydrocarbon receptor plasma agonist activity correlates with disease activity in progressive MS. Neurol Neuroimmunol Neuroinflamm. 2021;8(2):e933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rothhammer V, Borucki DM, Kenison JE, et al. Detection of aryl hydrocarbon receptor agonists in human samples. Sci Rep. 2018;8(1):4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothhammer V, Borucki DM, Garcia Sanchez MI, et al. Dynamic regulation of serum aryl hydrocarbon receptor agonists in MS. Neurol Neuroimmunol Neuroinflamm. 2017;4(4):e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaetani L, Boscaro F, Pieraccini G, et al. Host and microbial tryptophan metabolic profiling in multiple sclerosis. Front Immunol. 2020;11:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdullah A, Maged M, Hairul-Islam M I, et al. Activation of aryl hydrocarbon receptor signaling by a novel agonist ameliorates autoimmune encephalomyelitis. PLoS One. 2019;14(4):e0215981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173. [DOI] [PubMed] [Google Scholar]
- 15.Garrison PM, Tullis K, Aarts JM, Brouwer A, Giesy JP, Denison MS. Species-specific recombinant cell lines as bioassay systems for the detection of 2,3,7,8-tetrachlorodibenzo-p-dioxin-like chemicals. Fundam Appl Toxicol. 1996;30(2):194-203. [DOI] [PubMed] [Google Scholar]
- 16.He G, Tsutsumi T, Zhao B, et al. Third-generation Ah receptor-responsive luciferase reporter plasmids: amplification of dioxin-responsive elements dramatically increases CALUX bioassay sensitivity and responsiveness. Toxicol Sci. 2011;123(2):511-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanderson JT, Aarts JM, Brouwer A, Froese KL, Denison MS, Giesy JP. Comparison of Ah receptor-mediated luciferase and ethoxyresorufin-O-deethylase induction in H4IIE cells: implications for their use as bioanalytical tools for the detection of polyhalogenated aromatic hydrocarbons. Toxicol Appl Pharmacol. 1996;137(2):316-325. [DOI] [PubMed] [Google Scholar]
- 18.Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol. 2014;32:403-432. [DOI] [PubMed] [Google Scholar]
- 19.Quintana FJ. The aryl hydrocarbon receptor: a molecular pathway for the environmental control of the immune response. Immunology. 2013;138(3):183-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quintana FJ, Murugaiyan G, Farez MF, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107(48):20768-20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lanz TV, Williams SK, Stojic A, et al. Tryptophan-2,3-Dioxygenase (TDO) deficiency is associated with subclinical neuroprotection in a mouse model of multiple sclerosis. Sci Rep. 2017;7:41271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mancuso R, Hernis A, Agostini S, et al. Indoleamine 2,3 dioxygenase (IDO) expression and activity in relapsing-remitting multiple sclerosis. PLoS One. 2015;10(6):e0130715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu WH, Lu CZ, Huang YM, Link H, Xiao BG. A putative mechanism on remission of multiple sclerosis during pregnancy: estrogen-induced indoleamine 2,3-dioxygenase by dendritic cells. Mult Scler. 2007;13(1):33-40. [DOI] [PubMed] [Google Scholar]
- 24.Lamas B, Richard ML, Leducq V, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. 2016;22(6):598-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jangi S, Gandhi R, Cox LM, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. 2016;7:12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S, da Cunha AP, Rezende RM, et al. The host shapes the gut microbiota via fecal MicroRNA. Cell Host Microbe. 2016;19(1):32-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gutierrez-Vazquez C, Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity. 2018;48(1):19-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Platten M, Ho PP, Youssef S, et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science. 2005;310(5749):850-855. [DOI] [PubMed] [Google Scholar]
- 29.Bessede A, Gargaro M, Pallotta MT, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature. 2014;511(7508):184-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Junier MP. What role(s) for TGFalpha in the central nervous system?. Prog Neurobiol. 2000;62(5):443-473. [DOI] [PubMed] [Google Scholar]
- 31.Enholm B, Paavonen K, Ristimäki A, et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene. 1997;14(20):2475-2483. [DOI] [PubMed] [Google Scholar]
- 32.Lal N, Puri K, Rodrigues B. Vascular endothelial growth factor B and its signaling. Front Cardiovasc Med. 2018;5:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raja RH, Paterson AJ, Shin TH, Kudlow JE. Transcriptional regulation of the human transforming growth factor-alpha gene. Mol Endocrinol. 1991;5(4):514-520. [DOI] [PubMed] [Google Scholar]
- 34.Bates SE, Davidson NE, Valverius EM, et al. Expression of transforming growth factor alpha and its messenger ribonucleic acid in human breast cancer: its regulation by estrogen and its possible functional significance. Mol Endocrinol. 1988;2(6):543-555. [DOI] [PubMed] [Google Scholar]
- 35.Hoffman RM, Gilliland FD, Adams-Cameron M, Hunt WC, Key CR. Prostate-specific antigen testing accuracy in community practice. BMC Fam Pract. 2002;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwenkenbecher P, Sarikidi A, Wurster U, et al. McDonald criteria 2010 and 2005 compared: persistence of high oligoclonal band prevalence despite almost doubled diagnostic sensitivity. Int J Mol Sci. 2016;17(9):1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zang X, Zheng X, Hou Y, et al. Regulation of proinflammatory monocyte activation by the kynurenine-AhR axis underlies immunometabolic control of depressive behavior in mice. FASEB J. 2018;32(4):1944-1956. [DOI] [PubMed] [Google Scholar]
- 38.Seok SH, Ma ZX, Feltenberger JB, et al. Trace derivatives of kynurenine potently activate the aryl hydrocarbon receptor (AHR). J Biol Chem. 2018;293(6):1994-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Natividad JM, Agus A, Planchais J, et al. Impaired aryl hydrocarbon receptor ligand production by the gut microbiota is a key factor in metabolic syndrome. Cell Metab. 2018;28(5):737-749.e4. [DOI] [PubMed] [Google Scholar]
- 40.Schiering C, Wincent E, Metidji A, et al. Feedback control of AHR signalling regulates intestinal immunity. Nature. 2017;542(7640):242-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request to any qualified investigator.