

Abstract
Background
The developing hippocampus and cerebellum, unique among brain regions, exhibit a secondary surge in neurogenesis during the third trimester of pregnancy. Ethanol (EtOH) exposure during this period is results in a loss of tissue volume and associated neurobehavioral deficits. However, mechanisms that link EtOH exposure to teratology in these regions are not well understood. We therefore analyzed transcriptomic adaptations to EtOH exposure to identify mechanistic linkages.
Methods
Hippocampi and cerebella were microdissected at postnatal day (P)10, from control C57BL/6J mouse pups, and pups treated with 4 g/kg of EtOH from P4 to P9. RNA was isolated and RNA‐seq analysis was performed. We compared gene expression in EtOH‐ and vehicle‐treated control neonates and performed biological pathway‐overrepresentation analysis.
Results
While EtOH exposure resulted in the general induction of genes associated with the S‐phase of mitosis in both cerebellum and hippocampus, overall there was little overlap in differentially regulated genes and associated biological pathways between these regions. In cerebellum, EtOH additionally induced gene expression associated with the G2/M‐phases of the cell cycle and sonic hedgehog signaling, while in hippocampus, EtOH‐induced the pathways for ribosome biogenesis and protein translation. Moreover, EtOH inhibited the transcriptomic identities associated with inhibitory interneuron subpopulations in the hippocampus, while in the cerebellum there was a more pronounced inhibition of transcripts across multiple oligodendrocyte maturation stages.
Conclusions
These data indicate that during the delayed neurogenic period, EtOH may stimulate the cell cycle, but it otherwise results in widely divergent molecular effects in the hippocampus and cerebellum. Moreover, these data provide evidence for region‐ and cell‐type‐specific vulnerability, which may contribute to the pathogenic effects of developmental EtOH exposure.
Keywords: cerebellum, fetal alcohol spectrum disorders, hippocampus, postnatal ethanol, RNA‐seq
Comparative RNAseq analyses of developing mouse cerebellum and hippocampus identified common and unique actions of ethanol during their shared secondary neurogenic period for granule cell formation. In both regions, ethanol significantly induced cell‐cycle genes associated with S‐phase. However, ethanol preferentially altered oligodendrocyte lineage and microglia transcripts in cerebellum, while inhibiting inhibitory interneuron lineage genes in hippocampus. These effects, accompanied by effects on divergent molecular pathways, suggest significant region and cell‐type specific vulnerabilities to ethanol which may contribute to neurodevelopmental disorders.
INTRODUCTION
Heavy alcohol exposure during pregnancy is common (Bakhireva et al., 2017; SAMHSA, 2013) and can reprogram fetal development, resulting in a complex combination of anatomical anomalies and growth and neurobehavioral deficits that persist throughout an individual's lifetime (Wozniak, Riley, & Charness, 2019). These anomalies and deficits, collectively termed fetal alcohol spectrum disorders (FASD), range along a continuum of severity (Williams & Smith, 2015), and can be diagnosed in between 1% and 5% of school‐aged children in the United States (May et al., 2018). Moreover, FASD often results in the emergence of secondary disabilities (Moore & Riley, 2015), with great annual social, healthcare, and economic costs to individuals and society (Greenmyer et al., 2018).
Ethanol (EtOH) has profound effects on the developing central nervous system, resulting in widespread neuropathology, and consequently, deficits in learning and memory, impulse control, and motor function, as well as increased risk of addiction later in life (Carter et al., 2007; Glass et al., 2013; Norman et al., 2009; Streissguth et al., 2004). The vulnerability of the developing cerebellum and hippocampus to EtOH is well documented. Imaging studies demonstrate reduced cerebellum volume and white matter abnormalities which likely contribute to motor deficits including those in balance, coordination, and equilibrium as well as cerebellar‐dependent learning and memory deficits in individuals with FASD (Lebel et al., 2008; Norman et al., 2009; Riley & McGee, 2005). Similarly, EtOH reduces hippocampal volume in individuals with FASD, contributing to impaired hippocampal‐dependent learning and memory (Norman et al., 2009; Willoughby et al., 2008).
The cerebellum and hippocampus are unique among brain regions in that both undergo a late, secondary surge in histogenesis in the early postnatal period in rodents, which is equivalent to the third trimester of pregnancy in humans (Bayer et al., 1993; Clancy, Darlington, & Finlay, 2001). This period of development, characterized as the brain growth spurt, includes a number of critical maturation events, such as a late phase of neurogenesis, cell migration, and synaptogenesis, as well as oligodendrocyte maturation and myelination, all of which may be altered by EtOH exposure (Camarillo & Miranda, 2008; Rice & Barone, 2000; Santillano et al., 2005; Wilhelm & Guizzetti, 2015).
Rodent studies have shown that EtOH exposure during this early postnatal, third‐trimester‐equivalent period results in decreased cerebellar volume and Purkinje cell number and a delay in cellular development (Goodlett & Johnson, 1997; Napper & West, 1995), which is believed to contribute to motor function impairments, as well as cerebellum‐dependent learning and memory deficits (Diaz et al., 2014; Klintsova et al., 2002; Wagner et al., 2013). Early postnatal EtOH exposure to the hippocampus resulted in reduced hippocampal volume as well as loss of CA1, CA2, and CA3 pyramidal cells, along with a reduction in dentate gyrus granule cells (Barnes & Walker, 1981; Livy et al., 2003; West, Hamre, & Cassell, 1986). EtOH effects on the developing hippocampus were associated with hippocampal‐dependent learning and memory deficits in these animal models of FASD (Bonthius & West, 1991; Goodlett & Johnson, 1997; Zucca & Valenzuela, 2010).
Despite the importance of cerebellar and hippocampal pathology to the etiology of FASD, the mechanisms resulting in EtOH‐induced neuropathology in these late‐maturing brain regions are poorly understood. Therefore, in this study, we used a postnatal mouse model of third‐trimester‐equivalent EtOH exposure, followed by transcriptome sequencing and bioinformatic approaches to further identify pathogenic mechanisms. We report here that postnatal EtOH exposure both increased expression and decreased expression of genes in hippocampus and cerebellum. The hippocampus and cerebellum responded differentially to EtOH exposure, with pathways associated with cell cycle progression, Sonic Hedgehog, and cholesterol biosynthesis altered in the hippocampus while translation‐associated pathways were altered in the cerebellum. The expression of genes associated with the S‐phase of the cell cycle in both of these brain regions was increased. EtOH exposure also impaired the expression of genes associated with inhibitory interneuron lineages in the hippocampus and genes associated with the excitatory granule cell interneuron lineage in the cerebellum. Finally, EtOH decreased the expression of genes associated with multiple stages of oligodendrocyte maturation, but increased the expression of genes associated with microglia activation in the cerebellum. Collectively, these data indicate that despite ontogenetic similarities between hippocampus and cerebellum, the effects of developmental EtOH on these regions, with the exception of cell cycle induction, are remarkably different.
MATERIALS AND METHODS
Animals
Mice were housed in the federally approved Division of Laboratory Animal Medicine facility at the University of Arkansas for Medical Sciences (UAMS), and all animal use protocols were reviewed and approved by the UAMS Institutional Animal Care and Use Committee. Adult C57BL/6J mice were purchased from The Jackson Laboratory (stock #000664), and an in‐house breeding colony was established to produce animals for use in experiments. Individually housed pregnant dams were kept on a 10:14‐h light:dark cycle, were allowed access to food and water ad libitum for the duration of the experiments, and were checked daily for new litters with postnatal day 0 (P0) being designated as the day of birth. Experimental animals from a total of five litters were distributed among treatment groups, EtOH (E) or vehicle control (V), and were separated according to sex as evenly as possible. On P4‐9, intragastric gavage was utilized to administer EtOH at 4 g/kg/day in 20% intralipid (Fresenius Kabi) or 20% intralipid in which an equal volume of water was substituted for EtOH as a vehicle. As reported previously, peak blood EtOH concentrations using this model were 401 ± 16 (mean ± SD) mg/dl, at 90 min following EtOH administration (Drew et al., 2015). While these BECS are high, this EtOH treatment paradigm is commonly used in postnatal mouse models of FASD and importantly recapitulates much of the neuropathology observed in humans with FASD (Petrelli, Weinberg, & Hicks, 2018). Moreover, it should be noted that mice metabolize EtOH rapidly following gastric delivery, with a mean rate of elimination that is approximately five times higher than that observed for rats (Livy, Parnell, & West, 2003), another commonly used FASD model. Therefore, the functional effects of EtOH exposure are likely to be moderated in mice by rapid metabolism and elimination. Moreover, the literature documents that in humans, similar or significantly higher BECs can be attained and are well‐tolerated, at least among persons with chronic alcohol use disorders (Adachi et al., 1991), who are likely, as a group, to contribute substantially to the prevalence of FASD. Additionally, we previously demonstrated that no differences were observed in analogous endpoints in handled‐only animals, and thus, this control population was not included in this study (Drew et al., 2015; Kane et al., 2011). On P10, 24 h following the last E or V treatment, animals were anesthetized using isoflurane vapor followed by transcardial perfusion with phosphate‐buffered saline containing 5 U/ml heparin. The brain was removed, and the hippocampus and cerebellum were microdissected, flash‐frozen in liquid nitrogen, and stored at −80℃.
Isolation of RNA, RNA‐seq library preparation, and sequencing
Hippocampus and cerebellum were homogenized according to manufacturer specification using a BBX24B Bullet Blender homogenizer and 0.5‐mm glass beads (Next Advance). RNA was isolated using the RNeasy Mini Kit plus on‐column DNase Removal Kit (Qiagen). RNA quantity was assessed using the Qubit 3.0 fluorometer with the Qubit Broad‐Range RNA Assay Kit (Thermo Fisher Scientific), and an Agilent Fragment Analyzer with the Standard Sensitivity RNA Gel Kit (Agilent Technologies) was used to assess RNA quality. All samples had an RNA Quality No. ≥8.9 (Mean 9.7, Median 10.0). RNA‐seq libraries were prepared using the Illumina TruSeq mRNA Library Prep Kit with TruSeq unique dual‐indexed adapters (Illumina) and were quantified with the Qubit 1× dsDNA High‐Sensitivity NGS Gel Kit (Thermo Fisher). Libraries were further characterized for functionality with the KAPA Library Quantification Kit (Roche) and for fragment size using the Agilent Fragment Analyzer with the High‐Sensitivity NGS Gel Kit (Agilent). Library molarities were calculated followed by dilution and denaturation all according to manufacturer's specification for clustering. From the five litters treated, an initial set of three males (M) and two females (F) per treatment group were randomly selected for downstream analysis and a supplemental set of 1 M and 1 F per treatment group were taken from the same cohort of five litters for additional analysis (Litter 1, 1M/2F; Litter 2, 3M/1F; Litter 3, 1M/2F; Litter 4, 2M; and Litter 5, 1M/1F). The initial set of n = 5 vehicle and 5 EtOH‐exposed were clustered on a cBot and paired‐end sequenced on a HiSeq 3000 with a 150‐cycle SBS kit for 2X75 reads. A supplemental set of n = 2 vehicle and 2 EtOH‐exposed were clustered on a high‐output NextSeq 500 flow cell and paired‐end sequenced with a 150‐cycle SBS kit for 2X75 reads (Illumina). All data are deposited in NCBI GEO (GEO accession number GSE166196).
Bioinformatic analysis
Raw RNA‐sequence data were analyzed to identify significant differences in gene expression between the V and E treatment groups, sex‐dependent expression differences between these treatment groups, and the global biological pathways associated with disruption of these hippocampal and cerebellar genes. All reads were evaluated and trimmed of all adapter sequences and low‐quality bases using Trimmomatic read trimmer (Bolger, Lohse, & Usadel, 2014). Using Trimmomatic and the corresponding adapter sequence file for Illumina, reads were scanned with a sliding window of 5, cutting when the average quality per base drops below 20, then trimming reads at the beginning and end if base quality drops below 25, and finally dropping reads if the read length is less than 35. Reads were then mapped to the Mus musculus (mm10) genome assembly. Read mapping was performed using HISAT2 genomic analysis software platform version 2.1.0 (Kim, Langmead, & Salzberg, 2015). Transcript‐wise counts were generated using HTSeq (Anders, Pyl, & Huber, 2014). Differential gene expression tests were then performed using DESeq2 software version 2.1.8.3 following the guidelines recommended by Love and colleagues (Love, Huber, & Anders, 2014) using a sex x treatment experimental design. Batch correction was conducted in “R” statistical software using 2‐way ANOVA by including batch as a source of potential error. 20,590 genes had at least 1 read count in at least 1 sample and were processed for differential expression analysis using the regularized logs of normalized gene counts derived from DESeq2. Gene expression differences were considered significant at a Benjamini–Hochberg (BH)‐adjusted p‐value threshold of α = 0.05. We did observe a few genes whose expression was sex‐dependent, when controlling for the false discovery rate (see Table S1); however, there was no significant interaction effect between sex and treatment on differential gene expression. Therefore, our analyses focused on main effect of treatment on differential gene expression. All analyses were done on the Galaxy instance of the TAMU HPRC (https://hprcgalaxy.tamu.edu/).
Heat map and Principal Component Analysis (PCA) plots were generated from these processed data using “R” statistical software. The R‐based EnhancedVolcano package was used to make the volcano plots (Blighe et al., 2020). Pathway and network analysis were conducted using the R‐based ReactomePA package (Yu & He, 2016) and clusterProfiler (Yu et al., 2012).
To gain a better understanding of which cellular processes and cell types of the hippocampus and cerebellum are most sensitive to EtOH exposure during this time period, we leveraged publically available single‐cell RNA‐seq (scRNA‐seq) resources normally used to determine cell cycle phase of individual cells. Others have reported similar analyses using cell type‐specific markers obtained from scRNA‐seq studies in order to extrapolate cell composition of tissue used in bulk RNA‐seq (Jew et al., 2020). Cell cycle‐associated (S‐ and G2/M‐phase) gene lists were extracted from the “R” Seurat package (Stuart et al., 2019) and applied to our bulk RNA‐seq dataset. We calculated z‐scores across individual genes on the extracted gene lists and then averaged these individual gene z‐scores within each sample. The average z‐score of each sample was then used in 2‐way ANOVA analyses (sex x treatment) completed in “R.” This strategy allowed us to compare relative expression of differentially expressed genes (e.g., housekeeping genes are more highly expressed than other cell type‐specific genes) because our interest is not in absolute expression levels but rather relative expression levels as a whole for each gene list (e.g., do all genes go up or down due to treatment). This process was repeated for analysis of caspase transcripts (apoptosis‐related caspases, including caspase 2 (CASP2), CASP3/6/7/8/9/10 and the inflammation‐related caspases, CASP1/4/5/11/12; Shalini et al., 2015) and transcripts for cell type‐specific marker genes (Zeisel et al., 2018, 2015). In these analyses, we observed no effect of sex and no interaction effect between sex and treatment (see Tables S2 and S3). For this reason, we focused on main effect of treatment.
RESULTS
Differential gene expression in hippocampus and cerebellum following EtOH exposure
To gain an initial overview of transcriptomic changes induced by EtOH exposure, a PCA of the 500 most variant genes was performed on samples from the hippocampus (Figure 1A) and the cerebellum (Figure 1B) of P10 males and females. For the hippocampus, gene transcripts correlating and anticorrelating in both the first and the second principal components could distinguish those animals exposed to vehicle versus EtOH. For the cerebellum, gene transcripts correlating and anticorrelating within just the first principal component were enough to distinguish vehicle‐ versus EtOH‐exposed animals. Up‐ and downregulated genes were identified in both the P10 hippocampus and cerebellum that were common to both male and female pups (i.e., sex‐independent, males n = 4/group, females n = 3/group). In the hippocampus, RNA‐seq analysis identified 728 genes were significantly differentially regulated (B‐H‐adjusted p < 0.05), including 344 downregulated genes (47.25%) and 384 upregulated genes (52.75%; Figure 1C). In the cerebellum, RNA‐seq analysis identified 316 genes were significantly differentially regulated (B‐H‐adjusted p < 0.05), including 205 downregulated genes (64.87%) and 111 upregulated genes (35.13%; Figure 1D). We identified a group of 20 gene transcripts that were dysregulated in both the hippocampus and the cerebellum (Figure 1E, Venn diagram) and, 90% (18 out of 20) were dysregulated in a similar direction. Pathway analysis of these 18 genes identified a significantly associated network of “posttranslational modification, nucleic acid metabolism, small molecule biochemistry” (IPA score 55). Only 2 genes (Cmtm3 and Cpne2) were dysregulated in opposite directions (Figure 1F, union heatmap) in hippocampus and cerebellum.
FIGURE 1.
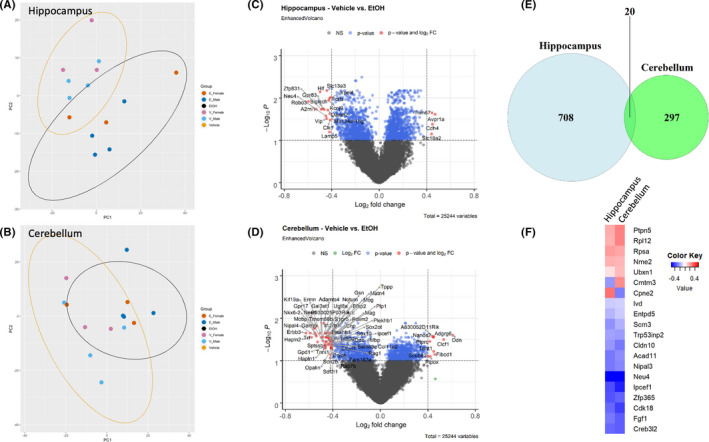
Differential gene expression in hippocampus and cerebellum. PCA of the top 500 genes contributing to the variance of the dataset in the hippocampus (A) and cerebellum (B). Volcano plots of log2 fold change and −log10 p‐value of all genes in the hippocampus (C) and cerebellum (D). Significantly altered genes (adjusted p < 0.05) with a fold change greater than or equal to ±0.4 are shown in red. (E) Venn diagram of differentially expressed genes from hippocampus and cerebellum. (F) Heatmap of the log2 fold change in the union set of differentially expressed genes from hippocampus and cerebellum. n = 4 males/treatment group, 3 females/treatment group
Clustering and functional profiling of genes differentially regulated by EtOH exposure
Next, hierarchical clustering analysis using Pearson correlation was conducted on those genes that were identified as significant, controlling for false discovery rate, hippocampus (Figure 2A), and cerebellum (Figure 3A). While pathway analysis using ReactomePA was unable to identify any significant networks associated with downregulated genes, a number of significant pathways were identified among the upregulated genes in both hippocampus and cerebellum.
FIGURE 2.
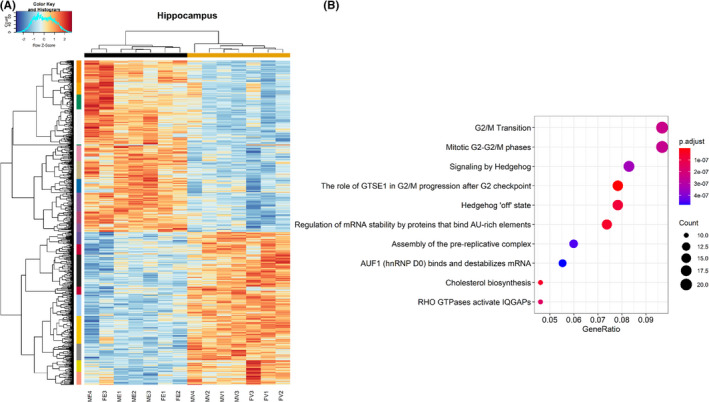
Clustering and pathway analysis of modules in the hippocampus. (A) Heatmap depicting relative gene expression across samples for significantly altered genes (adjusted p < 0.05). Hierarchical clustering dendrogram of identified modules in the hippocampus. (B) Dot plot depicting pathways related to significantly altered genes (adjusted p < 0.05) in the hippocampus as revealed by ReactomePA. n = 4 males/treatment group, 3 females/treatment group. (MV—male vehicle, FV—female vehicle, ME—male EtOH, FE—female EtOH)
FIGURE 3.
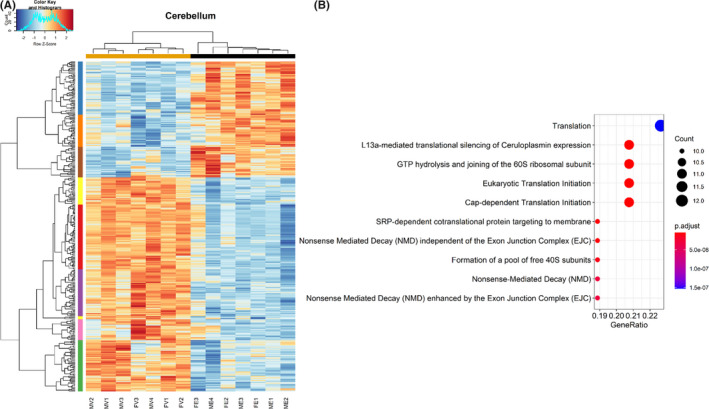
Clustering and pathway analysis of modules in the cerebellum. (A) Heatmap depicting relative gene expression across samples for significantly altered genes (adjusted p < 0.05). Hierarchical clustering dendrogram of identified modules in the cerebellum. (B) Dot plot depicting pathways related to significantly upregulated genes (adjusted p < 0.05) in the cerebellum as revealed by ReactomePA. n = 4 males/treatment group, 3 females/treatment group. (MV—male vehicle, FV—female vehicle, ME—male EtOH, FE—female EtOH)
In the hippocampus, pathways associated with the G2/M‐phase progression, Sonic Hedgehog, and cholesterol biosynthesis were significantly overrepresented among upregulated genes (Figure 2B). In contrast, pathway analysis of upregulated genes in the cerebellum identified processes associated with ribosomal biogenesis and protein translation (Figure 3B). To provide greater detail, including individual genes composing the pathways described above in Figures 2B and 3B, additional network analyses were conducted as described in Methods, which not only depict the individual genes that contribute to pathways shown in Figures 2B and 3B but also reveal the overlap between pathways. These network analyses are included in supplementary data (Figures S1 and S2).
EtOH exposure alters genes associated with cell cycle progression but not apoptosis in the hippocampus and cerebellum
Given that developmental EtOH exposure is known to perturb cell cycle progression (Anthony et al., 2008; Santillano et al., 2005), we next sought to determine whether genes associated with different phases of the cell cycle were altered. We extracted gene lists associated with S‐phase as well as G2/M‐phase of the cell cycle from the Seurat package in R. A 2‐way ANOVA, comparing the averages of z‐scores across all relevant genes, showed a significant increase in the expression of genes associated with S‐phase in both hippocampus, F (1, 10) = 20.213, p = 0.00115 (Figure 4A), and cerebellum, F (1, 10) = 7.144, p = 0.0234 (Figure 4B), of EtOH ‐exposed pups compared with vehicle control, independent of sex. However, there was no change in gene expression associated with G2/M‐phase in the hippocampus, F (1, 10) = 0.007, p = 0.934 (Figure 4C), and only a trend toward an effect of EtOH exposure in the cerebellum, F (1, 10) = 4.165, p = 0.0685 (Figure 4D), suggesting an exposure‐associated accumulation of cells in the S‐phase either as a result of an arrest at the S/G2 checkpoint or due to premature entry into S‐phase.
FIGURE 4.
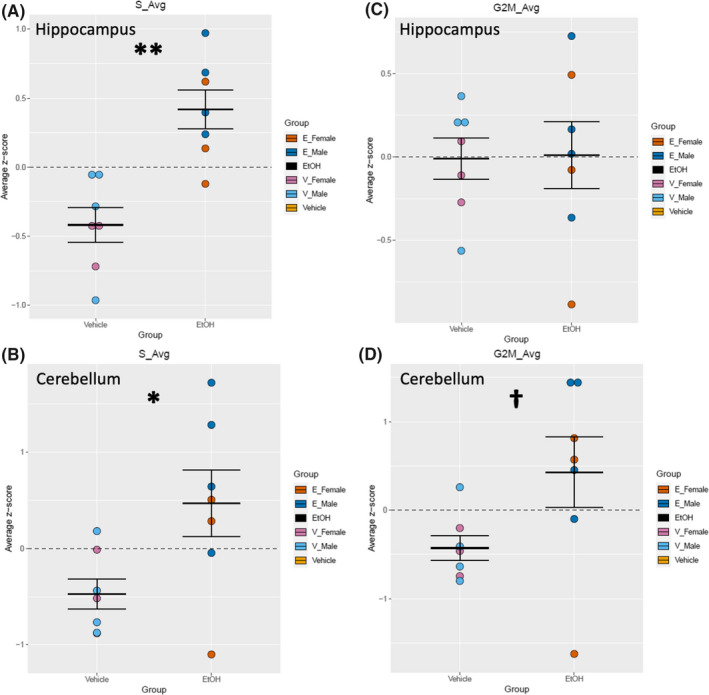
Cell cycle analysis‐associated genes in hippocampus and cerebellum. Quantification by average z‐score of S‐phase‐associated genes in the hippocampus (A) and cerebellum (B). Individual genes were z‐scored across samples, and then, average z‐score for each sample was calculated and used for analysis. Quantification by average z‐score of G2/M‐phase‐associated genes in the hippocampus (C) and cerebellum (D). Individual genes were z‐scored across samples, and then, average z‐score for each sample was calculated and used for analysis. n = 7; *, main effect of treatment, *p < 0.05, **p < 0.01, † p < 0.10
Next, regulation of the different phases of the cell cycle was examined in greater detail. Gene lists associated with positive and negative regulation of G1‐S transition, S‐phase progression, and G2‐M‐phase transition were extracted from the Mouse Genome Database Gene Ontology Browser (Bult et al., 2019). For the hippocampus, there was an increase in gene transcripts for proteins required for S‐phase function (e.g., DNA replication and centrosome duplication), F (1, 10) = 17.054, p = 0.00205 (Figure 5C). Additionally, there was an increase in genes associated with G2‐M‐phase transition, for both positive, F (1, 10) = 5.894, p = 0.0356 (Figure 5E), and negative, F (1, 10) = 12.046, p = 0.00601 (Figure 5F), regulators of phase transition. However, there were no alterations in the hippocampus in genes associated with positive, F (1, 10) = 2.718, p = 0.130 (Figure 5A), and negative, F (1, 10) = 1.002, p = 0.340 (Figure 5B), regulation of G1‐S transition or with negative S‐phase regulation, F (1, 10) = 1.378, p = 0.268 (Figure 5D). For the cerebellum, there was an increase in gene transcripts for proteins required for S‐phase, F (1, 10) = 7.096, p = 0.0237 (Figure 6C), as well as an increase in genes associated with negative regulation of G2‐M‐phase transition, F (1, 10) = 8.023, p = 0.0178 (Figure 6F). There was only a trend of increase in positive regulators of G2‐M transition in the cerebellum, F (1, 10) = 4.062, p = 0.0715 (Figure 6E). There was no difference between vehicle and EtOH in genes associated with positive, F (1, 10) = 3.246, p = 0.102 (Figure 6A), and negative, F (1, 10) = 0.389, p = 0.547 (Figure 6B), regulation of G1‐S transition or with negative S‐phase regulation, F (1, 10) = 0.427, p = 0.528 (Figure 6D).
FIGURE 5.
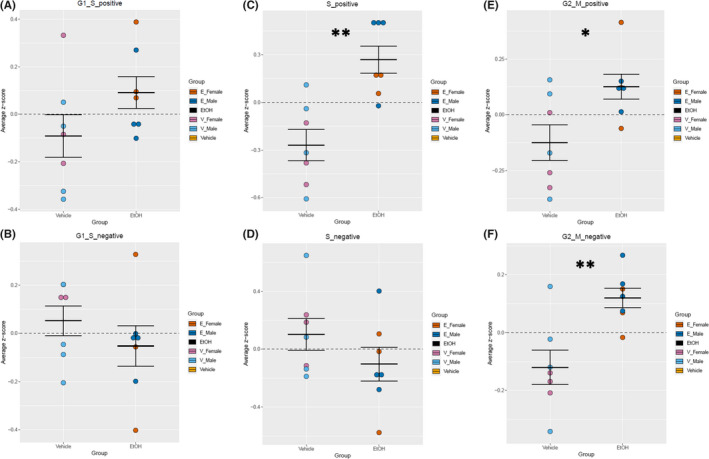
Analysis of cell cycle regulators in the hippocampus. Quantification by average z‐score of G1‐S transition‐positive regulators (A) and G1‐S transition‐negative regulators (B) in the hippocampus. Quantification by average z‐score of S‐phase‐positive regulators (C) and S‐phase‐negative regulators (D) in the hippocampus. Quantification by average z‐score of G2‐M transition‐positive regulators (E) and G2‐M transition‐negative regulators (F) in the hippocampus. Individual genes were z‐scored across samples, and then, average z‐score for each sample was calculated and used for analysis. n = 7; *, main effect of treatment, *p < 0.05, **p < 0.0.01
FIGURE 6.
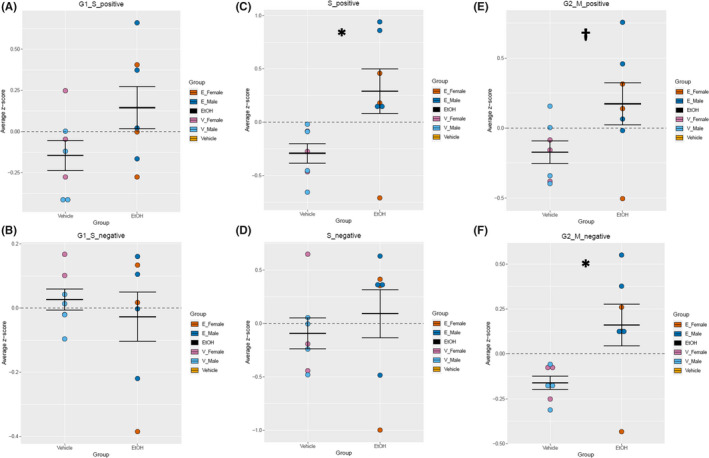
Analysis of cell cycle regulators in the cerebellum. Quantification by average z‐score of G1‐S transition‐positive regulators (A) and G1‐S transition‐negative regulators (B) in the cerebellum. Quantification by average z‐score of S‐phase‐positive regulators (C) and S‐phase‐negative regulators (D) in the cerebellum. Quantification by average z‐score of G2‐M transition‐positive regulators (E) and G2‐M transition‐negative regulators (F) in the cerebellum. Individual genes were z‐scored across samples, and then, average z‐score for each sample was calculated and used for analysis. n = 7; *, main effect of treatment, *p < 0.05, † p < 0.10
To determine whether developmental EtOH exposure also potentially influenced sensitization for apoptosis, we assessed the expression of gene transcripts associated with the execution phase of apoptosis (Fulda & Debatin, 2002; von Mering et al., 2001; Sabbagh et al., 2005), specifically the apoptosis‐related caspases, including caspase 2 (CASP2), CASP3/6/7/8/9/10 and the inflammation‐related caspases, CASP1/4/5/11/12 (Shalini et al., 2015). A 2‐way ANOVA, comparing the averages of z‐scores across all gene transcripts belonging to these classes, showed that there was no significant change in neither the apoptosis‐related caspases, hippocampus: F (1, 10) = 1.731, p = 0.554; cerebellum: F (1, 10) = 5.497, p = 0.113, nor in the inflammation‐related caspases, hippocampus: F (1, 10) = 0.554, p = 0.549; cerebellum: F (1, 10) = 0.608, p = 0.454.
EtOH exposure alters expression of genes associated with interneurons in the hippocampus
Previous studies have shown that early developmental EtOH exposure can reduce the number of GABAergic interneurons in the brain, including the hippocampus (Bird et al., 2018; Madden et al., 2020; Smiley et al., 2015). Because the interneuron population of the hippocampus is diverse (Maccaferri & Lacaille, 2003), we sought to determine whether EtOH exposure preferentially targets a specific subgroup of interneurons. To this end, we assessed publicly available single‐cell RNA‐seq (scRNA‐seq) data (Zeisel et al., 2018) for gene expression patterns that defined hippocampal inhibitory interneuron subpopulations (basket and bistratified cells, hippocamposeptal cells, cholecystokinin (CCK) interneurons, interneuron‐selective interneurons, medial ganglionic eminence (MGE)‐derived, and CGE‐derived neurogliaform cells). Multivariate analysis (by MANOVA) did not identify a global effect of EtOH on hippocampal interneurons, Pillai's trace statistic, F (1, 10) = 2.927, p = 0.129. However, univariate analysis did uncover a main effect of treatment, that is, that EtOH resulted in an overall decrease in the expression of genes associated with specific interneuron subpopulations, including basket and bistratified cells, F (1, 10) = 17.098, p‐adj = 0.006 (Figure 7A), hippocamposeptal cells, F (1, 10) = 6.952, p‐adj = 0.0373 (Figure 7B), CCK interneurons, F (1, 10) = 5.610, p‐adj = 0.0406 (Figure 7C), interneuron‐selective interneurons, F (1, 10) = 20.657, p‐adj = 0.00608 (Figure 7D), and both MGE‐derived, F (1, 10) = 7.082, p‐adj = 0.0373 (Figure 7E), and CGE‐derived, F (1, 10) = 5.527, p‐adj = 0.04057 (Figure 7F), neurogliaform cells. In contrast, there were no significant changes in these inhibitory interneuron‐associated genes in the cerebellum, potentially indicating that inhibitory interneuron subpopulations of the cerebellum are more resistant to EtOH exposure during this developmental window.
FIGURE 7.
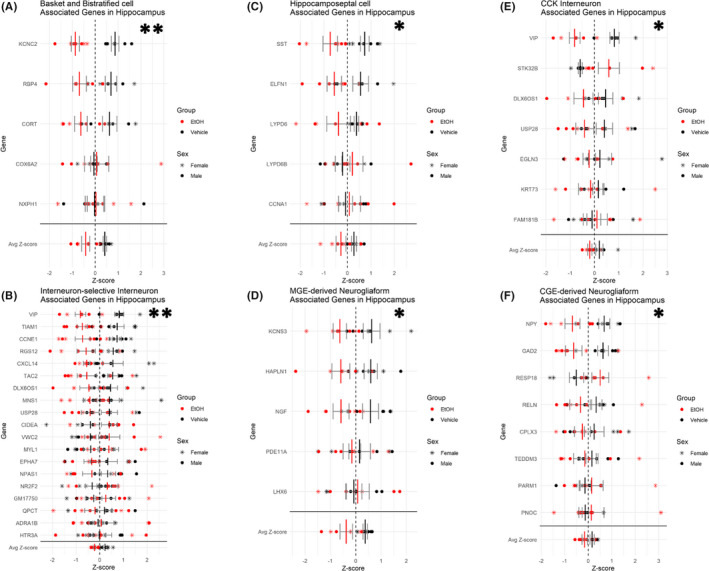
Downregulation of inhibitory interneuron‐associated genes in the hippocampus. Plot depicting relative expression of genes associated with different inhibitory interneuron subtypes in the hippocampus. Basket and bistratified cells (A), interneuron‐selective interneurons (B), hippocamposeptal cells (C), MGE‐derived neurogliaform (D), CCK interneuron (E), and CGE‐derived neurogliaform (F). Individual genes were z‐scored across samples, and then, average z‐score for each sample was calculated and used for analysis. Shown are values for each individual gene and then the average z‐score at the bottom. n = 7; *, main effect of treatment, *p < 0.05, **p < 0.01.
We also examined gene expression profiles that defined the excitatory class of granule cell interneurons in the hippocampus and the cerebellum. In the cerebellum, we observed a significant decrease in transcripts associated with granule cell interneurons, F (1, 10) = 8.348, p = 0.0161 (Figure 8A). However, we did not see a similar effect in the hippocampal granule cell interneurons, F (1, 10) = 0.607, p = 0.454 (Figure 8B), further suggesting that interneuron subpopulations are affected differently across brain regions.
FIGURE 8.
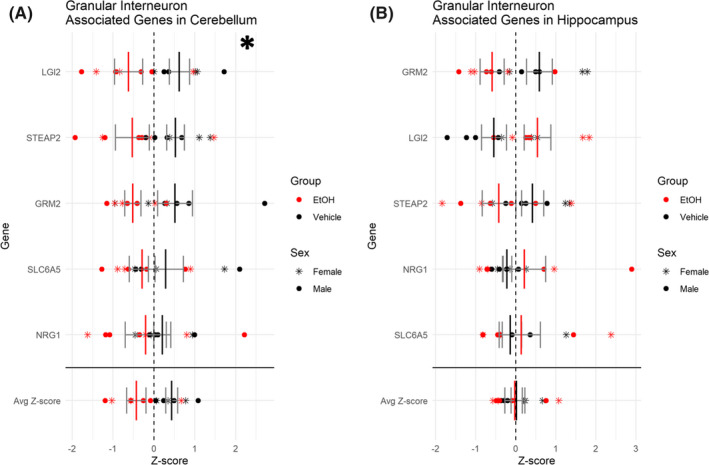
Downregulation of excitatory granule cell interneuron‐associated genes in the cerebellum with no change in hippocampus. Plots depicting relative expression of genes associated with the granule cell interneuron subtype in the cerebellum (A) and the hippocampus (B). Individual genes were z‐scored across samples, and then, average z‐score for each sample was calculated and used for analysis. Shown are values for each individual gene and then the average z‐score at the bottom. n = 7; *, main effect of treatment, *p < 0.05
EtOH exposure alters expression of genes associated with oligodendrocyte and microglia lineages in the cerebellum
Developmental EtOH exposure is documented to result in delayed expression of myelin basic protein (MBP; Chiappelli et al., 1991) and altered oligodendrocyte morphology and myelination (Dalitz et al., 2008) that may ultimately result in decreased cerebellar white matter (Archibald et al., 2001b). Additionally, a brain growth spurt occurs in rodents at approximately P9 (equivalent to late gestation in humans), that is, due, in part, to the genesis of oligodendrocyte precursors, their maturation into oligodendrocytes, and subsequently, initiation of myelination (Archibald et al., 2001b). To determine whether EtOH exposure during this time period influenced the progression of this growth spurt, we used publicly available scRNA‐seq data (Zeisel et al., 2018), to identify gene expression patterns that defined stages of oligodendrocyte maturation. Utilizing these stage‐selective gene expression patterns, we observed a main effect of treatment exemplified by a decrease in gene transcripts associated with committed oligodendrocyte precursors (COPs), F (1, 10) = 29.019, p‐adj = 0.00051 (Figure 9A), newly formed oligodendrocytes (NFOLs), F (1, 10) = 31.278, p‐adj = 0.00051 (Figure 9B), myelin‐forming oligodendrocytes (MFOLs), F (1, 10) = 32.076, p‐adj = 0.00051 (Figure 9C), and mature oligodendrocytes (MOLs), F (1, 10) = 24.853, p‐adj = 0.00069 (Figure 9D), but no change in oligodendrocyte precursors (OPCs), F (1, 10) = 2.112, p‐adj = 0.1768 (Figure 9E). Moreover, MANOVA revealed a global effect of EtOH exposure on the oligodendrocyte lineage, Pillai's trace statistic, F (1, 10) = 13.227, p = 0.00343. This suggests that the majority of stages of oligodendrocyte maturation are inhibited by EtOH exposure within the cerebellum. Additionally, as would be predicted from the loss of maturing oligodendrocyte populations, we observed a significant decrease in the expression of positive regulators of myelination as well as significantly decreased expression in myelination genes within the cerebellum (Table 1; Bult et al., 2019). There were also indications of the oligodendrocyte lineage being negatively impacted in the hippocampus, but at fewer stages of maturation, COPs‐ F (1, 10) = 11.080, p‐adj = 0.0191; NFOLs‐ F (1, 10) = 13.857, p‐adj = 0.0191.
FIGURE 9.
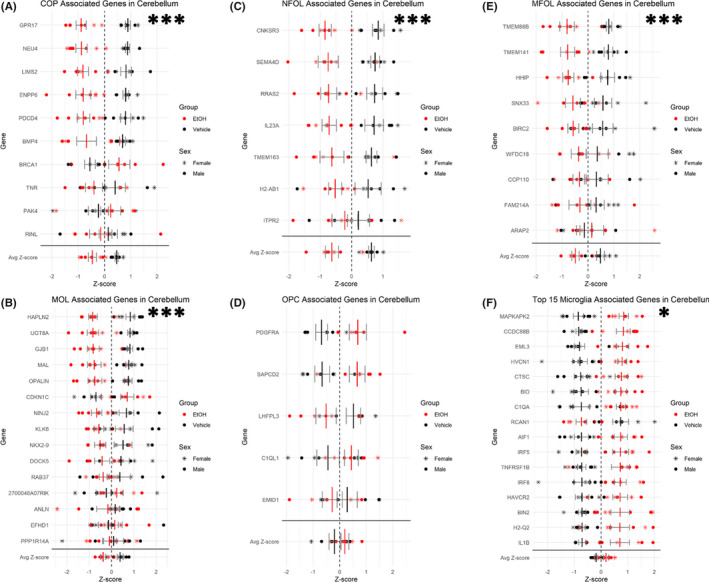
Downregulation of oligodendrocyte lineage‐associated genes and upregulation of microglia‐associated genes in the cerebellum. Plot depicting relative expression of genes associated with cell types of the oligodendrocyte lineage in the cerebellum. COPs (A), NFOLs (B), MFOLs (C), MOLs (D), and (OPCs) (E). (F) Plot depicting relative expression of microglia‐associated genes in the cerebellum. Individual genes were z‐scored across samples, and then, average z‐score for each sample was calculated and used for analysis. Shown are values for each individual caspase and then the average z‐score at the bottom. n = 7; *, main effect of treatment, *p < 0.05, ***p < 0.001
TABLE 1.
Genes involved in the regulation of myelination
| Gene | Fold change (Log2) | FDR p‐value |
|---|---|---|
| Positive regulators of myelination | ||
| Trf | −0.58955 | 0.022591 |
| Tppp | −0.43892 | 0.026530 |
| MAG | −0.43054 | 0.030783 |
| Sox10 | −0.41878 | 0.034345 |
| Myrf | −0.37515 | 0.045173 |
| Zfp488 | −0.32747 | 0.042248 |
| Myelination genes | ||
| Nkx6‐2 | −0.54354 | 0.024454 |
| Gal3st1 | −0.48336 | 0.034067 |
| Ugt8a | −0.46222 | 0.022591 |
| PlP1 | −0.45690 | 0.035326 |
| MOG | −0.45066 | 0.027676 |
| CNP | −0.44373 | 0.028693 |
| MBP | −0.43751 | 0.040057 |
| Tspan2 | −0.38537 | 0.042478 |
| Bcas1 | −0.35468 | 0.022243 |
| Arhgef10 | −0.33719 | 0.015223 |
| Mal | −0.31933 | 0.045683 |
| Olig2 | −0.31083 | 0.048980 |
| Jam3 | −0.26620 | 0.042762 |
| Mal2 | −0.22890 | 0.042029 |
| Adgrg6 | 0.44743 | 0.027274 |
Abbreviations: FDR, false discovery rate. Genes were selected based on FDR‐corrected p‐value < 0.05. Gene lists were obtained from the Mouse Genome Database (Bult et al., 2019).
Developmental EtOH exposure is also associated with increased microglia activation in the developing brain, including the cerebellum (Drew et al., 2015). We therefore examined the expression of genes associated with microglia as identified in a single‐cell RNA‐seq study (Zeisel et al., 2015). In both male and female cerebella, EtOH exposure resulted in an increase in the expression of genes associated with microglia, F (1, 10) = 8.505, p = 0.0154 (Figure 9F). In addition, our transcriptomic analysis demonstrated that EtOH significantly decreased the expression of astrocyte‐related genes in the hippocampus, F (1, 10) = 18.597, p = 0.0138 (Table S2).
DISCUSSION
Both the cerebellum and the hippocampus share a secondary and delayed wave of neurogenesis that results in the formation of granule cell interneurons. We examined the effects of EtOH exposure during this neurogenic wave, with the expectation that we would identify invariant, region‐independent mechanisms for EtOH teratogenesis. We did observe that EtOH exposure resulted in an overall transcriptomic pressure in both regions, toward increased accumulation of cells in S‐phase of cell cycle, consistent with previous observations (Li, Miller, & Luo, 2002; Santillano et al., 2005). It is likely that a significant increase in S‐phase‐associated genes reflects the synchronized behavior of a preponderance of the constituent cells within cerebellar and hippocampal tissue, reflecting an increase in S‐phase cells for the tissues as a whole. Granule cells are the most likely contributors to this, being the most numerous cell type and undergoing proliferation at this stage (Herculano‐Houzel, Mota, & Lent, 2006), though further more nuanced studies would need to be completed to confirm this hypothesis. Additionally, in cerebellum, there was also an increase in G2/M‐phase gene expression. However, in both cerebellum and hippocampus, there was no significant change in the expression of caspase transcripts, suggesting sensitization for apoptosis is not a significant feature of the persistent teratogenic response to EtOH in these regions, though it may indeed be a part of the acute response (McAlhany, West, & Miranda, 2000; Qi et al., 2014).
Regional differences in response, however, greatly outweighed similarities. Out of the approximately 1000 gene transcripts that were significantly regulated across both brain regions, less than 0.02% were shared in common. Not surprisingly, while Ephrin, Rho‐GTPase, and microtubule organization pathways were dysregulated in hippocampus, pathways such as ribosome biogenesis and protein translation initiation were upregulated by EtOH in the cerebellum. Interestingly, our observation that EtOH upregulated hippocampal ribosome biogenic pathways stands in contrast with previous reports that at least immediately following exposure, EtOH decreased transcripts associated with ribosomal biogenesis (Berres et al., 2017; Downing et al., 2012; Garic, Berres, & Smith, 2014; Green et al., 2007). Our data suggest that the acute phase of suppression may be followed by a rebound activation of pathways that support protein synthesis.
Other important findings were that interneuron subtype‐related gene expression profiles were broadly inhibited in hippocampus, while cerebellar inhibitory interneuron gene expression was largely unaffected, though in the case of the cerebellum, excitatory granule cell interneuron gene expression was significantly reduced. In contrast, the EtOH‐exposed cerebellum exhibited a more pronounced decrease in transcripts associated with multiple stages of oligodendrocyte genesis and maturation and myelin generation compared with the hippocampus where changes were present yet more subtle. Therefore, the transcriptomic effects and presumptive cell type targets of EtOH exposure in hippocampus and cerebellum are substantially different.
Numerous animal models have shown that interneurons are sensitive to EtOH in various brain regions, including the striatum, medial prefrontal cortex, cerebral cortex, orbitofrontal cortex, and hippocampus (Bird et al., 2018; De Giorgio et al., 2012; Hamilton et al., 2017; Kenton et al., 2020; Madden et al., 2020; Sadrian et al., 2014; Saito et al., 2018; Smiley et al., 2015). Consequently, developmental EtOH exposure has previously been characterized as resulting in “interneuronopathy” (Bird et al., 2018; Madden et al., 2020; Marguet et al., 2020; Skorput et al., 2015; Smiley et al., 2015). However, the effects of EtOH exposure are not always inhibitory. For example, prenatal exposure during mouse gestation has been shown to increase interneuron populations in the fetal cortex, medial prefrontal cortex, and orbitofrontal cortex, with a hypothesized increased migration from the medial and caudal ganglionic eminences (CGE; Cuzon et al., 2008; Kenton et al., 2020; Skorput et al., 2015; Skorput & Yeh, 2016). Previous studies using postnatal rodent models have shown that EtOH does decrease interneurons in the hippocampus (Bird et al., 2018; Madden et al., 2020; Sadrian et al., 2014), and moreover, EtOH‐treated human pluripotent stem cell‐derived neurons also exhibited decreased interneuron gene expression (Larsen et al., 2016). These studies suggest that EtOH may inhibit developmental programs for interneuron generation which is consistent with our current study in this postnatal FASD model. However, interneurons of the hippocampus are extremely heterogeneous with respect to developmental origin (Bayer & Altman, 1974; Tricoire et al., 2011), morphology (Booker & Vida, 2018), and gene expression profiles (Zeisel et al., 2015). To this end, we exploited the published data on gene expression profile diversity to assess changes in cell type representation due to EtOH exposure. This analysis resulted in a surprising observation that EtOH inhibited gene expression patterns across a diversity of inhibitory hippocampal interneuron types, suggesting that EtOH's effects were largely nonselective. Therefore, it was all the more surprising that identical interneuron subtype transcript patterns in the cerebellum were generally resistant to EtOH exposure. The mechanisms that contribute to the resiliency of the cerebellar inhibitory interneuron transcriptome, but the contrasting vulnerability of the excitatory granule cells, compared to the hippocampus are unknown and are an important target for future investigation to identify presumptive region‐specific and cell type selective neuroprotective mechanisms.
Regardless of mechanism mediating region‐specific sensitivity of interneuron populations, a perturbation in the maturation of hippocampal interneurons is likely to have significant repercussions on neurite growth (Groc et al., 2002) and synapse maturation (Colin‐Le Brun et al., 2004; Lauri et al., 2003). For example, spontaneous network activity driven by interneurons has been shown to play an important role in these processes, and EtOH exposure has been shown to disrupt this activity within interneurons (Galindo et al., 2005). Perturbation in interneuron maturation may also explain the emergence of deficits in hippocampal‐dependent learning and memory, such as those observed in animal models of FASD (Bonthius & West, 1991; Goodlett & Johnson, 1997; Zucca & Valenzuela, 2010).
Another key finding in our current study was that EtOH exposure in the cerebellum broadly inhibited gene transcripts associated with multiple stages of oligodendrocyte genesis and maturation as well as transcripts associated with myelination. In humans, oligodendrocyte maturation occurs primarily during late gestation and throughout the first two decades of life, and in rodents, oligodendrocyte maturation occurs primarily during the first two postnatal weeks (Dobbing & Sands, 1979; Lebel et al., 2008). Disruptions to, or delays in, myelination in the developing brain may result in deficient neuronal transmission, thereby contributing to neurocircuitry and behavioral dysfunctions associated with FASD (Guizzetti et al., 2014). Imaging studies of individuals with FASD have shown widespread abnormalities in white matter, suggesting that myelination and oligodendrocytes are particularly vulnerable to prenatal alcohol exposure (Wozniak, Riley, & Charness, 2019). White matter abnormalities in the cerebellum, for example, occur in children with FASD, and these abnormalities persist in adolescents and adults (Archibald et al., 2001a; Fan et al., 2015; Li et al., 2009; Ma et al., 2005; Mattson et al., 1992; Riley & McGee, 2005; Sowell et al., 2008; Treit et al., 2013; Wozniak & Muetzel, 2011). Early postnatal EtOH exposure in rats resulted in decreased cerebellar levels of MBP and MAG which are produced by MOLs, and this outcome persisted in adults (Zoeller et al., 1994). EtOH also decreased MBP expression and increased apoptosis in the rat cerebellum, which was potentiated in animals derived from iron‐deficient dams and associated with impaired eyeblink conditioning, a cerebellum‐dependent learning task (Rufer et al., 2012). Third‐trimester EtOH exposure in macaque monkeys resulted in increased apoptosis of oligodendrocytes (Creeley et al., 2013), and in the rat, EtOH exposure during the first two postnatal weeks resulted in a transient loss of corpus callosal oligodendrocytes and oligodendrocyte progenitor cells (Newville et al., 2017). In the current study, we demonstrated in the cerebellum that EtOH decreased the expression of gene transcripts associated with OPCs, NFOLs, MFOLs, and MOLs, but in contrast to the above study, not oligodendrocyte progenitor cells. These observations are novel not only because the effects of EtOH on different stages of oligodendrogliosis in FASD are largely unstudied, but also because our data suggest region‐specific patterns of vulnerability that appear to somewhat spare the hippocampus. It was surprising that though the hippocampus expressed an equivalent complement of oligodendrocyte‐stage‐specific gene transcripts as the cerebellum, hippocampal levels were less affected by EtOH exposure in this model. The basis for the differential effects of EtOH on oligodendrocyte linage cells in the developing cerebellum and hippocampus is unclear, yet intriguing. Myelination in the cerebellum occurs slightly earlier during development than in the hippocampus (van Tilborg et al., 2018). It is possible that EtOH differentially affects oligodendrocyte linage cells or oligodendrocyte development in these brain regions due to subtle regional differences in maturation stage during EtOH exposure in our postnatal model of FASD. Again, the source of region‐specific vulnerability and resilience is poorly understood and warrants further study, since loss of white matter is often documented in human imaging studies in individuals with FASD (Fan et al., 2016; Sowell et al., 2008).
Collectively, our data suggest that two brain regions which uniquely share a delayed neurogenic period nevertheless respond quite differently to EtOH exposure during this period. While increased expression of S‐phase‐associated genes was identified in both hippocampus and cerebellum, there was otherwise virtually no overlap in either molecular pathways or cell type‐specific patterns of vulnerability. An analysis of the reasons, why the inhibitory interneuron transcriptome was vulnerable to EtOH in the hippocampus, whereas the transcriptome for oligodendrocyte lineages was vulnerable in cerebellum, may provide clues to the presence of both resilience and risk factors in each of these brain regions that contribute to selective vulnerability to a common teratogen.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Fig S1
Fig S2
Table S1
Table S2
Table S3
Supplementary Material
ACKNOWLEDGMENTS
Next‐generation sequencing was performed by the UAMS Genomics Core Facility, which is supported by and located in the Winthrop P. Rockefeller Cancer Institute, at the University of Arkansas for Medical Sciences. Portions of this research were conducted with high‐performance research computing resources provided by Texas A&M University (https://hprc.tamu.edu). This work was supported by grants from the NIH, R01 AA024659 (RCM), F30 AA027698 (MRP), R01 AA024695 (PDD), R01 AA026665 (PDD), and R01 AA027111 (PDD).
Pinson, M. R. , Holloway, K. N. , Douglas, J. C. , Kane, C. J. M. , Miranda, R. C. , & Drew, P. D. (2021). Divergent and overlapping hippocampal and cerebellar transcriptome responses following developmental ethanol exposure during the secondary neurogenic period. Alcoholism: Clinical and Experimental Research, 45, 1408–1423. 10.1111/acer.14633
Marisa R. Pinson and Kalee N. Holloway contributed equally.
REFERENCES
- Adachi, J. , Mizoi, Y. , Fukunaga, T. , Ogawa, Y. , Ueno, Y. & Imamichi, H. (1991) Degrees of alcohol intoxication in 117 hospitalized cases. Journal of Studies on Alcohol, 52, 448–453. [DOI] [PubMed] [Google Scholar]
- Anders, S. , Pyl, P.T. & Huber, W. (2014) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony, B. , Zhou, F.C. , Ogawa, T. , Goodlett, C.R. & Ruiz, J. (2008) Alcohol exposure alters cell cycle and apoptotic events during early neurulation. Alcohol and Alcoholism, 43, 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archibald, S.L. , Fennema‐Notestine, C. , Gamst, A. , Riley, E.P. , Mattson, S.N. & Jernigan, T.L. (2001a) Brain dysmorphology in individuals with severe prenatal alcohol exposure. Developmental Medicine and Child Neurology, 43, 148–154. [PubMed] [Google Scholar]
- Archibald, S.L. , Fennema‐Notestine, C. , Gamst, A. , Riley, E.P. , Mattson, S.N. & Jernigan, T.L. (2001b) Brain dysmorphology in individuals with severe prenatal alcohol exposure. Developmental Medicine & Child Neurology, 43, 148–154. [PubMed] [Google Scholar]
- Bakhireva, L.N. , Sharkis, J. , Shrestha, S. , Miranda‐Sohrabji, T.J. , Williams, S. & Miranda, R.C. (2017) Prevalence of prenatal alcohol exposure in the State of Texas as assessed by phosphatidylethanol in newborn dried blood spot specimens. Alcoholism, Clinical and Experimental Research, 41, 1004–1011. [DOI] [PubMed] [Google Scholar]
- Barnes, D.E. & Walker, D.W. (1981) Prenatal ethanol exposure permanently reduces the number of pyramidal neurons in rat hippocampus. Brain Research, 227, 333–340. [DOI] [PubMed] [Google Scholar]
- Bayer, S.A. & Altman, J. (1974) Hippocampal development in the rat: cytogenesis and morphogenesis examined with autoradiography and low‐level X‐irradiation. The Journal of Comparative Neurology, 158, 55–79. [DOI] [PubMed] [Google Scholar]
- Bayer, S.A. , Altman, J. , Russo, R.J. & Zhang, X. (1993) Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology, 14, 83–144. [PubMed] [Google Scholar]
- Berres, M.E. , Garic, A. , Flentke, G.R. & Smith, S.M. (2017) Transcriptome profiling identifies ribosome biogenesis as a target of alcohol teratogenicity and vulnerability during early embryogenesis. PLoS One, 12, e0169351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird, C.W. , Taylor, D.H. , Pinkowski, N.J. , Jill Chavez, G. & Fernando Valenzuela, C. (2018) Long‐term reductions in the population of GABAergic interneurons in the mouse hippocampus following developmental ethanol exposure. Neuroscience, 383, 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blighe, K. , Rana, S. & Lewis, M. (2020). EnhancedVolcano: Publication‐ready volcano plots with enhanced colouring and labeling. https://github.com/kevinblighe/EnhancedVolcano
- Bolger, A.M. , Lohse, M. & Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthius, D.J. & West, J.R. (1991) Permanent neuronal deficits in rats exposed to alcohol during the brain growth spurt. Teratology, 44, 147–163. [DOI] [PubMed] [Google Scholar]
- Booker, S.A. & Vida, I. (2018) Morphological diversity and connectivity of hippocampal interneurons. Cell and Tissue Research, 373, 619–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult, C.J. , Blake, J.A. , Smith, C.L. , Kadin, J.A. & Richardson, J.E. (2019) Mouse Genome Database (MGD) 2019. Nucleic Acids Research, 47, D801–D806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarillo, C. & Miranda, R.C. (2008) Ethanol exposure during neurogenesis induces persistent effects on neural maturation: evidence from an ex vivo model of fetal cerebral cortical neuroepithelial progenitor maturation. Gene Expression, 14, 159–171. [PMC free article] [PubMed] [Google Scholar]
- Carter, R.C. , Jacobson, S.W. , Molteno, C.D. & Jacobson, J.L. (2007) Fetal alcohol exposure, iron‐deficiency anemia, and infant growth. Pediatrics, 120, 559–567. [DOI] [PubMed] [Google Scholar]
- Chiappelli, F. , Taylor, A.N. , Espinosa‐de‐los‐Monteros, A. & de‐Vellis, J. (1991) Fetal alcohol delays the developmental expression of myelin basic protein and transferrin in rat primary oligodendrocyte cultures. International Journal of Developmental Neuroscience, 9, 67–75. [DOI] [PubMed] [Google Scholar]
- Clancy, B. , Darlington, R.B. & Finlay, B.L. (2001) Translating developmental time across mammalian species. Neuroscience, 105, 7–17. [DOI] [PubMed] [Google Scholar]
- Colin‐Le Brun, I. , Ferrand, N. , Caillard, O. , Tosetti, P. , Ben‐Ari, Y. & Gaïarsa, J.L. (2004) Spontaneous synaptic activity is required for the formation of functional GABAergic synapses in the developing rat hippocampus. Journal of Physiology, 559, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creeley, C.E. , Dikranian, K.T. , Johnson, S.A. , Farber, N.B. & Olney, J.W. (2013) Alcohol‐induced apoptosis of oligodendrocytes in the fetal macaque brain. Acta Neuropathologica Communications, 1, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzon, V.C. , Yeh, P.W.L. , Yanagawa, Y. , Obata, K. & Yeh, H.H. (2008) Ethanol consumption during early pregnancy alters the disposition of tangentially migrating GABAergic interneurons in the fetal cortex. The Journal of Neuroscience, 28, 1854–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalitz, P. , Cock, M. , Harding, R. & Rees, S. (2008) Injurious effects of acute ethanol exposure during late gestation on developing white matter in fetal sheep. International Journal of Developmental Neuroscience, 26, 391–399. [DOI] [PubMed] [Google Scholar]
- De‐Giorgio, A. , Comparini, S.E. , Intra, F.S. & Granato, A. (2012) Long‐term alterations of striatal parvalbumin interneurons in a rat model of early exposure to alcohol. Journal of Neurodevelopmental Disorders, 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, M.R. , Vollmer, C.C. , Zamudio‐Bulcock, P.A. , Vollmer, W. , Blomquist, S.L. , Morton, R.A. et al. (2014) Repeated intermittent alcohol exposure during the third trimester‐equivalent increases expression of the GABA(A) receptor δ subunit in cerebellar granule neurons and delays motor development in rats. Neuropharmacology, 79, 262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing, J. & Sands, J. (1979) Comparative aspects of the brain growth spurt. Early Human Development, 3, 79–83. [DOI] [PubMed] [Google Scholar]
- Downing, C. , Balderrama‐Durbin, C. , Kimball, A. , Biers, J. , Wright, H. , Gilliam, D. & et al. (2012) Quantitative trait locus mapping for ethanol teratogenesis in BXD recombinant inbred mice. Alcoholism: Clinical and Experimental Research, 36, 1340–1354. [DOI] [PubMed] [Google Scholar]
- Drew, P.D. , Johnson, J.W. , Douglas, J.C. , Phelan, K.D. & Kane, C.J. (2015) Pioglitazone blocks ethanol induction of microglial activation and immune responses in the hippocampus, cerebellum, and cerebral cortex in a mouse model of fetal alcohol spectrum disorders. Alcoholism, Clinical and Experimental Research, 39, 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, J. , Jacobson, S.W. , Taylor, P.A. , Molteno, C.D. , Dodge, N.C. , Stanton, M.E. et al. (2016) White matter deficits mediate effects of prenatal alcohol exposure on cognitive development in childhood. Human Brain Mapping, 37, 2943–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, J. , Meintjes, E.M. , Molteno, C.D. , Spottiswoode, B.S. , Dodge, N.C. , Alhamud, A.A. et al. (2015) White matter integrity of the cerebellar peduncles as a mediator of effects of prenatal alcohol exposure on eyeblink conditioning. Human Brain Mapping, 36, 2470–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda, S. & Debatin, K.‐M. (2002) IFNγ sensitizes for apoptosis by upregulating caspase‐8 expression through the Stat1 pathway. Oncogene, 21, 2295–2308. [DOI] [PubMed] [Google Scholar]
- Galindo, R. , Zamudio, P.A. & Fernando Valenzuela, C. (2005) Alcohol is a potent stimulant of immature neuronal networks: implications for fetal alcohol spectrum disorders. Journal of Neurochemistry, 94, 1500–1511. [DOI] [PubMed] [Google Scholar]
- Garic, A. , Berres, M.E. & Smith, S.M. (2014) High‐throughput transcriptome sequencing identifies candidate genetic modifiers of vulnerability to fetal alcohol spectrum disorders. Alcoholism, Clinical and Experimental Research, 38, 1874–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, L. , Ware, A.L. , Crocker, N. , Deweese, B.N. , Coles, C.D. , Kable, J.A. et al. (2013) Neuropsychological deficits associated with heavy prenatal alcohol exposure are not exacerbated by ADHD. Neuropsychology, 27, 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlett, C.R. & Johnson, T.B. (1997) Neonatal binge ethanol exposure using intubation: timing and dose effects on place learning. Neurotoxicology and Teratology, 19, 435–446. [DOI] [PubMed] [Google Scholar]
- Green, M.L. , Singh, A.V. , Zhang, Y. , Nemeth, K.A. , Sulik, K.K. & Knudsen, T.B. (2007) Reprogramming of genetic networks during initiation of the Fetal Alcohol Syndrome. Developmental Dynamics, 236, 613–631. [DOI] [PubMed] [Google Scholar]
- Greenmyer, J.R. , Klug, M.G. , Kambeitz, C. , Popova, S. & Burd, L. (2018) A multicountry updated assessment of the economic impact of fetal alcohol spectrum disorders: costs for children and adults. Journal of Addiction Medicine, 12, 466–473. [DOI] [PubMed] [Google Scholar]
- Groc, L. , Petanjek, Z. , Gustafsson, B. , Ben‐Ari, Y. , Hanse, E. & Khazipov, R. (2002) In vivo blockade of neural activity alters dendritic development of neonatal CA1 pyramidal cells. European Journal of Neuroscience, 16, 1931–1938. [DOI] [PubMed] [Google Scholar]
- Guizzetti, M. , Zhang, X. , Goeke, C. & Gavin, D.P. (2014) Glia and neurodevelopment: focus on fetal alcohol spectrum disorders. Frontiers in Pediatrics, 2, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, G.F. , Hernandez, I.J. , Krebs, C.P. , Bucko, P.J. & Rhodes, J.S. (2017) Neonatal alcohol exposure reduces number of parvalbumin‐positive interneurons in the medial prefrontal cortex and impairs passive avoidance acquisition in mice deficits not rescued from exercise. Neuroscience, 352, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herculano‐Houzel, S. , Mota, B. & Lent, R. (2006) Cellular scaling rules for rodent brains. Proceedings of the National Academy of Sciences, 103, 12138–12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jew, B. , Alvarez, M. , Rahmani, E. , Miao, Z. , Ko, A. , Garske, K.M. et al. (2020) Accurate estimation of cell composition in bulk expression through robust integration of single‐cell information. Nature Communications, 11, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane, C.J.M. , Phelan, K.D. , Han, L. , Smith, R.R. , Xie, J. , Douglas, J.C. et al. (2011) Protection of neurons and microglia against ethanol in a mouse model of fetal alcohol spectrum disorders by peroxisome proliferator‐activated receptor‐γ agonists. Brain, Behavior, and Immunity, 25, S137–S145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenton, J.A. , Ontiveros, T. , Bird, C.W. , Fernando Valenzuela, C. & Brigman, J.L. (2020) Moderate prenatal alcohol exposure alters the number and function of GABAergic interneurons in the murine orbitofrontal cortex. Alcohol, 88, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. & Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nature Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klintsova, A.Y. , Scamra, C. , Hoffman, M. , Napper, R.M. , Goodlett, C.R. & Greenough, W.T. (2002) Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge‐like alcohol exposure in rats: II. A quantitative stereological study of synaptic plasticity in female rat cerebellum. Brain Research, 937, 83–93. [DOI] [PubMed] [Google Scholar]
- Larsen, Z.H. , Chander, P. , Joyner, J.A. , Floruta, C.M. , Demeter, T.L. & Weick, J.P. (2016) Effects of ethanol on cellular composition and network excitability of human pluripotent stem cell‐derived neurons. Alcoholism: Clinical and Experimental Research, 40, 2339–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri, S.E. , Lamsa, K. , Pavlov, I. , Riekki, R. , Johnson, B.E. , Molnar, E. et al. (2003) Activity blockade increases the number of functional synapses in the hippocampus of newborn rats. Molecular and Cellular Neurosciences, 22, 107–117. [DOI] [PubMed] [Google Scholar]
- Lebel, C. , Walker, L. , Leemans, A. , Phillips, L. & Beaulieu, C. (2008) Microstructural maturation of the human brain from childhood to adulthood. NeuroImage, 40, 1044–1055. [DOI] [PubMed] [Google Scholar]
- Li, L. , Coles, C.D. , Lynch, M.E. & Hu, X. (2009) Voxelwise and skeleton‐based region of interest analysis of fetal alcohol syndrome and fetal alcohol spectrum disorders in young adults. Human Brain Mapping, 30, 3265–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Miller, M.W. & Luo, J. (2002) Effects of prenatal exposure to ethanol on the cyclin‐dependent kinase system in the developing rat cerebellum. Brain Research. Developmental Brain Research, 139, 237–245. [DOI] [PubMed] [Google Scholar]
- Livy, D.J. , Miller, E.K. , Maier, S.E. & West, J.R. (2003) Fetal alcohol exposure and temporal vulnerability: effects of binge‐like alcohol exposure on the developing rat hippocampus. Neurotoxicology and Teratology, 25, 447–458. [DOI] [PubMed] [Google Scholar]
- Livy, D.J. , Parnell, S.E. & West, J.R. (2003) Blood ethanol concentration profiles: a comparison between rats and mice. Alcohol, 29, 165–171. [DOI] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. & Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, X. , Coles, C.D. , Lynch, M.E. , Laconte, S.M. , Zurkiya, O. , Wang, D. et al. (2005) Evaluation of corpus callosum anisotropy in young adults with fetal alcohol syndrome according to diffusion tensor imaging. Alcoholism, Clinical and Experimental Research, 29, 1214–1222. [DOI] [PubMed] [Google Scholar]
- Maccaferri, G. & Lacaille, J.‐C. (2003) Interneuron Diversity series: Hippocampal interneuron classifications—making things as simple as possible, not simpler. Trends in Neurosciences, 26, 564–571. [DOI] [PubMed] [Google Scholar]
- Madden, J.T. , Thompson, S.M. , Magcalas, C.M. , Wagner, J.L. , Hamilton, D.A. , Savage, D.D. et al. (2020) Moderate prenatal alcohol exposure reduces parvalbumin expressing GABAergic interneurons in the dorsal hippocampus of adult male and female rat offspring. Neuroscience Letters, 718, 134700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marguet, F. , Friocourt, G. , Brosolo, M. , Sauvestre, F. , Marcorelles, P. , Lesueur, C. et al. (2020) Prenatal alcohol exposure is a leading cause of interneuronopathy in humans. Acta Neuropathologica Communications, 8, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson, S.N. , Riley, E.P. , Jernigan, T.L. , Ehlers, C.L. , Delis, D.C. , Jones, K.L. et al. (1992) ‘Fetal alcohol syndrome: a case report of neuropsychological, MRI and EEG assessment of two children. Alcoholism: Clinical and Experimental Research, 16, 1001–1003. [DOI] [PubMed] [Google Scholar]
- May, P.A. , Chambers, C.D. , Kalberg, W.O. , Zellner, J. , Feldman, H. , Buckley, D. et al. (2018) Prevalence of fetal alcohol spectrum disorders in 4 US Communities. JAMA, 319, 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlhany, R.E. Jr. , West, J.R. & Miranda, R.C. (2000) Glial‐derived neurotrophic factor (GDNF) prevents ethanol‐induced apoptosis and JUN kinase phosphorylation. Developmental Brain Research, 119, 209–216. [DOI] [PubMed] [Google Scholar]
- von Mering, M. , Wellmer, A. , Michel, U. , Bunkowski, S. , Tłustochowska, A. , Brück, W. et al. (2001) Transcriptional regulation of caspases in experimental pneumococcal meningitis. Brain Pathology, 11, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, E.M. & Riley, E.P. (2015) What happens when children with fetal alcohol spectrum disorders become adults? Current Developmental Disorders Reports, 2, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napper, R.M. & West, J.R. (1995) Permanent neuronal cell loss in the cerebellum of rats exposed to continuous low blood alcohol levels during the brain growth spurt: a stereological investigation. The Journal of Comparative Neurology, 362, 283–292. [DOI] [PubMed] [Google Scholar]
- Newville, J. , Valenzuela, C.F. , Li, L. , Jantzie, L.L. & Cunningham, L.A. (2017) Acute oligodendrocyte loss with persistent white matter injury in a third trimester equivalent mouse model of fetal alcohol spectrum disorders. Glia, 65, 1317–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, A.L. , Crocker, N. , Mattson, S.N. & Riley, E.P. (2009) Neuroimaging and fetal alcohol spectrum disorders. Developmental Disabilities Research Reviews, 15, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli, B. , Weinberg, J. & Hicks, G.G. (2018) Effects of prenatal alcohol exposure (PAE): insights into FASD using mouse models of PAE. Biochemistry and Cell Biology, 96, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, Y. , Zhang, M. , Li, H. , Frank, J.A. , Dai, L. , Liu, H. et al. (2014) MicroRNA‐29b regulates ethanol‐induced neuronal apoptosis in the developing cerebellum through Sp1/Rax/PKR. Journal of Biological Chemistry, 289, 10201–10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, D. & Barone, S. Jr. (2000) Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environmental Health Perspectives, 108(Suppl. 3), 511–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley, E.P. & McGee, C.L. (2005) Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Experimental Biology and Medicine, 230, 357–365. [DOI] [PubMed] [Google Scholar]
- Rufer, E.S. , Tran, T.D. , Attridge, M.M. , Andrzejewski, M.E. , Flentke, G.R. & Smith, S.M. (2012) Adequacy of maternal iron status protects against behavioral, neuroanatomical, and growth deficits in fetal alcohol spectrum disorders. PLoS One, 7, e47499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbagh, L. , Bourbonnière, M. , Sékaly, R.P. & Cohen, L.Y. (2005) Selective up‐regulation of caspase‐3 gene expression following TCR engagement. Molecular Immunology, 42, 1345–1354. [DOI] [PubMed] [Google Scholar]
- Sadrian, B. , Lopez‐Guzman, M. , Wilson, D.A. & Saito, M. (2014) Distinct neurobehavioral dysfunction based on the timing of developmental binge‐like alcohol exposure. Neuroscience, 280, 204–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, M. , Smiley, J.F. , Hui, M. , Masiello, K. , Betz, J. , Ilina, M. et al. (2018) Neonatal ethanol disturbs the normal maturation of parvalbumin interneurons surrounded by subsets of perineuronal nets in the cerebral cortex: partial reversal by lithium. Cerebral Cortex, 29, 1383–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAMHSA (2013) "The NSDUH Report: 18 percent of pregnant women drink alcohol during early pregnancy." In NSDUH Report. Rockville, MD: Substance Abuse and Mental Health Services Administration. [Google Scholar]
- Santillano, D.R. , Kumar, L.S. , Prock, T.L. , Camarillo, C. , Tingling, J.D. & Miranda, R.C. (2005) Ethanol induces cell‐cycle activity and reduces stem cell diversity to alter both regenerative capacity and differentiation potential of cerebral cortical neuroepithelial precursors. BMC Neuroscience, 6, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalini, S. , Dorstyn, L. , Dawar, S. & Kumar, S. (2015) Old, new and emerging functions of caspases. Cell Death & Differentiation, 22, 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorput, A.G.J. , Gupta, V.P. , Yeh, P.W.L. & Yeh, H.H. (2015) Persistent interneuronopathy in the prefrontal cortex of young adult offspring exposed to ethanol in utero. The Journal of Neuroscience, 35, 10977–10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorput, A.G.J. & Yeh, H.H. (2016) Chronic gestational exposure to ethanol leads to enduring aberrances in cortical form and function in the medial prefrontal cortex. Alcoholism: Clinical and Experimental Research, 40, 1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley, J.F. , Saito, M. , Bleiwas, C. , Masiello, K. , Ardekani, B. , Guilfoyle, D.N. et al. (2015) Selective reduction of cerebral cortex GABA neurons in a late gestation model of fetal alcohol spectrum disorders. Alcohol, 49, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowell, E.R. , Johnson, A. , Kan, E. , Lu, L.H. , Van Horn, J.D. , Toga, A.W. et al. (2008) Mapping white matter integrity and neurobehavioral correlates in children with fetal alcohol spectrum disorders. Journal of Neuroscience, 28, 1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streissguth, A.P. , Bookstein, F.L. , Barr, H.M. , Sampson, P.D. , O'Malley, K. & Young, J.K. (2004) Risk factors for adverse life outcomes in fetal alcohol syndrome and fetal alcohol effects. Journal of Developmental and Behavioral Pediatrics, 25, 228–238. [DOI] [PubMed] [Google Scholar]
- Stuart, T. , Butler, A. , Hoffman, P. , Hafemeister, C. , Papalexi, E. , Mauck, W.M. et al. (2019) Comprehensive integration of single‐cell data. Cell, 177, 1888–902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tilborg, E. , de Theije, C.G.M. , van Hal, M. , Wagenaar, N. , de Vries, L.S. , Benders, M.J. et al. (2018) Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia, 66, 221–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treit, S. , Lebel, C. , Baugh, L. , Rasmussen, C. , Andrew, G. & Beaulieu, C. (2013) Longitudinal MRI reveals altered trajectory of brain development during childhood and adolescence in fetal alcohol spectrum disorders. Journal of Neuroscience, 33, 10098–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricoire, L. , Pelkey, K.A. , Erkkila, B.E. , Jeffries, B.W. , Yuan, X. & McBain, C.J. (2011) A blueprint for the spatiotemporal origins of mouse hippocampal interneuron diversity. Journal of Neuroscience, 31, 10948–10970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, J.L. , Klintsova, A.Y. , Greenough, W.T. & Goodlett, C.R. (2013) Rehabilitation training using complex motor learning rescues deficits in eyeblink classical conditioning in female rats induced by binge‐like neonatal alcohol exposure. Alcoholism, Clinical and Experimental Research, 37, 1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, J.R. , Hamre, K.M. & Cassell, M.D. (1986) Effects of ethanol exposure during the third trimester equivalent on neuron number in rat hippocampus and dentate gyrus. Alcoholism, Clinical and Experimental Research, 10, 190–197. [DOI] [PubMed] [Google Scholar]
- Wilhelm, C.J. & Guizzetti, M. (2015) Fetal alcohol spectrum disorders: an overview from the glia perspective. Frontiers in Integrative Neuroscience, 9, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, J.F. & Smith, V.C. (2015) Fetal alcohol spectrum disorders. Pediatrics, 136, e1395–e1406. [DOI] [PubMed] [Google Scholar]
- Willoughby, K.A. , Sheard, E.D. , Nash, K. & Rovet, J. (2008) Effects of prenatal alcohol exposure on hippocampal volume, verbal learning, and verbal and spatial recall in late childhood. Journal of the International Neuropsychological Society, 14, 1022–1033. [DOI] [PubMed] [Google Scholar]
- Wozniak, J.R. & Muetzel, R.L. (2011) What does diffusion tensor imaging reveal about the brain and cognition in fetal alcohol spectrum disorders? Neuropsychology Review, 21, 133–147. [DOI] [PubMed] [Google Scholar]
- Wozniak, J.R. , Riley, E.P. & Charness, M.E. (2019) Clinical presentation, diagnosis, and management of fetal alcohol spectrum disorders. The Lancet Neurology, 18, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, G. & He, Q.Y. (2016) ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Molecular BioSystems, 12, 477–479. [DOI] [PubMed] [Google Scholar]
- Yu, G. , Wang, L.G. , Han, Y. & He, Q.Y. (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel, A. , Hochgerner, H. , Lönnerberg, P. , Johnsson, A. , Memic, F. , van der Zwan, J. et al. (2018) Molecular architecture of the mouse nervous system. Cell, 174, 999–1014.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel, A. , Muñoz‐Manchado, A.B. , Codeluppi, S. , Lönnerberg, P. , La Manno, G. , Juréus, A. et al. (2015) Cell types in the mouse cortex and hippocampus revealed by single‐cell RNA‐seq. Science, 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zoeller, R.T. , Butnariu, O.V. , Fletcher, D.L. & Riley, E.P. (1994) Limited postnatal ethanol exposure permanently alters the expression of mRNAS encoding myelin basic protein and myelin‐associated glycoprotein in cerebellum. Alcoholism, Clinical and Experimental Research, 18, 909–916. [DOI] [PubMed] [Google Scholar]
- Zucca, S. & Valenzuela, C.F. (2010) Low concentrations of alcohol inhibit BDNF‐dependent GABAergic plasticity via L‐type Ca2+ channel inhibition in developing CA3 hippocampal pyramidal neurons. Journal of Neuroscience, 30, 6776–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Table S2
Table S3
Supplementary Material