

Abstract
As p53-loss of function confers high-level drug resistance in neuroblastoma, p53-independent therapies might have superior activity in recurrent neuroblastoma. We tested the activity of vorinostat, a histone deacetylase inhibitor, and flavopiridol, a pan-Cdk inhibitor, in a panel of multidrug-resistant neuroblastoma cell lines that included lines with wild-type (wt) and transcriptionally active TP53 (n = 3), mutated (mt) and loss of function (LOF) TP53 (n = 4) or p14ARF deletion (n = 1). The combination of vorinostat and flavopiridol was synergistic and significantly more cytotoxic (p<0.001) in cell lines with p53-LOF and in the clones stably transfected with dominant negative p53 plasmids. Cell cycle analysis by flow cytometry demonstrated prominent cell cycle arrest in G2/M for a cell line with wt TP53 (SK-N-RA) at 16–20 hours (37%), while cells with mt TP53 (CHLA-90) slipped into subG1 at 6–24 hours (25–40% specific cell death). The morphological hallmarks of mitotic cell death including defective spindle formation and abnormal cytokinesis were detected by confocal microscopy after the treatment with the vorinostat + flavopiridol combination in CHLA-90. The combination caused reduction in the expression of G2/M proteins (cyclin B1, Mad2, MPM2) in two cell lines with mt TP53, but not in those with wt TP53. Plk1 expression was reduced in all treated lines. siRNA knockdown of Mad2 and cyclin B1 or Plk1 synergistically reduced the clonogenicity of CHLA-90 cells. The combination of HDAC inhibitor and flavopiridol may be a unique approach to treating neuroblastomas with p53-LOF, one that evokes induction of mitotic failure.
Keywords: pediatric tumors, drug resistance, histone deacetylase inhibitors, cdk inhibitors, synergy
INTRODUCTION
Neuroblastoma is the most common extracranial solid tumor of childhood (1). Myeloablative cytotoxic therapy and 13 cis-retinoic acid (2) or anti-GD2 immunotherapy (3) has improved the outcome for high-risk neuroblastoma patients; however, most of these patients develop progressive disease which is refractory to additional cycles of chemotherapy. We have previously shown that the mechanism of such resistance is often through the loss of p53 function due to mutations in TP53 or to MDM2 amplification (4).
The tumor suppressor p53 is the most frequently altered gene in a wide variety of tumors. Inactivation of p53 protein occurs through mutations in the p53 gene or through alterations in genes whose products regulate p53, such as genomic amplification of the negative regulator MDM2, or inactivation of downstream cell cycle regulator p14ARF. Alteration of p53 activity contributes to an aggressive chemo or radiotherapy-resistant phenotype, and consequently, p53 is a target of research aimed at development of novel anticancer treatments. Among the chief functions of p53 are DNA damage-induced checkpoints in G1 and G2 phases of the cell cycle (5). The mechanisms by which p53 regulates the G2/M transition involve regulation of Cdk1 and transcriptional regulation of Cdc25C, 14-3-3σ, cyclin B1 and Cdk1 genes. Moreover, p53 has been implicated as an upstream regulator of BubR1 (6) and Mad1 (7). Blastocysts with simultaneous deletion of Mad2 and p53 show accelerated progression through mitosis (8). Mad1, Mad2, and BubR1 are components of the mitotic spindle-assembly checkpoint that prevents the onset of anaphase until all chromosomes are properly aligned at the metaphase plate.
It has also been shown that Plk1 associates with the DNA-binding domain of p53 (9), and that Plk1 depletion in systems with p53-loss of function results in cell death (10). However, the role of p53 in Plk1 inactivation remains to be clarified (11). Plk1 is a serine/threonine kinase with an indispensible set of functions regulating mitosis: centrosome maturation, bipolar spindle formation, activation of the Cdk1/cyclin B1 complex and execution of cytokinesis.
Vorinostat (Zolinza, suberoylanilide hydroxamic acid, or SAHA, Figure 1A) is an inhibitor of histone deacetylases (HDAC). The anti-tumor activity of vorinostat occurs through induction of differentiation, growth arrest, or apoptosis, as documented in a broad range of cancer cell lines. Anti-tumor activity has also been demonstrated in a number of in vivo models. Vorinostat received Food and Drug Administration (FDA) approval in 2006 for treatment of advanced cutaneous T-cell lymphoma.
Figure 1.
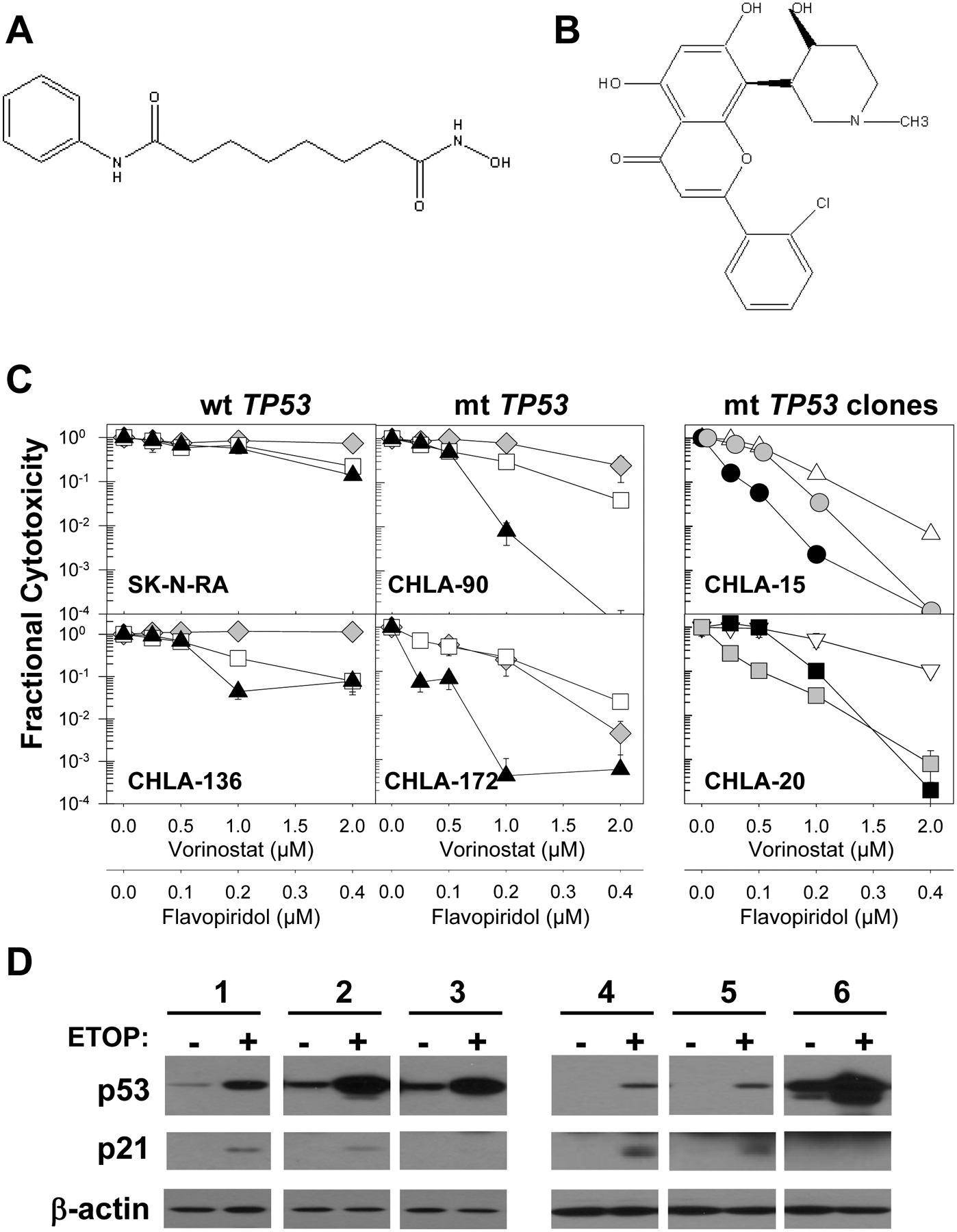
Effect of the vorinostat and flavopiridol drug combination in neuroblastoma cell lines. A. Chemical structure of vorinostat. B. Chemical structure of flavopiridol. C. Neuroblastoma cells were pre-treated with vorinostat and 24 hrs later flavopiridol was added for an additional 4 days. Dose-response curves of representative multidrug-resistant neuroblastoma cell lines are shown. CHLA-136 and SK-N-RA cells carry wt and transcriptionally active p53; CHLA-90 and CHLA-172 carry mt TP53 with loss of function. Dose-response curves for vorinostat (◆), flavopiridol (□), and vorinostat + flavopiridol (▲) were obtained using the DIMSCAN assay. CHLA-15 (△) and CHLA-20 (∇) cells transfected with empty vector, or mt p53 (pR175H or pR273H) expression vectors were tested with the vorinostat + flavopiridol combination. Clones with mt TP53 are designated as CHLA-15175A (●), CHLA-15175B (●), CHLA-20273C (■), and CHLA-20273D (■). D. Failure to induce p21 in response to etoposide (5 μg/ml for 16 hrs) confirms p53-loss of function in CHLA-15175A, CHLA-15175B, CHLA-20273C, and CHLA-20273D clones in immunoblot analyses. Samples: 1 – CHLA-15 empty vector control; 2 – clone CHLA-15175A, 3 – clone CHLA-15175B; 4 –CHLA-20 empty vector control; 5 – clone CHLA-20273C; and 6 – clone CHLA-20273D. Equal loading of protein was confirmed by β-actin expression.
Flavopiridol (NSC 649890, Figure 1B) is a novel cyclin-dependent kinase (CDK) inhibitor (12) that has exhibited potent growth-inhibitory activity against a number of human tumor cell lines, both in vitro and in xenografts. Flavopiridol as a single agent has produced 40% durable partial responses in refractory chronic lymphocytic leukemia (CLL) patients in a phase I trial given as a 30-min loading dose followed by 4 hours of infusion administered weekly for 4 weeks (13,14).
Flavopiridol and HDAC inhibitors have been shown to synergize in leukemia, breast, lung, esophageal cancer and pleural mesothelioma models (15–20). Synergistic cell kill has been attributed to flavopiridol-mediated transcriptional repression of the anti-apoptotic genes p21, Mcl-1, XIAP, and NF-κB. We tested the anti-tumor activity of the vorinostat and flavopiridol combination in representative multidrug-resistant neuroblastoma cell lines with mutant (mt) or wild-type (wt) TP53.
MATERIALS AND METHODS
Cell Lines.
We used 11 neuroblastoma cell lines: CHLA-15, CHLA-20, SK-N-BE(1), SK-N-BE(2), SK-N-RA, LA-N-6, CHLA-136, CHLA-79, CHLA-119, CHLA-90, CHLA-172. the neuroblastoma origin of these cell lines has been confirmed previously (21).
SK-N-RA cells were maintained in complete medium consisting of RPMI (Mediatech, Inc, Manassas, VA) supplemented with 10% heat inactivated fetal bovine serum (Omega Scientific, Tarzana, CA). All other cell lines were cultured in complete medium consisting of Iscove’s modified Dulbecco’s medium (Bio Whittaker, Walkersville, MD) supplemented with 3 mM L-glutamine (Gemini Bioproducts, Inc., Calabasas, CA); 5 μg/mL insulin, 5 μg/mL transferrin, and 5 ng/mL of selenous acid (ITS™ Culture Supplement; Collaborative Biomedical Products, Bedford, MA); and 20% heat-inactivated fetal bovine serum. All neuroblastoma cell lines were used in this study before passage 40. The cell lines were cultured without antibiotics in a humidified incubator (95% air–5% CO2) at 37 °C. All cell lines tested negative for mycoplasma. Cell lines were not selected for drug resistance in vitro. The identities of all cell lines were confirmed by the short tandem repeat (STR) assay. Unique short tandem repeats were identified for all cell lines, except between CHLA-15 and CHLA-20, and between SK-N-BE(1) and SK-N-BE(2), pairs of cell lines that were derived from the same patient and had identical short tandem repeats, as expected.
Drugs, Chemicals, Antibodies, and Plasmids.
Vorinostat and flavopiridol were obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, National Cancer Institute (Bethesda, MD). Fluorescein diacetate was purchased from Eastman Kodak Company (Rochester, NY), and eosin Y was from Sigma Chemical Co (St. Louis, MO). Antibodies were purchased from various manufacturers: anti-53 (DO7) from BDPhramingen™ (San Diego, CA); anti-Plk1 (208G4), caspase-9 mouse mAb (C9), and caspase-3 rabbit mAb (8G10) from Cell Signaling Technology (Boston, MA); anti-p21 (C-19) and anti-MAD2 (sc-28261) from Santa Cruz Biotechnology Inc (Santa Cruz, CA); anti-MPM2 (ab5506) from Abcam Inc (Cambridge, MA); and horse radish peroxidase–conjugated secondary antibodies from Santa Cruz Biotechnology. Plasmids containing a mutant p53 R175H or R273H gene were a kind gift from Dr. B. Vogelstein (The Johns Hopkins Medical Institute, and Howard Hughes Medical Institute).
Cytotoxicity Assay.
We determined the cytotoxicity of vorinostat and flavopiridol alone or in combination using DIMSCAN, a semiautomatic fluorescence-based digital image microscopy system that quantifies viable cells in tissue culture multi-well plates on the basis of their selective accumulation of fluorescein diacetate. DIMSCAN is capable of measuring cytotoxicity over a 4-log dynamic range by quantifying total fluorescence per well which is proportional to the number of viable, clonogenic cells after eliminating background fluorescence with digital thresholding and eosin Y quenching.
To conduct formal quantitative analysis of the interaction between vorinostat and flavopiridol, we employed a fixed-ratio analysis for combination cytotoxicity assays; drugs were tested on a linear scale at concentration ranges that included clinically achievable blood plasma levels. A fixed ratio of 5:1 was chosen after selecting the maximally tested drug concentrations of vorinostat and flavopiridol. Cells were exposed to 0 – 2 μM vorinostat because it was reported to be in the range of clinically achievable when administered orally (22). The mean steady-state concentrations of flavopiridol attainable in patients and inhibitory to Cdks (23) were tested (0 – 0.4 μM) in our experiments.
Cell lines were seeded into 96-well plates in 150 μL of complete medium (3,000–5,000 cells per well) and incubated overnight. Vorinostat (at various concentrations) in 100 μL of complete medium was added to each well (12 replicate wells per each concentration of vorinostat). Flavopiridol (0 to 0.4 μM) was added to individual wells 24 hours later. Each drug concentration was tested in 12 replicate wells. Cell lines were incubated in the presence of flavopiridol for 5 days, after which fluorescein diacetate in 50 μL of 0.5% eosin Y (final concentration of fluorescein diacetate 10 μg/mL) was added to each well and cells were incubated for an additional 25 minutes at 37 °C. Total fluorescence was then measured using the DIMSCAN system and results were expressed as surviving fractions of treated cells compared with control cells.
Transfections.
pR175H and pR273H contain 1.8kb of full length p53 cDNA cloned into pCMV-neo-Bam under transcriptional control of the immediate early enhancer/promoter of human cytomegalovirus (CMV). pR175H and pR273H encode mutant forms of p53 and differ from wild type TP53 by a single point mutation at codons 175 or 273, respectively. CHLA-15 (passage 22) and CHLA-20 (passage 23) cells were transfected using lipofectamine 2000 Regent (Invitrogen, Carlsbad, CA) with 4 μg of DNA from the control pCMV-neo-Bam vector or the pR175H and pR273H vectors. After selection in serum-free IMDM with 300 μg/ml G418 (Gemini Bio-Products, West Sacramento, CA), mass cultures were established and expanded in selective medium.
Gene knock-down by siRNA.
We used Lipofectamine™ 2000 reagent (Invitrogen, Carlsbad, CA) to transfect cells with small interfering RNAs (siRNA) targeted against Plk1 (Qiagen, Valencia, CA, Cat. #SI02223837), Mad2 (Santa Cruz Biotechnology, Inc, Santa Cruz, CA, Cat. #sc-35837), and Cyclin B1 (Thermo Scientific, Lafayette, CO) genes. For Cyclin B1 silencing, the mixture of two target sequences was used (1:1 ratio): CAACAUUACCUGUCAUAUA and UGCACUAGUUCAAGAUUUA. Control cells were transfected with an siRNA containing a scrambled sequence (Qiagen, Valencia, CA). For Mad2 and cyclin B1 or Plk1 co-transfection experiments, 5×105 CHLA-90 cells were seeded into 6-well plates; gene silencing of transfected cells was confirmed by immunoblotting of cell lysates assayed at 48 hrs, clonogenicity was assayed at 72 hrs using the DIMSCAN assay.
Cell cycle analysis by flow cytometry.
Prior to drug exposures cells were synchronized in FBS-free medium for 24 hrs. Flow cytometry was performed on a BD LSR II flow cytometer (BD Biosciences, San Jose, CA) and data was acquired and analyzed using BD FACSDiva software (San Jose, CA). Cell cycle phases were quantified employing MultiCycle software (Phoenix Flow Systems, San Diego, CA).
Apoptosis.
The APO-DIRECT™ kit (Cat #: 556381, BD Pharmingen, San Jose, CA) was used to identify DNA fragmentation by two-color flow cytometry using propidium iodide (PI) and FITC-dUTP added to DNA fragments by terminal deoxynucleotidyltransferase (TdT). Staining with the APO-DIRECT™ kit was performed according to the manufacturer’s instructions. The percentage of cells showing TdT-mediated fluorescent staining was determined using band pass filters of 525 ± 25 nm for FITC and 610 ± 10 nm for PI.
The pan-caspase inhibitor BOC-d-fmk was used to block the apoptotic component of cell death in CHLA-90 cells. Cells were pre-incubated with 40 μM BOC-d-fmk 2 hrs prior to the addition of 2 μM vorinostat, 0.2 μM flavopiridol or their combination, and an additional 40 μM was added 24 hrs later.
Immunoblotting.
20 μg of total protein per lane was fractionated on 4–20% Tris-Glycine gels (Invitrogen, Carlsbad, CA), transferred to PVDF Filter Paper (Invitrogen), and probed with primary and then HRP-labeled secondary antibodies. Proteins were visualized using SuperSignal® West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL).
Immunofluorescence.
10,000 CHLA-90 cells were seeded on chamber slides and treated with 2 μM vorinostat (36 hrs), flavopiridol (0.2 μM, 12 hrs), or their combination. Cells were then fixed in 3.65% formaldehyde in PBS for 20 mins, permeabilized with 0.25% Triton-X, and blocked with 3% BSA in PBS for 1 hr at room temperature. Next, coverslips were labeled with 5 μg/mL anti-tubulin and 0.1 μg/mL 4’6-diamino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA) for 1 hr at room temperature, washed in PBS and air-dried. Coverslips were mounted with Prolong® Gold antifade reagent (Invitrogen, Carlsbad, CA). Images were visualized using the axiovert200 microscope (Carl Zeiss MicroImaging, Inc.) equipped with 100x Zeiss Plan-Apochromat objective lens and Hamamtsu C4742-80 camera. IPLab 4.0.8 (BD Biosciences Bioimaging, Rockville, MD) software was used for image acquisition, processing, measurement, and analysis.
Statistical Analysis.
LC90 (i.e., the drug concentration that was lethal for 90% of the cell population) values were calculated using a previously described algorithm.
One-way analysis of variance (ANOVA) was used to investigate the difference in cytotoxicity of each of the drugs or drug combinations (flavopiridol, vorinostat, vorinostat + flavopiridol) in neuroblastoma cell lines with wt TP53 or p53-LOF. For each of the drugs or drug combinations, the mean number of logs of cell kill was calculated and then compared between cell lines with p53-LOF and cell lines with wt TP53. Analysis was conducted at the two highest doses tested in the experiments. Analyses were performed using SAS version 9.1 software (SAS Institute, Inc., Cary, NC, USA).
Dose-response curves of the vorinostat + flavopiridol combination for parental cells with wild-type TP53 vs. clones transfected with mt p53 were compared. The difference in the cytotoxic effect was examined by comparing the area between the dose-response curve and the 100% survival line. Statistical analysis assumed that the difference between the untreated controls and the highest drug concentration was 1. The fixed ratio design of the cytotoxicity experiments allowed the concentration increments to be quantitated at 1/8, 1/4, 1/2, and 1. The area between the survival curves for each treatment group and the 100% survival line was viewed as the sum of areas of four trapezoids, computed as:
Where were the mean numbers of logs of cell kill at the 0 th, 1st, 2nd, 3rd and 4th drug conditions. Finally, the contrast was computed as:
and tested the difference between the two dose-response curves, where were the mean number of logs of cell kill at each concentration of the parental cells, and were the mean number of logs of cell kill for cells transfected with mt TP53. The significance was determined using contrast analysis in analysis of variance, with p values corrected by Bonferroni method. Data were analyzed with SAS Version 9.1 software.
Poisson regression analysis was used to compare the difference in the number of abnormal mitoses between control and the vorinostat + flavopiridol treated groups as evaluated by confocal microscopy.
To compare difference in the fluorescence scores for cells expressing Mad2, cyclin B1, Plk1, Mad2 + cyclin B1 or Mad2 + Plk1 siRNAs, a multiple linear regression model was first fit using data from all the groups, then the difference in fluorescence scores between two different groups was evaluated by testing the appropriate linear combinations using the STATA LINCOM command. The fluorescence scores were transformed to the base 10 logarithmic scale before analysis was performed.
RESULTS
Cytotoxicity of the vorinostat and flavopiridol combination.
Vorinostat and flavopiridol were tested sequentially at the fixed ratio 5:1 in 1 drug-sensitive [SK-N-BE(1)] and 8 multidrug-resistant cell lines [SK-N-RA, CHLA-79, CHLA-136, LA-N-6, SK-N-BE(2), CHLA-90, CHLA-119, and CHLA-172] (Table 1, Figure 1A, Supplemental Figure 1). The drug concentrations lethal for 90% or 99% of treated cells (LC90 and LC99) were calculated from dose-response curves using the DIMSCAN assay, which provides a 4 log dynamic range. Synergy (CI ≤ 0.7), assayed at LC90 and LC99, was observed in one cell line with wild-type (wt) TP53 (CHLA-136), and in all four cell lines with mutant (mt) TP53 [SK-N-BE(2), CHLA-90, CHLA-119, and CHLA-172]. We have previously shown these cell lines to be resistant to the commonly used chemotherapeutic drugs melphalan, carboplatin, and/or etoposide (21). Our panel included a pair of cell lines one member of which was established from a patient at diagnosis [SK-N-BE(1)] and the other member established later from the same patient after disease progression following multi-agent chemoradiotherapy [SK-N-BE(2)] (21). Interestingly, combination of vorinostat + flavopiridol was more active in the SK-N-BE(2) cell line than in drug-sensitive SK-N-BE(1) cells (Supplemental Figure 1, Table 1). In addition to sequential drug exposures, vorinostat and flavopiridol were administered simultaneously in CHLA-90 cells, but no synergy was observed with this timing schedule (data not shown).
Table 1.
LC90 and LC99 values for vorinostat, flavopiridol and their combination
| Cell Lines | LC90 (μM) | CI IC90* | LC99 (μM) | CI IC99 | ||||
|---|---|---|---|---|---|---|---|---|
| V | FL | V + FL | V | FL | V + FL | |||
| Wild-type TP53 | ||||||||
| SK-N-BE(1) | 1.8 | 0.2 | 0.7 + 0.1 | 1.0 ± 0.3 | >2.0 | 1.0 | >2.0 + >0.4 | ND |
| SK-N-RA | >2.0¶ | >0.4 | >2.0 + >0.4 | ND§ | >2.0 | >0.4 | >2.0 + >0.4 | ND |
| CHLA-79 | >2.0 | 0.4 | 1.4 + 0.3 | 0.8 ± 0.3 | >2.0 | 0.4 | 1.7 + 0.3 | 0.8 ± 0.4 |
| CHLA-136 | >2.0 | 0.4 | 1.3 + 0.3 | 0.7 ± 0.2 | >2.0 | >0.4 | >2.0 + >0.4 | 0.5 ± 0.3 |
| Mutant TP53 | ||||||||
| SK-N-BE(2) | >2.0 | 0.3 | 1.1 + 0.2 | 0.7 ± 0.2 | >2.0 | 0.4 | 1.5 + 0.3 | 0.5 ± 0.2 |
| CHLA-119 | >2.0 | 0.3 | 0.6 + 0.1 | 0.5 ± 0.2 | >2.0 | >0.4 | 0.9 + 0.2 | 0.2 ± 0.2 |
| CHLA-90 | >2.0 | 0.4 | 0.6 + 0.1 | 0.3 ± 0.1 | >2.0 | >0.4 | 0.5 + 0.3 | 0.2 ± 0.1 |
| CHLA-172 | 0.8 | 0.2 | 0.4 + 0.05 | 0.5 ± 0.3 | 2.0 | >0.4 | 0.6 + 0.1 | 0.4 ± 0.2 |
| p14 deletion | ||||||||
| LA-N-6 | >2.0 | 0.3 | 0.8 + 0.2 | 0.7 ± 0.3 | >2.0 | >0.4 | 1.5 + 0.3 | 0.4 ± 0.3 |
| Transfected clones | ||||||||
| CHLA-15EV | 1.0 + 0.2 | 2.0 + 0.4 | ||||||
| CHLA-15175a | 0.3 + 0.06 | 0.7 + 0.1 | ||||||
| CHLA-15175b | 0.6 + 0.1 | 1.0 + 0.2 | ||||||
| CHLA-20EV | >2.0 + >0.4 | >2.0 + >0.4 | ||||||
| CHLA-20273c | 1.0 + 0.2 | 2.0 + 0.4 | ||||||
| CHLA-20273d | 1.1 + 0.2 | 1.4 + 0.3 | ||||||
If 90% or 99% cytotoxicity was not achieved at the tested concentrations, then LC90 or LC99 values were reported as “> the highest tested concentration”. The highest concentrations tested were 2 μM for vorinostat, and 0.4 μM for flavopiridol. Similarly, if 90% or 99% cytotoxicity was achieved at concentrations lower than tested, then LC90 or LC99 values were reported as “< the lowest tested concentration”. The lowest concentrations tested were 0.025 μM for vorinostat, and 0.05 μM for flavopiridol.
The combination index (CI) correlates the cytotoxicity of drug combinations to the single-agent activities. Standard deviations (±) for CI values are also included.
CI was not determined (ND) if cytotoxicity was not achieved within the concentration range tested. Interpretation of CI values: 0.1–0.3 = strong synergism, >0.3 – 0.7 = synergism, >0.7–0.9 = slight synergism, >0.9 –1.0 = additive effect, more than 1.1 = antagonism.
We conducted one-way analysis of variance (ANOVA) to determine the difference in the cytotoxic effect of the vorinostat + flavopiridol combination in cell lines with wt TP53 vs mt TP53. The analysis was conducted for two drug concentrations: vorinostat 1 μM + flavopiridol 0.2 μM, and vorinostat 2 μM + flavopiridol 0.4 μM. Statistically significant cytotoxicity was confirmed in cell lines with wt or mt TP53 at the two highest tested concentrations (p<0.0001). However, vorinostat + flavopiridol was significantly more cytotoxic in cell lines with mt TP53 than in cell lines with wt TP53 (p<0.001).
Effect of p53 mutations on the activity of the vorinostat + flavopiridol combination.
To examine whether the vorinostat + flavopiridol combination is predominantly active in neuroblastomas carrying mt p53, we transfected CHLA-15 and CHLA-20 cells with the pCMV-Neo-Bam empty vector as a control or with mutant p53 (pR175H or pR273H) expression vectors, and two G418-resistant clones were selected from each cell line. The clones were designated as CHLA-15175A, CHLA-15175B, CHLA-20273C, and CHLA-20273D. Compared to other neuroblastoma cell lines, CHLA-15 and CHLA-20 clump to a lesser extent and thus were chosen for these experiments to yield higher transfection efficiency. The sensitivities of mtp53 transduced clones, parental cell lines, and empty vector transduced controls to the vorinostat + flavopiridol combination were compared using the DIMSCAN assay (Figure 1B, Table 1). Examining the activity of mt p53 in transduced clones with the p53-specific monocolonal antibody DO7, we detected increased basal levels of p53 protein in three of four clones transduced with mt p53 expression vector. In addition, reduced p53 activity was confirmed by the lack of induction of p21 in etoposide-challenged samples in clones CHLA-15175B and CHLA-20273D and modest induction of p21 in clones CHLA-15175A and CHLA-20273C using immunoblotting (Figure 1C). Transfection with p53 mutant expression vectors sensitized both cell lines to the vorinostat + flavopiridol combination (Figure 1B). LC90 values of CHLA-15 clones (CHLA-15175A and CHLA-15175B) were 1.6 to 3-times lower relative to empty vector controls (designated as CHLA-15EV). Similarly, LC90 values of the CHLA-20273 clones were >2–times lower relative to CHLA-20EV (Table 1).
Contrast analysis of variance, corrected with the Bonferroni method, demonstrated statistically significant sensitivity (p<0.001) for clones expressing mt p53 relative to empty vector controls in response to the tested combination.
Induction of apoptosis by the vorinostat + flavopiridol combination.
Addressing whether apoptosis has a role in cell death induced by the combination, we detected increased apoptosis by the TUNEL assay in mt TP53 CHLA-90 cells (Figure 2A). Flavopiridol and especially the vorinostat + flavopiridol combination increased apoptosis, which was partially reversed by pre-treatment with the pan-caspase inhibitor BOC-d-fmk (Figure 2A). In addition, activation of caspase-3 and −9 was measured by immunoblotting in 2 cell lines with wt TP53 (CHLA-136 and CHLA-15) as well as 2 cell lines with mt TP53 (CHLA-90 and CHLA-172) (Figure 2B, Supplemental Figure 2). Activation of caspase-3 and −9 was sustained throughout exposure to the combination in CHLA-136 and CHLA-15 cell lines. In CHLA-90 and CHLA-172 cell lines, caspase-3 and -9 activation was detectable for 12 hrs.
Figure 2.
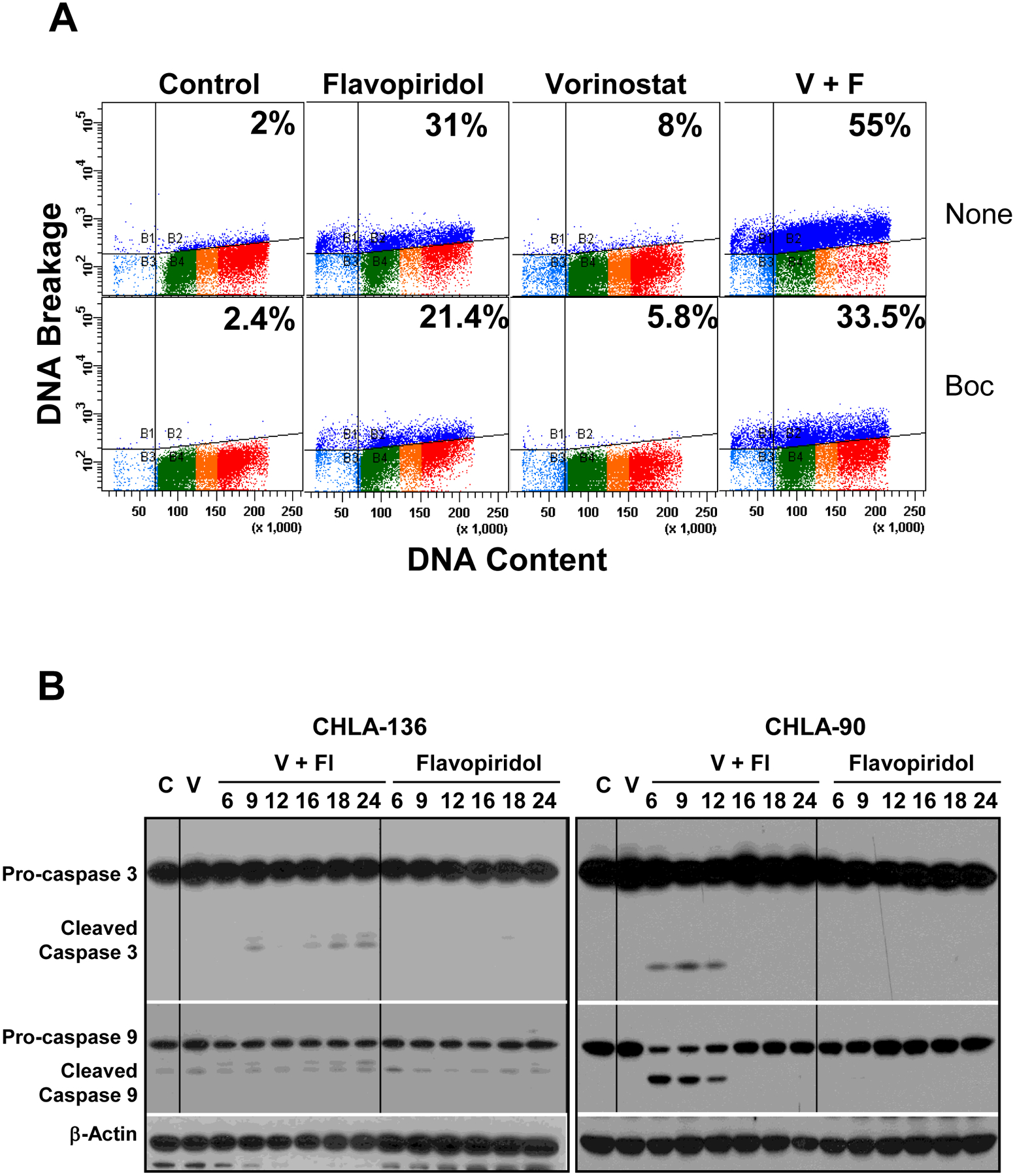
Induction of apoptosis by the vorinostat + flavopiridol drug combination. A. Induction of apoptosis in the CHLA-90 cell line measured by TUNEL assay. Cells were pre-treated with vorinostat (2 μM) for 24 hrs and then flavopiridol (0.2 μM) was added for an additional 24 hrs. Apoptosis induced by vorinostat alone was assayed after a 48-hr exposure. For apoptosis inhibition, the pan-caspase BOC-d-fmk (40 μM) was added 2 hrs prior to vorinostat and an additional 40 μM was added 24 hrs later according to the manufacturer’s instructions. B. Immunoblot analysis of caspase-3 (35 kDa) and caspase-9 (47 kDa) is shown in representative cell lines with wt TP53 (CHLA-136) or mt TP53 (CHLA-90). Drug effects were determined according to activation of caspase-3 to a cleaved 19 kDa form, and caspase-9 to a cleaved 35 kDa form. Equal loading of protein was confirmed by β-actin expression. Cells were pre-treated with 2 μM vorinostat (V) for 24 hrs, and then 0.2 μM flavopiridol (Fl) was added for additional an 6, 9, 12, 16, 18, or 24 hrs. C – Vehicle treated controls.
Collectively, our data suggests that this drug combination induces cell death that may be both caspase-dependent and caspase-independent.
Effect of vorinostat + flavopiridol on cell cycle distribution.
Various HDAC inhibitors and flavopiridol have been shown to modulate cell cycle progression. HDAC inhibitors of various classes induce G1 and/or G2/M arrest, or premature sister chromatid separation (24) through alterations of BubR1 (25), p55Cdc/Cdc20 (26), p21,cyclins D1, E, A, cdc25A (27) and other targets. Treatment with flavopiridol in mitosis can result in premature mitotic exit (28). As predominant cytotoxicity in our cell lines was seen in the presence of mt TP53, and since p53 controls G1 and G2 checkpoints, we measured cell cycle in CHLA-90 (mt TP53) and SK-N-RA (wt TP53) lines treated with the drug combination at regular time intervals for 24 hrs (Figure 3). Cells were pre-treated with or without 2 μM vorinostat and exposed to 0.2 μM flavopiridol. Accumulation in G2 was prominent in wt TP53 SK-N-RA cells while mt TP53 CHLA-90 cells slipped into subG0/G1 after treatment with the combination (Figure 3). This observation suggested that cell death mediated by the tested combination may occur through failed mitosis in neuroblastoma lines with mt TP53, due to abrogation of the G2/M checkpoint in mutant cells.
Figure 3.
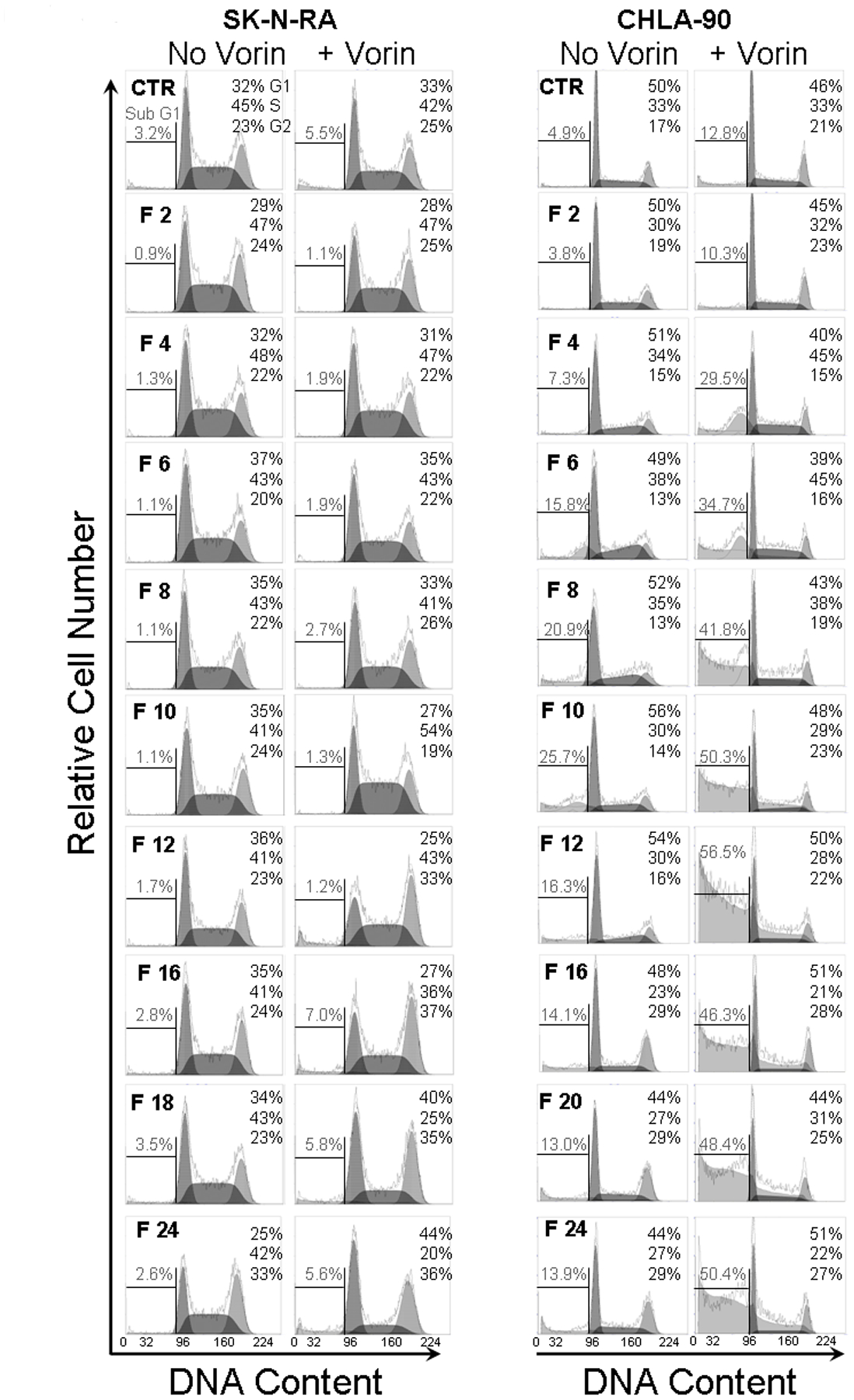
Cell cycle anlysis in cell lines with wt TP53 and mt TP53. SK-N-RA (wt TP53) and CHLA-90 (mt TP53) cells were pre-treated with 2 μM vorinostat (Vorin) for 24 hrs, and then 0.2 μM flavopiridol (F) was added for an additional 2, 4, 6, 8, 10, 12, 16, 18, or 24 hrs. G1, S, and G2/M phases of the cell cycle were then measured for permeabilized cells stained with propidium iodide using flow cytometry. Frequencies of intact cells in G1, S, and G2/M phases are given in the upper right of each histogram. Frequencies of sub-G1 events, indicative of dead cells, is shown on the left of each histogram.
Effect of vorinostat + flavopiridol on mitosis.
To confirm our hypothesis that the combination mediates cell death of mt TP53 neuroblastomas through failed mitosis, CHLA-90 cells were examined by immunofluorescence to visualize α-tubulin and DNA for abnormal spindle formation and DNA condensation (Figure 4A). Confocal microscopy of the stained cells showed that, while DMSO-treated cells go through various phases of mitosis without any abnormality, cells treated with the combination exhibited profound abnormalities in spindle formation and cytokinesis (Figure 4B).
Figure 4.
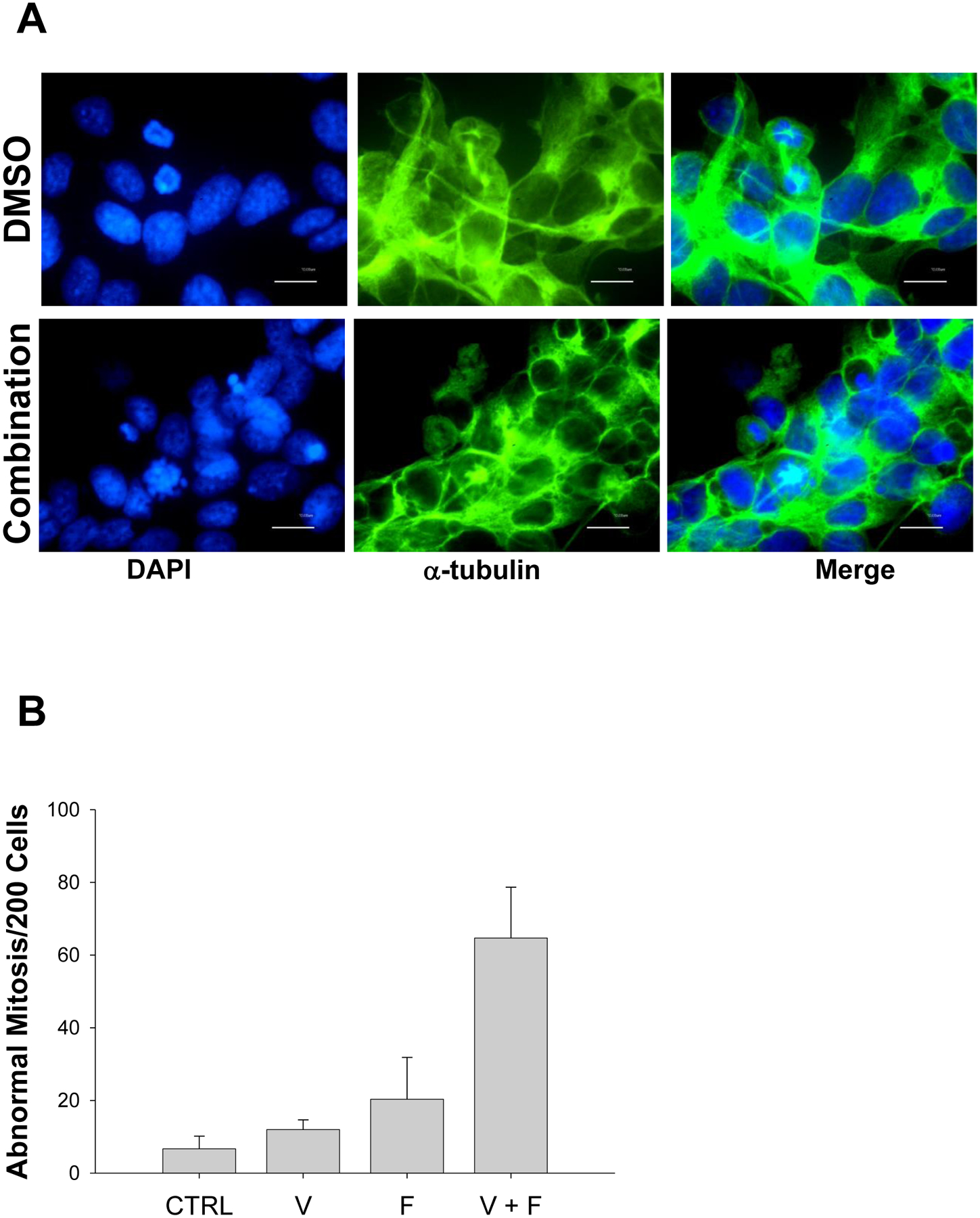
The vorinostat + flavopiridol drug combination causes abnormal mitosis. A. CHLA-90 cells were pre-treated with 2 μM vorinostat for 24 hrs and then 0.2 μM flavopiridol was added for an additional 12 hrs. DNA was visualized with DAPI and microtubules were examined with an anti-α-tubulin antibody. B. Cells exhibiting abnormal cytokinesis, condensed and fragmented chromatin, or cells without nuclei were counted manually in the presence or absence of the vorinostat and flavopiridol combination. A total of 200 events were counted for each condition. Results represent means ± st. dev. from three separate experiments. CTRL – cells treated with vehicle solutions; V – cells treated with vorinostat; F – cells treated with flavopiridol; V + F – cells treated with the combination of vorinostat and flavopiridol.
G2 checkpoint activation by the vorinostat + flavopiridol combination.
To characterize activation of the combination-induced G2 checkpoint, we examined the time-kinetic expression of cdk1, pCdk1 Thr14/Tyr15, and cyclin B1 by immunoblotting in 2 cell lines with mt TP53 (CHLA-90 and CHLA-172) and 2 cell lines with wt TP53 (CHLA-136 and SK-N-RA) (Figure 5A, Supplemental Figure 3A). Dephosphorylation of Cdk1 on Thr14 and Tyr15 by Cdc25C is shown to result in activation of Cdk1, and entry into anaphase depends on the loss of cyclin B1/Cdk1 activity. Cdk1 expression was maintained in CHLA-136, SK-N-RA and CHLA-90 cells throughout the drug exposures; however, pCdk1 Thr14/Tyr15 expression was down-regulated in all tested cell lines by flavopiridol and the combination except SK-N-RA in which we were unable to detect pCdk1 Thr14/Tyr15. We observed dramatic downregulation of cyclin B1 in response to flavopiridol and more so to the combination in cell lines with mt TP53 (in CHLA-90, 3-fold by flavopiridol and >7-fold by the combination, in CHLA-172 3-fold with either flavopiridol or the combination), compared to those with wt TP53 (in CHLA-136 induction by flavopiridol, 1.3-fold down-regulation by the combination; in SK-N-RA 1.5-fold by flavopiridol and 1.8-fold by the combination).
Figure 5.
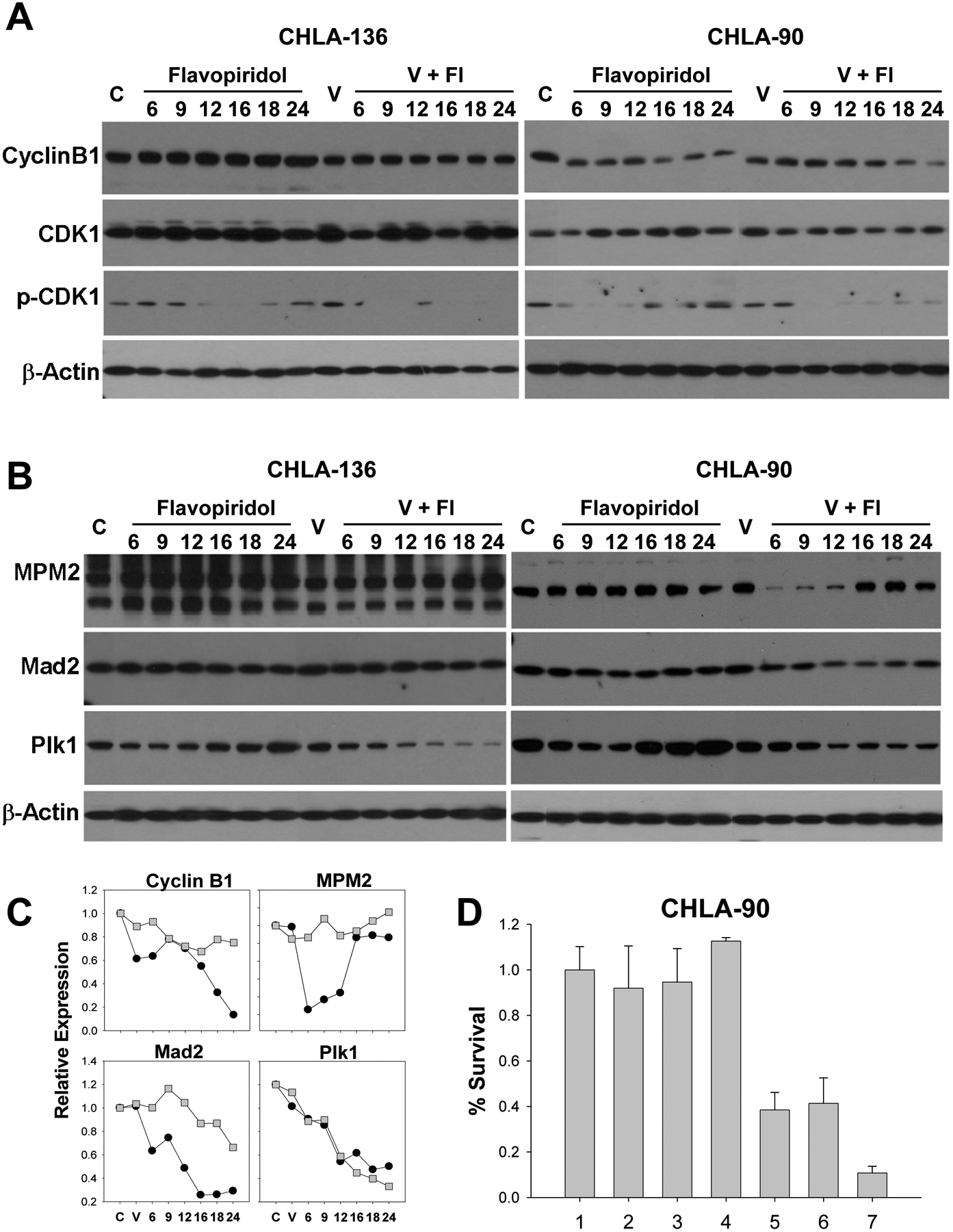
Effect of vorinostat (V) + flavopiridol (Fl) on expression of G2/M proteins. Representative neuroblastoma cell lines with wt TP53 (CHLA-136) and mt TP53 (CHLA-90) are shown. Cells were pre-treated with 2 μM vorinostat (V) for 24 hrs, and then 0.2 μM flavopiridol (Fl) was added for an additional 6, 9, 12, 16, 18, or 24 hrs. A. Immunoblot analysis of cyclin B1 (55 kDa), Cdk1 (34 kDa), and pCdk1 Thy15/Tyr15 (34 kDa) protein expression. Equal loading of protein was confirmed by β-actin expression. B. Immunoblot analysis of MPM2 (84 kDa), Mad2 (24 kDa), and Plk1 (62 kDa) protein expression. Equal loading of protein was confirmed by β-actin expression. C. Quantitative analysis of expression of cyclin B1, MPM2, Mad2, and Plk1 proteins as the ratio to β-actin in CHLA-136 (■) and CHLA-90 (●) cell lines treated with vorinostat + flavopiridol at 6, 9, 12, 16, 18, and 24 hrs. C – Vehicle treated controls, V – treatment with 2 μM vorinostat. D. CHLA-90 cells were transfected with 200 pMol total (100 pMol of target siRNA + 100 pMol scrambled siRNA for single targets and 100 pMol of each in co-transfection experiments) of scrambled, Mad2, cyclin B1, and Plk1 siRNAs. Forty eight hrs later, transfection efficacy was confirmed by immunoblotting (data not shown), and 72 hrs later cell clonogenicity was determined using the DIMSCAN assay. The DIMSCAN assay was performed in 3 separate experiments. Lanes: 1 – control; 2 – scrambled siRNA; 3 – Mad2 siRNA; 4 – cyclin B1; 5 – co-expression of Mad2 and cyclin B1 siRNAs; 6 – Plk1 siRNA; 7 - co-expression of Mad2 + Plk1 siRNA.
The vorinostat + flavopiridol combination affects mitosis exit markers in mt TP53 neuroblastoma cell lines.
We measured MPM2, Mad2 and Plk1 in protein lysates obtained from cells treated with vorinostat and/or flavopiridol at frequent time intervals by immunoblotting (Figure 5B). These studies were carried out on 2 cell lines with mt TP53 (CHLA-90 and CHLA-172) and 2 cell lines with wt TP53 (CHLA-136 and SK-N-RA).
During normal mitosis, MPM2 staining is intense in prophase and diminishes during interphase; weakly positive MPM2 expression in interphase is associated with recognition of centriolar proteins (29). MPM2 expression is considered a marker of mitosis (30) and disappearance of MPM2 is commonly used to assay mitotic exit. In wt TP53 CHLA-136 and SK-N-RA cells, MPM2 expression was maintained throughout exposure to the vorinostat + flavopiridol combination. In contrast, mt TP53 CHLA-90 and CHLA-172 cells exhibited dramatic decrease in MPM2 expression: 4 to 18-fold at 6 to 12 hrs in CHLA-90, and 2.4 to 4.8-fold at 6 to 16 hrs in CHLA-172.
We measured expression of Plk1, an essential kinase regulating both mitotic progression at multiple points and also mitotic exit (31). Temporary downregulation of Plk1 was achieved by flavopiridol treatment, but exposure to the combination caused sustained downregulation in all cell lines (Figure 5B, C, Supplemental Figure 3B).
We assayed expression of Mad2, a major downstream effector of the spindle assembly checkpoint whose degradation is a requirement for the onset of anaphase. In SK-N-RA cells, Mad2 expression was maintained throughout exposure to the vorinostat + flavopiridol combination (Supplemental Figure 3B). In CHLA-136, Mad2 expression decreased to 67% of basal level at 24 hrs of the treatment (Figure 5B). In CHLA-90 and CHLA-172 cells, flavopiridol led to temporary Mad2 down-regulation, but the combination caused the continuous decrease in Mad2 expression, dropping to 26% (CHLA-90) and 23% (CHLA-172) of basal levels at 24 hrs.
Our data indicate that cells with mt TP53 are induced to exit from mitosis when treated with the drug combination.
Depletion of G2/M proteins is lethal for a multidrug-resistant neuroblastoma cell line.
To confirm that aberrant mitosis can be used as a therapeutic strategy against p53 loss-of-function neuroblastomas, we examined the clonogenicity of CHLA-90 cells that were transfected with Mad2 siRNA in the absence or presence of cyclin B1 or Plk1 siRNA. CHLA-90 cells were chosen for this siRNA transfection experiment as these cells grow in single suspension and accommodate higher transfection efficiency. Mad2 was chosen for co-transfection experiments with cyclin B1 and Plk1 due to selective down-regulation of this protein by the drug combination in mt TP53 cell lines. Transient expression of Mad2, cyclin B1, or Plk1 siRNA produced robust depletion (70% for Mad2, 80% for cyclin B1, and 85% for Plk1) of target proteins as measured at 48-hrs by immunoblotting, whereas mock siRNA did not affect levels of any of these proteins. Simultaneous reduction of Mad2 and cyclin B1 or Plk1 levels acted synergistically with significant (p < 0.001) growth inhibition/cell death of CHLA-90 cell line as measured by the DIMSCAN assay at 72-hrs (Figure 5D). Our data indicate that the silencing of G2/M genes can sensitize multidrug-resistant neuroblastoma cells with p53-loss of function.
DISCUSSION
We have previously shown that relapsed neuroblastomas manifest resistance to multiple drugs that is sustained in vitro (21), and such drug resistance is often caused by loss of TP53 function (4). In addition to mutations, TP53 loss of function in neuroblastomas has been ascribed to MDM2 amplification (4) or p14ARF inactivation (32).
TP53, due to its central role in the normal cellular stress response and frequent inactivation in tumors, is an appealing target for drug development. Several approaches are being pursued to target altered TP53: selective expression of replication-deficient viruses that can selectively propagate in p53-deficient cells (33), reconstitution of the wild-type (wt) TP53 gene (34), or restoration of the transactivation function to mutant (mt) p53 protein with the use of small molecules (35). Alternatively, the defective checkpoint status of cells with inactive TP53 has been exploited with drugs that can inhibit Chk2, Plk1, Aurora Kinases, Wee1 and other proteins that regulate the G2/M checkpoint (36–38).
Our study was undertaken to determine the in vitro activity of vorinostat and flavopiridol in well characterized multidrug-resistant neuroblastoma cell lines (21). Our interest in vorinostat is derived from our previous observation that histone deacetylase 1 (HDAC1) was highly expressed in multidrug-resistant neuroblastoma cell lines relative to drug-sensitive cell lines, and inhibition of HDAC1 with siRNA or a small molecule, depsipeptide, sensitized highly drug-resistant cell lines to commonly used chemotherapeutic agents (39). Vorinostat as a single agent demonstrated limited activity in pediatric in vitro and in vivo models (40), however due to its broad range of mechanisms of action vorinostat remains the drug of interest for development with other drugs in combinations and evaluation in children is ongoing (www.clinicaltrials.gov); most recently a phase I trial of vorinostat and 13 cis-retinoic acid was completed (41). In pediatric patients, phase I clinical trial of flavopiridol has not shown objective responses (42) signifying the importance of identifying active combinations for this drug. Both vorinostat and flavopiridol possess distinct mechanisms of action from traditional chemotherapeutic drugs, consequently they may prove effective against neuroblastomas that acquired resistance during treatment.
Both vorinostat and flavopiridol are known to have G2/M inhibitory effects and were combined to induce cell death through cell cycle perturbation in multidrug-resistant cell lines with mt TP53 or wt TP53. In our study, greater cytotoxicity was observed in cell lines with mt TP53 (SK-N-BE(2), CHLA-90, CHLA-119, and CHLA-172), or p14ARF deletion (LA-N-6) (43), compared to those with functional p53/wt TP53. We confirmed the requirement for p53 loss of function in the activity of the vorinostat + flavopiridol combination by demonstrating enhanced cytotoxicity in clones of wt TP53 cells (CHLA-15 and CHLA-20) stably expressing mt TP53. Moreover, the multidrug-resistant SK-N-BE(2) cell line with mt TP53 was more susceptible to the combination than the exquisitely drug sensitive SK-N-BE(1) line. SK-N-BE(1) and SK-N-BE(2) represent a pair of cell lines, one member of which was established from a patient at diagnosis (SK-N-BE(1)) and carries wt TP53, and another member was established from the same patient after disease progression following multi-agent chemoradiotherapy (SK-N-BE(2)) (21).
Previous studies of the vorinostat + flavopiridol combination, conducted in breast cancer and leukemia cells of myeloid and/or lymphoid lineage, have shown that the combination can induce apoptosis through marked increase in mitochondrial damage (44) or downregulation of XIAP and Mcl-1 (18,20). We focused on examination of G2/M perturbation and associated molecular targets as the mechanism of synergy in highly drug resistant neuroblastoma cell lines that often fail to undergo apoptosis in response to standard and investigational drugs. We demonstrated differential cell cycle progression for cell lines with wt TP53 (SK-N-RA) or mt TP53 (CHLA-90) when treated with vorinostat + flavopiridol. SK-N-RA cells arrested in G2 phase, while CHLA-90 cells failed to arrest following genomic insult and instead proceeded to aberrant mitosis (Figure 3). Respectively, cell lines with wt TP53 and mt TP53 displayed differential expression of cyclin B1, Mad2, and MPM2 proteins following vorinostat + flavopiridol treatments (Figure 5A,B, Supplemental Figure 3) relative to wt TP53 cells.
Mad2 is central to the spindle assembly checkpoint, a cellular mechanism that monitors the accuracy of chromosome segregation. Severely reduced levels of Mad2 provokes extensive cell death due to suppression of mitotic checkpoint signaling (45,46), a phenomenon more pronounced in models lacking TP53 (8). However, transient down-regulation of Mad2 through the use of Mad2 siRNA did not affect clonogenic survival of mt TP53 neuroblastoma cells (CHLA-90) (Figure 5D). We co-expressed Mad2 with targets also down-regulated by the vorinostat + flavopiridol combination: cyclin B1 or Plk1 siRNA. The Cyclin B/cdk1 complex is generally thought to prevent activation of the anaphase promoting complex (APC), whose activity begins to rise during anaphase and thereby initiates mitotic exit (47), although premature degradation of cyclin B can promote early APC activation (45). Co-expression of Mad2 siRNA with cyclin B1 siRNA resulted in significant (p < 0.001) reduction in clonogenic survival relative to single target knockdown (Figure 5D). Similar results were obtained by combined reduction of Mad2 and Plk1 levels (Figure 5D).
Plk1 is a member of the polo-like kinases (Plk) family of serine/threonine kinases with fundamental functions in cell cycle progression such as activation of Cdk1, chromosome segregation, centrosome maturation, bipolar spindle formation, regulation of APC, and execution of cytokinesis. Several investigations using p53 deficient cells have demonstrated that Plk1-depletion was sufficient to induce apoptosis or DNA damage, while cells with intact TP53 were less sensitive to Plk1 depletion (10). More recently, the Plk1 inhibitor, BI-2536, was shown to be selectively effective against colorectal cancer cells without wt TP53, both in vitro and in vivo (48). In our studies, the combination of vorinostat + flavopiridol caused Plk1 depletion in all examined cell lines irrespective of their TP53 status. However, transient expression of Plk1 siRNA in neuroblastoma cell lines was predominantly lethal to cells with mt TP53 (CHLA-90) compared to wt TP53 (CHLA-136) (data not shown); thus, our data is consistent with the findings of other investigators, and affirms that Plk1 can be considered as a therapeutic target in neuroblastomas with p53 loss-of-function.
A recently completed phase I pharmacokinetic study of pulse-dose vorinostat and split-dose flavopiridol did not demonstrate objective responses in adults with advanced solid tumors (49). Nonetheless, it was hypothesized that the combination has the potential to be highly active; it is conceivable that, as we have found in neuroblastomas, the vorinostat + flavopiridol combination can be selectively active in tumors with defective TP53 and the lack of objective responses in the study may partly be attributable to the selection of the patient population.
Our data demonstrate that the vorinostat + flavopiridol combination is predominantly cytotoxic to neuroblastomas with p53-loss of function via TP53 mutations or p14ARF deletion and that such cytotoxicity is mediated through aberrant G2/M progression. Thus, development of therapies targeting G2 and the spindle checkpoint may provide a promising strategy against drug-resistant neuroblastomas with p53-loss of function.
Supplementary Material
Acknowledgments
Vorinostat and Flavopiridol were generously provided by Merck Sharp & Dohme Corp. and Sanofi Aventis, respectively, and the National Cancer Institute, NIH. The authors would like to express their gratitude to Dr. C. Patrick Reynolds (TTUHSC) for providing cell lines, Dr. B. Vogelstein (The Johns Hopkins Medical Institute) for providing plasmids with mutant p53 R175H and R273H.
Supported in part by National Cancer Institute Grant CA81403 (N. Keshelava, M. Sheard, R. L. Ji, Sposto); Children’s Cancer Research Fund (N. Keshelava); the Neil Bogart Memorial Laboratories of the T.J. Martell Foundation for Leukemia, Cancer, and AIDS Research (N. Keshelava).
Footnotes
No financial or personal relationships to disclose.
References:
- 1.Crist WM and Kun LE Common solid tumors of childhood. N.Engl.J.Med 1991;324:461–71. [DOI] [PubMed] [Google Scholar]
- 2.Matthay KK, Villablanca JG, Seeger RC et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N.Engl.J.Med 1999;341:1165–73. [DOI] [PubMed] [Google Scholar]
- 3.Yu AL, Gilman AL, Ozkaynak MF et al. A phase III randomized trial of the chimeric anti-GD2 antibody ch14.18 with GM-CSF and IL2 as immunotherapy following dose intensive chemotherapy for high-risk neuroblastoma: Children’s Oncology Group (COG) study ANBL0032. ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2009;27:15s: [Google Scholar]
- 4.Keshelava N, Zuo JJ, Chen P et al. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res 2001;61:6185–93. [PubMed] [Google Scholar]
- 5.Bunz F, Dutriaux A, Lengauer C et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;20;282:1497–501. [DOI] [PubMed] [Google Scholar]
- 6.Oikawa T, Okuda M, Ma Z et al. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol.Cell Biol 2005;25:4046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chun AC and Jin DY Transcriptional regulation of mitotic checkpoint gene MAD1 by p53. J.Biol.Chem 2003;278:37439–50. [DOI] [PubMed] [Google Scholar]
- 8.Burds AA, Lutum AS, and Sorger PK Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc Natl.Acad.Sci.U.S.A 2005;102:11296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ando K, Ozaki T, Yamamoto H et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J.Biol.Chem 2004;279:25549–61. [DOI] [PubMed] [Google Scholar]
- 10.Liu X and Erikson RL Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl.Acad.Sci.U.S.A 2003;100:5789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan JH, Feng Y, Fisher RH, Maloid S, Longo DL, and Ferris DK Polo-like kinase 1 inactivation following mitotic DNA damaging treatments is independent of ataxia telangiectasia mutated kinase. Mol.Cancer Res 2004;2:417–26. [PubMed] [Google Scholar]
- 12.Sedlacek HH Mechanisms of action of flavopiridol. Crit Rev.Oncol.Hematol 2001;38:139–70. [DOI] [PubMed] [Google Scholar]
- 13.Phelps MA, Lin TS, Johnson AJ et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 2009;113:2637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrd JC, Lin TS, Dalton JT et al. Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood 2007;109:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen DM, Schrump WD, Tsai WS et al. Enhancement of depsipeptide-mediated apoptosis of lung or esophageal cancer cells by flavopiridol: activation of the mitochondria-dependent death-signaling pathway. J.Thorac.Cardiovasc.Surg 2003;125:1132–42. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen DM, Schrump WD, Chen GA et al. Abrogation of p21 expression by flavopiridol enhances depsipeptide-mediated apoptosis in malignant pleural mesothelioma cells. Clin.Cancer Res 2004;10:1813–25. [DOI] [PubMed] [Google Scholar]
- 17.Almenara J, Rosato R, and Grant S Synergistic induction of mitochondrial damage and apoptosis in human leukemia cells by flavopiridol and the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA). Leukemia. 2002;16:1331–43. [DOI] [PubMed] [Google Scholar]
- 18.Gao N, Dai Y, Rahmani M, Dent P, and Grant S Contribution of disruption of the nuclear factor-kappaB pathway to induction of apoptosis in human leukemia cells by histone deacetylase inhibitors and flavopiridol. Mol.Pharmacol 2004;66:956–63. [DOI] [PubMed] [Google Scholar]
- 19.Rosato RR, Almenara JA, Yu C, and Grant S Evidence of a functional role for p21WAF1/CIP1 down-regulation in synergistic antileukemic interactions between the histone deacetylase inhibitor sodium butyrate and flavopiridol. Mol.Pharmacol 2004;65:571–81. [DOI] [PubMed] [Google Scholar]
- 20.Rosato RR, Almenara JA, Kolla SS et al. Mechanism and functional role of XIAP and Mcl-1 down-regulation in flavopiridol/vorinostat antileukemic interactions. Mol.Cancer Ther 2007;6:692–702. [DOI] [PubMed] [Google Scholar]
- 21.Keshelava N, Seeger RC, Groshen S, and Reynolds CP Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Res 1998;58:5396–405. [PubMed] [Google Scholar]
- 22.Kelly WK, O’Connor OA, Krug LM et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J.Clin.Oncol 2005;23:3923–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan AR, Yang X, Berman A et al. Phase I trial of the cyclin-dependent kinase inhibitor flavopiridol in combination with docetaxel in patients with metastatic breast cancer. Clin.Cancer Res 2004;10:5038–47. [DOI] [PubMed] [Google Scholar]
- 24.Magnaghi-Jaulin L, Eot-Houllier G, Fulcrand G, and Jaulin C Histone deacetylase inhibitors induce premature sister chromatid separation and override the mitotic spindle assembly checkpoint. Cancer Res 2007;67:6360–7. [DOI] [PubMed] [Google Scholar]
- 25.Dowling M, Voong KR, Kim M, Keutmann MK, Harris E, and Kao GD Mitotic spindle checkpoint inactivation by trichostatin a defines a mechanism for increasing cancer cell killing by microtubule-disrupting agents. Cancer Biol.Ther 2005;4:197–206. [PubMed] [Google Scholar]
- 26.Iacomino G, Medici MC, Napoli D, and Russo GL Effects of histone deacetylase inhibitors on p55CDC/Cdc20 expression in HT29 cell line. J.Cell Biochem 2006;99:1122–31. [DOI] [PubMed] [Google Scholar]
- 27.Abramova MV, Pospelova TV, Nikulenkov FP, Hollander CM, Fornace AJ, and Pospelov VA G1/S arrest induced by HDAC inhibitor sodium butyrate in E1A+Ras transformed cells is mediated through down-regulation of E2F activity and stabilization of beta -catenin. J.Biol.Chem 2006;281:21040–51. [DOI] [PubMed] [Google Scholar]
- 28.Potapova TA, Daum JR, Pittman BD et al. The reversibility of mitotic exit in vertebrate cells. Nature 2006;440:954–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vandre DD, Davis FM, Rao PN, and Borisy GG Phosphoproteins are components of mitotic microtubule organizing centers. Proc Natl.Acad.Sci.U.S.A 1984;81:4439–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tapia C, Kutzner H, Mentzel T, Savic S, Baumhoer D, and Glatz K Two mitosis-specific antibodies, MPM-2 and phospho-histone H3 (Ser28), allow rapid and precise determination of mitotic activity. Am.J.Surg.Pathol 2006;30:83–9. [DOI] [PubMed] [Google Scholar]
- 31.van Vugt MA and Medema RH Getting in and out of mitosis with Polo-like kinase-1. Oncogene 2005;24:2844–59. [DOI] [PubMed] [Google Scholar]
- 32.Carr J, Bell E, Pearson AD et al. Increased frequency of aberrations in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines established at relapse. Cancer Res. 2006;66:2138–45. [DOI] [PubMed] [Google Scholar]
- 33.O’Shea CC, Johnson L, Bagus B et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 2004;6:611–23. [DOI] [PubMed] [Google Scholar]
- 34.Wiman KG Restoration of Wild-Type p53 Function in Human Tumors: Strategies for Efficient Cancer Therapy. Adv.Cancer Res 2007;97:321–38. [DOI] [PubMed] [Google Scholar]
- 35.Bykov VJ, Issaeva N, Shilov A et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat.Med 2002;8:282–8. [DOI] [PubMed] [Google Scholar]
- 36.Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, and O’Connor PM UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J.Natl.Cancer Inst 1996;88:956–65. [DOI] [PubMed] [Google Scholar]
- 37.Mizuarai S, Yamanaka K, Itadani H et al. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol.Cancer 2009;8:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dar AA, Belkhiri A, Ecsedy J, Zaika A, and El-Rifai W Aurora kinase A inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells. Cancer Res 2008;68:8998–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keshelava N, Davicioni E, Wan Z et al. Histone Deacetylase 1 Gene Expression and Sensitization of Multidrug-Resistant Neuroblastoma Cell Lines to Cytotoxic Agents by Depsipeptide. J.Natl.Cancer Inst 2007; [DOI] [PubMed] [Google Scholar]
- 40.Keshelava N, Houghton PJ, Morton CL et al. Initial testing (stage 1) of vorinostat (SAHA) by the pediatric preclinical testing program. Pediatr.Blood Cancer 2009;53:505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fouladi M, Park JR, Stewart CF et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children’s Oncology Group phase I consortium report. J.Clin.Oncol 2010;28:3623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitlock JA, Krailo M, Reid JM et al. Phase I clinical and pharmacokinetic study of flavopiridol in children with refractory solid tumors: a Children’s Oncology Group Study. J.Clin.Oncol 2005;23:9179–86. [DOI] [PubMed] [Google Scholar]
- 43.Thompson PM, Maris JM, Hogarty MD et al. Homozygous deletion of CDKN2A (p16INK4a/p14ARF) but not within 1p36 or at other tumor suppressor loci in neuroblastoma. Cancer Res. 2001;61:679–86. [PubMed] [Google Scholar]
- 44.Almenara J, Rosato R, and Grant S Synergistic induction of mitochondrial damage and apoptosis in human leukemia cells by flavopiridol and the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA). Leukemia 2002;16:1331–43. [DOI] [PubMed] [Google Scholar]
- 45.Michel L, az-Rodriguez E, Narayan G, Hernando E, Murty VV, and Benezra R Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl.Acad.Sci.U.S.A 2004;101:4459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kops GJ, Foltz DR, and Cleveland DW Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl.Acad.Sci.U.S.A 2004;101:8699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peters JM The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol.Cell 2002;9:931–43. [DOI] [PubMed] [Google Scholar]
- 48.Sur S, Pagliarini R, Bunz F et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl.Acad.Sci.U.S.A 2009;106:3964–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dickson MA, Rathkopf DE, Carvajal RD et al. A phase I pharmacokinetic study of pulse-dose vorinostat with flavopiridol in solid tumors. Invest New Drugs 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.