

SUMMARY
BAF chromatin remodeling complexes play important roles in chromatin regulation and cancer. Here, we report that Ewing sarcoma cells are dependent on the autocrine signaling mediated by NELL2, a secreted glycoprotein that has been characterized as an axon guidance molecule. NELL2 uses Robo3 as the receptor to transmit critical growth signaling. NELL2 signaling inhibits cdc42 and upregulates BAF complexes and EWS-FLI1 transcriptional output. We demonstrate that cdc42 is a negative regulator of BAF complexes, inducing actin polymerization and complex disassembly. Furthermore, we identify NELL2highCD133highEWS-FLI1high and NELL2lowCD133lowEWS-FLI1low populations in Ewing sarcoma, which display phenotypes consistent with high and low NELL2 signaling, respectively. We show that NELL2, CD133, and EWS-FLI1 positively regulate each other and upregulate BAF complexes and cell proliferation in Ewing sarcoma. These results reveal a signaling pathway regulating critical chromatin remodeling complexes and cancer cell proliferation.
Graphical Abstract
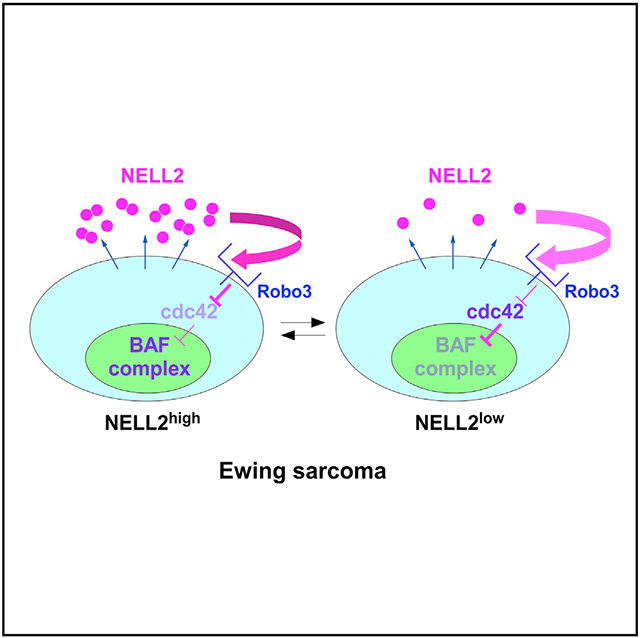
In brief
BAF complexes play important roles in chromatin remodeling and cancers, but the signaling pathways regulating these complexes are poorly understood. Jayabal et al. report that NELL2-cdc42 signaling regulates BAF complexes and Ewing sarcoma cell growth.
INTRODUCTION
Slit-Robo signaling plays a pivotal role in axon guidance (Blockus and Chédotal, 2016). Binding of Slit ligand to Robo1/2 receptor on the surface of axons results in repulsion of the axons (Brose et al., 1999; Long et al., 2004; Zou et al., 2000). Mammalian Robo3, a divergent member of the Robo family, does not bind Slits (Mambetisaeva et al., 2005; Zelina et al., 2014), but is, nonetheless, required for midline crossing of commissural axons (Chen et al., 2008; Sabatier et al., 2004). The search for a ligand for Robo3 identified NELL2 (neural epidermal growth factor-like-like 2) (Jaworski et al., 2015). NELL2 is a secreted glycoprotein predominantly expressed in neural tissues and was shown to repel axons through Robo3 (Jaworski et al., 2015). A chicken homolog of NELL2, Nel (neural epidermal growth factor [EGF]-like), also functions as an inhibitory axon guidance cue (Jiang et al., 2009; Nakamura et al., 2012).
The BAF (Brg/Brahma-associated factors) or mammalian SWI/SNF complexes are ATP-dependent chromatin remodeling complexes implicated in a variety of cancers (Hodges et al., 2016; Kadoch and Crabtree, 2015; St Pierre and Kadoch, 2017). The 15 BAF subunits encoded by 29 genes are collectively mutated in >20% of all human cancers (Hodges et al., 2016; Kadoch and Crabtree, 2015; St Pierre and Kadoch, 2017). Recurrent inactivating mutations of key BAF subunits in cancers suggested a tumor suppressor role for the BAF complexes. In addition, cancer-specific alteration or re-targeting of BAF complexes was proposed to play a tumor-promoting role in certain cancer types (Boulay et al., 2017; Kadoch and Crabtree, 2013). Despite the critical importance of BAF complexes in chromatin remodeling and cancers, the signaling pathways regulating these complexes are poorly understood.
Ewing sarcoma is a cancer of bone and soft tissue in children that is characterized by a chromosomal translocation generating a fusion between EWS and an Ets family transcription factor, most commonly FLI1 (Jedlicka, 2010; Lawlor and Sorensen, 2015; Lessnick and Ladanyi, 2012; Mackintosh et al., 2010; Toomey et al., 2010). EWS-FLI1 fusion accounts for 85% of the cases. EWS-FLI1 regulates the expression of a number of genes important for cell proliferation and tumor progression (Hancock and Lessnick, 2008), can transform mouse cells (González et al., 2007; May et al., 1993), and is required for proliferation and tumorigenicity of Ewing sarcoma cells. Therefore, EWS-FLI1 is considered a causative oncoprotein for Ewing sarcoma. Concerning the mechanism of gene activation by EWS-FLI1, it was recently demonstrated that EWS-FLI1 recruits the BAF complexes to the target genes to activate their expression (Boulay et al., 2017), suggesting a tumor-promoting role for the BAF complexes in this cancer.
In this study, we describe a NELL2 cytokine signaling pathway that regulates the BAF complexes and Ewing sarcoma growth. Our results indicate that NELL2 signaling inhibits cdc42, which induces actin polymerization and BAF complex disassembly.
RESULTS
Secretome proteomics identifies NELL2 as a target of EWS-FLI1
While the gene expression program regulated by EWS-FLI1 has been well studied, the effect of EWS-FLI1 on protein secretion has not been systematically analyzed. We used small hairpin RNA (shRNA)-mediated silencing of EWS-FLI1 and secretome proteomics to address this question (Figure 1A). A-673 Ewing sarcoma cells were infected with lentiviruses expressing an shRNA against the FLI1 C-terminal region or luciferase (control). After verifying the silencing of EWS-FLI1 protein (Figure 1B), the proteins secreted in the conditioned medium were identified by mass spectrometry and quantified by spectral counting. The complete list of proteins identified with high confidence and their abundance ratios between EWS-FLI1-silenced and control cells is shown in Table S1.
Figure 1. Ewing sarcoma is dependent on NELL2, a target of EWS-FLI1.

(A) Outline of secretome proteomic analysis. Note that EWS-FLI1 (FLI1 C terminus) shRNA could also silence untranslocated FLI1, which is expressed at low levels.
(B) shRNA-mediated silencing of EWS-FLI1 in A-673 cells.
(C) EWS-FLI1 silencing reduces NELL2 protein levels in A-673 secretome. The quantification based on spectral counting by mass spectrometry (left); the immunoblotting data (right).
(D) EWS-FLI1 silencing reduces NELL2 RNA levels in A-673 cells. *p < 0.05 (n = 3).
(E) EWS-FLI1 induces NELL2 RNA expression in human mesenchymal stem cells (hMSCs), the putative cells of origin of Ewing sarcoma. The quantitative real-time RT-PCR data (left). *p < 0.05 (n = 3); the immunoblotting data (right).
(F) EWS-FLI1 binds to the NELL2 gene promoter. Chromatin immunoprecipitation analysis for EWS-FLI1 binding to the promoter of NELL2 and known EWS-FLI1 target genes (NR0B1, GLI-1, and FOXO1), as well as control (GAPDH), with and without EWS-FLI1 silencing (n = 3).
(G) NELL2 is highly expressed in Ewing sarcoma tumors and cell lines. NELL2 RNA expression was analyzed by qRT-PCR and was normalized to the levels in hMSCs (n = 3).
(H) NELL2 silencing inhibits Ewing sarcoma proliferation. NELL2 was silenced by siRNAs and cell proliferation was assessed by the IncuCyte live-cell imaging system. NELL2 silencing was verified by immunoblotting (bottom).
(I) Recombinant NELL2 rescues proliferation arrest induced by NELL2 silencing. A-673 cells were transfected with NELL2 or control siRNAs and were treated with or without recombinant NELL2 (250 ng/ml).
(J) NELL2 siRNAs have little effect on the proliferation of 293 and HeLa cells.
(K) NELL2 RNA expression in A-673, 293, and HeLa cells, which was normalized to the levels in hMSCs (n = 3).
(L) siRNA-mediated silencing of NELL2 RNA in 293 and HeLa cells (n = 3).
(M) NELL2 silencing inhibits anchorage-independent growth of A-673 cells. shRNA-mediated silencing of NELL2 was verified by immunoblotting (right). *p < 0.05 (6 independent experiments).
(N) NELL2 silencing inhibits xenograft tumorigenicity of A-673 cells (n = 5, p < 0.05).
(O) Ewing sarcoma depends on autocrine signaling by NELL2, an EWS-FLI1 target.
One of the high-confidence proteins that displayed a significant alteration in abundance upon EWS-FLI1 silencing was NELL2, which exhibited a nearly 10-fold decrease after silencing EWS-FLI1 (Figure 1C; Table S1). NELL2 is predominantly expressed in neural tissues and has been studied as an axon guidance molecule (Jaworski et al., 2015; Jiang et al., 2009; Nakamura et al., 2012).
We found that NELL2 transcript levels are significantly reduced upon EWS-FLI1 silencing (Figure 1D). Conversely, lentiviral expression of EWS-FLI1 in human mesenchymal stem cells (hMSCs), the putative cells of origin of Ewing sarcoma, increased NELL2 transcript and protein levels (Figure 1E). Using chromatin immunoprecipitation (ChIP), we determined that endogenous EWS-FLI1 binds to the NELL2 gene promoter in A-673 cells, and this binding was abolished by EWS-FLI1 silencing (Figure 1F), suggesting that NELL2 is a direct transcriptional target of EWS-FLI1. Consistent with NELL2 being an EWS-FLI1 target, NELL2 is highly expressed in Ewing sarcoma tumors and cell lines compared with MSCs (Figure 1G).
Ewing sarcoma is dependent on NELL2
To elucidate the role of NELL2 in Ewing sarcoma, we tested the effects of manipulation of NELL2 expression. Silencing of NELL2 by a pool of small interfering RNAs (siRNAs) strongly inhibited the proliferation of all 15 Ewing sarcoma cell lines tested (A-673, EW8, TC71, TC32, SK-N-MC, CHLA-9, ES1, ES2, ES3, ES6, ES7, ES8, SK-NEP-1, SK-ES-1, and RD-ES; Figures 1H and S1). Importantly, the proliferation inhibition by NELL2 silencing was completely rescued by the addition of purified recombinant NELL2 protein to the culture medium (Figure 1I), indicating that Ewing sarcoma is dependent on extracellular NELL2. NELL2 siRNAs had little effect on the proliferation of 293/HEK293 and HeLa cells (Figure 1J). NELL2 RNA expression was lower in 293 and HeLa cells than in A-673 cells (Figure 1K), and NELL2 siRNAs reduced NELL2 RNA levels in 293 and HeLa cells (Figure 1L).
We then used lentivirus-mediated shRNA expression to stably silence NELL2. shRNA-mediated silencing of NELL2 severely impaired the anchorage-independent growth (Figure 1M) and xenograft tumorigenicity (Figure 1N) of Ewing sarcoma cells. These results suggest that Ewing sarcoma is dependent on NELL2, an EWS-FLI1 target (Figure 1O).
Robo3 serves as the NELL2 receptor in Ewing sarcoma
NELL2 was previously identified as the ligand for the axon guidance receptor, Robo3 (Jaworski et al., 2015). NELL2 repels axons through Robo3 and contributes to commissural axon guidance to the midline (Jaworski et al., 2015). We wished to determine whether Robo3 also serves as the NELL2 receptor in Ewing sarcoma.
Robo3 expression was readily detectable in Ewing sarcoma tumors and cell lines, but undetectable in MSCs (Figure 2A). We tested the role of Robo3 as the NELL2 receptor in Ewing sarcoma using ligand-binding assays (Figure 2B). C-terminally FLAG-tagged NELL2 was produced from transfected 293T/HEK293T cells and was incubated with A-673 cells that were transfected with Robo3 siRNA pool or control siRNA pool. After washing the cells to remove unbound NELL2-FLAG, a whole-cell lysate was prepared and analyzed by immunoblotting to assess the binding of NELL2-FLAG to A-673 cells. As shown in Figure 2B, NELL2-FLAG bound to control siRNA transfected A-673 cells, and this binding was abolished by the silencing of Robo3, indicating that NELL2 binds Robo3 in Ewing sarcoma. The specificity of the NELL2-Robo3 interaction was further confirmed by ligand-binding assays using alkaline phosphatase-fused NELL2 (NELL2-AP) (Figure 2C). NELL2-AP bound to COS cells expressing Robo3, but not Robo1 or vector (Figure 2C). NELL2 silencing resulted in the accumulation of Robo3-FLAG on the A-673 cell surface, which was reversed by the addition of recombinant NELL2 (Figure 2D).
Figure 2. Robo3 serves as the NELL2 receptor in Ewing sarcoma.

(A) Robo3 RNA expression in Ewing sarcoma tumors and cell lines (n = 3).
(B) NELL2 ligand-binding assays in A-673 cells. NELL2-FLAG bound to control siRNA transfected cells, and the binding was abolished by Robo3 silencing.
(C) NELL2 ligand-binding assays in COS cells. NELL2 fused to alkaline phosphatase (NELL2-AP) or AP alone was produced from transfected 293T cells and was incubated with COS cells that were transfected with vector, Robo3, or Robo1. The binding of NELL2-AP or AP to cells was visualized by AP reaction. NELL2-AP specifically bound to Robo3-expressing COS cells. Scale bars: 60 μm.
(D) NELL2 silencing results in accumulation of lentivirally expressed Robo3-FLAG on A-673 cell surface, which was reversed by recombinant NELL2. The quantification of the fraction of cells with surface Robo3-FLAG staining, based on the counting of >200 cells, is shown on the right. *p < 0.05; scale bars: 10 μm.
(E) Robo3 silencing inhibits Ewing sarcoma proliferation. A-673, EW8, TC32, and TC71 cells were transfected with Robo3 siRNA pool or control siRNA pool, and cell proliferation was assessed by IncuCyte (top). Robo3 silencing was verified by immunoblotting (bottom).
(F) Robo3 siRNAs targeting different regions of Robo3 inhibit A-673 cell proliferation. A-673 cells were transfected with 6 Robo3 siRNAs that target different regions of Robo3. Cell proliferation was assessed by IncuCyte (top). Robo3 silencing was verified by immunoblotting (bottom).
(G) NELL2 requires Robo3 to simulate Ewing sarcoma proliferation. A-673 cells were transfected with NELL2 siRNAs, Robo3 siRNAs, and/or control siRNAs and were treated with or without recombinant NELL2 (250 ng/mL) as indicated. Cell proliferation was assessed by IncuCyte.
(H) Robo3 serves as the NELL2 receptor in Ewing sarcoma.
We then examined the role of Robo3 in Ewing sarcoma by siRNA-mediated silencing. Robo3 silencing by siRNA pool strongly inhibited the proliferation of Ewing sarcoma cell lines (Figure 2E). This was further confirmed by six individual siRNAs targeting different regions of Robo3 (Figure 2F), which all reduced Robo3 protein levels and inhibited cell proliferation. Subsequently, we tested whether NELL2 requires Robo3 to stimulate Ewing sarcoma cell proliferation. Recombinant NELL2 was able to rescue the proliferation of cells transfected with NELL2 siRNAs, but not cells that were co-transfected with NELL2 siRNAs and Robo3 siRNAs (Figure 2G), demonstrating that NELL2 requires Robo3 to transmit growth signaling. These results indicate that Robo3 functions as the NELL2 receptor in Ewing sarcoma (Figure 2H).
NELL2 signaling downregulates cdc42
In Slit-Robo1/2 signaling, the binding of Slit ligand to Robo1/2 receptor activates Slit-Robo guanosine triphosphatase (GTPase)-activating proteins (srGAPs), which inactivate Rho family G proteins, primarily cdc42, resulting in axon repulsion (Wong et al., 2001). Unlike Robo1/2, mammalian Robo3 does not bind Slit and, instead, uses NELL2 as the ligand (Jaworski et al., 2015). While the signaling downstream of the NELL2-Robo3 interaction has not been studied, the binding site for srGAPs, conserved cytoplasmic motif 3 (Wong et al., 2001), is also present in Robo3, which led us to hypothesize that NELL2-Robo3 signaling inhibits cdc42 through srGAPs.
We found that the silencing of srGAP1/2 strongly inhibits the proliferation of Ewing sarcoma cells (Figure 3A). We note the cross-silencing of srGAP1 and srGAP2 (Figure 3A, right), which may reflect their hetero-oligomerization (Müller et al., 2020). Silencing of either NELL2 or Robo3 in Ewing sarcoma cells resulted in increased filopodia (Figure 3B), which is primarily regulated by cdc42 (Mattila and Lappalainen, 2008). Using the pull-down of cell lysate with GST-PAK1, which selectively interacts with GTP-bound active cdc42 and active Rac, we showed that NELL2 silencing robustly increases the levels of active cdc42 and active Rac in Ewing sarcoma cells (Figure 3C). Activation of cdc42 and Rac by NELL2 silencing was abolished by the addition of recombinant NELL2 (Figure 3D, lanes 1–3). The silencing of srGAP1 and srGAP2 increased the levels of active cdc42 and active Rac, which were not affected by NELL2 silencing or recombinant NELL2 (Figure 3D, lanes 4–6). These results indicate that srGAPs act downstream of NELL2 to inhibit cdc42 and Rac. Furthermore, proliferation inhibition of Ewing sarcoma cells induced by silencing of either NELL2 or Robo3 was abolished by a cdc42-specific inhibitor, ML141 (Figure 3E), which does not inhibit Rac, Ras, or Rab (Surviladze et al., 2010). In addition, siRNA-mediated silencing of cdc42 abrogated the proliferation inhibition by NELL2 silencing (Figure 3F). These results suggest that NELL2 signaling primarily inhibits cdc42, the normal function of which is to stimulate filopodia formation and inhibit the proliferation of Ewing sarcoma cells (Figure 3G).
Figure 3. NELL2 signaling downregulates cdc42 and upregulates the BAF complexes.

(A) Silencing of srGAPs inhibits Ewing sarcoma proliferation. srGAP1 and srGAP2 were silenced by siRNAs, and cell proliferation was assessed by IncuCyte (left). The silencing of srGAP1 and srGAP2 was verified by immunoblotting (right).
(B) NELL2 silencing or Robo3 silencing increases filopodia in Ewing sarcoma cells. The effect of NELL2 silencing or Robo3 silencing on actin cytoskeleton was assessed by phalloidin staining (red). Cell nuclei were stained by DAPI (blue). The quantification of filopodia using the filopodia image analysis software (Saha et al., 2016) is shown at the bottom. For each sample, 10 randomly chosen fields containing a total of 100–250 cells were analyzed. *p < 0.05 compared with control siRNA transfected cells. Scale bars: 10 μm.
(C) NELL2 silencing activates cdc42 and Rac in Ewing sarcoma. A-673 and EW8 cells were transfected with NELL2 siRNAs or control siRNAs. Two days after transfection, the levels of GTP-bound, active cdc42, Rac, and RhoA were examined by GST-PAK1 (for cdc42 and Rac1) or GST-Rhotekin-RBD (for Rho A) pull-down of whole-cell lysate followed by anti-cdc42, Rac, and Rho A immunoblotting.
(D) The regulation of cdc42 and Rac activities by NELL2 requires srGAPs.
(E) A selective cdc42 inhibitor, ML141, abrogates the proliferation inhibition by NELL2 silencing or Robo3 silencing in Ewing sarcoma. A-673 and EW8 cells were transfected with NELL2 siRNAs, Robo3 siRNAs, or control siRNAs and were treated with or without 5 μM ML141. Cell proliferation was assessed by IncuCyte.
(F) cdc42 silencing abrogates the proliferation inhibition by NELL2 silencing. The silencing of NELL2 and cdc42 was verified by immunoblotting (right).
(G) NELL2 signaling inhibits cdc42, the normal function of which is to promote filopodia formation and inhibit cell proliferation in Ewing sarcoma.
(H) NELL2 silencing reduces EWS-FLI1 target gene expression in Ewing sarcoma. A-673 and EW8 cells were transfected with NELL2 siRNAs or control siRNAs, and the RNA expression of indicated genes was examined by qRT-PCR and is presented after normalization to the levels in control siRNAs transfected cells (blue). The expression of EWS-FLI1 target genes is reduced in NELL2-silenced cells (red) (n = 3).
(I) NELL2 silencing reduces the protein levels of some of the BAF subunits in Ewing sarcoma. Ewing sarcoma cells were transfected with NELL2 siRNAs or control siRNAs, and the levels of indicated BAF subunits were examined by immunoblotting.
(J) Recombinant NELL2 restores the protein levels of BAF subunits in NELL2-silenced cells. A-673 and EW8 cells were transfected with NELL2 siRNAs or control siRNAs and were treated with recombinant NELL2 (250 ng/mL) for the indicated time (hours). The levels of BAF subunits were assessed by immunoblotting.
(K) NELL2 signaling upregulates the BAF complexes and enhances the transcriptional output of EWS-FLI1.
NELL2 signaling upregulates the BAF chromatin remodeling complexes and EWS-FLI1 transcriptional output
Analysis of gene expression changes induced by NELL2 silencing revealed the reduced expression of EWS-FLI1 target genes in Ewing sarcoma (Figure 3H). As EWS-FLI1 was recently shown to recruit the BAF chromatin remodeling complexes to activate its target genes (Boulay et al., 2017), we examined the effect of NELL2 silencing on the levels of the BAF complex subunits in five Ewing sarcoma cell lines and found that NELL2 silencing selectively reduces BRG1, BRM, BAF250A/ARID1A, BAF155, and BAF47 (Figure 3I). In addition, a few other subunits such as BAF53A and BRD9 were affected by NELL2 silencing in a cell line-specific manner. NELL2 silencing reduced the protein levels of BRG1, BAF155, and BAF47 (Figure 3I), but not their RNA levels (Figures 3H and S4A). This suggests that NELL2-cdc42 signaling regulates the protein stability of these BAF subunits, which was confirmed in Figure S2D.
Importantly, the reduced BAF subunit levels in NELL2 siRNA-transfected cells were rescued by the addition of recombinant NELL2 to the culture medium (Figure 3J). These results suggest that extracellular NELL2 signals to increase the protein levels of key BAF subunits and enhances the transcriptional output of EWS-FLI1 (Figure 3K).
cdc42 downregulates the BAF complexes
Because NELL2 signaling affects both cdc42 and BAF complexes, we next investigated the link between cdc42 and BAF. While NELL2 silencing normally results in reduced BRG1, BAF250A, BAF155, and BAF47 in Ewing sarcoma cells, the cdc42 inhibitor, ML141, abolished this response (Figure S2A), suggesting that cdc42 acts downstream of NELL2 to regulate the BAF complexes. The role of cdc42 in BAF downregulation was further supported by activating cdc42. A 4-h treatment with a cdc42/Rac/Rho A activator, CN04, significantly reduced the levels of BRG1, BAF250A, BAF155, and BAF47 in A-673 cells (Figure S2B). Furthermore, transfection of a constitutively active cdc42 Q61L mutant reduced BRG1, BRM, BAF250A, BAF155, and BAF47 in A-673 cells (Figure S2C). Reduced protein levels of BRG1, BAF250A, BAF155, and BAF47 upon CN04 treatment were restored by a proteasome inhibitor, MG-132 (Figure S2D), suggesting that CN04 induces the proteasomal degradation of these BAF subunits. Importantly, the BAF downregulation by cdc42 is not limited to Ewing sarcoma cells. CN04 treatment reduced BRG1, BAF155, and BAF47, but not BAF60B, in 293 (embryonic kidney), HeLa (cervical carcinoma), HCT116 (colon adenocarcinoma), and IMR-90 (fibroblast) cells (Figure S2E). CN04 also reduced BRG1 and BAF155 in BT-12 malignant rhabdoid tumor cells (Figure S2E), which lack BAF47 expression due to inactivating mutations, suggesting that BAF47 is not required for the downregulation of BRG1 and BAF155 by CN04. By transfecting active and inactive mutants of cdc42, Rac1, and Rho A in 293T cells, we found that active cdc42 (Q61L), active Rac1 (Q61L), and, to a lesser extent, active Rho A (Q63L) reduce endogenous BRG1, BAF155, and BAF47 (Figure S2F) or co-transfected GFP-BRG1 (Figure S2G). These results indicate that cdc42 and other Rho family G proteins downregulate the BAF complexes (Figure S2H).
NELL2-cdc42 signaling regulates actin polymerization and assembly of BAF complexes
The BAF complexes contain β-actin monomer as a subunit, which is bound to the ATPase subunit, BRG1 (Zhao et al., 1998). β-Actin monomer as well as an actin-related subunit, BAF53, is required for the maximal ATPase activity of BRG1 and for the association of the BAF complexes with the nuclear matrix (Zhao et al., 1998). Because cdc42 is a well-established activator of actin polymerization, we analyzed the status of actin in the BAF complexes upon cdc42 activation.
Anti-BRG1 IP of A-673 cell nuclear extract revealed that CN04 treatment significantly increases the abundance of β-actin and, to a lesser extent, BAF53A in the BAF complexes (Figure 4A). Similar results were obtained by IP with anti-BAF57 antibody (Figure S3B). The BAF complexes immunopurified by anti-BRG1 antibody were also analyzed by dot blot for the binding to biotin-conjugated phalloidin, which selectively binds actin polymers. CN04 treatment increased the phalloidin-binding of BAF complexes (Figure 4B). Similar results were obtained by immunopurifying the BAF complexes using anti-BAF57 antibody (Figure S3C).
Figure 4. NELL2-cdc42 signaling regulates actin polymerization and assembly of the BAF complexes.
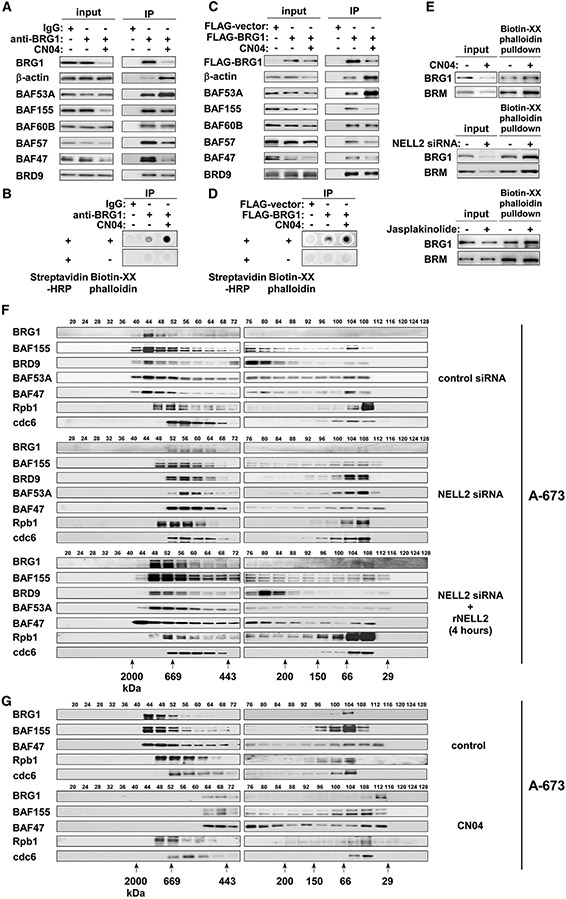
(A) CN04 treatment increases the abundance of actin in the BAF complexes in A-673 cells. A-673 cells were treated or not with 1 μg/mL CN04 for 4 h, and the nuclear extract was immunoprecipitated with anti-BRG1 antibody or control immunoglobulin G (IgG). The abundance of indicated BAF subunits in the BAF complexes was examined by immunoblotting.
(B) CN04 treatment increases the phalloidin reactivity of BAF complexes in A-673 cells. The BAF complexes immunopurified as in (A) were analyzed for the binding to Biotin-XX-Phalloidin by dot blot.
(C) CN04 treatment increases the abundance of actin in the BAF complexes in 293T cells. 293T cells were transfected with FLAG-BRG1 or FLAG-vector and were treated or not with 1 μg/mL CN04 for 4 h, as indicated. FLAG-BRG1-containing BAF complexes were isolated by anti-FLAG immunoprecipitation, and the abundance of indicated BAF subunits was examined by immunoblotting.
(D) CN04 treatment increases the phalloidin reactivity of BAF complexes in 293T cells. The BAF complexes immunopurified as in C were analyzed for the binding to Biotin-XX-Phalloidin by dot blot.
(E) The amount of BRG1 and BRM pulled down from A-673 nuclear extract with Biotin-XX-Phalloidin increases with CN04 treatment (1 μg/mL for 4 h, top), NELL2 silencing (center), and Jasplakinolide treatment (100 nM for 1 h, bottom).
(F) NELL2 signaling reversibly regulates the assembly of BAF complexes in A-673 cells. Nuclear extract of A-673 cells treated as indicated was analyzed by gel filtration chromatography and immunoblotting.
(G) CN04 treatment disassembles the BAF complexes in A-673 cells.
We also transfected FLAG-tagged BRG1 in 293T cells and isolated the BAF complexes by anti-FLAG IP of nuclear extract. CN04 treatment increased the abundance of β-actin and BAF53A in the FLAG-BRG1-containing BAF complexes (Figure 4C). The phalloidin dot blot analysis revealed that FLAGBRG1-containing BAF complexes purified from CN04-treated 293T cells react more strongly with phalloidin than those from untreated cells (Figure 4D).
The pull-down of A-673 cell nuclear extract with biotin-conjugated phalloidin revealed that more BRG1 and BRM are pulled down by biotin-conjugated phalloidin upon CN04 treatment (Figure 4E, top), NELL2 silencing (Figure 4E, center), and treatment with Jasplakinolide, a pharmacological inducer of actin polymerization (Figure 4E, bottom).
Collectively, these results suggest that polymerized actin is bound to the BAF complexes upon cdc42 activation or NELL2 silencing.
We then used gel filtration chromatography of A-673 cell nuclear extract to evaluate the assembly of the BAF complexes in response to NELL2 signaling (Figure 4F). In control siRNA-transfected cells, the BAF subunits mostly co-eluted with BRG1 in high-molecular-weight complexes (fractions 40–52). NELL2 silencing caused a size shift of the BAF complexes to a lower molecular weight range (fractions 52–64). Importantly, 4-h treatment with recombinant NELL2 largely restored the size of the BAF complexes (Figure 4F, bottom). NELL2 silencing did not affect the elution profiles of Rpb1 and cdc6, a component of RNA polymerase II and replication pre-initiation complex, respectively (Figure 4F). These results indicate that extracellular NELL2 signals to regulate the assembly of the BAF complexes.
We also used gel filtration chromatography to assess the effect of cdc42 activation on the BAF complex assembly. We found that CN04 treatment reduces the size of BAF complexes in A-673 cells (Figure 4G) and 293T cells (Figure S3A). Furthermore, transfection of the cdc42 Q61L active mutant similarly reduced the size of BAF complexes in 293T cells (Figure S3A). These results demonstrate that NELL2 silencing or cdc42 activation results in the disassembly of the BAF complexes.
SW13 adrenal carcinoma cells do not express BRG1 and BRM (Muchardt and Yaniv, 1993); thus, actin is not associated with the BAF complexes in this cell line (Zhao et al., 1998). Importantly, CN04 treatment barely affected the size of BAF complexes in vector-transfected SW13 cells, whereas in BRG1-transfected SW13 cells, CN04 treatment significantly reduced the size of BAF complexes (Figure S3D). Furthermore, CN04 treatment did not affect the protein levels of BAF155 and BAF47 in vector-transfected SW13 cells, but reduced these BAF subunits in BRG1-transfected SW13 cells (Figure S3E). These results suggest that cdc42 induces actin polymerization, leading to BAF complex disassembly and degradation of specific BAF subunits.
NELL2highCD133highEWS-FLI1high and NELL2lowCD133lowEWS-FLI1low populations in Ewing sarcoma display phenotypes consistent with high and low NELL2 signaling, respectively
Immunohistochemical staining of Ewing sarcoma tumors revealed that NELL2 expression is heterogeneous (Figure 5A). The heterogeneity of NELL2 expression was also observed in Ewing sarcoma cell lines. Immunofluorescent staining of A-673 cells detected the cells that express NELL2 and the cells that do not detectably express NELL2, and these correlated well with the expression of EWS-FLI1 (Figure 5B). shRNA-mediated silencing of EWS-FLI1 abolished the staining of EWS-FLI1 in A-673 cells (Figure S4B), indicating the specificity of staining. Furthermore, we found that NELL2 expression also correlates with the expression of CD133 (Figure 5C), a transmembrane protein used as a marker of stem cells in some normal tissues and cancers (Glumac and LeBeau, 2018).
Figure 5. NELL2highCD133highEWS-FLI1high and NELL2lowCD133lowEWS-FLI1low populations in Ewing sarcoma display phenotypes consistent with high and low NELL2 signaling, respectively.

(A) NELL2 expression is heterogeneous in Ewing sarcoma tumor. NELL2 expression in surgically resected Ewing sarcoma tumor was examined by immunohistochemistry (brown signals). Scale bar: 60 μm.
(B) NELL2 expression is heterogeneous in Ewing sarcoma cells and correlates with EWS-FLI1 expression. NELL2 and EWS-FLI1 expression in A-673 cells was assessed by anti-NELL2 (red) and anti-FLI1 C terminus (green) immunofluorescent staining. The nuclei were stained with DAPI. Scale bar: 10 μm.
(C) NELL2 expression correlates with CD133 expression in Ewing sarcoma cells. NELL2 and CD133 expression in A-673 cells was assessed by anti-NELL2 (green) and anti-CD133 (AC133, red) immunofluorescent staining. The nuclei were stained with DAPI. Scale bar: 10 μm.
(D) The CD133low population displays lower NELL2, EWS-FLI1, BRG1, BAF250A, and BAF155 levels than the CD133high population. A-673 cells were incubated with anti-CD133 (AC133) antibody and were sorted into the CD133high and CD133low populations. The expression of indicated proteins was assessed by immunoblotting.
(E) The CD133low population displays reduced EWS-FLI1 target gene expression. The RNA expression of indicated genes was assessed by qRT-PCR and is presented after normalization to the levels in the CD133high population (blue). The expression of EWS-FLI1 target genes is reduced in the CD133low population (red) (n = 3).
(F) The CD133low population displays slower growth than the CD133high population.
(G) The CD133low population displays reduced NELL2 and EWS-FLI1 expression. A-673, CHLA-9, EW8, TC71, TC32, and SK-N-MC Ewing sarcoma cells were sorted into the CD133high and the CD133low populations, and the RNA expression of CD133, NELL2, and EWS-FLI1 was assessed by qRT-PCR (n = 3).
(H) The CD133low population displays increased filopodia. Actin cytoskeleton was visualized by phalloidin staining (red). Cell nuclei were stained by DAPI (blue). Scale bars: 10 μm. The quantification of filopodia is shown in Figure S4D.
(I) NELL2 silencing inhibits the proliferation of both CD133high and CD133low populations.
(J) The CD133low population displays reduced sphere formation; *p < 0.05 (3 independent experiments).
(K) The CD133low population displays reduced xenograft tumorigenicity (n = 5, p < 0.05).
(L) The CD133low population displays reduced migration; *p < 0.05 (3 independent experiments).
(M) Prolonged cisplatin or doxorubicin treatment enriches the CD133low population.
Using anti-CD133 (AC133) antibody, we sorted Ewing sarcoma cells into the CD133high and CD133low populations, which revealed that the CD133low population displays a lower expression of NELL2, EWS-FLI1, BRG1, BAF250A, and BAF155 than the CD133high population (Figures 5D, 5G, and S4C). There was comparable expression of Robo3 in the two populations (Figure 5D). The CD133low population also exhibited a lower expression of EWS-FLI1 target genes (Figure 5E), slower proliferation (Figure 5F), and more filopodia (Figures 5H and S4D) than the CD133high population. These results suggest that NELL2 expression is heterogeneous in Ewing sarcoma and that the NELL2highCD133highEWS-FLI1high and NELL2lowCD133lowEWS-FLI1low populations display phenotypes consistent with high and low NELL2 signaling, respectively.
Although the NELL2lowCD133lowEWS-FLI1low population expresses lower levels of NELL2 than the NELL2highCD133highEWS-FLI1high population and displays phenotypes that are consistent with lower NELL2 signaling, NELL2 silencing inhibited the proliferation of both populations (Figure 5I), indicating that both populations are dependent on NELL2. When we continued the culture of the CD133high and CD133low populations for a longer time, we observed that the CD133high population generated the CD133low population and vice versa (Figure S4E), demonstrating the interconversion of the two populations. We also dissociated cells from a patient-derived xenograft (PDX) tumor of Ewing sarcoma (NCH-EWS-1), which was never propagated by in vitro culture, and sorted the cells into the CD133high and CD133low populations, which recapitulated the phenotypes of cell line-derived CD133high and CD133low populations (Figure S5). We assessed the purity of tumor cells derived from the PDX tumor by staining for CD99, a widely used marker for Ewing sarcoma, and found that both CD133high and CD133low cells derived from the PDX tumor are predominantly positive for CD99 (Figures S5F and S5G).
The CD133high population exhibited higher tumor sphere formation (Figure 5J), higher xenograft tumorigenicity (Figures 5K and S5D), and higher migration rate (Figures 5L and S5E) than the CD133low population. Interestingly, however, the CD133low population is more resistant to the chemotherapeutic drugs cisplatin and doxorubicin than the CD133high population (Figure S6). Prolonged treatment with cisplatin or doxorubicin enriched the CD133low population (Figure 5M), further supporting the chemoresistance of CD133low cells.
NELL2, CD133, and EWS-FLI1 positively regulate each other and increase the BAF subunits and cell proliferation in Ewing sarcoma
Because the NELL2lowCD133lowEWS-FLI1low population displays phenotypes consistent with low NELL2 signaling, we tested whether increasing the levels of NELL2 alters the phenotypes of this population. We found that the addition of recombinant NELL2 rescues the slow growth of the CD133low population in a dose-dependent manner (Figure 6A). Furthermore, recombinant NELL2 increased CD133, BRG1, BAF250A, BAF155, and BAF47 in the CD133low population in a dose- and time-dependent fashion (Figure 6B). While recombinant NELL2 at ≥250 ng/mL was able to rescue the slow growth of the CD133low population (Figure 6A), the ELISA measurements determined the NELL2 concentration in the culture supernatant of Ewing sarcoma cell lines to be ~50 ng/mL (Figure 6C), which is insufficient to rescue the slow growth of the CD133low population (Figure 6A). This suggests that cross-feeding of NELL2 from the CD133high population would not rescue the slow growth of the CD133low population in a mixed culture.
Figure 6. NELL2, CD133, and EWS-FLI1 positively regulate each other and increase the BAF subunits and cell proliferation in Ewing sarcoma.

(A) Slow growth of the CD133low population can be rescued by recombinant NELL2. The CD133low population was treated with the indicated concentration of recombinant NELL2, and cell proliferation was assessed by IncuCyte in comparison with the CD133high population.
(B) Recombinant NELL2 increases CD133 and BAF subunits in the CD133low population. The CD133low population was treated with the indicated concentration of recombinant NELL2 for 4 or 24 h, and the protein levels of CD133, BRG1, BAF250A, BAF155, and BAF47 were assessed by immunoblotting.
(C) NELL2 concentration in the culture supernatant of Ewing sarcoma cell lines. NELL2 concentration in the culture supernatant of 5 Ewing sarcoma cell lines, DMEM medium, and RPMI1640 medium was determined by ELISA (3 independent experiments).
(D) Increasing CD133 in the CD133low population results in increased NELL2, EWS-FLI1, BRG1, BAF250A, BAF155, and BAF47. The CD133low population was infected with CD133-expressing lentivirus, and the expression of indicated proteins was assessed by immunoblotting in comparison with uninfected CD133low population and CD133high population.
(E) Increasing CD133 in the CD133low population results in increased cell proliferation. The proliferation of cells in (D) was assessed by IncuCyte.
(F) CD133 silencing results in reduced NELL2, EWS-FLI1, BRG1, and BAF155. A-673, EW8, TC71, and CHLA-9 cells were transfected with CD133 siRNAs (+) or control siRNAs (−), and the expression of indicated proteins was assessed by immunoblotting.
(G) CD133 silencing results in reduced cell proliferation. A-673 cells were transfected with CD133 siRNAs or control siRNAs and cell proliferation was assessed by IncuCyte.
(H) NELL2 silencing results in reduced CD133. A-673 and EW8 cells were transfected with NELL2 siRNAs or control siRNAs, and the levels of NELL2 and CD133 were assessed by immunoblotting.
(I) EWS-FLI1 silencing results in reduced NELL2 and CD133. A-673 and EW8 cells were infected with lentiviruses expressing FLI1 C terminus shRNA (+) or control shRNA (−) and were selected with puromycin. The expression of EWS-FLI1, NELL2, and CD133 was assessed by immunoblotting.
(J) EWS-FLI1 binds to the P2 and P6 promoters of the CD133 gene. Chromatin immunoprecipitation was performed as in Figure 1F (n = 3).
(K) EWS-FLI1 silencing results in reduced CD133 transcript levels in A-673 cells; *p < 0.05 (n = 3).
(L) EWS-FLI1 expression results in increased CD133 transcript levels in human mesenchymal stem cells; *p < 0.05 (n = 3).
(M) NELL2, CD133, and EWS-FLI1 positively regulate one another in Ewing sarcoma.
We also tested the effect of increasing the levels of CD133 in the CD133low population. We used lentivirus to express CD133 in the CD133low population, which increased CD133 to the levels comparable to those of the CD133high population (Figure 6D). Increasing CD133 in the CD133low population resulted in increased NELL2, EWS-FLI1, BRG1, BAF250A, BAF155, and BAF47 protein levels (Figure 6D), as well as increased cell proliferation (Figure 6E). Conversely, silencing of CD133 in Ewing sarcoma cells resulted in reduced NELL2, EWS-FLI1, BRG1, and BAF155 protein levels (Figure 6F) and reduced cell proliferation (Figure 6G). In addition, silencing of NELL2 resulted in reduced CD133 in Ewing sarcoma cells (Figure 6H).
Finally, shRNA-mediated silencing of EWS-FLI1 resulted in reduced NELL2 and CD133 in Ewing sarcoma cells (Figure 6I). Six different promoters have been identified in the upstream region of the human CD133 gene (Sompallae et al., 2013). ChIP assays demonstrated strong binding of EWS-FLI1 to the P2 and P6 promoters of the CD133 gene (Figure 6J). The silencing of EWS-FLI1 reduced CD133 transcript levels in A-673 cells (Figure 6K), while exogenous EWS-FLI1 expression induced CD133 transcript levels in hMSCs (Figure 6L). These results indicate that CD133 is a transcriptional activation target of EWS-FLI1.
Collectively, these findings suggest that NELL2, CD133, and EWS-FLI1 positively regulate each other (Figure 6M), increasing the BAF subunits and cell proliferation in Ewing sarcoma.
The silencing of NELL2 in A-673 cells abolished the binding of the BAF complexes to the EWS promoter (Figure S7A) and suppressed the expression of EWS and EWS-FLI1 (Figure S7B). The silencing of NELL2 in A-673 cells also abolished the binding of EWS-FLI1 and the BAF complexes to the P2 and P6 promoters of CD133 (Figures S7C and S7D) and suppressed CD133 expression (Figure S7E).
The binding of the BAF complexes to the EWS promoter was detected in the CD133high population, but not in the CD133low population of A-673 cells (Figure S7F). Increasing CD133 in the CD133low population induced the binding of the BAF complexes to the EWS promoter (Figure S7F) and induced EWS and EWS-FLI1 expression (Figure S7G).
The binding of EWS-FLI1 and the BAF complexes to the NELL2 promoter was detected in the CD133high population, but not in the CD133low population of A-673 cells (Figures S7H and S7I). Increasing CD133 in the CD133low population induced the binding of EWS-FLI1 and the BAF complexes to the NELL2 promoter (Figures S7H and S7I) and induced NELL2 expression (Figure S7J).
As demonstrated earlier, EWS-FLI1 directly activates the transcription of NELL2 (Figures 1D-1F) and CD133 (Figures 6J-6L).
These results clarify the mechanistic basis for the mutually enhancing relationship among NELL2, EWS-FLI1, and CD133 as summarized in Figure 6M.
CD133 signaling downregulates cdc42 and upregulates the BAF complexes
Regarding the signaling downstream of CD133, we found that the CD133low population displays higher levels of active cdc42 and active Rac than the CD133high population and that increasing CD133 in the CD133low population results in reduced active cdc42 and active Rac (Figure 7A), suggesting that CD133 signals to inhibit cdc42 and Rac. The treatment of the CD133low population with the cdc42 inhibitor ML141 increased BRG1, BAF155, and BAF47 protein levels (Figure 7B) and increased cell proliferation (Figure 7C), suggesting that cdc42 mediates CD133 signaling. The C-terminal cytoplasmic tail of CD133 directly binds and activates Src (Liu et al., 2016), and we found that the CD133low population displays reduced active Src, which was restored by increasing CD133 levels (Figure 7D).
Figure 7. CD133 signaling downregulates cdc42 and upregulates the BAF complexes.

(A) The CD133low population displays increased active cdc42 and active Rac levels, which are reduced by lentiviral expression of CD133.
(B) A cdc42 inhibitor, ML141, increases the levels of BRG1, BAF155, and BAF47 in the CD133low population.
(C) ML141 increases the proliferation of the CD133low population.
(D) The CD133low population displays reduced Src Y419 phosphorylation and reduced caveolin-1 Y14 phosphorylation, which were restored by lentiviral expression of CD133.
(E) A Src inhibitor, dasatinib, reduces Src Y419 phosphorylation, caveolin-1 Y14 phosphorylation, BRG1, BAF155, and BAF47 in A-673 cells.
(F) Caveolin-1 silencing reduces BRG1, BAF155, and BAF47 in A-673 cells.
(G) Dasatinib increases the levels of active cdc42 and active Rac in unsorted A-673 cells and in CD133-restored CD133low population.
(H) Caveolin-1 silencing increases the levels of active cdc42 and active Rac in unsorted A-673 cells and in CD133-restored CD133low population.
(I) GDF6 silencing does not induce phosphorylation of caveolin-1 or suppression of cdc42 and Rac.
(J) CD133 inhibits cdc42 and increases BAF subunits in non-Ewing sarcoma cells. Lentiviral expression of CD133 in 5 non-Ewing sarcoma cell lines resulted in increased Src Y419 phosphorylation, increased caveolin-1 Y14 phosphorylation, reduced cdc42 activity, reduced Rac activity, and increased BAF subunits (BRG1, BAF155, and BAF47).
(K) NELL2 regulates the BAF subunit levels independently of Src activity. In the presence of dasatinib, which depleted phosphorylated active Src, NELL2 silencing reduced the levels of BRG1, BAF155, and BAF47, which were restored by the addition of recombinant NELL2 (250 ng/mL) to the culture medium.
(L) NELL2 and CD133 downregulate cdc42 and upregulate the BAF complexes by distinct mechanisms.
One of the well-established substrates of Src is caveolin-1 (Glenney and Zokas, 1989), a component of plasma membrane invaginations called caveolae. Caveolin-1 is highly expressed in Ewing sarcoma and promotes cell proliferation and tumorigenicity (Tirado et al., 2006). Caveolin-1 functions as an inhibitor (guanine nucleotide dissociation inhibitor) of cdc42 (Nevins and Thurmond, 2006), and this activity of caveolin-1 appears to be enhanced when caveolin-1 is tyrosine phosphorylated (Cheng et al., 2010). We found that the CD133low population displays reduced tyrosine phosphorylation of caveolin-1, which was restored by increasing CD133 levels (Figure 7D).
Treatment of A-673 cells with a Src inhibitor, dasatinib, resulted in dose-dependent inhibition of Src, tyrosine de-phosphorylation of caveolin-1, and downregulation of BRG1, BAF155, and BAF47 (Figure 7E). Dasatinib treatment also increased active cdc42 and active Rac in unsorted A-673 cells and in CD133-overexpressing CD133low populations (Figure 7G). The latter data suggest that Src inhibition counteracts the downregulation of cdc42/Rac by CD133.
The silencing of caveolin-1 in A-673 cells reduced tyrosine-phosphorylated caveolin-1, BRG1, BAF155, and BAF47 (Figure 7F). Caveolin-1 silencing also activated cdc42 and Rac in unsorted A-673 cells and re-activated cdc42 and Rac in the CD133-overexpressing CD133low population (Figure 7H). These results suggest that CD133 downregulates cdc42 through Src and caveolin-1 (Figure 7L).
We have recently reported that the prodomain of a BMP family cytokine, GDF6, binds CD99 and recruits C-terminal Src kinase (CSK) to the cytoplasmic domain of CD99, leading to the inhibition of Src in Ewing sarcoma (Zhou et al., 2020). We therefore tested the effect of GDF6 silencing on caveolin-1, cdc42, and Rac. We found that GDF6 silencing results in the activation of Src, but does not induce the phosphorylation of caveolin-1 or the suppression of cdc42 and Rac (Figure 7I). CD133 and GDF6-CD99-CSK may target distinct pools of Src, and only the former affects caveolin-1 and cdc42/Rac.
Lentiviral expression of CD133 in 293, 293T, HeLa, HCT116, and Aska (synovial sarcoma) cells resulted in increased active Src, increased tyrosine phosphorylation of caveolin-1, diminished cdc42 and Rac activity, and increased BAF subunits (BRG1, BAF155, and BAF47) (Figure 7J). This suggests that CD133-BAF signaling also occurs in non-Ewing sarcoma cells.
Because both NELL2 signaling and CD133-Src signaling inhibit cdc42 and upregulate the BAF complexes, we tested whether NELL2 signaling acts independently of CD133-Src signaling. In the presence of dasatinib, which depleted phosphorylated active Src, the silencing of NELL2 resulted in further reduced levels of BRG1, BAF155, and BAF47, which was rescued by recombinant NELL2 treatment (Figure 7K). This indicates that, unlike CD133 signaling, NELL2 signaling does not depend on Src activity.
These results suggest that NELL2 and CD133 downregulate cdc42 and upregulate the BAF complexes by distinct mechanisms (Figure 7L).
DISCUSSION
NELL2-cdc42 signaling appears to affect the levels of only a subset of the BAF subunits, BRG1, BRM, BAF250A, BAF155, and BAF47 (Figure 3I). Some of these subunits were shown to affect each other’s protein levels (siRNA-mediated silencing of one subunit results in the reduced protein levels of a few other subunits, but not the remaining subunits) (Watanabe et al., 2014). In addition, mouse BAF155 (SRG3) was shown to bind and stabilize BRG1, BAF47, and BAF60A by attenuating their proteasomal degradation (Sohn et al., 2007). We note that the BAF subunits regulated by NELL2-cdc42 signaling or reported to display interdependent protein stability do not necessarily correlate with the proposed BAF complex assembly pathway (Mashtalir et al., 2018). It is likely that NELL2-cdc42 signaling influences the level of the module within the BAF complexes that displays interdependent protein stability. Because the NELL2-cdc42-regulated module contains essential catalytic subunits (BRG1 and BRM), BAF47, which is required for BAF-chromatin association (Nakayama et al., 2017), and BAF250A, which is the most frequently mutated BAF subunit in cancers (Hodges et al., 2016; Kadoch and Crabtree, 2015; St Pierre and Kadoch, 2017), we surmise that NELL2 signaling upregulates the activity of the BAF complexes. This is supported by the reduced EWS-FLI1 transcriptional output upon NELL2 silencing in Ewing sarcoma (Figure 3H).
BAF250A and EWS-FLI1 physically and functionally interact in Ewing sarcoma (Selvanathan et al., 2015; Selvanathan et al., 2019), and these two proteins stabilize each other (Selvanathan et al., 2019). The silencing of NELL2 reduced both the RNA and the protein levels of BAF250A in Ewing sarcoma (Figures 3I and S4A). Thus, it is possible that, in addition to the disassembly and destabilization of the BAF complexes, the destabilization of the EWS-FLI1 protein by reduced BAF250A contributes to the reduced EWS-FLI1 transcriptional output upon NELL2 silencing. Reduced BAF250A upon NELL2 silencing may also contribute to the destabilization of the BAF complexes. In addition, it is possible that NELL2-BAF signaling and other modulators of chromatin remodeling such as RING1B (Sánchez-Molina et al., 2020) cooperatively regulate EWS-FLI1 transcriptional output in Ewing sarcoma.
While NELL2 signaling is tumor promoting in Ewing sarcoma, where the BAF complexes are used by EWS-FLI1 to activate the target genes and are thus playing a tumor-promoting role, it is possible that NELL2 signaling mediates tumor suppression in other cancers. The BAF complexes have both tumor-promoting and tumor-suppressing roles, depending on the type of cancer. Different BAF subunits are recurrently inactivated by mutations in different cancers, suggesting that the BAF complexes act as tumor suppressors in these cancers. By upregulating the BAF complexes, NELL2 signaling can also mediate tumor suppression in cancers other than Ewing sarcoma. In addition, it will be interesting to see whether the alteration of the BAF complexes by NELL2 signaling also plays a role in axon guidance and neural development. The study that identified NELL2 as a repulsion-inducing ligand for Robo3 did not analyze downstream signaling (Jaworski et al., 2015). It is possible that neurons respond to NELL2 signaling both by the reorganization of actin cytoskeletons, which is considered to mediate axon repulsion, and by chromatin remodeling through BAF complexes. Developmental stage-specific BAF complexes play important roles in mammalian neural development, and a large number of mutations of the BAF subunits are found in a variety of human neurological disorders (Son and Crabtree, 2014).
We identified NELL2highCD133highEWS-FLI1high and NELL2lowCD133lowEWS-FLI1low populations in Ewing sarcoma, which display phenotypes consistent with high and low NELL2 signaling, respectively (Figure 5). Previous studies found that CD133 expression is heterogeneous in Ewing sarcoma tumors and cell lines (Cornaz-Buros et al., 2014; Jiang et al., 2010; Suvà et al., 2009) and the CD133+ population was suggested to represent the cancer stem cells in Ewing sarcoma (Cornaz-Buros et al., 2014; Suvà et al., 2009). Recent studies also found that EWS-FLI1 activity is heterogeneous in Ewing sarcoma cell lines and tumors (Aynaud et al., 2020; Franzetti et al., 2017). Our study uncovered that NELL2, CD133, and EWS-FLI1 positively regulate each other and increase BAF subunits and cell proliferation in Ewing sarcoma (Figures 6 and S7). It should be noted that the CD133high cells in our study are defined by the high levels of the AC133 epitope, which faithfully reflects the cancer stem cell state in colon cancer and glioblastoma (Kemper et al., 2010; Lathia et al., 2015).
Compared with the NELL2lowCD133lowEWS-FLI1low population, the NELL2highCD133highEWS-FLI1high population displayed higher proliferation, higher tumor sphere formation, higher xenograft tumorigenicity, and higher migration rate but was more sensitive to chemotherapeutic drug treatment (Figures 5, S5, and S6), suggesting that there are two populations of cells in Ewing sarcoma, each responsible for different aspects of tumor progression: growth versus chemoresistance. This is somewhat different from the tumor heterogeneity model established in epithelial carcinomas, in which stemness, tumor-initiating capability, migration/invasiveness, and chemotherapy resistance tend to reside in the same or overlapping carcinoma sub-populations. The tumor heterogeneity in Ewing sarcoma and possibly other sarcomas may work differently from that in carcinomas. It will be important to further dissect the roles of the NELL2highCD133highEWS-FLI1high and NELL2lowCD133lowEWS-FLI1low populations in Ewing sarcoma growth, invasion, drug resistance, and recurrence. While these two populations display contrasting phenotypes, both are dependent on NELL2 (Figure 5I), supporting the notion that NELL2 signaling is a promising therapeutic target.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yuzuru Shiio (shiio@uthscsa.edu).
Materials availability
Plasmids generated in this study are available from the lead contact.
Data and code availability
The mass spectrometry data have been deposited to the PRIDE database and assigned the identifier, PXD016052.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Female 5 - 6 week old C.B.17SC scid−/− mice were used. All mice were housed in a pathogen-free vivarium in the University of Texas Health Science Center at San Antonio. Mice were randomly allocated to treatment groups. Blinding of the researcher measuring tumor size was employed. The animal research method was reviewed and approved for humaneness by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio.
Cell lines
A-673, SK-N-MC, 293, 293T, HeLa, HCT116, IMR-90, and Aska cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. EW8, TC32, TC71, CHLA-9, ES1, ES2, ES3, ES6, ES7, ES8, RD-ES, RH4, RH5, RD, and RMS13 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum. SK-NEP-1 and SK-ES-1 cells were cultured in McCoy’s 5a medium supplemented with 15% fetal bovine serum. A-673, SK-N-MC, SK-NEP-1, SK-ES-1, RD-ES, RD, RMS13, 293, 293T, HeLa, HCT116, and IMR-90 cells were from ATCC. TC71 cells were from the Coriell Institute for Medical Research. EW8, TC32, and CHLA-9 cells were from Dr. Patrick Grohar. RH4 and RH5 cells were from Dr. Javed Khan. The cell lines were STR-authenticated and were routinely tested for the absence of mycoplasma. Cord blood-derived human mesenchymal stem cells were purchased from Vitro Biopharma (Golden, CO) and were cultured in low-serum MSC-GRO following the manufacturer’s procedure.
METHOD DETAILS
Transfection and viral infection
Calcium phosphate co-precipitation was used for transfection of 293T cells. Lentiviruses were prepared by transfection in 293T cells following System Biosciences’ protocol, and the cells infected with lentiviruses were selected with 2 μg/mL puromycin for 48 hours. The target sequences for shRNAs are as follows: FLI1 C terminus shRNA, AACGATCAGTAAGAATACAGAGC; luciferase shRNA, GCACTCTGATTGACAAATACGATTT; NELL2 shRNA-1, CGAGAATTTGAGTCCTGGATA; and NELL2 shRNA-2, CCTACTTTGAAG GAGAAAGAA. The following siRNAs were used: human NELL2 siRNA SMARTpool (M-012185-00; Dharmacon), human Robo3 siRNA SMARTpool (M-026504-00; Dharmacon), human Robo3 siRNAs NM_022370 (SASI_Hs01_00099511, SASI_Hs01_00099512, SASI_Hs01_00099513, SASI_Hs01_00099514, SASI_Hs01_00099515 and SASI_Hs01_00099516; MilliporeSigma), human cdc42 siRNA SMARTpool (M-005057-01; Dharmacon), human CD133 siRNA SMARTpool (M-010630-01; Dharmacon), human srGAP1 siRNA SMARTpool (M-026974-00; Dharmacon), human srGAP2 siRNA SMARTpool (M-021531-03; Dharmacon), human caveolin-1 siRNA SMARTpool (M-003467-01; Dharmacon), human GDF6 siRNA SMARTpool (M-0330055-01; Dharmacon), and Non-Targeting siRNA Pool #2 (D-001206-14-05; Dharmacon). siRNA transfection of cell lines was performed using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher).
Protein sample preparation and proteomic analysis
The preparation of secreted protein samples, mass spectrometry analysis, and proteomics data processing were performed essentially as described (Elzi et al., 2016; Jayabal et al., 2017). A-673 cells were infected with lentiviruses expressing an shRNA against FLI1 C-terminal region or luciferase (control) and were selected with 2 μg/ml puromycin for two days. Cells were washed six times with DMEM without serum. Subsequently, cells were cultured in DMEM without serum for 24 hours and the culture supernatant was harvested. The supernatant was centrifuged, filtered through a 0.45-μm filter (Millipore), and concentrated using a 3,000-Da cutoff Amicon Ultra Centrifugal Filter Units (Millipore). The proteins in each sample were fractionated by SDS-PAGE and visualized by Coomassie blue. Each gel lane was divided into six slices, and the proteins in each slice were digested in situ with trypsin (Promega modified) in 40 mM NH4HCO3 overnight at 37°C. The resulting tryptic peptides were analyzed by HPLC-ESI-tandem mass spectrometry on a Thermo Fisher LTQ Orbitrap Velos Pro mass spectrometer. The Xcalibur raw files were converted to mzXML format and were searched against the UniProtKB/Swiss-Prot human protein database (UniProt release 2016_04) using X! TANDEM CYCLONE TPP (2011.12.01.1 - LabKey, Insilicos, ISB). Methionine oxidation was considered as a variable modification in all searches. Up to one missed tryptic cleavage was allowed. The X! Tandem search results were analyzed by the Trans-Proteomic Pipeline, version 4.3. Peptide/protein identifications were validated by the Peptide/ProteinProphet software tools (Keller et al., 2002; Nesvizhskii et al., 2003). Relative quantification is based on the ratio of the number of the spectra assigned to a protein and the total number of spectra in A-673/luciferase shRNA versus A-673/EWS-FLI1 shRNA samples.
RNA samples and real-time quantitative RT-PCR
De-identified Ewing sarcoma tumor RNA samples were obtained from the Cooperative Human Tissue Network. Total cellular RNA was isolated using TRIzol reagent (Invitrogen). Reverse transcription was performed using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher) as per manufacturer’s instructions. Quantitative PCR was performed using PowerUp SYBR Green Master Mix (Thermo Fisher) on Applied Biosystems ViiA 7 Real-Time PCR System. Each sample was analyzed in triplicate. The following primers were used: NELL2 forward, 5′-CAGAATGCACTGTTACCTGGA-3′, NELL2 reverse, 5′-GCTGATCCCAATTCTCTTTCA-3′; Robo3 forward, 5′-CCGGACGACAGATATTACAACG-3′, Robo3 reverse, 5′-GCTGGGTCAATGGTGCTATAG-3′; NR0B1 forward, 5′-AGGGGACCGTGCTCTTTAAC-3′, NR0B1 reverse, 5′-CTGAGTTCCCCACTGGAGTC-3′; NKX2-2 forward, 5’-CAGCGACAACCCGTACAC-3′, NKX2-2 reverse, 5′-GACTTGGAGCTTGAGTCCTGA-3′; EZH2 forward, 5′-TGGGAAAGTACACGGGGATA-3′, EZH2 reverse, 5′-TATTGACCAAGGGCATTCAC-3′; NPY1R forward, 5′-CCATCGGACTCTCATAGGTTGTC-3′, NPY1R reverse, 5′-GACCTGTACTTATTGTCTCTCATC-3′; NGFR forward, 5′-CCTCATCCCTGTCTATTGCTCC-3′, NGFR reverse, 5′-GTTGGCTCCTTGCTTGTTCTGC-3′; PAPPA forward, 5′-CAGAATGCACTGTTACCTGGA-3′, PAPPA reverse, 5′-GCTGATCCCAATTCTCTTTCA-3′; CD133 forward, 5′-GACCGACTGAGACCCAACAT-3′, CD133 reverse, 5′-TGGTTTGGCGTTGTACTCTG-3′; BRG1 forward, 5′-AGCGATGACGTCTCTGAGGT-3′, BRG1 reverse, 5′-GTACAGGGACACCAGCCACT-3′; β-actin forward, 5′-AGAGCTACGAGCTGCCTGAC-3′, β-actin reverse, 5′-AGCACTGTGTTGGCGTACAG-3′; STAT3 forward, 5′-GGCATTCGGGAAGTATTGTCG-3′, STAT3 reverse, 5′-GGTAGGCGCCTCAGTCGTATC-3′; MDR1 forward, 5′-CACGTGGTTGGAAGCTAACC-3′, MDR1 reverse, 5′-GAAGGCCAGAGCATAAGATGC-3′; EWS-FLI1 forward, 5′-GGCAGCAGAACCCTTCTTAT-3′, EWS-FLI1 reverse, 5′-GGCCGTTGCTCTGTATTCTTA-3′; GAPDH forward, 5′-GGTGTGAACCATGAGAAGTATGA-3′, GAPDH reverse, GAGTCCTTCCACGATACCAAAG; and EWS forward, 5′-CCACGGATTACAGTACCTATAGC-3′, EWS reverse, 5′-GGCTGTCCATAGGTTCCATAG-3′.
Immunoblotting
Unless otherwise noted in the figure legends, 15 μg of whole-cell lysate or 20 μg of conditioned medium was separated by SDS-PAGE and analyzed by immunoblotting as described (Jayabal et al., 2017). The following antibodies were used: rabbit polyclonal anti-FLI1 (ab15289, Abcam); mouse monoclonal anti-FLAG M2 (F1804, MilliporeSigma); rabbit monoclonal anti-NELL2 (ab181376, Abcam); goat polyclonal anti-PGK1 (sc-17943, Santa Cruz Biotechnology); rabbit polyclonal anti-Robo3 (LS-C345713, LSBio); goat polyclonal anti-Robo3 (PA5-18714, Thermo Fisher Scientific); mouse monoclonal anti-cdc42 (ACD03, Cytoskeleton); rabbit polyclonal anti-Rac1/2/3 (2465, Cell Signaling Technologies); rabbit polyclonal anti-Rho A (2117, Cell Signaling Technologies); goat polyclonal anti-BRG1 (A303-877A, Bethyl Laboratories); rabbit monoclonal anti-BRG1 (49360, Cell Signaling Technologies); rabbit polyclonal anti-BRM (A301-014A-T, Bethyl Laboratories); mouse monoclonal anti-ARID1A/BAF250 (sc-32761, Santa Cruz Biotechnology); rabbit polyclonal anti-BAF170 (A301-039A-T, Bethyl Laboratories); rabbit monoclonal anti-BAF155 (11956, Cell Signaling Technologies); rabbit polyclonal anti-BAF60B (A301-596A-T, Bethyl Laboratories); rabbit polyclonal anti-BAF57 (A300-810A-T, Bethyl Laboratories); rabbit polyclonal anti-BAF53A (A301-391A-T, Bethyl Laboratories); rabbit monoclonal anti-BAF47 (8745, Cell Signaling Technologies); rabbit polyclonal anti-BRD9 (A303-781A-T, Bethyl Laboratories); rabbit monoclonal anti-SS18 (21792, Cell Signaling Technologies); rabbit monoclonal anti-CD133 (64326, Cell Signaling Technologies); rabbit polyclonal anti-srGAP1 (A301-286A-T, Bethyl Laboratories); rabbit polyclonal anti-srGAP2 (GTX130797, GeneTex); mouse monoclonal anti-tubulin (DM1A, Thermo Fisher Scientific); rabbit polyclonal anti-β-actin (4967, Cell Signaling Technologies); mouse monoclonal anti-Rpb1 (2629, Cell Signaling Technologies); rabbit monoclonal anti-cdc6 (3387, Cell Signaling Technologies); rabbit monoclonal anti-caveolin-1 (3267, Cell Signaling Technologies); rabbit polyclonal anti-phospho-caveolin-1 (3251, Cell Signaling Technologies); rabbit monoclonal anti-Src (2123, Cell Signaling Technologies); rabbit monoclonal anti-phospho-Src Family (6943, Cell Signaling Technologies); and rabbit polyclonal anti-GDF6 (NBP1-91934, Novus Biologicals). The following HRP-conjugated secondary antibodies were used: goat anti-rabbit (7074) and goat anti-mouse (7076) (Cell Signaling Technologies); donkey anti-goat (A50-201P, Bethyl Laboratories).
Immunoprecipitation
Cells were washed three times with PBS, resuspended in 5 mL of buffer A (10 mm HEPES-KOH (pH 7.9), 1.5 mM MgCl2,10 mM KCl, 0.5 mM DTT), and Dounce homogenized 10 times using a tight pestle. Dounce homogenized nuclei were centrifuged at 228 × g for 5 min at 4°C. The nuclear pellet was resuspended in 3 mL of 0.25 mM sucrose, 10 mM MgCl2; layered over 3 mL of 0.35 mM sucrose, 0.5 mM MgCl2; and centrifuged at 1,430 × g for 5 min at 4°C. The nuclear pellet was resuspended in 1 mL of IP buffer (20mM HEPES (pH 7.9), 350mM NaCl, 0.1% NP-40, 1mM DTT, 0.2mM PMSF, 2mg/ml leupeptin and 2 mg/ml aprotinin) and sonicated. The following antibodies were used for immunoprecipitation: rabbit monoclonal anti-BRG1 (ab110641, Abcam), rabbit monoclonal anti-BAF57 (33360, Cell Signaling Technologies), and mouse monoclonal anti-FLAG M2 (F1804, MilliporeSigma). The immunoprecipitates were washed four times with IP buffer and analyzed by immunoblotting.
Immunofluorescence
Cells grown on coverslips were fixed with 4% paraformaldehyde for 15 minutes at room temperature, washed with PBS, and permeabilized with 0.2% pre-chilled Triton X-100/PBS for 5 minutes. The samples were blocked with culture medium for one hour and incubated with the primary antibody for three hours followed by the secondary antibody for one hour. The following primary antibodies were used: rabbit polyclonal anti-NELL2 (NBP1-82527, Novus Biologicals); mouse monoclonal anti-FLAG M2 (F1804, MilliporeSigma); mouse monoclonal anti-CD133 AC133 (130-090-422, Miltenyi Biotec); rabbit polyclonal anti-FLI1 (ab15289, Abcam); and mouse monoclonal anti-CD99 (MS-1633-P0, Thermo Fisher Scientific). The following secondary antibodies were used: Alexa Fluor 488, goat anti-rabbit IgG (A11034, Thermo Fisher Scientific); Alexa Fluor 568, goat anti-rabbit IgG (A11036, Thermo Fisher Scientific); Alexa Fluor 488, goat anti-mouse IgM (A21042, Thermo Fisher Scientific); and Alexa Fluor 594, goat anti-mouse IgG (A11032, Thermo Fisher Scientific). Nuclei were stained with DAPI. DyLight 554 Phalloidin (13054, Cell Signaling Technologies) was used to visualize filopodia. The images were collected with a FluoView FV3000 confocal laser scanning microscope (Olympus).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described (Carey et al., 2009a) using rabbit polyclonal anti-FLI1 antibody (ab15289, Abcam), rabbit monoclonal anti-BRG1 antibody (ab110641, Abcam), or control rabbit IgG (ab37415, Abcam). The primer sequences used for ChIP are as follows: NELL2 forward, 5′-CTCTCTCTCTCTCTCTAACCATCTC-3′, NELL2 reverse, 5′-TCTCTGGGACCAGCATACA-3′; FOXO1 forward, 5′-GGAAGAGGTTCCCACGGAGGGCAT-3′, FOXO1 reverse, 5′-CCGGCGACACTTTGTTTACT-3′; GLI-1 forward, 5′-AGAGCCTGGGGGTGAGACAT-3′, GLI-1 reverse, 5′-GCCTCTTCAACTTAACCGCATGA-3′; NR0B1 forward, 5′-GTTTGTGCCTTCATGGGAAATGGTTATTC-3′; NR0B1 reverse, 5′-CTAGTGTCTTGTGTGTCCCTAGGG-3′; GAPDH forward, 5′-TCCTCCTGTTTCATCCAAGC-3′; GAPDH reverse, 5′-TAGTAGCCGGGCCCTACTTT-3′;CD133 P1 forward, 5′-GAACTGCGGGGAGAGCGTGGTG-3′; P1 reverse, 5′- TCCCCGAGAGCGAGTCCGAAGTC-3′; CD133 P2 forward, 5′-CGACCACAGCGGGAGTAG-3′; P2 reverse, 5′-GCGAGAGGCTGGGAAGGT-3′; CD133 P3 forward, 5′-GACCGGACAACAAAGAGGAG-3′; P3 reverse, 5′-CTCCAGACACGGGCTTTC-3′; CD133 P4 forward, 5′-CCGCCCCGCCGCTCATTC-3′; P4 reverse, 5′-GCTTCCCCGCCCTTTACCTC-3′; CD133 P5 forward, 5′-TATGGCTTTATGCTGTTTTTCAA-3′; P5 reverse, 5′-CTCATCCCGGCCGCATTAGAC-3′; CD133 P6 forward, 5′-GTGCTTCCTGCTCCTCTTC-3′; P6 reverse, 5′-GCTAGCAAGATCCTCCAAACA-3′; and EWS forward, 5′-TCCCAAAGTGCTGGGATTAC-3′, EWS reverse, 5′-TCTCATGGTTTCCCTTTCTAGC-3′. The EWS primers amplify an 87 bp sequence approximately 3 kb from the EWS transcription start site. The genomic locations of the primers are: EWS forward primer, chromosome 22, 29265034 – 29265054 and EWS reverse primer, chromosome 22, 29265098 – 29265120.
GST pull-down assays
Cells were lysed in TNE buffer (10 mM Tris, pH 7.4, 150 mM, NaCl, 1% NP-40, 1 mM EDTA, and protease inhibitors). The active form of cdc42, Rac, and Rho A was analyzed by pull-down of cell lysate with GST-PAK1 (which selectively binds active cdc42 and active Rac) and GST-RBD (which selectively binds active Rho A) followed by immunoblotting for cdc42, Rac, and Rho A as described (Ren et al., 1999).
Gel filtration chromatography
Nuclear extract was prepared as described (Carey et al., 2009b). The extract (5 mg) was loaded onto HiPrep 16/60 Sephacryl S-400 HR column (GE Healthcare) equilibrated with a gel filtration buffer (150 mM NaCl, 20 mM HEPES- KOH, 10 mM KCl, 1 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, pH 7.5). Proteins were eluted at 1 ml/min and 1 mL fractions were collected. Every fourth fraction was concentrated and analyzed by immunoblotting.
Phalloidin dot blot assays
The BAF complexes were immuno-purified from nuclear extract using anti-BRG1 or anti-BAF57 (for endogenous BAF) or anti-FLAG antibody (for FLAG-BRG1-containing BAF) and were spotted onto a nitrocellulose membrane (sample amount normalized by cell number). The membrane was allowed to dry at room temperature for 30 minutes. The membrane was rinsed with TBST (150mM NaCl, 20 mM Tris, pH7.4, 0.05% Tween-20), blocked with 5% BSA in TBST, and incubated with Biotin-XX Phalloidin (Biotium) either overnight at 4°C or for two hours at room temperature followed by three washes with TBST. The membrane was incubated with High Sensitivity Streptavidin-HRP (21130, Pierce/Thermo Fisher) for one hour and was washed three times with TBST, followed by detection with ECL reagent.
Cell proliferation and xenograft tumorigenicity assays
Anchorage-dependent cell proliferation was assessed by IncuCyte live-cell imaging system (Essen BioScience). The IncuCyte system monitors cell proliferation by analyzing the occupied area (% confluence) of cell images over time. At least four fields from four wells were assayed for each experimental condition. The cell seeding density was 2000 cells per well in a 96-well plate. For each assay, biological replicates were performed to confirm the reproducibility of results. Anchorage-independent cell proliferation was evaluated by soft agar colony formation assays. A-673 cells were infected with lentiviruses expressing shRNAs against NELL2 or scrambled shRNA and were selected with 2 μg/ml puromycin. Four days after infection, 4 × 103 cells were plated in soft agar. The soft agar cultures were composed of two layers: a base layer [4 mL in a 60-mm dish; DMEM/10% fetal bovine serum/0.6% noble agar (A5431, MilliporeSigma)/penicillin/streptomycin] and a cell layer (2 mL in a 60-mm dish; DMEM/10% fetal bovine serum/0.3% noble agar/penicillin/ streptomycin). Colonies were grown for three weeks and counted. Colonies (> 50 cells) were scored by randomly counting 10 fields per dish. Sphere formation assays were done as described (Dasgupta et al., 2017) using ultra-low attachment 6-well plates (Corning; seeding density 1 × 104 cells/well) and DMEM/F-12 medium supplemented with B27, human recombinant epidermal growth factor (20 ng/ml), and basic fibroblast growth factor (20 ng/ml). For xenograft tumorigenicity assays, cells were subcutaneously injected into the flanks of SCID mice (2 × 106 cells/injection, five mice/group). Tumor growth was monitored weekly using a caliper. While it is not possible to predict the effect size, we chose the sample size of five mice per group based on our prior experience with xenograft experiments. Mice were randomly allocated to treatment groups. Blinding of the researcher measuring tumor size was employed. The animal research method was reviewed and approved for humaneness by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio.
Migration assays
Migration assays were performed in modified Boyden chambers (Corning® Transwell®, 8 μm pore size, MilliporeSigma). Cells were seeded at 5 × 104 cells/200 μL serum free-DMEM/F-12 medium in the upper chamber. The lower chamber was filled with DMEM/F-12 supplemented with 10% FBS. After 20-hour incubation, the membrane was gently removed from the chamber and the cells on the upper surface were removed using cotton swabs. Cells on the lower surface that migrated through the membrane were fixed with 50% methanol and stained with 0.1% crystal violet. The migrated cells were counted from eight randomly chosen fields.
Flow cytometry
Cells were trypsinized, washed with FACS wash buffer (PBS, 0.5% BSA, 2 mM EDTA), and incubated with PE-conjugated human CD133/1 antibody (clone AC133, Miltenyi Biotec; 1:100 in FACS wash buffer) for 20 minutes at 4°C. Cells were washed three times with FACS wash buffer and the CD133high and CD133low cell populations were sorted by using BD FACSAria (Becton Dickinson). The FACSDiva 6.1.3 software (Becton Dickinson) was used for sample analysis.
Dissociation of a patient-derived xenograft tumor
A patient-derived xenograft tumor was washed and dissociated into single-cell suspensions using the tumor dissociation kit (Miltenyi Biotec 130–095-929) and gentleMACS Octo-dissociator (Miltenyi Biotec 130-096-427), following the manufacturer’s protocol. Dissociated cells were cultured in DMEM/F-12 medium supplemented with 10% FBS.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical analyses were performed with Prism (GraphPad Software) with a two-tailed Student’s t test. Data are expressed as mean ± SEM. The results were considered significant when p < 0.05. The number of replicates, independent samples, and animals is indicated in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit polyclonal anti-FLI1 | Abcam | ab15289 |
| mouse monoclonal anti-FLAG M2 | MilliporeSigma | F1804 |
| rabbit monoclonal anti-NELL2 | Abcam | ab181376 |
| rabbit polyclonal anti-Robo3 | LSBio | LS-C345713 |
| goat polyclonal anti-Robo3 | Thermo Fisher Scientific | PA5-18714 |
| mouse monoclonal anti-cdc42 | Cytoskeleton | ACD03 |
| rabbit polyclonal anti-Rac1/2/3 | Cell Signaling Technologies | 2465 |
| rabbit polyclonal anti-Rho A | Cell Signaling Technologies | 2117 |
| goat polyclonal anti-BRG1 | Bethyl Laboratories | A303-877A |
| rabbit monoclonal anti-BRG1 | Cell Signaling Technologies | 49360 |
| rabbit polyclonal anti-BRM | Bethyl Laboratories | A301-014A-T |
| mouse monoclonal anti-ARID1A/BAF250 | Santa Cruz Biotechnology | sc-32761 |
| rabbit polyclonal anti-BAF170 | Bethyl Laboratories | A301-039A-T |
| rabbit monoclonal anti-BAF155 | Cell Signaling Technologies | 11956 |
| rabbit polyclonal anti-BAF60B | Bethyl Laboratories | A301-596A-T |
| rabbit polyclonal anti-BAF57 | Bethyl Laboratories | A300-810A-T |
| rabbit polyclonal anti-BAF53A | Bethyl Laboratories | A301-391A-T |
| rabbit monoclonal anti-BAF47 | Cell Signaling Technologies | 8745 |
| rabbit polyclonal anti-BRD9 | Bethyl Laboratories | A303-781A-T |
| rabbit monoclonal anti-SS18 | Cell Signaling Technologies | 21792 |
| rabbit monoclonal anti-CD133 | Cell Signaling Technologies | 64326 |
| rabbit polyclonal anti-srGAP1 | Bethyl Laboratories | A301-286A-T |
| rabbit polyclonal anti-srGAP2 | GeneTex | GTX130797 |
| mouse monoclonal anti-tubulin | Thermo Fisher Scientific | DM1A |
| rabbit polyclonal anti-β-actin | Cell Signaling Technologies | 4967 |
| mouse monoclonal anti-Rpb1 | Cell Signaling Technologies | 2629 |
| rabbit monoclonal anti-cdc6 | Cell Signaling Technologies | 3387 |
| rabbit monoclonal anti-caveolin-1 | Cell Signaling Technologies | 3267 |
| rabbit polyclonal anti-phospho-caveolin-1 | Cell Signaling Technologies | 3251 |
| rabbit monoclonal anti-Src | Cell Signaling Technologies | 2123 |
| rabbit monoclonal anti-phospho-Src Family | Cell Signaling Technologies | 6943 |
| rabbit polyclonal anti-GDF6 | Novus Biologicals | NBP1-91934 |
| HRP-conjugated goat anti-rabbit | Cell Signaling Technologies | 7074 |
| HRP-conjugated goat anti-mouse | Cell Signaling Technologies | 7076 |
| HRP-conjugated donkey anti-goat | Bethyl Laboratories | A50-201P |
| rabbit monoclonal anti-BRG1 | Abcam | ab110641 |
| rabbit monoclonal anti-BAF57 | Cell Signaling Technologies | 33360 |
| rabbit polyclonal anti-NELL2 | Novus Biologicals | NBP1-82527 |
| mouse monoclonal anti-CD133 AC133 | Miltenyi Biotec | 130-090-422 |
| mouse monoclonal anti-CD99 | Thermo Fisher Scientific | MS-1633-P0 |
| Alexa Fluor 488, goat anti-rabbit IgG | Thermo Fisher Scientific | A11034 |
| Alexa Fluor 568, goat anti-rabbit IgG | Thermo Fisher Scientific | A11036 |
| Alexa Fluor 488, goat anti-mouse IgM | Thermo Fisher Scientific | A21042 |
| Alexa Fluor 594, goat anti-mouse IgG | Thermo Fisher Scientific | A11032 |
| control rabbit IgG | Abcam | ab37415 |
| Bacterial and virus strains | ||
| DH10B | Thermo Fisher Scientific | 12331013 |
| NEB® Stable Competent E. coli | New England Biolabs | C3040H |
| Biological samples | ||
| De-identified Ewing sarcoma tumor RNA samples | Cooperative Human Tissue Network | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher Scientific | 13778150 |
| Recombinant NELL2 protein | R&D Systems | 8946-NL-050 |
| ML141 | Selleck Chemicals | S7686 |
| Rho/Rac/Cdc42 Activator 1 (CN04) | Cytoskeleton | CN04-A |
| puromycin dihydrochloride | MilliporeSigma | P8833 |
| doxorubicin hydrochloride | MilliporeSigma | D1515 |
| MG-132 | MilliporeSigma | 474790 |
| Biotin-XX-Phalloidin | Biotium | 00028 |
| jasplakinolide | Cayman Chemical | 11705 |
| cisplatin | Cayman Chemical | 13119 |
| dasatinib | Apexbio | A3017 |
| Critical commercial assays | ||
| NELL2 ELISA kit | Aviva Systems Biology | OKCD01073 |
| Deposited data | ||
| The mass spectrometry data | the PRIDE database | PXD016052 |
| Experimental models: Cell lines | ||
| A-673 | ATCC | CRL-1598 |
| SK-N-MC | ATCC | HTB-10 |
| 293 | ATCC | CRL-1573 |
| 293T | ATCC | CRL-11268 |
| HeLa | ATCC | CCL-2 |
| HCT116 | ATCC | CCL-247 |
| IMR-90 | ATCC | CCL-186 |
| Aska-SS | RIKEN Bioresource Center | RCB3576 |
| EW8 | Dr. Patrick Grohar | N/A |
| TC32 | Dr. Patrick Grohar | N/A |
| TC71 | Coriell Institute | GM11654 |
| CHLA-9 | Dr. Patrick Grohar | N/A |
| ES1 | Dr. Peter Houghton | N/A |
| ES2 | Dr. Peter Houghton | N/A |
| ES3 | Dr. Peter Houghton | N/A |
| ES6 | Dr. Peter Houghton | N/A |
| ES7 | Dr. Peter Houghton | N/A |
| ES8 | Dr. Peter Houghton | N/A |
| RD-ES | ATCC | HTB-166 |
| RH4 | Dr. Javed Khan | N/A |
| RH5 | Dr. Javed Khan | N/A |
| RD | ATCC | CCL-136 |
| RMS13 | ATCC | CRL-2061 |
| SK-NEP-1 | ATCC | HTB-48 |
| SK-ES-1 | ATCC | HTB-86 |
| Cord blood-derived human mesenchymal stem cells | Vitro Biopharma | SC00A1 |
| Experimental models: Organisms/strains | ||
| C.B.17SC scid−/− mice, 5-6 weeks old | Taconic Biosciences | CB17SC |
| Oligonucleotides | ||
| PCR primers (Table S2) | this study | N/A |
| Recombinant DNA | ||
| pcDNA3.1 vector | Invitrogen/Thermo Fisher Scientific | V79020 |
| pCDH1-MCS1-EF1-Puro vector | System Biosciences | CD510A-1 |
Highlights.
The autocrine signaling mediated by NELL2 maintains Ewing sarcoma cell growth
NELL2 signaling inhibits cdc42 and enhances EWS-FLI1 transcriptional output
NELL2-cdc42 signaling regulates the assembly and the stability of BAF complexes
Ewing sarcoma harbors cell populations displaying high and low NELL2 signaling
ACKNOWLEDGMENTS
We thank Robert Eisenman for helpful discussions and critical review of the manuscript. We thank Patrick Grohar and Javed Khan for cell lines. We thank the Cooperative Human Tissue Network for Ewing sarcoma tumor RNA samples, the University of Texas Health Science Center at San Antonio (UTHSCSA) Institutional Mass Spectrometry Laboratory (Sammy Pardo ad Dana Molleur) for mass spectrometry analysis, the UTHSCSA flow cytometry facility for flow cytometry analysis, and the UTHSCSA Histology/Immunohistochemistry Laboratory for immunohistochemistry analysis. This work was supported by the National Cancer Institute; the National Institutes of Health (CA202485 to Y.S. and CA165995 to P.J.H.); the Cancer Prevention and Research Institute of Texas (RP160487, RP160841, and RP190385 to Y.S. and RP160716 to P.J.H.); the Owens Medical Research Foundation (to Y.S.); the National Center for Advancing Translational Sciences; National Institutes of Health, through the Clinical and Translational Science Award (CTSA) UL1 TR001120; the Mays Cancer Center P30 Cancer Center Support Grant from the National Cancer Institute (CA054174) for the mass spectrometry and the flow cytometry shared resources; and the National Institutes of Health for purchase of the Orbitrap mass spectrometer (1S10RR025111-01 to S.T.W.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109254.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aynaud MM, Mirabeau O, Gruel N, Grossetete S, Boeva V, Durand S, Surdez D, Saulnier O, Zaidi S, Gribkova S, et al. (2020). Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep. 30, 1767–1779.e6. [DOI] [PubMed] [Google Scholar]
- Blockus H, and Chédotal A (2016). Slit-Robo signaling. Development 143, 3037–3044. [DOI] [PubMed] [Google Scholar]
- Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, Awad ME, Rengarajan S, Volorio A, McBride MJ, et al. (2017). Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 171, 163–178.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose K, Bland KS, Wang KH, Arnott D, Henzel W, Goodman CS, Tessier-Lavigne M, and Kidd T (1999). Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 96, 795–806. [DOI] [PubMed] [Google Scholar]
- Carey MF, Peterson CL, and Smale ST (2009a). Chromatin immunoprecipitation (ChIP). Cold Spring Harbor Protoc. 2009, pdb.prot5279. [DOI] [PubMed] [Google Scholar]
- Carey MF, Peterson CL, and Smale ST (2009b). Dignam and Roeder nuclear extract preparation. Cold Spring Harbor Protoc. 2009, pdb.prot5330. [DOI] [PubMed] [Google Scholar]
- Chen Z, Gore BB, Long H, Ma L, and Tessier-Lavigne M (2008). Alternative splicing of the Robo3 axon guidance receptor governs the midline switch from attraction to repulsion. Neuron 58, 325–332. [DOI] [PubMed] [Google Scholar]
- Cheng ZJ, Singh RD, Holicky EL, Wheatley CL, Marks DL, and Pagano RE (2010). Co-regulation of caveolar and Cdc42-dependent fluid phase endocytosis by phosphocaveolin-1. J. Biol. Chem 285, 15119–15125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornaz-Buros S, Riggi N, DeVito C, Sarre A, Letovanec I, Provero P, and Stamenkovic I (2014). Targeting cancer stem-like cells as an approach to defeating cellular heterogeneity in Ewing sarcoma. Cancer Res. 74, 6610–6622. [DOI] [PubMed] [Google Scholar]
- Dasgupta A, Trucco M, Rainusso N, Bernardi RJ, Shuck R, Kurenbekova L, Loeb DM, and Yustein JT (2017). Metabolic modulation of Ewing sarcoma cells inhibits tumor growth and stem cell properties. Oncotarget 8, 77292–77308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzi DJ, Song M, Blackman B, Weintraub ST, López-Terrada D, Chen Y, Tomlinson GE, and Shiio Y (2016). FGF19 functions as autocrine growth factor for hepatoblastoma. Genes Cancer 7, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzetti GA, Laud-Duval K, van der Ent W, Brisac A, Irondelle M, Aubert S, Dirksen U, Bouvier C, de Pinieux G, Snaar-Jagalska E, et al. (2017). Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 36, 3505–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenney JR Jr., and Zokas L (1989). Novel tyrosine kinase substrates from Rous sarcoma virus-transformed cells are present in the membrane skeleton. J. Cell Biol 108, 2401–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glumac PM, and LeBeau AM (2018). The role of CD133 in cancer: a concise review. Clin. Transl. Med 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González I, Vicent S, de Alava E, and Lecanda F (2007). EWS/FLI-1 oncoprotein subtypes impose different requirements for transformation and metastatic activity in a murine model. J. Mol. Med. (Berl.) 85, 1015–1029. [DOI] [PubMed] [Google Scholar]
- Hancock JD, and Lessnick SL (2008). A transcriptional profiling meta-analysis reveals a core EWS-FLI gene expression signature. Cell Cycle 7, 250–256. [DOI] [PubMed] [Google Scholar]
- Hodges C, Kirkland JG, and Crabtree GR (2016). The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med 6, a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski A, Tom I, Tong RK, Gildea HK, Koch AW, Gonzalez LC, and Tessier-Lavigne M (2015). Operational redundancy in axon guidance through the multifunctional receptor Robo3 and its ligand NELL2. Science 350, 961–965. [DOI] [PubMed] [Google Scholar]
- Jayabal P, Houghton PJ, and Shiio Y (2017). EWS-FLI-1 creates a cell surface microenvironment conducive to IGF signaling by inducing pappalysin-1. Genes Cancer 8, 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedlicka P (2010). Ewing Sarcoma, an enigmatic malignancy of likely progenitor cell origin, driven by transcription factor oncogenic fusions. Int. J. Clin. Exp. Pathol 3, 338–347. [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Obama H, Kuan SL, Nakamura R, Nakamoto C, Ouyang Z, and Nakamoto M (2009). In vitro guidance of retinal axons by a tectal lamina-specific glycoprotein Nel. Mol. Cell. Neurosci 41, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Gwye Y, Russell D, Cao C, Douglas D, Hung L, Kovar H, Triche TJ, and Lawlor ER (2010). CD133 expression in chemo-resistant Ewing sarcoma cells. BMC Cancer 10, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, and Crabtree GR (2013). Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, and Crabtree GR (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci. Adv 1, e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, and Aebersold R (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, Zeilstra J, Pals ST, Mehmet H, Stassi G, and Medema JP (2010). The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 70, 719–729. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, and Rich JN (2015). Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor ER, and Sorensen PH (2015). Twenty Years on: What Do We Really Know about Ewing Sarcoma and What Is the Path Forward? Crit. Rev. Oncog 20, 155–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessnick SL, and Ladanyi M (2012). Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annu. Rev. Pathol 7, 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Xing Y, Cao B, Yang F, Yang T, Ai Z, Wei Y, and Jiang J (2016). The Interaction between Cancer Stem Cell Marker CD133 and Src Protein Promotes Focal Adhesion Kinase (FAK) Phosphorylation and Cell Migration. J. Biol. Chem 291, 15540–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Sabatier C, Ma L, Plump A, Yuan W, Ornitz DM, Tamada A, Murakami F, Goodman CS, and Tessier-Lavigne M (2004). Conserved roles for Slit and Robo proteins in midline commissural axon guidance. Neuron 42, 213–223. [DOI] [PubMed] [Google Scholar]
- Mackintosh C, Madoz-Gúrpide J, Ordóñez JL, Osuna D, and Herrero-Martín D (2010). The molecular pathogenesis of Ewing’s sarcoma. Cancer Biol. Ther 9, 655–667. [DOI] [PubMed] [Google Scholar]
- Mambetisaeva ET, Andrews W, Camurri L, Annan A, and Sundaresan V (2005). Robo family of proteins exhibit differential expression in mouse spinal cord and Robo-Slit interaction is required for midline crossing in vertebrate spinal cord. Dev. Dyn 233, 41–51. [DOI] [PubMed] [Google Scholar]
- Mashtalir N, D’Avino AR, Michel BC, Luo J, Pan J, Otto JE, Zullow HJ, McKenzie ZM, Kubiak RL, St Pierre R, et al. (2018). Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1288.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PK, and Lappalainen P (2008). Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol 9, 446–454. [DOI] [PubMed] [Google Scholar]
- May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, Zucman J, Thomas G, and Denny CT (1993). Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc. Natl. Acad. Sci. USA 90, 5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchardt C, and Yaniv M (1993). A human homologue of Saccharomyces cerevisiae SNF2/SWI2 and Drosophila brm genes potentiates transcriptional activation by the glucocorticoid receptor. EMBO J. 12, 4279–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller PM, Rademacher J, Bagshaw RD, Wortmann C, Barth C, van Unen J, Alp KM, Giudice G, Eccles RL, Heinrich LE, et al. (2020). Systems analysis of RhoGEF and RhoGAP regulatory proteins reveals spatially organized RAC1 signalling from integrin adhesions. Nat. Cell Biol 22, 498–511. [DOI] [PubMed] [Google Scholar]
- Nakamura R, Nakamoto C, Obama H, Durward E, and Nakamoto M (2012). Structure-function analysis of Nel, a thrombospondin-1-like glycoprotein involved in neural development and functions. J. Biol. Chem 287, 3282–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, Ku WL, Teng M, Cui K, Williams RT, et al. (2017). SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat. Genet 49, 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Nevins AK, and Thurmond DC (2006). Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic beta-cells. J. Biol. Chem 281, 18961–18972. [DOI] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, and Schwartz MA (1999). Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18, 578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier C, Plump AS, Le Ma, Brose K, Tamada A, Murakami F, Lee EY, and Tessier-Lavigne M (2004). The divergent Robo family protein rig-1/Robo3 is a negative regulator of slit responsiveness required for midline crossing by commissural axons. Cell 117, 157–169. [DOI] [PubMed] [Google Scholar]
- Saha T, Rathmann I, Viplav A, Panzade S, Begemann I, Rasch C, Klingauf J, Matis M, and Galic M (2016). Automated analysis of filopodial length and spatially resolved protein concentration via adaptive shape tracking. Mol. Biol. Cell 27, 3616–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Molina S, Figuerola-Bou E, Blanco E, Sánchez-Jiménez M, Táboas P, Gómez S, Ballaré C, García-Domínguez DJ, Prada E, Hontecillas-Prieto L, et al. (2020). RING1B recruits EWSR1-FLI1 and cooperates in the remodeling of chromatin necessary for Ewing sarcoma tumorigenesis. Sci. Adv 6, eaba3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvanathan SP, Graham GT, Erkizan HV, Dirksen U, Natarajan TG, Dakic A, Yu S, Liu X, Paulsen MT, Ljungman ME, et al. (2015). Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. Proc. Natl. Acad. Sci. USA 112, E1307–E1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvanathan SP, Graham GT, Grego AR, Baker TM, Hogg JR, Simpson M, Batish M, Crompton B, Stegmaier K, Tomazou EM, et al. (2019). EWS-FLI1 modulated alternative splicing of ARID1A reveals novel oncogenic function through the BAF complex. Nucleic Acids Res. 47, 9619–9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn DH, Lee KY, Lee C, Oh J, Chung H, Jeon SH, and Seong RH (2007). SRG3 interacts directly with the major components of the SWI/SNF chromatin remodeling complex and protects them from proteasomal degradation. J. Biol. Chem 282, 10614–10624. [DOI] [PubMed] [Google Scholar]
- Sompallae R, Hofmann O, Maher CA, Gedye C, Behren A, Vitezic M, Daub CO, Devalle S, Caballero OL, Carninci P, et al. (2013). A comprehensive promoter landscape identifies a novel promoter for CD133 in restricted tissues, cancers, and stem cells. Front. Genet 4, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son EY, and Crabtree GR (2014). The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am. J. Med. Genet. C. Semin. Med. Genet 166C, 333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Pierre R, and Kadoch C (2017). Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr. Opin. Genet. Dev 42, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surviladze Z, Waller A, Strouse JJ, Bologa C, Ursu O, Salas V, Parkinson JF, Phillips GK, Romero E, Wandinger-Ness A, et al. (2010). A Potent and Selective Inhibitor of Cdc42 GTPase. In Probe Reports from the NIH Molecular Libraries Program (National Center for Biotechnology Information; ). [PubMed] [Google Scholar]
- Suvà ML, Riggi N, Stehle JC, Baumer K, Tercier S, Joseph JM, Suvà D, Clément V, Provero P, Cironi L, et al. (2009). Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res. 69, 1776–1781. [DOI] [PubMed] [Google Scholar]
- Tirado OM, Mateo-Lozano S, Villar J, Dettin LE, Llort A, Gallego S, Ban J, Kovar H, and Notario V (2006). Caveolin-1 (CAV1) is a target of EWS/FLI-1 and a key determinant of the oncogenic phenotype and tumorigenicity of Ewing’s sarcoma cells. Cancer Res. 66, 9937–9947. [DOI] [PubMed] [Google Scholar]
- Toomey EC, Schiffman JD, and Lessnick SL (2010). Recent advances in the molecular pathogenesis of Ewing’s sarcoma. Oncogene 29, 4504–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Ui A, Kanno S, Ogiwara H, Nagase T, Kohno T, and Yasui A (2014). SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 74, 2465–2475. [DOI] [PubMed] [Google Scholar]
- Wong K, Ren XR, Huang YZ, Xie Y, Liu G, Saito H, Tang H, Wen L, Brady-Kalnay SM, Mei L, et al. (2001). Signal transduction in neuronal migration: roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell 107, 209–221. [DOI] [PubMed] [Google Scholar]
- Zelina P, Blockus H, Zagar Y, Péres A, Friocourt F, Wu Z, Rama N, Fouquet C, Hohenester E, Tessier-Lavigne M, et al. (2014). Signaling switch of the axon guidance receptor Robo3 during vertebrate evolution. Neuron 84, 1258–1272. [DOI] [PubMed] [Google Scholar]
- Zhao K, Wang W, Rando OJ, Xue Y, Swiderek K, Kuo A, and Crabtree GR (1998). Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell 95, 625–636. [DOI] [PubMed] [Google Scholar]
- Zhou F, Elzi DJ, Jayabal P, Ma X, Chiu YC, Chen Y, Blackman B, Weintraub ST, Houghton PJ, and Shiio Y (2020). GDF6-CD99 Signaling Regulates Src and Ewing Sarcoma Growth. Cell Rep. 33, 108332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Stoeckli E, Chen H, and Tessier-Lavigne M (2000). Squeezing axons out of the gray matter: a role for slit and semaphorin proteins from midline and ventral spinal cord. Cell 102, 363–375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data have been deposited to the PRIDE database and assigned the identifier, PXD016052.