

ABSTRACT
T cell receptor (TCR)-redirected T cells target intracellular antigens such as Wilms’ tumor 1 (WT1), a tumor-associated antigen overexpressed in several malignancies, including acute myeloid leukemia (AML). For both chimeric antigen receptor (CAR)- and TCR-redirected T cells, several clinical studies indicate that T cell subsets with a less-differentiated phenotype (e.g. stem cell memory T cells, TSCM) survive longer and mediate superior anti-tumor effects in vivo as opposed to more terminally differentiated T cells. Cytokines added during in vitro and ex vivo culture of T cells play an important role in driving the phenotype of T cells for adoptive transfer. Using the OP9-DL1 co-culture system, we have shown previously that we are able to generate in vitro, starting from clinically relevant stem cell sources, T cells with a single tumor antigen (TA)-specific TCR. This method circumvents possible TCR chain mispairing and unwanted toxicities that might occur when introducing a TA-specific TCR in peripheral blood lymphocytes. We now show that we are able to optimize our in vitro culture protocol, by adding IL-21 during maturation, resulting in generation of TA-specific T cells with a less-differentiated phenotype and enhanced in vitro anti-tumor effects. We believe the favorable TSCM-like phenotype of these in vitro generated T cells preludes superior in vivo persistence and anti-tumor efficacy. Therefore, these TA-specific T cells could be of use as a valuable new form of patient-tailored T cell immunotherapy for malignancies for which finding a suitable CAR-T target antigen is challenging, such as AML.
KEYWORDS: acute myeloid leukemia (AML), T cell immunotherapy, common gamma chain cytokines, hematopoietic stem cells, OP9-DL1
Introduction
Despite the successes seen with adoptive transfer of chimeric antigen receptor (CAR) T cells targeting CD19 in B cell malignancies,1 a suitable target surface antigen has yet to be discovered for acute myeloid leukemia (AML). This target antigen should be present on both leukemic blasts and leukemic stem cells (LSC), the latter often deemed responsible for relapse,2 and absent from healthy tissues and hematopoietic stem and progenitor cells (HSPC), avoiding intolerable toxicities. Wilms’ tumor 1 (WT1), an intracellular antigen, is overexpressed in the majority of AML patients on both leukemic blasts and LSC.3 Clinical trials using T cells engineered to express a WT1-specific T cell receptor (TCR) have shown promising results.4 However, TCR-engineered T cells are typically generated from peripheral blood lymphocytes (PBL) that already express an endogenous TCR, which can lead to mispairing between the endogenous and introduced TCRα and TCRβ chains, and consequently off-target toxicities.5,6
In an effort to circumvent both mispairing of TCR chains and extensive ex vivo culturing of T cells, our group developed a method to generate tumor antigen (TA)-specific T cells with a single TCR and naive-like characteristics from TCR-transduced HSPC isolated from postnatal thymus,7 cord blood,8 and mobilized peripheral blood or bone marrow from both healthy donors and AML patients.9 This strategy is based on the OP9-DL1 in vitro co-culture system and agonist selection to induce T cell differentiation from HSPC. In this culture protocol, interleukin-7 (IL-7) is added during agonist selection and IL-2 during feeder expansion.7
Several clinical studies demonstrated that less-differentiated T cell subsets, i.e. naive (TN) and central memory (TCM) T cells, mediate superior antitumor effects and survive longer as compared to more differentiated subsets, i.e. effector memory (TEM) and effector (TEFF) T cells.10–12 These studies indicate that objective responses correlate with the degree of in vivo persistence of the adoptively transferred T cells, which is higher for less-differentiated subsets.10,11 Furthermore, prolonged ex vivo culture has been shown to lead to a more terminally differentiated phenotype.11,13 In 2011, Gattinoni and colleagues described a population of stem cell memory T cells (TSCM), with a naive-like phenotype and characteristics similar to conventional memory T cells.14 Cieri et al. were able to generate TSCM-like cells in vitro, by activating TN with anti-CD3/CD28 beads in the presence of exogenous IL-7 and IL-15.15 In vivo mouse experiments by both research groups suggested longer in vivo persistence, increased proliferative capacity and superior antitumor activity of TSCM as compared to other memory T cell subsets, indicating that TSCM might be the ideal T cell subset for adoptive T cell transfer.
These and other experiments show that cytokines added during in vitro or ex vivo culture of T cells before adoptive transfer play an important role in phenotype and consequently in in vivo efficacy of the resulting T cells.15–18 Therefore, we investigated the effect of (combinations of) the common gamma-chain cytokines IL-2, IL-7, IL-15 and IL-21 on proliferation, phenotype and functionality of our in vitro generated TA-specific T cells. We show that by adapting the cytokine mix added during different steps of our in vitro culture protocol, maturation is accelerated or T cells with a favorable less-differentiated phenotype and enhanced interferon-gamma (IFN-γ) production and target cell lysis could be generated.
Materials and methods
Isolation of human CD34+ cells
Umbilical cord blood (UCB) was obtained and used following guidelines of the Medical Ethical Committee of the Ghent University Hospital. Informed consent was obtained in accordance with the Declaration of Helsinki. Mononuclear cells were isolated using density gradient centrifugation (Lymphoprep; Axis-Shield, 1114547). CD34+ cells were enriched by magnetic activated cell sorting with anti-CD34 magnetic beads (MACS beads; Miltenyi, 130–046-703) to a purity of >95%.
Culture of cell lines
The T2 cell line, a human HLA-A2+ cell line deficient for transporter associated with antigen processing (TAP), and JY cell line, a human HLA-A2+ EBV-immortalized B-cell line, were obtained from the American Type Culture Collection (ATCC) and cultured in Iscove’s Modified Dulbecco’s Medium (IMDM; Thermo Fisher Scientific, 12440053) supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine (Thermo Fisher Scientific, 25030–081), 100 IU/ml penicillin and 100 IU/ml streptomycin (Thermo Fisher Scientific, 15140–122) (complete IMDM, cIMDM). The HL-60-A2 cell line, a kind gift from Dr B. Depreter (University Hospital Brussels, Belgium;19), was also cultured in cIMDM. The THP-1 cell line, a human monocytic cell line derived from a patient with acute monocytic leukemia, a kind gift from Dr N. Lambrechts (VITO, Mol, Belgium), was cultured in Roswell Park Memorial Institute medium (RPMI; Thermo Fisher Scientific, 52400–025) supplemented with 10% FCS, 2 mM L-glutamine, 100 IU/ml penicillin, 100 IU/ml streptomycin and 0.05 mM 2-mercaptoethanol (Sigma-Aldrich, M6250). The OP9-DL1 cell line, a mouse stromal cell line genetically modified to express the human Notch ligand Delta-like 1 (DL1), was a kind gift from Dr J. C. Zúñiga-Pflücker (University of Toronto, Canada). This cell line was cultured in Minimum Essential Medium alpha (MEMα; Thermo Fisher Scientific, 12561–056) supplemented with 20% FCS, 2 mM L-glutamine, 100 IU/ml penicillin and 100 IU/ml streptomycin. OP9-DL1 cells were grown near confluence, harvested and split every 2–3 days.
OP9-DL1 co-cultures
OP9-DL1 co-cultures were carried out as described earlier.7 In brief, CD34+ HSPC isolated from UCB were cultured on a subconfluent OP9-DL1 monolayer in co-culture medium consisting of α-MEM (Thermo Fisher Scientific, 12000–063) supplemented with 20% FCS, 2 mM L-glutamine, 100 IU/ml penicillin and 100 IU/ml streptomycin (OP9-DL1 co-culture medium). For lymphoid commitment prior to TCR transduction, 50 ng/ml stem cell factor (SCF; PeproTech, 300–07), 20 ng/ml FMS-like tyrosine kinase 3 ligand (Flt3L; Miltenyi, 130–096-474) and 10 ng/ml interleukin-7 (IL-7; R&D Systems, 207-IL-025) were added. After transduction, cells were maintained in the same OP9-DL1 co-culture medium, with cytokine concentrations of 5 ng/ml SCF, 10 ng/ml Flt3L and 10 ng/ml IL-7. Cells were harvested and co-cultured with fresh OP9-DL1 every 3–4 days.
Isolation and stimulation of peripheral blood lymphocytes
Mononuclear cells were isolated as described above. A total of 1 × 106 were seeded per well in a 24-well culture plate (BD Biosciences) in 1 ml cIMDM supplemented with 10 ng/ml IL-12 (PeproTech, 200–12). ImmunoCultTM Human CD3/CD28/CD2 T cell activator (StemCell Technologies, 10990) was added as described by the manufacturer. After 2 days, cells were harvested and retrovirally transduced.
Retroviral transfection and transduction
Retroviral supernatants were produced after transfection of the Phoenix-A packaging cell line (gift from Dr P Achacoso and Dr G. P. Nolan, Stanford University School of Medicine, Canada) with a retroviral construct containing the Wilms’ Tumor 1 (WT1)-specific T cell receptor (TCR), which recognizes the HLA-A2-restricted RMFPNAPYL (126–134) peptide from WT1, and is described in.7 A total of 250 000 cells were seeded per well on a retronectin (Takara, T100A)-coated 24-well culture plate (BD Biosciences) in the presence of viral supernatant and cytokines, followed by a centrifugation step for 90 minutes at 2300 rpm and 32°C. For OP9-DL1 co-culture cells, cytokines added during retroviral transduction were SCF, Flt3L and IL-7. For transduction of PBL, 2.5 ng/ml IL-2 (Miltenyi, 130–097-748), 10 ng/ml IL-7, 10 ng/ml IL-15 (Miltenyi, 130–095-765), 10 ng/ml IL-21 (PeproTech, 200–21), 10 ng/ml IL-7 + 10 ng/ml IL-15 or 10 ng/ml IL-15 + 10 ng/ml IL-21 was/were added.
Agonist peptide stimulation
Cells were harvested from OP9-DL1 co-culture and seeded in tissue culture plates (BD Biosciences) in cIMDM with 10 µg/ml relevant WT1126−134 agonist peptide (Anaspec by Eurogentec, custom peptide). Cytokines were added in the following concentrations and combinations: 2.5 ng/ml IL-2, 10 ng/ml IL-7, 10 ng/ml IL-15, 10 ng/ml IL-21, 10 ng/ml IL-7 + 10 ng/ml IL-15 or 10 ng/ml IL-15 + 10 ng/ml IL-21. Cells were harvested after 5–6 days and assessed by flow cytometry.
T cell expansion
Cells were seeded on a mixture of 106 40-Gy irradiated peripheral blood mononuclear cells and 105 50-Gy irradiated JY cells per 24-well in cIMDM supplemented with 2 µg/ml phytohemagglutinin (PHA; Remel, R30852801). Fresh medium with cytokines (concentrations identical to those described above) was added every 3–4 days. After 10–14 days, cells were harvested. Prior to expansion, PBL were sorted on eGFP+ using the FACSAria cell sorter (BD Biosciences), to retain only WT1 TCR-transduced T cells.
Flow cytometry and antibodies
Staining of surface markers was performed in phosphate buffered saline (PBS; Thermo Fisher Scientific, 10010023) by adding fluorescent antibodies at concentrations as recommended by the supplier. The following list contains the anti-human monoclonal antibodies that were used. Fluorescein isothiocyanate (FITC)-conjugated: HLA-A2 (BD Biosciences, 551285); phycoerythrin (PE)-conjugated: CD7 (BioLegend, 343106), CD62L (BioLegend, 304806), CD95 (Miltenyi, 130–099-094), CD184 (BD Biosciences, 555974), CD279 (Miltenyi, 130–120-388), CD314 (Biolegend, 320808); allophycocyanin (APC)-conjugated: CD8 (Biolegend, 344722), CD45RA (Biolegend, 304112), CD62L (Biolegend, 304810), CD183 (Biolegend, 353707); pacific blue/eFluor 450/V450-conjugated: CD1a (Thermo Fisher Scientific, 48–0019-42); APC-Cy7/APC-eFluor780/APC-Fire750-conjugated: CD8α (Biolegend, 344746), CD27 (Biolegend, 302816); PE-Cy7: CD3 (Biolegend, 300420), CD5 (Thermo Fisher Scientific, 25–0059-42), CD8β (Thermo Fisher Scientific, 25–5273-42); peridinin chlorophyll protein complex (PerCP)-Cy5.5/BB700-conjugated: streptavidin (Thermo Fisher Scientific, 45–4317-80); amcyan/BV510-conjugated: CD45 (BD Biosciences, 563204); biotin-conjugated: CD45RO (homemade). Exclusion of dead cells was based on propidium iodide (Thermo Fisher Scientific, P1304MP) staining. Flow cytometric analysis was performed on the LSR II (BD Biosciences). Flow cytometry data were analyzed using FACS DIVA software (BD Biosciences) and FlowJo software (TreeStar Inc).
Flow cytometric determination of IFN-γ production
T2 cells were loaded overnight with relevant (WT1126−134) or irrelevant (influenza matrix M1, Eurogentec, AS-28310) peptides at a concentration of 10 µg/ml.
For stimulation, effector (E) and target (T) cells were added to a 96-well tissue culture plate at an E/T ratio of 1/2 in 100 µl of cIMDM. For positive controls, 96-well tissue culture plates were coated with 10 µg/ml anti-CD3 antibody (OKT3, ATCC, CRL-8001) in PBS and anti-CD28 antibody (BD Biosciences, 555725) was added to effector cells at a final concentration of 2 µg/ml. After 1 h of stimulation, brefeldin A (Golgiplug; BD Biosciences, 555029) was added and an additional 16 hours later, cells were harvested and analyzed by flow cytometry.
Staining of surface markers was performed in Dulbecco’s PBS (DPBS; Lonza, 17–512 F), supplemented with 1% FCS by adding fluorescent antibodies at concentrations as recommended by the supplier. Intracellular staining for IFN-γ was performed using Fix & Perm kit (BD Biosciences, 554714), according to the supplier’s instructions. Anti-human monoclonal antibody used for intracellular staining: IFN-γ PE (BD Biosciences, 554552). Viable cells were gated based on negative signal for Fixable Viability Dye (eFluor 506; Thermo Fisher Scientific, 65–0866-18). Flow cytometric analysis was performed on the LSR II and data was analyzed using FACS DIVA software.
51Chromium release assay
A total of 0.5 × 106 target cells were labeled with 50 µCi Na251CrO4 (Perkin Elmer, NEZ030005MC). After 90 minutes incubation at 37°C, cells were washed three times and 1 000 target cells were added per well to varying numbers of effector cells in 96-well V-bottom plates (NUNC, Thermo Fisher Scientific, 249662), to obtain different E/T ratios. After 4 hours of co-incubation, 70 µl of supernatant was harvested and added to 230 µl of liquid scintillation cocktail (OptiPhase HiSafe 3; Perkin Elmer, 1200–437) for liquid scintillation counting in the 1450 LSC & Luminescence Counter (Wallac Microbeta Trilux; Perkin Elmer). Percentage specific lysis was calculated for duplicate wells as follows:
Spontaneous release was determined in culture medium, and Triton X-100 (Sigma-Aldrich) was added to determine maximum release.
Real-time quantitative PCR (RT-qPCR)
Total RNA was extracted from cell lines, using the miRNeasy minikit (Qiagen, 217004). cDNA was synthesized using the Superscript III reverse transcriptase kit (Thermo Fisher Scientific, 18080–044). RT-qPCR with the SYBR Green I technology was performed using the LightCycler 480 SYBR Green I Master kit (Roche, 04707516001) on a LightCycler 480 II (Roche) according to the manufacturer’s protocol. Primers used were the following: WT1 FW 5ʹ-ACACAACGCCCATCCTCTGCG-’3, WT1 REV 5ʹ-GGCACACGTCGCACATCCTGAA-3ʹ; YWHAZ FW 5ʹ-ACTTTTGGTACATTGTGGCTTCAA-3ʹ, YWHAZ REV 5ʹ-CCGCCAGGACAAACCAGTAT-3ʹ; GAPDH FW 5ʹ-TGCACCACCAACTGCTTAGC-3ʹ, GAPDH REV 5ʹ-GGCATGGACTGTGGTCATGAG-3ʹ (Integrated DNA Technologies). Results were analyzed using the ΔΔCt method.
Statistics
Statistical analyses were performed in Prism v9.0.2 (GraphPad Software), using applicable statistical tests as indicated in figure legends. Results were considered statistically significant when the p-value was less than 0.05.
Results
Cytokine mix composition during agonist selection can accelerate in vitro T cell maturation from stem cells
CD34+ HSPC were isolated from HLA-A2+ cord blood (CB) and cultured for 14 days on OP9-DL1 to obtain T cell lineage-committed cells (Figure 1), as evidenced by the combined surface expression of CD5 and CD7 (Fig S1A). At this point, cells were transduced to express a TA-specific TCR, which recognizes the HLA-A2 restricted WT1126−134 peptide. At this point in culture, cells do not express CD4 yet (Fig S1B). Additional OP9-DL1 co-culture induces further T-lineage differentiation of TCR-transduced cells to CD4+ CD8+ double positive (DP) cells (Fig S1B+C and (7)). Expression of eGFP, as a marker for transduced cells, gradually increased during culture (Fig S2A). At the DP stage, eGFP+ cells are mainly CD3+ TCRαβ+ (Fig S3) and CD1a+ CD27− (Figure 2(a), left). Maturation to single positive (SP) T cells was obtained by agonist selection, as described by Snauwaert et al.7 Since HSPC were isolated from HLA-A2+ CB samples, addition of the cognate HLA-A2-restricted peptide to the DP cells resulted in cross-presentation and induced maturation, as indicated by upregulation of CD27 expression and downregulation of CD1a (Figure 2(a), right). After agonist selection, cells remained eGFP+ (Fig S2B) and SP cells were mainly CD8αβ+ (as described in7 and shown in Fig S4).
Figure 1.
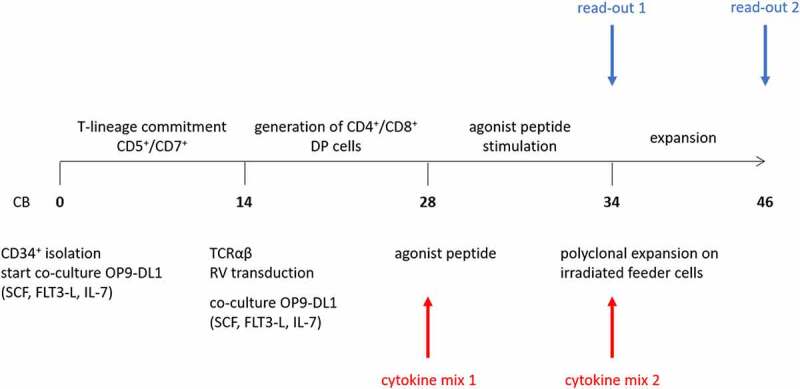
Culture protocol to obtain phenotypically and functionally mature T cells from CD34+ HSPC. Numbers in bold indicate time (in days) of in vitro culture. Red arrows (bottom) indicate points of addition of different cytokine mixes, i.e. cytokine mix 1 during agonist selection and cytokine mix 2 during polyclonal feeder expansion. Blue arrows (top) indicate read-out timepoints, i.e. after agonist selection (read-out 1) and after polyclonal feeder expansion (read-out 2). Abbreviations: CB, cord blood; SCF, stem cell factor; FLT3-L, FMS-like tyrosine kinase 3 ligand; IL-7, interleukin-7; DP, double positive; RV, retroviral
Figure 2.
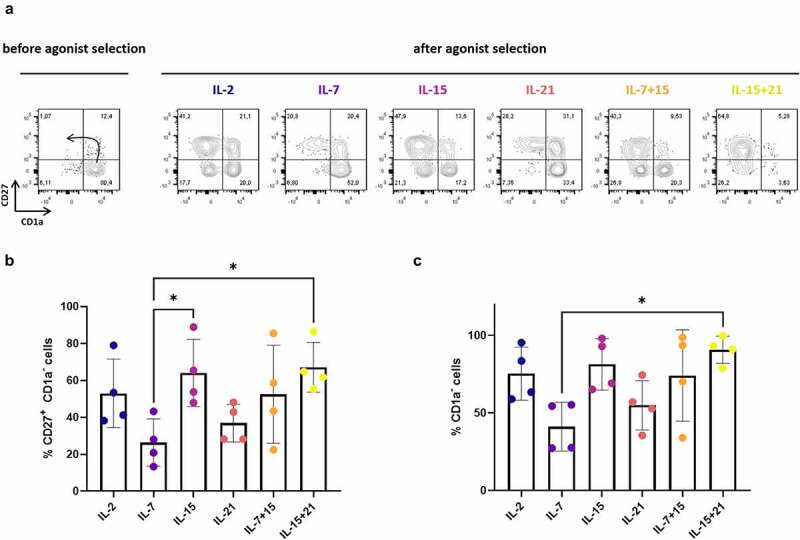
Effect of cytokine mix composition during agonist selection on in vitro T cell maturation. (a) Maturation from CD1a+ CD27− to CD1a− CD27+ cells after agonist selection. Representative plots from one in vitro culture (of a total of 4 donors). The arrow in the first plot marks the expected differentiation: from CD1a+ CD27−, to CD1a+ CD27+ after upregulation of CD27 and finally CD1a− CD27+ after CD1a downregulation. Percentage of CD27+ CD1a− cells (b) and CD1a− cells (c) after agonist selection. Cells were agonist selected with the cognate peptide in the presence of cytokine(s) as shown. Gated on eGFP+ transduced cells. For (b) and (c) individual values and mean ± SD of 4 donors is shown. Results were compared using Kruskal–Wallis test followed by Dunn’s multiple comparisons test. p-value < 0.05 (*)
In our in vitro T cell generation protocol, IL-7 is typically added during agonist selection.7 Since it has been shown that cytokines have differential effects on T cell maturation, expansion and functionality,15–18 we first wanted to investigate the effect of the common gamma-chain cytokines IL-2, IL-7, IL-15 and IL-21 during the agonist selection process. For the remainder of the manuscript, we will annotate cytokines added during agonist selection as “cytokine mix 1”, whereas cytokines added during later feeder expansion will be described as “cytokine mix 2”. Furthermore, two different timepoints of read-out are used, after agonist selection (“read-out 1”) and after feeder expansion (“read-out 2”) (Figure 1). During T cell maturation, we observed accelerated differentiation kinetics when adding IL-2 or IL-15 (alone or in combination with IL-7 or IL-21) instead of IL-7 (i.e. in cytokine mix 1), as shown by a higher percentage of CD1a− cells and CD27+ CD1a− cells after 6 days of agonist peptide stimulation (i.e. at read-out 1) (Figure 2(a–c)). Due to high donor variability, statistical significance was only reached when comparing the percentage of CD27+ CD1a− cells between IL-7 and IL-15 (alone or in combination with IL-21) conditions and the total percentage of CD1a− cells between IL-7 and IL-15 + IL-21 conditions (Figure 2b+C). Conversely, substituting IL-7 for IL-21 did not accelerate T cell maturation (Figure 2(a–c)).
Before agonist selection, already a small percentage (5.63–15.6%) of CD1a- CD27+ cells are present (Figure 2(a)), probably due to endogenous presence of WT1 in co-culture cells and cross-presentation of the WT1 peptide by HLA-A2,7 leading to minor spontaneous induction of maturation. Previously, we have shown that maturation after agonist peptide stimulation arises from CD27− CD1a+ cells and is not attributed to expansion of already present CD27+ CD1a+ cells.9
Cytokine mix composition during agonist selection and feeder expansion has an impact on proliferation
Besides maturation kinetics, we also investigated the effect of the common gamma-chain cytokines IL-2, IL-7, IL-15 and IL-21 on proliferation of maturing T cells during in vitro T cell differentiation. After agonist selection (i.e. at read-out 1), a 2- to 5-fold higher mean expansion was observed when IL-7, alone or in combination with IL-15, was added together with the agonist peptide, as compared to IL-2, IL-21 or IL-15 alone (i.e. in cytokine mix 1) (Figure 3(a)). Also, combining IL-15 and IL-21 showed a trend toward a higher fold expansion during agonist selection as compared to IL-15 or IL-21 alone (Figure 3(a)).
Figure 3.
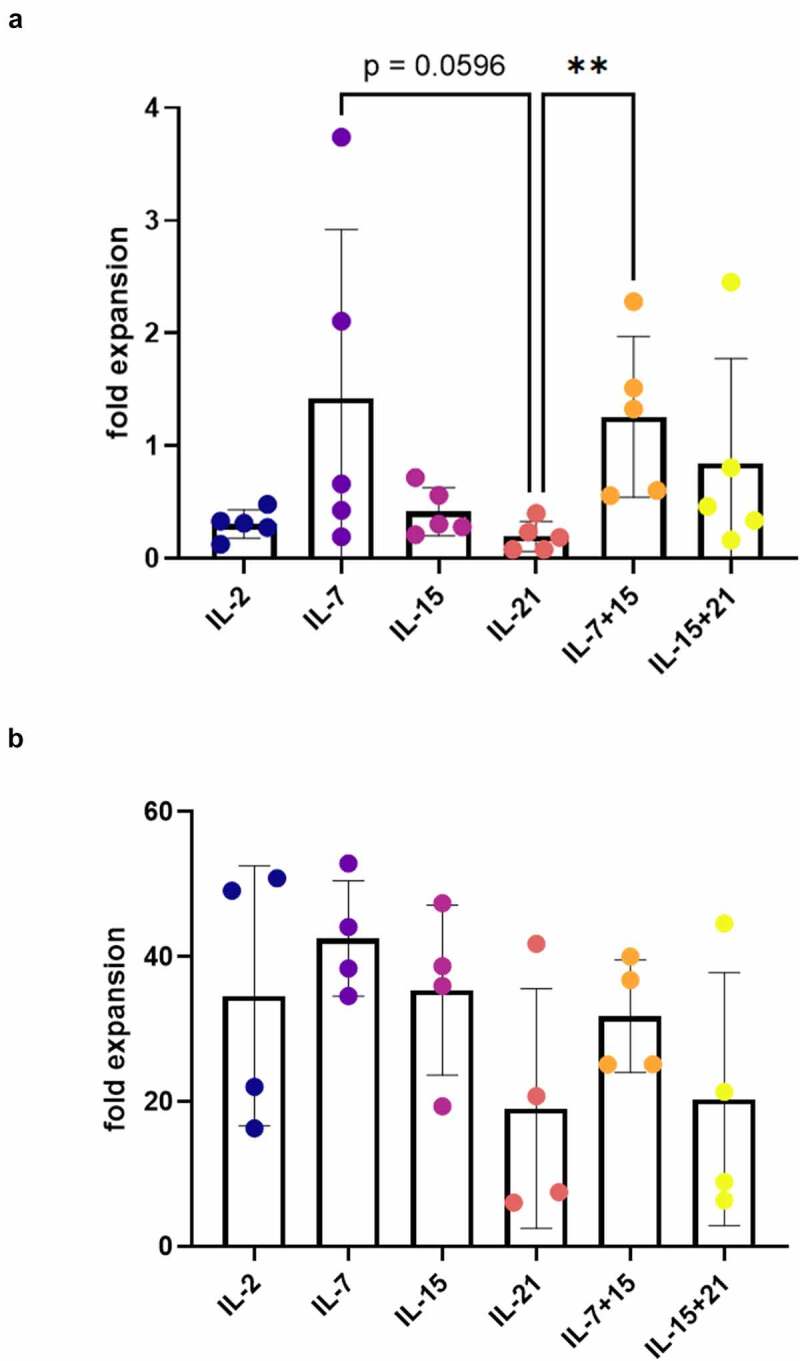
Effect of cytokine mix composition on proliferation during agonist selection and feeder expansion. (a) Fold expansion during agonist selection. Cells were agonist selected with the cognate peptide in the presence of cytokine(s) as shown. Individual values and mean ± SD of 5 donors. (b) Fold expansion during polyclonal feeder expansion. Cytokine(s) as shown was/were added during both agonist selection and feeder culture (i.e. in both cytokine mix 1 and 2). Individual values and mean ± SD of 4 donors. Results were compared using Kruskal–Wallis test followed by Dunn’s multiple comparisons test. p-value < 0.01 (**), p indicates p-value
Taking together the maturation and expansion data, we obtained the highest absolute number of CD1a− CD27+ and CD1a− mature cells after agonist selection with IL-7 or IL-7 in combination with IL-15 (i.e. at read-out 1) (Fig S5A+B). Also, combining IL-15 and IL-21 resulted in a mean absolute number of CD1a− CD27+ and CD1a− cells comparable to IL-7 and IL-7 + IL-15, but variation between cultures was high (Figure 3(a) and S5A+B). Notably, addition of IL-21 alone during agonist selection resulted in a significantly lower expansion rate and lower absolute number of mature cells, as compared to addition of IL-7 + IL-15 (Figure 3(a) and S5A+B).
After agonist selection, cells were polyclonally expanded on irradiated feeder cells to obtain functionally mature TA-specific T cells (i.e. at read-out 2). During feeder expansion, every 3–4 days fresh culture medium was added with either IL-2, IL-7, IL-15, IL-21, or a combination of IL-7 and IL-15, or IL-15 and IL-21 (cytokine mix 2). Addition of IL-21, either alone or in combination with IL-15, showed a trend toward a lower mean expansion rate (Figure 3(b)). Also, when comparing IL-15 alone to IL-15 in combination with IL-21, we observed a potential inhibitory effect of IL-21 on expansion induced by IL-15 (Figure 3(b)).
In vitro T cell maturation and polyclonal expansion in the presence of IL-21 leads to mature T cells with a less-differentiated phenotype
Naive T cell subsets (TN, TSCM and TCM) have been shown to be superior to more differentiated subsets (TEM and TEFF) for use in adoptive T cell immunotherapy.11,12,14 The effect of common gamma-chain cytokines IL-2, IL-15 and IL-21 on the phenotype of adoptively transferred T cells has been studied elaborately.16–18 Other studies have indicated that addition of IL-7 and IL-15 during ex vivo expansion results in T cells with a stem cell memory (TSCM) phenotype, which is considered as the ideal subset for adoptive T cell transfer.14,15 Since it is known that extensive in vitro culture and expansion might lead to more differentiated and exhausted T cells,10–13,20 and we observed differences in expansion rate depending on the cytokine mix, we investigated the phenotype of the resulting in vitro differentiated T cells. Therefore, we evaluated the expression of various differentiation and exhaustion markers on our cells, including CD62L, CXCR3, CXCR4, NKG2D, CD95, CD45RA and CD45RO, using flow cytometry.
After agonist selection (i.e. at read-out 1), we analyzed the phenotype of CD1a− mature T cells. Cells that were agonist selected in the presence of IL-7 (i.e. in cytokine mix 1), as was typically done in our co-culture protocol,7 displayed a high percentage of less-differentiated CD45RA+ CD62L+ cells (Figure 4(a)). A similar high percentage of CD45RA+ CD62L+ cells was obtained when adding IL-21 during agonist selection, but not if IL-2 or IL-15 (alone or in combination with IL-7 or IL-21) were added (Figure 4(a)). Comparing IL-7 and IL-21 conditions, no significant difference could be observed in the proportion of CD62L+ cells (Figure 4(b)), but IL-21-exposed cells had a more immature phenotype, based on the higher MFI for CD62L within the CD62L+ population (Figure 4(c)). Although this difference was not significant when comparing all cytokine conditions reciprocally (Figure 4(c)), in a head-to-head comparison between IL-7 and IL-21, the CD62L MFI was significantly higher for IL-21 (p < 0.05, data not shown). In addition, the percentage of CD62L+ cells and CD62L MFI was significantly higher in the IL-21 condition as compared to IL-15 conditions (Figure 4b+C). Additionally, we stained for the chemokine receptors CXCR3, associated with effector T cell trafficking and function21 and downregulated upon exhaustion,22,23 and CXCR4, important in homeostatic migration and predominantly expressed on TN and TCM,15,24 and both previously described to be expressed on TSCM.14,15 No significant differences were observed in CXCR3 expression between cytokine conditions (minimum mean 14.9% CXCR3+ cells for IL-15, maximum mean 37.6% CXCR3+ cells for IL-15 + IL-21), whereas CXCR4 expression was significantly lower for cells which were agonist selected in the presence of IL-21 (Fig S7A+B). We also investigated the expression of NKG2D and CD95, both markers typically seen on activated T cells.25 Additionally, CD95 is a known hallmark of memory T cell subsets.23,26 After agonist selection in the presence of IL-21, expression of both NKG2D and CD95 was lower (Figure 4(d–f)), although expression of CD95 was highly donor-dependent in our in vitro cultures (figure 4(f) and Fig S8). To further elucidate these results, we combined CD62L, CD45RA, CD95 and CXCR3 staining to identify T cell subpopulations,23 for a representative in vitro culture. We observed a higher percentage of naive T cells (TN, CD45RA+ CD62L+ CXCR3− CD95−) in IL-7 and IL-21 conditions (Fig S7C). Also, TSCM (CD45RA+ CD62L+ CXCR3+ CD95+) were only present in the IL-21 condition, as opposed to the other conditions (Fig S7C). Conditions with IL-2 and IL-15 (alone or in combination with IL-7 or IL-21) mostly gave rise to T cells with a more differentiated effector phenotype (TEFF, CD45RA+ CD62L− CXCR3− CD95+) (Fig S7C).
Figure 4.
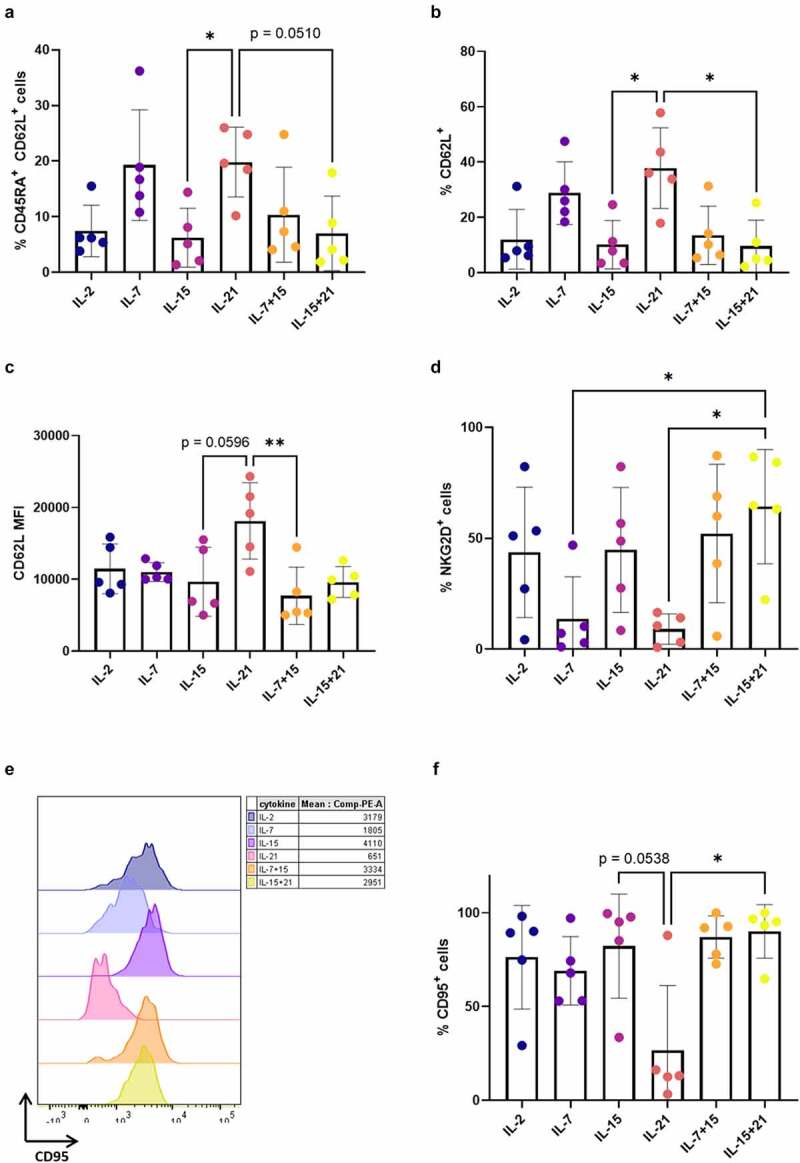
Addition of IL-21 during maturation induces mature T cells with a less-differentiated phenotype. Phenotype of cells after agonist selection, gated on eGFP+ CD1a− transduced mature cells, or eGFP+ CD1a− CD62L+ cells for (c). Gating strategy is shown in Fig S6. Cells were agonist selected with the cognate peptide in the presence of cytokine(s) as shown. Percentage of CD45RA+ CD62L+ (a), CD62L+ (b), NKG2D+ (d), CD95+ (f) cells. (c) Mean fluorescence intensity (MFI) for CD62L. For (a–d and f) individual values and mean ± SD of 5 donors is shown. Results were compared using Kruskal–Wallis test followed by Dunn’s multiple comparisons test. p-value < 0.05 (*), p-value < 0.01 (**), p indicates p-value. (e) Expression (left) and MFI (right) for CD95 for one representative in vitro culture (of a total of 5 donors). CD95 expression and MFI for the other 4 donors are shown in Supplementary Figure S8
Following polyclonal feeder expansion (i.e. at read-out 2), it became increasingly clear that agonist selected cells, which were further polyclonally expanded in the presence of IL-21 (IL-21 only, in both cytokine mix 1 and 2), showed a more immature phenotype. A significantly higher percentage of these IL-21-expanded cells expressed CD62L and the MFI for CD62L within these CD62L+ cells was also significantly higher as compared to IL-15 or IL-7 + IL-15 (Figure 5a+B and Fig S9). Examining CD45RA and CD45RO expression, addition of IL-21 led to a lower percentage of CD45RO+ CD45RA− cells, and a higher percentage of CD45RA+ CD45RO− cells (Figure 5(c)). Analysis of T cell subsets for a representative in vitro culture indicated that cells with a TSCM-like phenotype were only present in the IL-21 condition (Figure 5(d)).
Figure 5.
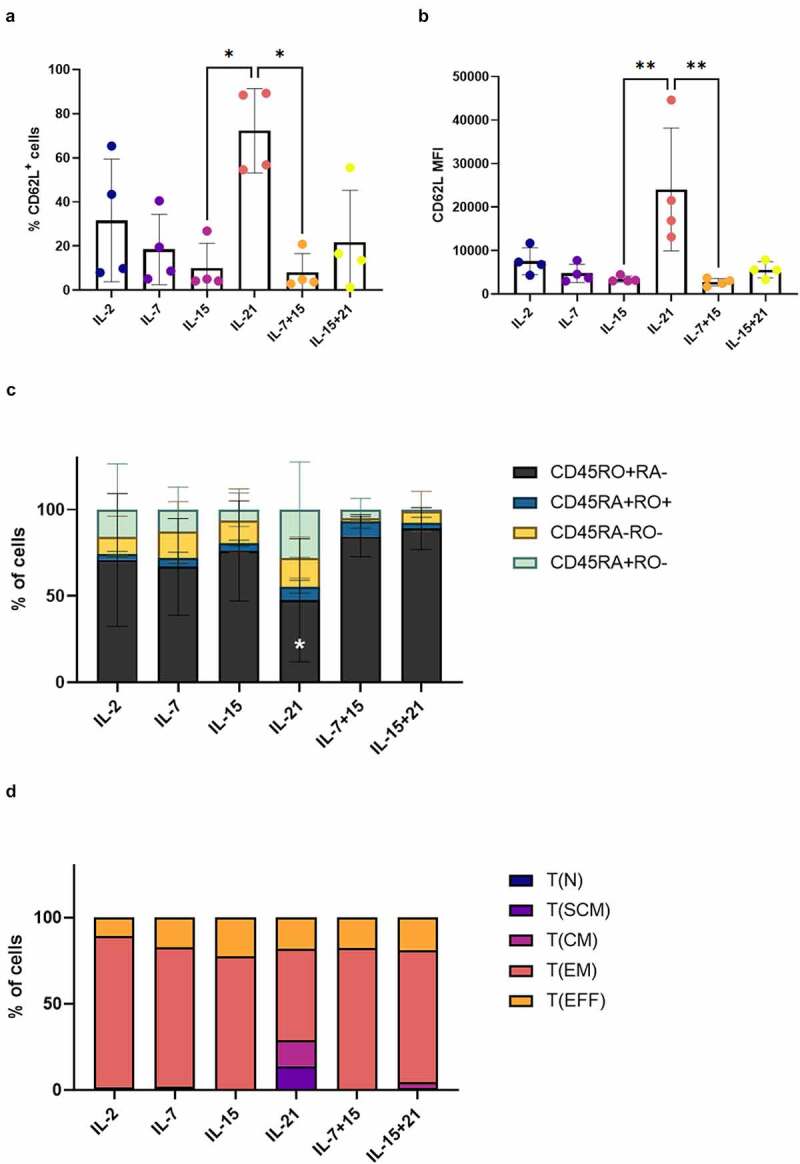
Addition of IL-21 during feeder expansion results in T cells with a less-differentiated phenotype. Phenotype of in vitro differentiated T cells after polyclonal feeder expansion, gated on eGFP+ transduced cells, or eGFP+ CD62L+ cells for (b). Cytokine(s) as shown was/were added during both agonist selection and feeder culture. Percentage of CD62L+ cells (a) and CD62L MFI (b). Individual values and mean ± SD of 4 donors. Results were compared using Kruskal–Wallis test followed by Dunn’s multiple comparisons test. p-value < 0.05 (*), p-value < 0.01 (**). (c) Percentage of cells in CD45RA/CD45RO quadrants of dotplots. Mean ± SD of 4 donors. Results were compared using two-way ANOVA followed by Dunnett’s multiple comparisons test. CD45RO+RA− p-value < 0.05 (*) for IL-21 vs IL-7 + 15 and p-value 0.01 (**) for IL-21 vs IL-15 + 21. (d) Percentages of T cell subsets. T(N), naive T cells (CD45RA+ CD62L+ CXCR3− CD95−); T(SCM), stem cell memory T cells (CD45RA+ CD62L+ CXCR3+ CD95+); T(CM), central memory T cells (CD45RA− CD62L+ CXCR3+ CD95+); T(EM), effector memory T cells (CD45RA− CD62L− CXCR3− CD95+); T(EFF), effector T cells (CD45RA+ CD62L− CXCR3− CD95+). Results from a representative co-culture
We also evaluated the expression of PD-1, a known marker for T cell exhaustion.27 The percentage of cells positive for PD-1 expression after polyclonal feeder expansion was low in all conditions (effect of cytokine mix 1 and 2 at read-out 2) (Fig S10A). IL-21 (alone or in combination with IL-15) seemed to induce a small upregulation of PD-1 expression, reflected in the percentages of PD-1+ cells (Fig S10A). Within PD-1+ cells, the MFI for PD-1 was highest in the condition where IL-15 + IL-21 was added (Fig S10B). Overall, PD-1 expression remained negligible, with only a minority of cells expressing PD-1 in conditions where IL-21 was added (Fig S10A).
Next, we investigated if IL-21 had the same effect on the phenotype of in vitro cultured peripheral blood lymphocytes (PBL). For this, we transduced PBL with the WT1 TCR and expanded them on irradiated feeder cells after sorting of eGFP+ TCR-transduced cells. The same cytokine mixes as described for in vitro differentiated T cells were added during both transduction and expansion. After expansion, we observed high percentages of CD62L+ cells in the IL-7 and IL-21 conditions (Figure 6a+B), with a trend toward higher CD62L MFI for PBL cultured in the presence of IL-21 as compared to IL-7 (Figure 6a+C), confirming the results obtained with our in vitro differentiated T cells. Due to high donor variability (with one donor being entirely CD45RA+, data not shown), differences in percentages of CD45RA+ CD62L+ cells (Fig S11A) and CD45RA/RO expression were not significant (Fig S11B).
Figure 6.
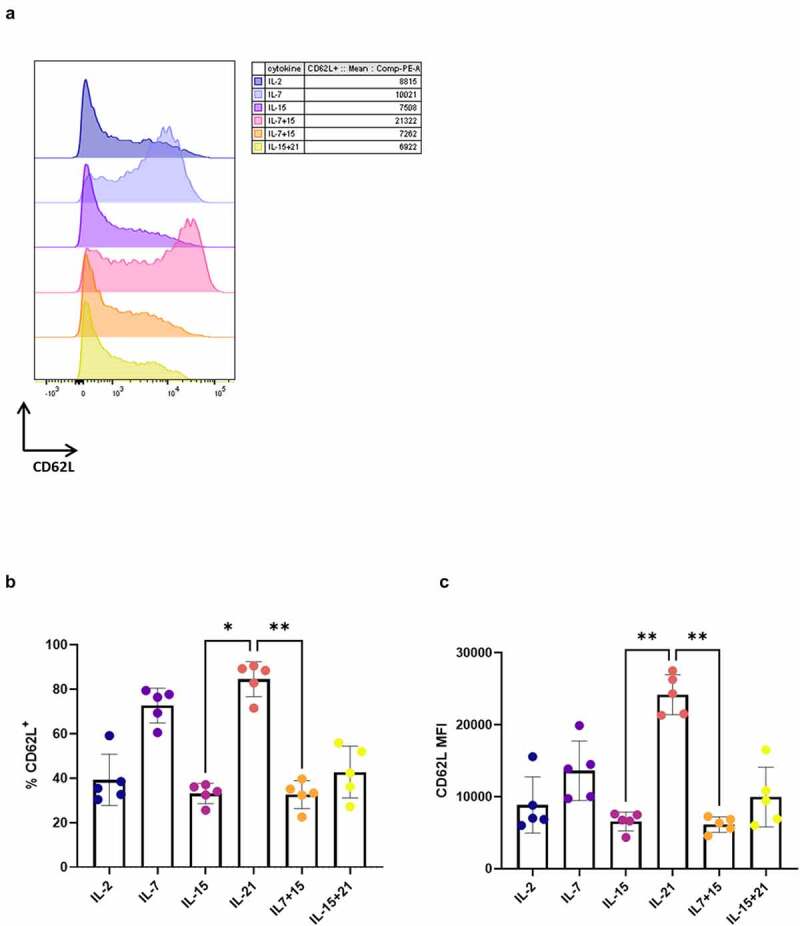
Addition of IL-21 during expansion of TCR-transduced PBL results in T cells with high CD62L expression. CD62L expression after polyclonal feeder expansion of WT1 TCR-transduced PBL, gated on eGFP+ transduced cells for (a) and (b), or eGFP+ CD62L+ cells for (c). (a) Expression (left) and MFI (right) for CD62L for a representative PBL donor (of a total of 5 donors). Percentage of CD62L+ cells (b) and MFI for CD62L within these cells (c). Individual values and mean ± SD of 5 donors is shown. Results were compared using Kruskal–Wallis test followed by Dunn’s multiple comparisons test. p-value < 0.05 (*), p-value < 0.01 (**)
Addition of IL-21 only during feeder expansion is insufficient to obtain mature T cells with a less-differentiated phenotype
It was previously described that extensive in vitro expansion results in an exhausted T cell phenotype.11,28 We observed the opposite, as cells matured and expanded in the presence of IL-21 (i.e. IL-21 only as both cytokine mix 1 and 2) showed less proliferation and accordingly a less-differentiated phenotype at both read-out 1 and 2. However, these low expansion rates might lead to a (too) low number of T cells at the end of the in vitro culture. Therefore, we investigated if the beneficial effect of IL-21 on the phenotype of in vitro generated TA-specific T cells (i.e. at read-out 2) would still be apparent when only added during feeder expansion (i.e. IL-21 addition only in cytokine mix 2, not in cytokine mix 1). Cells were stimulated with the agonist peptide in the presence of IL-7 (i.e. IL-7 as cytokine mix 1), as described in our co-culture protocol,7 and afterward polyclonally expanded in the presence of IL-21 (i.e. IL-21 as cytokine mix 2), instead of the previously used IL-2. Adding IL-21 only during feeder expansion did not result in high CD62L expression (both in percentages and MFI), as seen when IL-21 was added during both agonist selection and feeder expansion (cytokine mix 1 and 2) (Figure 7a+B). Furthermore, CD45RA and CD45RO percentages were similar between conditions where IL-7 was added during agonist selection (i.e. in cytokine mix 1), regardless of the cytokine added during feeder expansion (i.e. IL-7 or IL-21 in cytokine mix 2), whereas addition of IL-21 during both agonist selection and feeder expansion (i.e. IL-21 in cytokine mix 1 and 2) resulted in a higher percentage of CD45RA+CD45RO− cells and a lower percentage of CD45RO+CD45RA− cells (Figure 7(c)). These results indicate that IL-21 probably exerts an important part of its effects on the phenotype of the resulting T cells during agonist selection.
Figure 7.
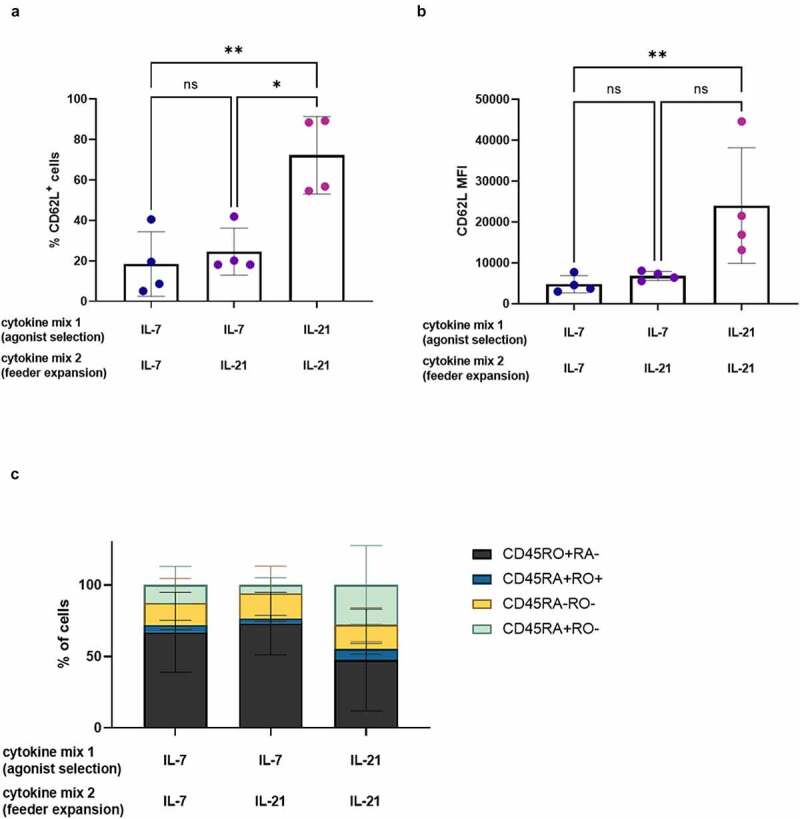
IL-21 exerts its effect on phenotype during agonist selection. Phenotype of cells after polyclonal feeder expansion, gated on eGFP+ transduced cells, or eGFP+ CD62L+ cells for (b). Cytokine(s) as shown was/were added during both agonist selection (cytokine mix 1) and feeder culture (cytokine mix 2). Percentage of CD62L+ cells (a) and CD62L MFI (b). Individual values and mean ± SD of 4 donors. Results were compared using Kruskal–Wallis test with uncorrected Dunn’s test. p-value < 0.05 (*), p-value < 0.01 (**), ns not significant. (c) Percentage of cells in CD45RA/CD45RO quadrants of dotplots. Mean ± SD of 4 donors. Results were compared using two-way ANOVA with uncorrected Fisher’s LSD. Differences were not significant
Polyclonal expansion in the presence of IL-21 leads to superior interferon-gamma production
Several clinical studies have reported that less-differentiated T cell subsets (TN and TCM) possess superior antitumor potency as compared to the more differentiated TEM and TEFF.10,12 Furthermore, in vivo experiments comparing TSCM to other memory T cell subsets indicated superior antitumor activity for TSCM.14,15 As we observed that cytokine(s) added during maturation and expansion (cytokine mix 1 and 2, respectively) can alter the phenotype of the obtained T cells, we also wanted to investigate the effect of these cytokines on the functionality of the in vitro generated TA-specific T cells. Polyclonal feeder expansion is a necessary step in our in vitro culture protocol to obtain functionally mature T cells capable of exerting effector functions.7 Therefore, functionality of T cells was assessed only after polyclonal feeder expansion (i.e. at read-out 2).
During polyclonal expansion, the percentage of eGFP+ TCR-transduced cells further increased, to reach nearly 100% at read-out 2 (Fig S2C+D). At this point, cells were CD8+ in all cytokine conditions (Fig S12A+B) and both CD8αβ+ and CD8αα+ cells were present in an approximately 50/50 ratio (as described in7 and shown in Fig S12A+C).
Interferon-gamma (IFN-γ) production after recognition of WT1-expressing target cells was measured through intracellular staining. For this, in vitro generated WT1-specific T cells were cultured with THP-1 and HL-60-A2, both HLA-A2+ WT1+ cell lines, HLA-A2+ T2 cells pulsed with relevant (WT1) or irrelevant (influenza) peptide, or JY cells (HLA-A2+ WT1−). WT1 and HLA-A2 expression of cell lines is shown in Fig S13. As a positive control, T cells were stimulated with aCD3/aCD28, whereas culture medium was added as a negative control. T cells which were agonistically selected and expanded in the presence of IL-21 (i.e. IL-21 in both cytokine mix 1 and 2), gave rise to the largest amount of IFN-γ–producing cells (on average 6.5%, range 0.9–15%) after recognition of HLA-A2+ WT1+ target cells, which was significantly higher when compared to cells cultured in the presence of IL-2, IL-7, IL-15, or IL-7 + IL-15 (Figure 8(a)). While a very small percentage of cells in the IL-21 and IL-15 + IL-21 condition was CD4+ after polyclonal expansion (Fig S12), CD8+ cells were the main IFN-γ-producing subset (Fig S14). Antigen-independent stimulation with anti-CD3 and anti-CD28 resulted in the highest percentages of IFN-γ-producing cells in conditions where IL-21 was added, with an average percentage of 20.6% IFN-γ+ cells for IL-21 (range 6.2–37.8%) and 26.9% for IL-15 and IL-21 combined (range 20.3–35.9%), which was significantly higher than the other cytokine culture conditions (Figure 8(a)). Next to the proportion of IFN-γ-producing cells, we also investigated if culture in the presence of IL-21 resulted in a higher production of IFN-γ on a per cell basis. The geometric mean fluorescence intensity (gMFI) for IFN-γ+ cells showed a trend toward higher numbers in the IL-21 and IL-15 + IL-21 conditions, although differences were small due to necessary exclusion of certain conditions and/or donors (Fig S15A-C).
Figure 8.
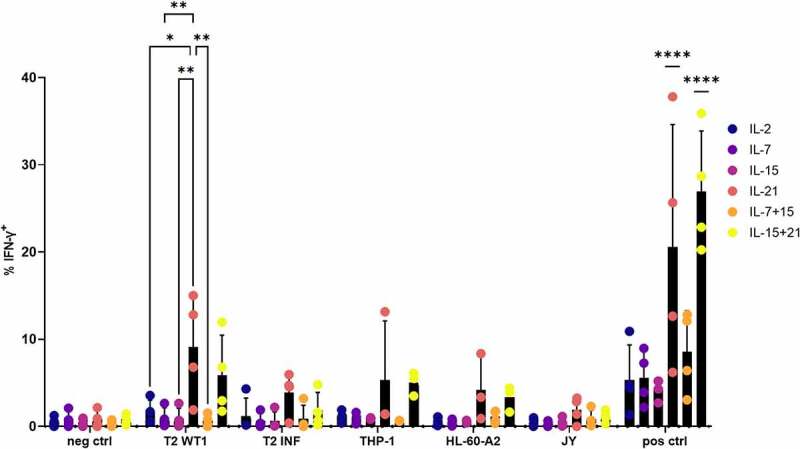
Polyclonal expansion of in vitro differentiated T cells in the presence of IL-21 leads to superior IFN-γ production. Intracellular staining of in vitro generated T cells for IFN-γ, after co-culture of in vitro differentiated T cells with T2 cells pulsed with relevant (WT1) or irrelevant (INF) peptide (10 µg/ml), THP-1 or HL-60-A2 (both HLA-A2+ WT1+), or JY (HLA-A2+ WT1−) cells. Culture medium was used as a negative control (neg ctrl), stimulation with aCD3/aCD28 as a positive control (pos ctrl). Effector/target ratio 1/2. Gating on eGFP+ TCR-transduced cells. Cytokine(s) as shown was/were added during both agonist selection and feeder culture. Individual values and mean ± SD of 4 donors, unless otherwise indicated. Results were compared using two-way ANOVA followed by Tukey’s multiple comparisons test. p-value < 0.05 (*), p-value < 0.01 (**), p-value < 0.0001 (****). IL-21 and IL-15 + IL-21 were significantly different from the other cytokine conditions, not from each other
As shown above, the addition of IL-21 early in in vitro cultures (during agonist selection, i.e. in cytokine mix 1) rather than during feeder expansion, significantly impacted the phenotype of the obtained T cells. Also, functionally mature T cells obtained after maturation and expansion in the presence of IL-21 (alone or in combination with IL-15) (in both cytokine mix 1 and 2) consisted of the highest percentage of IFN-γ-producing cells, indicating addition of IL-21 also impacted functionality. Therefore, we investigated whether IL-21 was already necessary during agonist selection to obtain these high IFN-γ+ cell percentages. We found that addition of IL-21 during feeder expansion only (i.e. IL-21 addition only in cytokine mix 2, not in cytokine mix 1) was sufficient, as percentages of IFN-γ+ cells after antigen-independent stimulation with aCD3/aCD28 were not significantly different between cells agonist selected in the presence of IL-7 and polyclonally expanded with IL-21 (i.e. IL-7 in cytokine mix 1, IL-21 in cytokine mix 2) and cells exposed to IL-21 during both culture steps (Fig S16 condition 2 vs condition 3, respectively). Also, changing the cytokine added during feeder expansion (i.e. in cytokine mix 2) from IL-2, which was typically done in our co-culture protocol,7 to IL-21 resulted in a higher percentage of IFN-γ-producing cells, even when the cytokine added during agonist selection remained the same (i.e. IL-7 in cytokine mix 1) (Fig S16, condition 1 vs condition 2). Therefore, we could illustrate that the effect of IL-21 on functionality is also apparent when IL-21 is added during feeder expansion only.
As PBL expanded in the presence of IL-21 showed a less-differentiated phenotype similar to our in vitro differentiated T cells, we investigated if IL-21 had a similar effect on IFN-γ production as seen with in vitro differentiated T cells. In contrast with what we observed for our in vitro differentiated T cells, expansion in the presence of IL-21 resulted in a high percentage of CD8+ T cells, which was significantly higher than the percentage of CD4+ T cells obtained with this cytokine (Figure 9(a)). Comparing IL-21 to the other cytokine conditions, the percentage of CD8+ T cells was also significantly higher as to when PBL were expanded in the presence of IL-2 or IL-7 (Figure 9(a)). In addition, the percentage of CD4+ T cells was significantly lower when PBL were expanded in the presence of IL-21 as compared to IL-7 (Figure 9(a)). As PBL were sorted on eGFP+ before expansion, the average percentage of eGFP+ WT1 TCR-transduced T cells of total PBL used for functionality tests was 96.2% (range 93.2–98.4%) (data not shown). In agreement with the functionality data obtained with our in vitro differentiated T cells, WT1 TCR-transduced PBL expanded in the presence of IL-21 contained the highest proportion of IFN-γ+ cells after recognition of T2 cells pulsed with WT1 peptide or antigen-independent stimulation with aCD3/aCD28 (Figure 9(b)). On the other hand, and in contrast with in vitro differentiated T cells, expansion of PBL in the presence of IL-15 + IL-21 did not lead to a high percentage of IFN-γ+ cells, and the percentage in this condition was even significantly lower as compared to IL-21 alone, after stimulation with aCD3/aCD28 (Figure 9(b)). As a higher proportion of CD8+ T cells were generated when PBL were expanded in the presence of IL-21, we also gated on CD4+ and CD8+ cells separately. We found a higher percentage of IFN-γ+ cells within both the CD4+ and CD8+ cells for PBL expanded in the presence of IL-21 after stimulation with aCD3/aCD28 (Fig S17A+B). Furthermore, IFN-γ production after recognition of T2 cells pulsed with WT1 peptide was mediated by CD8+ T cells (Fig S17A+B), as observed with our in vitro differentiated T cells (Fig S14). The gMFI for IFN-γ+ cells was significantly higher for WT1 TCR-transduced PBL expanded in the presence of IL-21 (alone or in combination with IL-15) (Figure 9(c)), which corroborates the data for in vitro differentiated T cells, and indicates that in vitro culture in the presence of IL-21 generates T cells with a higher inherent capacity for IFN-γ production.
Figure 9.
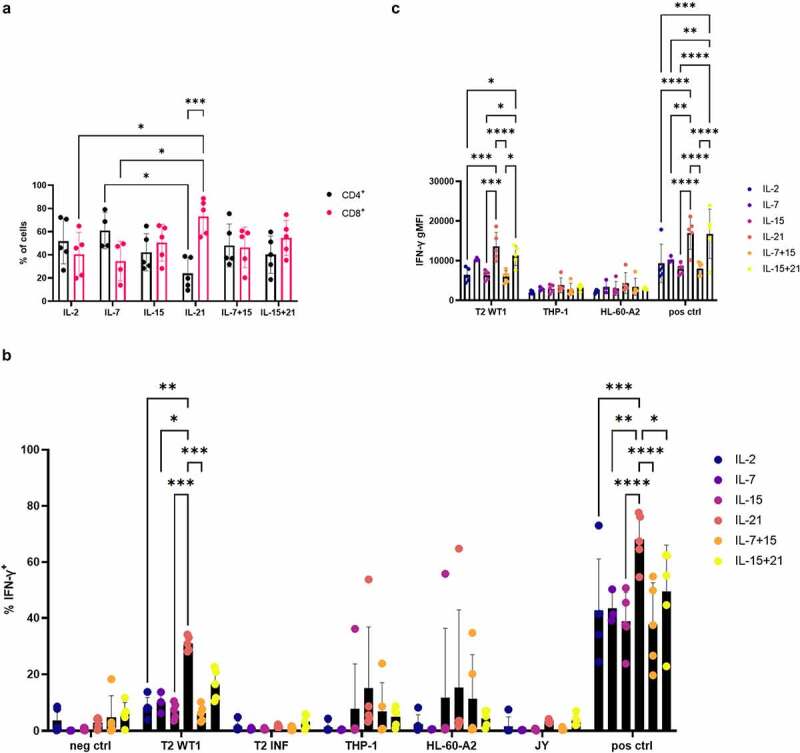
Expansion of TCR-transduced PBL in the presence of IL-21 leads to superior IFN-γ production. (a) Percentage of CD4+ and CD8+ T cells after expansion of TCR-transduced PBL in the presence of cytokines as indicated. Results were compared using two-way ANOVA followed by Tukey’s multiple comparisons test for comparison between cytokine conditions or followed by Šídák’s multiple comparisons test for comparison of percentages of CD4+ and CD8+ T cells within one cytokine condition. (b) Intracellular staining of in vitro expanded TCR-transduced PBL for IFN-γ, after co-culture of T cells with T2 cells pulsed with relevant (WT1) or irrelevant (INF) peptide (10 µg/ml), THP-1 or HL-60-A2 (both HLA-A2+ WT1+), or JY (HLA-A2+ WT1−) cells. Culture medium was used as a negative control (neg ctrl), stimulation with aCD3/aCD28 as a positive control (pos ctrl). Effector/target ratio 1/2. Results were compared using two-way ANOVA followed by Dunnett’s multiple comparisons test. (c) Geometric mean fluorescence intensity (gMFI) for IFN-γ+ cells from the experiment as described for (b). Results were compared using two-way ANOVA followed by Tukey’s multiple comparisons test. Gating on eGFP+ TCR-transduced cells for (a) and (b), and on eGFP+ IFN-γ+ cells for (c). Cytokine(s) as shown in figures was/were added during both transduction and expansion. Individual values and mean ± SD of 5 donors are shown for (a–c). p-value < 0.05 (*), p-value < 0.01 (**), p-value < 0.001 (***), p-value < 0.0001 (****)
Combining IL-15 and IL-21 leads to enhanced killing of tumor antigen-expressing target cell lines by in vitro differentiated T cells
Next to interferon-gamma production, we also assessed the effect of the different cytokines on specific lysis of target cell lines using a 51chromium release assay. Addition of IL-15 + IL-21 during both agonist selection and feeder expansion (i.e. in cytokine mix 1 and 2) leads to superior lysis of T2 cells pulsed with WT1 peptide (on average 53.9% specific lysis, range 27.3–76.6%) (Figure 10(a)). As we observed some background lysis of T2 cells pulsed with irrelevant influenza peptide (average 6.3% over all conditions, range 0–17.8%) (Figure 10(a)), we calculated the actual specific lysis of T2 WT1 (i.e. recognition of WT1 peptide presented in the context of HLA-A2) by subtracting the background signal from the original calculated specific lysis signal. This resulted in the same overall trend, with addition of IL-15 + IL-21 still leading to the highest percentage of target cell lysis, although differences were not significant anymore (Fig S18). Specific lysis in the IL-21 condition was reduced to a greater extent as compared to the other conditions, due to higher background lysis of T2 INF (Figure 10(a) and Fig S18). For lysis of the HL-60-A2 cell line (WT1+ HLA-A2+), highest killing percentages were obtained when IL-15 in combination with IL-7 (on average 29% specific lysis, range 10.9–49.3%) or in combination with IL-21 (on average 27.4% specific lysis, range 10.6–53.3%) was added during both agonist selection and feeder expansion (i.e. in cytokine mix 1 and 2) (Figure 10(a)).
Figure 10.
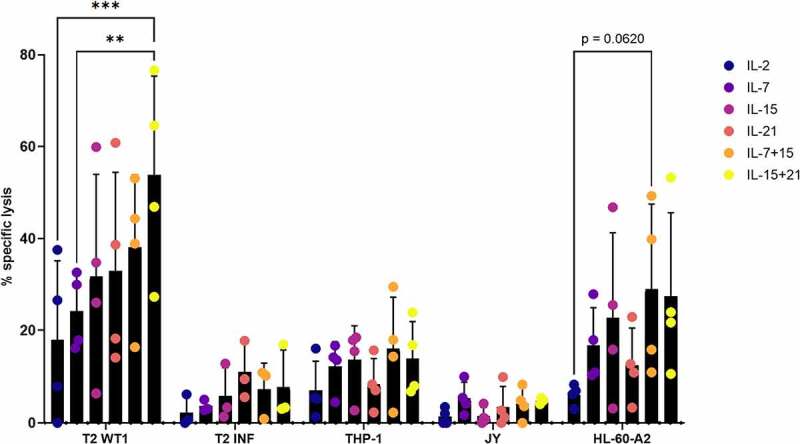
Combining IL-15 and IL-21 leads to enhanced killing of tumor antigen-expressing target cell lines by in vitro differentiated T cells. Percentage specific lysis determined via 4-hour 51chromium release assay after co-culture of in vitro generated T cells with T2 cells pulsed with relevant (WT1) or irrelevant (INF) peptide (10 µg/ml), THP-1 or HL-60-A2 (both HLA-A2+ WT1+), or JY (HLA-A2+ WT1−) cells. Effector/target ratio 10/1. Cytokine(s) as shown was/were added during both agonist selection and feeder culture. Individual values and mean ± SD of 4 donors or 3 donors for T2 INF. Results were compared using two-way ANOVA followed by Tukey’s multiple comparisons test. p-value < 0.01 (**), p-value < 0.001 (***), p indicates p-value
When performing a 51chromium release assay with WT1 TCR-transduced PBL, which were expanded in the presence of the same cytokine mixes, the highest lysis percentages were obtained in the IL-21 and IL-15 + IL-21 conditions for T2 pulsed with WT1 peptide (on average 62.5% specific lysis, range 49.7–72.0% and on average 64.6% specific lysis, range 40.7–81.2%, respectively) (Fig S19A). Strikingly, nearly no lysis of WT1+ HLA-A2+ target cell lines was observed (Fig S19A). Again, we observed background lysis of T2 cells pulsed with irrelevant INF peptide (Fig S19A). Subtracting this background signal from the original signal resulted in loss of significant differences between cytokine conditions for lysis of T2 cells pulsed with relevant peptide (Fig S19B).
Discussion
Adoptive transfer of TA-specific T cells is a promising treatment strategy for hematological malignancies. In 2019, Chapuis and colleagues demonstrated that treating high-risk AML patients with WT1-specific T cells following allogeneic hematopoietic stem cell transplantation (alloHSCT) prevented relapse in 100% of patients, as compared to a control group with only 54% relapse-free survival,4 underlining the efficacy of adoptive transfer of TCR-engineered T cells. The investigators isolated T cells with known specificity (EBV) from mobilized peripheral blood of healthy stem cell donors, for introduction of TCRα and TCRβ chain genes of the WT1-specific TCR, to minimize the risk of allogeneic graft-versus-host disease (GVHD) and enhance survival of transferred T cells.4,29,30 In a superior strategy to prevent GVHD and mispairing of endogenous and introduced TCRα and TCRβ chains,5,6 our group developed a method for in vitro generation of TA-specific T cells with a single TCR, starting from HSPC, which have not yet rearranged their endogenous TCR chains, using the OP9-DL1 co-culture protocol. In our classical in vitro culture protocol,7 IL-7 is added together with the cognate peptide recognized by the TCR, to induce agonist selection and induce maturation of CD4+ CD8+ DP cells. After agonist selection, cells are polyclonally expanded on feeder cells with PHA in the presence of IL-2. We here show that we are able to optimize the phenotype and functionality of the resulting in vitro generated TA-specific T cells by changing the cytokines added during the different culture steps to IL-21.
We investigated the effect of multiple common gamma-chain cytokines, including IL-2, IL-7, IL-15 and IL-21 (and combinations of IL-7 + IL-15 and IL-15 + IL-21). IL-2 is known to enhance the antitumor activity of T cells and is therefore often used for ex vivo expansion of tumor-infiltrating lymphocytes (TILs) or other T cells used for adoptive transfer. However, high levels of IL-2 are also known to induce terminal differentiation and activation-induced cell death (AICD), whereas lower levels induce and maintain regulatory T cells (Treg).31,32 Therefore, IL-2 might not be the ideal cytokine for expansion of adoptive T cells aimed to treat malignancies. With our in vitro differentiated T cells, addition of IL-2 resulted in only modest expansion, also related to a high percentage of cell death (data not shown). Furthermore, only low percentages of IFN-γ-producing cells and low percentages of target cell lysis were seen with addition of IL-2. As opposed to IL-2, IL-15 inhibits AICD and induces the generation and maintenance of long-lived memory CD8+ T cells.31,32 Results published by Klebanoff et al. and Hinrichs et al. demonstrate that IL-15 is able to improve the in vivo anti-tumor efficacy of adoptively transferred murine T cells.16,17 We observed that addition of IL-15 resulted in the highest target cell lysis. However, IL-15 is a known potent inducer of NK cell development,31 and in OP9-DL1 co-cultures it has been described that IL-15 induces NK cell-like killing by T cells.33 This could explain why killing was higher in conditions where IL-15 was added. Recently, our group described an in vivo agonist selected population which acquires cytotoxic functionality and NK-like properties when cultured in the presence of IL-15.34
In our hands, combining IL-15 and IL-21 resulted in increased expansion and target cell lysis as compared to IL-15 or IL-21 alone, indicating a synergistic effect of IL-15 and IL-21. These results are in line with findings by Zeng et al. and Pouw et al., describing the synergistic effect of IL-15 and IL-21 on murine T cells, resulting in enhanced proliferation, increased IFN-γ production and cytotoxicity.18,35 The synergistic effects of both cytokines could be attributed to activation of distinct downstream mediators, amplifying cytokine production and cytotoxicity. IL-21 signals predominantly through Jak3, STAT1 and STAT3, whereas downstream mediators of IL-15 are STAT5 and ERK1/2.17,18,36 As a possible explanation for the cooperative effect of IL-15 and IL-21 on IFN-γ production, Pouw et al. mention the presence of regulatory sites in the IFN-γ promoter which are responsive to both STAT- and ERK-dependent transcription factors.35 Furthermore, different signal transduction pathways initiated by distinct cytokines also explain differences in expression of phenotypic markers as observed in our experiments.17,18,36
In 2011, Gattinoni and colleagues described a less-differentiated memory T cell population, i.e. TSCM, which displays a naive-like phenotype (CD45RA+ CD45RO− CD62L+ CCR7+ CD27+ CD28+ IL-7Rα+) but also expresses CD95 and IL-2Rβ (CD122), similar to conventional memory T cells.14 Also, higher expression levels of BCL-2, LFA-1, CXCR3 and CXCR4 and lower levels of CD38 and CD31 were observed when compared to TN. Cieri and colleagues attempted and succeeded at generating TSCM-like T cells in vitro, by activating TN with anti-CD3/CD28 beads in the presence of IL-7 and IL-15.15 In their hands, this population expresses a CD62L+ CCR7+ CD45RA+ CD45RO+ IL-7Rα+ CD95+ phenotype. As TSCM are believed to be the ideal T cell subset for adoptive T cell transfer, we evaluated the presence of cells with a TSCM-like phenotype in our in vitro cultures. Although we observed a high percentage of CXCR4+ cells, we could not observe TSCM-like cells (CD45RA+ CD62L+ CXCR3+ CD95+) after agonist selection in the presence of IL-7 and IL-15. On the other hand, agonist selection in the presence of IL-21 did generate TSCM-like cells, although average CD95 expression was lower as compared to the other cytokine conditions. Moreover, we observed only minimal expansion in the presence of IL-21, which might be explained by the quiescent nature of TSCM under non-lymphopenic conditions.37 The generation of TSCM-like T cells in the presence of IL-21 has been described before.38,39 As opposed to the results of Cieri et al., Chen and colleagues, who compared differentiation of TN to TSCM in the presence of IL-2, IL-7, IL-15 or IL-21 after aCD3/CD28 activation, showed that IL-21 has the highest potential to induce TSCM,40 which is in line with our observations. Discrepancies in TSCM phenotype indicate differences between TSCM-like cells generated in vitro from HSPC in OP9-DL1 co-cultures (and subsequent agonist selection with the cognate peptide in the presence of cytokine(s), as compared to TSCM-like cells as generated in vitro starting from TN by Cieri et al. and Chen et al. (by activating TN with aCD3/aCD28 in the presence of cytokine(s)), and TSCM cells as isolated directly from healthy donors by Gattinoni et al.14,15,40
In their report, Vizcardo et al. state that CD8+ SP T cells generated in OP9-DL1 co-cultures are ‘abnormal’, with high expression of NK cell markers, indicating innate-like features, and upregulation of PTCRA, RAG1, RAG2 and RORC, indicating an immature phenotype.41 However, we demonstrated that addition of the agonist peptide to induce maturation from DP to CD8+ SP T cells in OP9-DL1 co-cultured cells results in downregulation of RAG1, RAG2, RORC and PTCRA. Furthermore, no NK cell markers are observed.7 On the other hand, and in line with the observations by Vizcardo et al., CD8αα T cells emerged in our cultures (as also described by Snauwaert et al).7 However, expression of CD8β was only lost after polyclonal feeder expansion, not after agonist selection. We do acknowledge that, as our cells are agonist selected cells, they might not fit into one of the conventional T cell differentiation states (TN, TSCM, TCM, TEM or TEFF) but rather represent an unconventional T cell subset. At the mRNA level, expression of conventional T cell markers (e.g. NELL2 and LRRN3) are absent in both the in vivo and in vitro agonist selected cells.7,42 At the protein level, unconventional agonist selected T cells, both in human thymus and CB, express high levels of CD95 and CXCR3, similar to our in vitro generated agonist selected T cells. Human unconventional agonist selected T cells are characterized by PD-1 expression in thymus, which is gradually lost in the corresponding population in CB, and also after IL-15-induced proliferation.34,42 PD-1 expression on our CB-derived in vitro generated T cells was, however, low (when IL-21 alone or in combination with IL-15 was added) or absent (with the other cytokine mixes). On the other hand, the previously described unconventional T cells express only low levels of CD62L that are even more downregulated after IL-15-induced proliferation,34,42 which we also observed after feeder expansion when IL-15 was added, but not with IL-21. These distinct phenotypes mark differences between in vitro agonist selected T cells and unconventional agonist selected T cells isolated directly from human thymus or CB. Furthermore, the cytokine(s) added during in vitro agonist selection also impact(s) the phenotype and functionality of the resulting in vitro generated agonist selected T cells.
Feeder expansion is a necessary step in our in vitro culture protocol to obtain functionally mature T cells capable of IFN-γ production and target cell lysis in vitro.7 In our hands, after feeder expansion, cells cultured in the presence of IL-21 still showed a high CD62L expression and partly TSCM-like phenotype. However, as we expect our T cells to expand in vivo after injection, we would inject them before in vitro feeder expansion, to avoid extensive culture leading to a skewed TEM/TEFF phenotype, as we previously demonstrated.9 Several studies indicate that lymphodepletion prior to adoptive transfer of T cells results in increased availability of endogenous cytokines important for homeostasis of naive and memory T cells (i.e. IL-7, IL-15 and IL-21), supporting in vivo persistence and anti-tumor efficacy.10,15,43–46 As we observed only minor expansion during agonist selection in the presence of IL-21, the number of T cells generated at this point in our in vitro culture might be too low to confer effective in vivo antitumor efficacy. In that case, we could opt to add co-stimulation during agonist selection, as this leads to increased proliferation, while exerting no other effects on the agonist selection process, as was previously shown by our group.7
Enhanced CD62L expression has been shown to be associated with augmented in vivo efficacy of adoptively transferred T cells,12,20 and results by Chen and others indicate TSCM generated in the presence of IL-21 mediate superior in vivo antitumor responses by both TA-stimulated T cells and CAR-transduced T cells.40,47,48 Also, Chapuis and colleagues elegantly demonstrated that addition of IL-21 during pre-treatment of TA-specific T cells results in a less-differentiated memory phenotype of these T cells, leading to prolonged in vivo survival and efficacy after adoptive transfer.49 Therefore, we speculate that addition of IL-21 during agonist selection, which resulted in markedly higher CD62L expression and TSCM-like cells in our in vitro culture system, will result in generation of T cells with long-term in vivo persistence and superior in vivo efficacy.
The protocol we describe here for generating TA-specific T cells could be clinically applicable, once GMP-compliant procedures have been developed. Alternatives for the use of murine xenogeneic OP9-DL1 cells for T cell commitment of HSPC are plate-bound DL1-Fc50–52 and Notch ligand bound to microbeads.53 Also, serum-free culture methods, as described by multiple groups,50,52,54 should be applied. As shown in Figure 1, our current in vitro culture protocol to generate mature T cells from CB-derived HSPC takes up to 34 days until after agonist selection. Starting from 1 × 106 CD34+ cells (approximate cell number obtained from 1 CB), 1.4 × 108 up to 3.4 × 1010 cells are generated after agonist selection, depending on the cytokine added during agonist selection (Fig S20). For clinical applications, clinically relevant stem cell sources will be used, as described.9
In conclusion, we have shown here that we are able to enhance the efficacy of in vitro generated TA-specific T cells by generating T cells with a less-differentiated phenotype through adapting the cytokines added during in vitro culture. Addition of IL-21 during agonist selection results in mature T cells with a less-differentiated TSCM-like phenotype, including increased CD62L expression, and high IFN-γ production after polyclonal feeder expansion. We believe that these TA-specific T cells could be a very valuable form of patient-tailored T cell immunotherapy, once the necessary preclinical analyses have been carried out.
Supplementary Material
Acknowledgments
The authors would like to thank Dr C. Matthys of the Cord Blood Bank for providing CB samples. We are indebted to Dr B. Descamps and Prof. Dr C. Vanhove of the Infinity Lab of Ghent University for help with irradiation of feeder cells and S. Vermaut for help with flow cytometry.
Funding Statement
This research was supported by the Research Foundation – Flanders (Fonds voor Wetenschappelijk Onderzoek Vlaanderen, FWO) under Grant 27958 2016-2021; and Stichting tegen Kanker under Grant 2014-166 c/2014/230. SDM, MP, TT and TK are supported by the Research Foundation – Flanders (FWO). JI is supported by the Special Research Fund (BOF) of Ghent University.
Disclosure statement
The authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jäger U, Jaglowski S, Andreadis C, Westin JR, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-Cell lymphoma. New Engl J Med. 2019;380(1):45–17. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 2.Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, Medeiros JJF, Rao-Bhatia A, Jaciw-Zurakowsky I, Marke R, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547(7661):104-+. doi: 10.1038/nature22993. [DOI] [PubMed] [Google Scholar]
- 3.Rosenfeld C, Cheever MA, Gaiger A.. WT1 in acute leukemia, chronic myelogenous leukemia and myelodysplastic syndrome: therapeutic potential of WT1 targeted therapies. Leukemia. 2003;17(7):1301–1312. doi: 10.1038/sj.leu.2402988. [DOI] [PubMed] [Google Scholar]
- 4.Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, Voillet V, Gottardo R, Ragnarsson GB, Bleakley M, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. 2019;25(7):1064-+. doi: 10.1038/s41591-019-0472-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser ADM, Pouw N, Debets R, Kieback E, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16(5):565–U98. doi: 10.1038/nm.2128. [DOI] [PubMed] [Google Scholar]
- 6.van Loenen MM, De Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, Van Rood JJ, Falkenburg JHF, Heemskerk MHM.. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci U S A. 2010;107(24):10972–10977. doi: 10.1073/pnas.1005802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snauwaert S, Verstichel G, Bonte S, Goetgeluk G, Vanhee S, Van Caeneghem Y, De Mulder K, Heirman C, Stauss H, Heemskerk MHM. In vitro generation of mature, naive antigen-specific CD8+ T cells with a single T-cell receptor by agonist selection. Leukemia. 2014;28(4):830–841. doi: 10.1038/leu.2013.285. [DOI] [PubMed] [Google Scholar]
- 8.Van Caeneghem Y, De Munter S, Tieppo P, Goetgeluk G, Weening K, Verstichel G, Bonte S, Taghon T, Leclercq G, Kerre T, et al. Antigen receptor-redirected T cells derived from hematopoietic precursor cells lack expression of the endogenous TCR/CD3 receptor and exhibit specific antitumor capacities. Oncoimmunology. 2017;6(3):e1283460. doi: 10.1080/2162402X.2017.1283460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonte S, De Munter S, Goetgeluk G, Ingels J, Pille M, Billiet L, Taghon T, Leclercq G, Vandekerckhove B, Kerre T, et al. T-cells with a single tumor antigen-specific T-cell receptor can be generated in vitro from clinically relevant stem cell sources. Oncoimmunology. 2020;9(1):1727078. doi: 10.1080/2162402X.2020.1727078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou JH, Shen XL, Huang JP, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175(10):7046–7052. doi: 10.4049/jimmunol.175.10.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu ZY, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8(+) T cells. J Clin Invest. 2005;115(6):1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–U325. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, Bondanza A, Bordignon C, Peccatori J, Ciceri F, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121(4):573–584. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 16.Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T Cells. Proc Natl Acad Sci U S A. 2004;101(7):1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, Klebanoff CA, Rosenberg SA, Leonard WJ, Restifo NP, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8(+) T cells for adoptive immunotherapy. Blood. 2008;111(11):5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng R, Spolski R, Finkelstein SE, Oh SK, Kovanen PE, Hinrichs CS, Pise-Masison CA, Radonovich MF, Brady JN, Restifo NP, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. 2005;201(1):139–148. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Depreter B, Weening KE, Vandepoele K, Essand M, De Moerloose B, Themeli M, Cloos J, Hanekamp D, Moors I, D’hont I, et al. TARP is an immunotherapeutic target in acute myeloid leukemia expressed in the leukemic stem cell compartment. Haematologica. 2020;105(5):1306–1316. doi: 10.3324/haematol.2019.222612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, et al. Central memory self/tumor-reactive CD8(+) T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102(27):9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. 2011;317(5):620–631. doi: 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, Harty JT, Badovinac VP. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity. 2010;33(1):128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. 2017;23(1):18–27. doi: 10.1038/nm.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolfl M, Merker K, Morbach H, Van Gool SW, Eyrich M, Greenberg PD, Schlegel PG. Primed tumor-reactive multifunctional CD62L(+) human CD8(+) T cells for immunotherapy. Cancer Immunol Immun. 2011;60(2):173–186. doi: 10.1007/s00262-010-0928-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verneris MR, Karimi M, Baker J, Jayaswal A, Negrin RS. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. 2004;103(8):3065–3072. doi: 10.1182/blood-2003-06-2125. [DOI] [PubMed] [Google Scholar]
- 26.Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. Eur J Immunol. 2013;43(11):2797–2809. doi: 10.1002/eji.201343751. [DOI] [PubMed] [Google Scholar]
- 27.Barber DL, Wherry EJ, Masopust D, Zhu BG, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 28.Huang JP, Khong HT, Dudley ME, El-Gamil M, Li YF, Rosenberg SA, Robbins PF. Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. J Immunother. 2005;28(3):258–267. doi: 10.1097/01.cji.0000158855.92792.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GMA, Papagno L, Ogg GS, King A, Lechner F, Spina CA. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8(4):379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 30.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118(1):294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dwyer CJ, Knochelmann HM, Smith AS, Wyatt MM, Rivera GOR, Arhontoulis DC, Bartee E, Li Z, Rubinstein MP, Paulos CM. Fueling cancer immunotherapy with common gamma chain cytokines. Front Immunol. 2019;10:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pilipow K, Roberto A, Roederer M, Waldmann TA, Mavilio D, Lugli E. IL15 and T-cell Stemness in T-cell–Based Cancer Immunotherapy. Cancer Res. 2015;75(24):5187–5193. doi: 10.1158/0008-5472.CAN-15-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao YB, Parkhurst MR, Zheng Z, Cohen CJ, Riley JP, Gattinoni L, Restifo NP, Rosenberg SA, Morgan RA. Extrathymic generation of tumor-specific T cells from genetically engineered human hematopoletic stem cells via notch signaling. Cancer Res. 2007;67(6):2425–2429. doi: 10.1158/0008-5472.CAN-06-3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Billiet L, Goetgeluk G, Bonte S, De Munter S, De Cock L, Pille M, Ingels J, Jansen H, Weening K, Van Nieuwerburgh F. Human thymic CD10(+) PD-1(+) intraepithelial lymphocyte precursors acquire interleukin-15 responsiveness at the CD1a(-) CD95(+) CD28(-) CCR7(-) developmental stage. Int J Mol Sci. 2020;21(22):8785. doi: 10.3390/ijms21228785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pouw N, Treffers-Westerlaken E, Kraan J, Wittink F, Ten Hagen T, Verweij J, Debets R. Combination of IL-21 and IL-15 enhances tumour-specific cytotoxicity and cytokine production of TCR-transduced primary T cells. Cancer Immunol Immun. 2010;59(6):921–931. doi: 10.1007/s00262-010-0818-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The molecular basis of IL-21-mediated proliferation. Blood. 2007;109(10):4135–4142. doi: 10.1182/blood-2006-10-054973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turtle CJ, Swanson HM, Fujii N, Estey EH, Riddell SR, Distinct A. Subset of self-renewing human memory CD8(+) T cells survives cytotoxic chemotherapy. Immunity. 2009;31(5):834–844. doi: 10.1016/j.immuni.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alvarez-Fernandez C, Escriba-Garcia L, Vidal S, Sierra J, Briones J. A short CD3/CD28 costimulation combined with IL-21 enhance the generation of human memory stem T cells for adoptive immunotherapy. J Transl Med. 2016;14. doi: 10.1186/s12967-016-0973-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Cong YN, Jia MM, He QQ, Zhong HQ, Zhao Y, Li H, Yan M, You J, Liu J, et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat Commun. 2021;12(1):951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen YS, Yu F, Jiang YW, Chen JL, Wu K, Chen XX, Lin Y, Zhang H, Li L, Zhang Y, et al. Adoptive transfer of interleukin-21-stimulated human CD8+ T memory stem cells efficiently inhibits tumor growth. J Immunother. 2018;41(6):274–283. doi: 10.1097/CJI.0000000000000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vizcardo R, Klemen ND, Islam SMR, Gurusamy D, Tamaoki N, Yamada D, Koseki H, Kidder BL, Yu Z, Jia L, et al. Generation of tumor antigen-specific iPSC-derived thymic emigrants using a 3D thymic culture system. Cell Rep. 2018;22(12):3175–3190. doi: 10.1016/j.celrep.2018.02.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verstichel G, Vermijlen D, Martens L, Goetgeluk G, Brouwer M, Thiault N, Van Caeneghem Y, De Munter S, Weening K, Bonte S, et al. The checkpoint for agonist selection precedes conventional selection in human thymus. Sci Immunol. 2017;2(8):eaah4232. doi: 10.1126/sciimmunol.aah4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wrzesinski C, Restifo NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Curr Opin Immunol. 2005;17(2):195–201. doi: 10.1016/j.coi.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lizee G, Overwijk WW, Radvanyi L, Gao JJ, Sharma P, Hwu P. Harnessing the power of the immune system to target cancer. Annu Rev Med. 2013;64:71–90. [DOI] [PubMed] [Google Scholar]
- 45.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou JH, Huang J, Powell DJ, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173(12):7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, Mi T, Maiti S, Kebriaei P, Lee DA, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011;71(10):3516–3527. doi: 10.1158/0008-5472.CAN-10-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sabatino M, Hu JH, Sommariva M, Gautam S, Fellowes V, Hocker JD, Dougherty S, Qin H, Klebanoff CA, Fry TJ, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128(4):519–528. doi: 10.1182/blood-2015-11-683847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, Duerkopp N, Roberts IM, Pogosov GL, Ho WY, et al. Transferred WT1-reactive CD8(+) T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. 2013;5(174). doi: 10.1126/scitranslmed.3004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandez I, Ooi TP, Roy K. Generation of functional, antigen- specific CD81 human T cells from cord blood stem cells using exogenous notch and tetramer- TCR signaling. Stem Cells. 2014;32(1):93–104. doi: 10.1002/stem.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varnum-Finney B, Brashem-Stein C, Bernstein ID. Combined effects of Notch signaling and cytokines induce a multiple log increase in precursors with lymphoid and myeloid reconstituting ability. Blood. 2003;101(5):1784–1789. doi: 10.1182/blood-2002-06-1862. [DOI] [PubMed] [Google Scholar]
- 52.Delaney C, Varnum-Finney B, Aoyama K, Brashem-Stein C, Bernstein ID. Dose-dependent effects of the Notch ligand Delta1 on ex vivo differentiation and in vivo marrow repopulating ability of cord blood cells. Blood. 2005;106(8):2693–2699. doi: 10.1182/blood-2005-03-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taqvi S, Dixit L, Roy K. Biomaterial-based notch signaling for the differentiation of hematopoietic stem cells into T cells. J Biomed Mater Res A. 2006;79a(3):689–697. doi: 10.1002/jbm.a.30916. [DOI] [PubMed] [Google Scholar]
- 54.Seet CS, He CB, Bethune MT, Lie SW, Chick B, Gschweng EH, Zhu Y, Kim K, Kohn DB, Baltimore D, et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Methods. 2017;14(5):521-+. doi: 10.1038/nmeth.4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.