

Abstract
A novel coronavirus disease (COVID-19), caused by SARS-CoV-2, has spread over more than 100 countries all over the world. The World Health Organization has recognized Coronavirus as a pandemic and finding an effective drug for this infectious disease is of high importance. In this study, we have explored the potent inhibitors of COVID-19 main protease from Tinospora cordifolia an Ayurvedic herb locally called as Amrita meaning ‘immortality’ and two other Ayurveda plants namely Cinnamomum zeylanicum and Myristica fragrans. Saponarin, a phytochemical present in Tinospora cordifolia showed a very promising result with the binding affinity of − 8.75 kcal/mol. Remdesivir and Favipiravir, the experimental drugs that are known to show inhibitory activity towards COVID-19 are used as a control. The Docking results were verified by the means of molecular dynamic analysis. This study suggests that Saponarin can be a potential inhibitor for the main protease of the COVID-19.
Keywords: COVID-19, Molecular docking, Phytochemicals, Tinospora cordifolia
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) belongs to the genus Betacoronavirus of the family Coronaviridae. It is an airborne positive-sense single-stranded RNA virus causing coronavirus disease 2019 (COVID-19). The symptoms of this infection range from runny nose and cough to severe acute respiratory syndrome and kidney failure [1]. Being recognized as a pandemic, timely and extensive research is of high importance in finding an effective drug for this disease [2].
Phytomedicines are been used since times immemorable and such traditions are still practiced today, namely Ayurveda. In recent times, alongside drug repurposing [3, 4], the phytochemicals are been used for evidence-based drug development.
T. cordifolia, an herbaceous vine of the family Menispermaceae locally known as amrita literally meaning immortality is native to India, Myanmar and Sri Lanka. In Ayurvedic medicine, it is considered to be one of the most divine herbs mainly constituting Terpenoids, Alkaloids, Lignans and Steroids [5]. This plant has been known since long to cure a vast variety of diseases. Recently, this plant gained great interest in the field of drug discovery owing to its medicinal properties. The plant is reported to show anti-allergic, anti-arthritic, anti-diabetic, anti-inflammatory, anti-leprotic, anti-malarial, anti-neoplastic, anti-oxidant, antiperiodic, antispasmodic, anti-stress, hepatoprotective and immunomodulatory activities [6]. Myristica fragrans is medicinal plant of India which are known to manage the Kapha diseases such as tastelessness of tongue and cough [7] while Cinnamon zeylanicum is also a medicinal plant which is known to show medicinal activities towards influenza, cough and bronchitis [8].
Owing to their medicinal activity towards the symptoms of the coronavirus [9], these three medicinal plants from India are selected for this study to determine their inhibitory activity towards the main protease [10, 11] of the COVID-19 using computational techniques namely molecular docking and molecular dynamics studies.
Methods
Preparation of the protein structure
The three-dimensional crystal structure of COVID-19 main protease (PDB ID: 6LU7) was retrieved from the protein data bank. The protein structure was prepared using the protein preparation wizard of the Schrödinger suite using Maestro.
Identification and preparation of ligands
The phytochemicals of T. cordifolia, Cinnamomum zeylanicum and Myristica fragrans were identified through the review of the literature [5, 6, 12–14]. The structures of the phytochemicals were either downloaded through PubChem or drawn using Maestro. The phytochemical structures were then prepared for docking studies using LigPrep module of the Schrödinger suite.
Molecular docking and binding energy estimation
The docking simulations were carried out using Glide, a receptor-ligand docking module of Schrödinger suite.
Remdesivir and Favipiravir [15–17], the compounds that have shown activity against COVID-19 were used as a control in docking studies. The peptide-like inhibitor already available in the obtained PDB file is used as a reference to determine the active site of the protein.
Molecular dynamics
The top ligand from the docking studies was subjected to molecular dynamics simulations to study the stability and conformational flexibility. GROMACS (Version 2018.2) was used for the same. As the CHARMM36 force field [18–20] that was used for MD simulations lack force field parameters of the ligand studied, SwissParam [21] web server was used to generate its topology. the proteins and the ligands were then merged, solvated, minimized and equilibrated. TIP3P water model was used to solvate the protein-ligand complex. To neutralize the system, Cl- ions and Na+ ions were added accordingly before minimization. The energy minimization step was performed using the steepest descent algorithm until the maximum force is less than 10.0 kJ/mol. The NVT and NPT ensembles were performed for 100ps each to equilibrate the system with the reference pressure and temperature of 1 Bar and 310 K respectively. After equilibration, MD simulation was performed for 100ns at constant temperature and pressure using the leapfrog algorithm. Trajectory snapshots were stored at every 100ps during the simulation period to aid in analysis.
Trajectory analysis
The root means square deviation (RSMD) from the initial structure during the simulation period was analyzed using GROMACS analysis toolkit utilities and a graph was generated for the same. The analysis toolkit was also used to further analyse the simulation.
MM/PBSA analysis
The binding free energy between the COVID-19 main protease and Saponarin complex is calculated using the Molecular Mechanic/Poisson–Boltzmann surface area (MM/PBSA) by the polar & apolar solvation energy calculations and potential energy calculations in vacuum. The energies are calculated for every 1 ns and the potential energy in vacuum, polar and apolar solvation energies are summated to determine the binding energy between the COVID-19 main protease and Saponarin using the MM/PBSA tool [22].
Further, to understand the contribution of each amino acid towards the binding energy, the interaction energies are decomposed using the MM-PBSA method considering the van der Waals, electrostatic, polar and apolar contribution of each protein residue.
Results and discussion
Since ancient times it is known that Ayurveda, a plant medical methodology has been proven to show inhibitory activity against various diseases including viral pathogens in host cells. Phytochemicals that have been reported in various studies for their presence in T.cordifolia, C.zeylanicum and M.fragrans were identified and screened for their inhibitory activity against COVID-19 main protease. All the phytochemicals were prepared using LigPrep wizard of Schrödinger and were further subjected to molecular docking analysis using Glide with considering Remdesivir and Favipiravir, the drugs that were studied to show inhibitory activity towards COVID-19 as a control. The top phytochemical from the docking analysis was further analyzed by the means of all-atom molecular dynamics and MMPBSA to study its stability as a complex with the COVID-19 main protease.
Docking analysis
As stated earlier, Remdesivir and Favipiravir were used as a reference compound in docking studies. According to the Glide results, Saponarin showed better binding affinity than the reference compounds. Table 1 reveals the docking scores (Kcal/mol) of the ligands as calculated by Glide along with that of Remdesivir and Favipiravir, the reference compounds. To get an insight into the interactions between ligand and protein, LigPlot + [23] was used to generating 2D interaction plots. Docking pose analysis revealed that the interactions of the Saponarin were similar to that of both Remdesivir as well as the inhibitory peptide-like molecule already present in the protein structure. The same can be seen in Figure 1. Examining the interaction pattern, the ligand that is studied can be a potent inhibitor of the COVID-19 main protease. The common interactions between the peptide-like inhibitor and the other ligands are circled in red.
Table 1.
Docking scores of the ligands as calculated by Glide
| Tinospora cordifolia | Cinnamomum zeylanicum | Myristica fragrans | |||
|---|---|---|---|---|---|
| Name | Score (kcal/mol) | Name | Score (kcal/mol) | Name | Score (kcal/mol) |
| Saponarin | − 8.76 | Cinnamyl Acetate | − 5.38 | Alpha- Thujen | − 5.98 |
| Cordifoline | − 8.67 | Caryophyllene Oxide | − 5.25 | p- Menth 2-en-1-ol | − 5.74 |
| Tinosponone | − 7.35 | Alpha-Copaene | − 4.76 | 3- Carene | − 5.69 |
| Syringin | − 7.31 | Caryophyllene | − 4.72 | Alpha- Terpinolene | − 5.68 |
| Arabinogalactan | − 6.83 | Camphor | − 4.70 | Beta Pinene | − 5.67 |
| 3_4-di_hydroxy-3-methoxy-benzyl_-4-_4-hydroxy3-methoxy-benzyl-tetrahydrofuran | − 6.82 | Eugenol | − 4.55 | Gamma- Terpinene | − 5.55 |
| Beta-Ecdysterone | − 6.49 | Trans-alpha-Bergamotene | − 4.31 | Alpha Fellandrene | − 5.50 |
| Palmatoside | − 6.17 | Cinnamaldehyde | − 4.17 | Alpha- Terpinene | − 5.43 |
| Palmatine | − 6.04 | Alpha-Bergamotene | − 4.06 | p- Cymene | − 5.43 |
| 3_4-dihydroxy-3-methoxybenzyl_4-hydroxy-3-methoxybenzyl | − 5.98 | Alpha- Pinene | − 5.30 | ||
| Beta-Sitosterol | − 5.94 | Alpha- Terpineol | − 5.28 | ||
| Tinosporinone | − 5.86 | Control | p- Menth 1-en-4-ol | − 5.23 | |
| Tinocordifolioside | − 5.81 | Name | Score (kcal/mol) | Limonene | − 5.01 |
| Tinocordiside | − 5.73 | Remdesivir | − 8.65 | Elermicin | − 4.93 |
| Tinocordioside | − 5.71 | Favipiravir | − 5.76 | Camphene | − 4.86 |
| Mustakone | − 5.60 | Safrole | − 4.86 | ||
| Berberine | − 5.32 | Myristicin | − 4.74 | ||
| 1-Methyl-2 pyrrolidinone | − 5.08 | Linalool | − 4.30 | ||
| Choline | − 4.32 | Beta- Ocimene | − 3.45 | ||
| 1-Octacosanol | − 3.04 | Beta Myrcene | − 2.91 | ||
| 1-Heptacosanol | − 2.09 | ||||
Fig. 1.

Interaction diagrams. a The interactions between the protein and peptide-like inhibitor present in the protein structure. b The interactions between the protein and Saponarin. c The interactions between the protein and Remdesivir
Molecular dynamics analysis
To study the structural and conformational behavior of the COVID-19 main protease and the ligand, Molecular Dynamic simulation was incorporated. The MD simulations were performed for 100ns for the protein-ligand complex. RMSD of the protein and ligand was calculated using the initial structure as the reference and a graph was generated to study the stability of the complex. RMSD is a global measure of structural fluctuations that helps in studying the dynamic stability of the protein-ligand complex. After studying the RMSD graph, it was observed that there were no significant fluctuations in the structure. The RMSD graph of the protein and ligand is shown in Fig 2. To further confirm the stability of the complex, the timeline analysis of the evolution of the secondary structure was plotted using VMD [24]. The timeline graph is shown in Fig 3. The secondary structure timeline suggests that the structure is stable including the residues that were constantly fluctuating confirming the stability of the complex. To quantify the strength of the interaction between the ligand and receptor, interaction energy was calculated based on short-range Lennard- Jones potential. The Lennard- Jones potential states the nonbonded interaction energy between the molecules. The Interaction energy graph suggests that Saponarin shows stable interaction with minor fluctuations conforming the complex stability. The graph for the same is shown in Fig 4. The RMSD, secondary structure evolution and the nonbonded interaction energy of the protein and ligand did not show any major fluctuations and were in the acceptable range signifying high stability.
Fig. 2.

RMSD plot for Protein and Saponarin
Fig. 3.

RMSD plot for Protein and Saponarin
Fig. 4.

Nonbonded (Lennard–Jones) interaction energy graph between Protein and Saponarin
MM/PBSA analysis
The average per-residue contribution of the protein over the 100 ns simulation time towards the binding affinity between the protein and the ligand is shown as the residue decomposition plot (MM-PBSA) in Fig. 5. This plot suggests that the amino acids identified to be responsible for the interactions during the docking studies show high stability and play an important role in inhibitory activity of the COVID-19 protease.
Fig. 5.
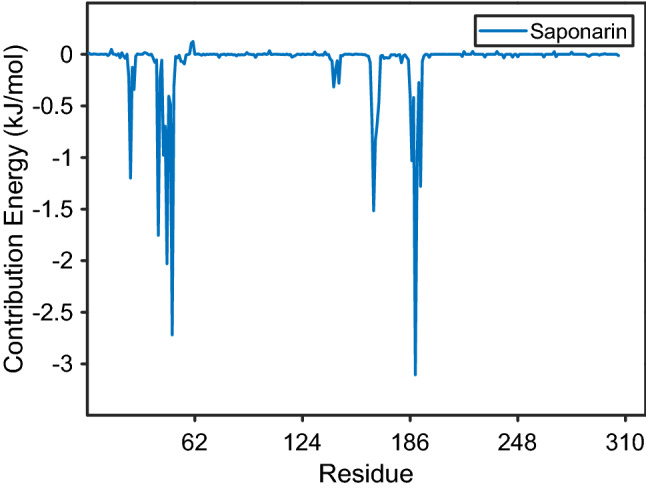
The residue decomposition plot of the interactions between the Protein and the Saponarin
In summary, this work sets out to discover non-peptide compounds from traditional Indian plants namely T. cordifolia, C. zeylanicum and M. fragrans with inhibitory activity towards SARS-CoV-2. Incorporating virtual screening methodology, COVID-19 main protease was subjected to molecular docking studies. The compound with highest docking score was considered to show promising results against of COVID-19 main protease after analyzing the interaction diagram in reference with the peptide-like protease inhibitor of COVID-19 main protease, Remdesivir and Favipiravir. The protein-ligand stability was then examined by the means of 100ns molecular dynamics simulation depicting the dynamic behavior of the complex. Results show that Saponarin, the phytochemical with highest docking score is stable throughout the simulation without showing any significant fluctuation. Further, the MMPBSA analysis conformed that the protein residues responsible for the interactions during docking studies show stable interactions with the COVID-19 main protease at the active site. Therefore, in terms of inhibition of COVID-19 main protease, Saponarin can show remarkable results summarizing it as an effective inhibitor of COVID-19 main protease.
Acknowledgements
The research work was carried out in the laboratory of the Bioinformatics, Department of Applied Sciences, Indian Institute of Information Technology Allahabad, Prayagraj-India. The Molecular Dynamic simulation results reported in this work were performed on the Central Computing Facility of IIITA, Prayagraj. Viswajit Mulpuru is grateful to the Ministry of Human Resource Development, Government of India and Indian Institute of Information Technology Allahabad for the scholarship to pursue a doctoral degree.
Declarations
Conflict of interest
The authors declare no competing financial interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Viswajit Mulpuru, Email: pbi2017001@iiita.ac.in.
Nidhi Mishra, Email: nidhimishra@iiita.ac.in.
References
- 1.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakraborty C, Sharma AR, Sharma G, Bhattacharya M, Saha RP, Lee S-S. Extensive partnership, collaboration, and teamwork is required to stop the COVID-19 outbreak. Arch Med Res. 2020;51:728–730. doi: 10.1016/j.arcmed.2020.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saha RP, Sharma AR, Singh MK, Samanta S, Bhakta S, Mandal S, et al. Repurposing drugs, ongoing vaccine, and new therapeutic development initiatives against COVID-19. Front Pharmacol. 2020;11:1258. doi: 10.3389/fphar.2020.01258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saha A, Sharma AR, Bhattacharya M, Sharma G, Lee S-S, Chakraborty C. Tocilizumab: a therapeutic option for the treatment of cytokine storm syndrome in COVID-19. Arch Med Res. 2020;51:595–597. doi: 10.1016/j.arcmed.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sinha K, Mishra NP, Singh J, Khanuja SP. Tinospora cordifolia (Guduchi): a reservoir plant for therapeutic applications: a review. Indian J Trad Knowl. 2004;3:257–270. [Google Scholar]
- 6.Saha S, Ghosh S. Tinospora cordifolia: one plant, many roles. Anc Sci Life. 2012;31:151–159. doi: 10.4103/0257-7941.107344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chunekar K. Bhaavprakash Nighantu of Sri Bhavamisra. Pandey G, editor. Varanasi: Chaukhamba Bharati Academy; 2002.
- 8.Adarsh A, Chettiyar B, Kanthesh BM, Raghu N. Phytochemical screening and antimicrobial activity of “Cinnamon zeylanicum”. Int J Pharm Res Innov. 2020;13:22–33. [Google Scholar]
- 9.Chakraborty C, Sharma AR, Sharma G, Bhattacharya M, Lee SS. SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): diagnostic and proposed therapeutic options. Eur Rev Med Pharmacol Sci. 2020;24:4016–4026. doi: 10.26355/eurrev_202004_20871. [DOI] [PubMed] [Google Scholar]
- 10.Maurya AK, Mishra N. In silico validation of coumarin derivatives as potential inhibitors against Main Protease, NSP10/NSP16-Methyltransferase, Phosphatase and Endoribonuclease of SARS CoV-2. J Biomol Struct Dyn. 2020;1–16. [DOI] [PMC free article] [PubMed]
- 11.Maurya SK, Maurya AK, Mishra N, Siddique HR. Virtual screening, ADME/T, and binding free energy analysis of anti-viral, anti-protease, and anti-infectious compounds against NSP10/NSP16 methyltransferase and main protease of SARS CoV-2. J Recept Signal Transduction. 2020;40:605–612. doi: 10.1080/10799893.2020.1772298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uppuluri S, Ali SL, Nirmala T, Babu SD. A review on Tinospora cordifolia. International Curr Pharm Rev Res. 2013;4:61–68. [Google Scholar]
- 13.Rao PV, Gan SH. Cinnamon: a multifaceted medicinal plant. Evidence-Based Complement Altern Med. 2014;2014:1–12. doi: 10.1155/2014/642942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis SK, James B, Varughese S, Nair MS. Phytochemical investigation on Myristica fragrans stem bark. Nat Prod Res. 2019;33:1204–1208. doi: 10.1080/14786419.2018.1457670. [DOI] [PubMed] [Google Scholar]
- 15.Saha A, Sharma AR, Bhattacharya M, Sharma G, Lee S-S, Chakraborty C. Probable molecular mechanism of remdesivir for the treatment of COVID-19: need to know more. Arch Med Res. 2020;51:585–586. doi: 10.1016/j.arcmed.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pilkington V, Pepperrell T, Hill A. A review of the safety of favipiravir—a potential treatment in the COVID-19 pandemic? J Virus Erad. 2020;6:45–51. doi: 10.1016/S2055-6640(20)30016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J, MacKerell AD. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J Comput Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 20.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 21.Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: A fast force field generation tool for small organic molecules. J Comput Chem. 2011;32:2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 22.Kumari R, Kumar R. Open Source Drug Discovery Consortium, Lynn A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J Chem Inf Model. 2014;54:1951–62. [DOI] [PubMed]
- 23.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 24.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]