

ABSTRACT
Poxviruses are exceptional in having a complex entry-fusion complex (EFC) that is comprised of 11 conserved proteins embedded in the membrane of mature virions. However, the detailed architecture is unknown and only a few bimolecular protein interactions have been demonstrated by coimmunoprecipitation from detergent-treated lysates and by cross-linking. Here, we adapted the tripartite split green fluorescent protein (GFP) complementation system in order to analyze EFC protein contacts within living cells. This system employs a detector fragment called GFP1-9 comprised of nine GFP β-strands. To achieve fluorescence, two additional 20-amino-acid fragments called GFP10 and GFP11 attached to interacting proteins are needed, providing the basis for identification of the latter. We constructed a novel recombinant vaccinia virus (VACV-GFP1-9) expressing GFP1-9 under a viral early/late promoter and plasmids with VACV late promoters regulating each of the EFC proteins with GFP10 or GFP11 attached to their ectodomains. GFP fluorescence was detected by confocal microscopy at sites of virion assembly in cells infected with VACV-GFP1-9 and cotransfected with plasmids expressing one EFC-GFP10 and one EFC-GFP11 interacting protein. Flow cytometry provided a quantitative way to determine the interaction of each EFC-GFP10 protein with every other EFC-GFP11 protein in the context of a normal infection in which all viral proteins are synthesized and assembled. Previous EFC protein interactions were confirmed, and new ones were discovered and corroborated by additional methods. Most remarkable was the finding that the small, hydrophobic O3 protein interacted with each of the other EFC proteins.
IMPORTANCE Poxviruses are enveloped viruses with a DNA-containing core that enters cells following fusion of viral and host membranes. This essential step is a target for vaccines and therapeutics. The entry-fusion complex (EFC) of poxviruses is unusually complex and comprised of 11 conserved viral proteins. Determination of the structure of the EFC is a prerequisite for understanding the fusion mechanism. Here, we used a tripartite split green fluorescent protein assay to determine the proximity of individual EFC proteins in living cells. A network connecting components of the EFC was derived.
KEYWORDS: green fluorescent protein, membrane proteins, multiprotein complex, proximity analysis, vaccinia virus, virus entry
INTRODUCTION
Poxviruses are large, enveloped, DNA viruses that replicate entirely in the cytoplasm of infected cells (1). Entry is dependent on the fusion of viral and cellular membranes at the cell surface or internally following endocytosis, depending on the virus strain and cell type (2–5). Entry of the core into the cytoplasm occurs by a two-step mechanism consisting of lipid mixing of the viral and cellular membranes, followed by pore formation (6). The process is mediated by 11 conserved poxviral proteins that form the entry-fusion complex (EFC) (7, 8). The EFC proteins are unglycosylated and, in the case of vaccinia virus (VACV), vary from approximately 4 to 43 kDa with a combined mass of 232 kDa, assuming each component is present only once (Table 1). The EFC is embedded in the membrane of the mature virion (MV) via N- or C-terminal hydrophobic domains of the proteins, which are unstable and degraded when viral membrane formation is prevented (9–11). Super-resolution microscopy indicates that the EFC is localized at the tips of virions (12). The organization of the EFC has not yet been determined, though some bimolecular component interactions have been identified (13–16), and the crystal structures of the ectodomains of two VACV proteins, L1 and F9, have been reported (17, 18).
TABLE 1.
EFC proteins
| Gene | Paralog | SIa | kDa | HDb | S-Sc | ASd | Reference(s) |
|---|---|---|---|---|---|---|---|
| A16L | G9R, J5L | G9 | 43 | C | + | 14, 31 | |
| A21L | 14 | N | + | 32 | |||
| A28L | H2 | 16 | N | + | 33, 34 | ||
| F9L | L1R | 24 | C | + | + | 18, 35 | |
| G3L | L5 | 13 | N | 7, 36 | |||
| G9R | A16L, J5L | A16 | 39 | C | + | 14, 37 | |
| H2R | A28 | 22 | N | + | 26 | ||
| J5L | A16L, G9R | 15 | C | + | 7 | ||
| L1R | F9L | 27 | C | + | + | 17, 38 | |
| L5R | G3 | 15 | C | + | 39 | ||
| O3L | 4 | N | 40 |
SI, subunit interaction.
HD, hydrophobic domains. C, C-terminal; N, N-terminal.
S-S, intramolecular disulfide bonds.
AS, atomic structure solved.
Although the EFC is membrane-associated, the component protein interactions have been analyzed mainly following solubilization with detergent. In the present study, we employed the tripartite split superfolder green fluorescent protein (GFP) complementation system of Cabantous and colleagues (19, 20) to analyze EFC protein interactions in living infected cells. An earlier bipartite complementation system required the interaction of a large detector GFP1-10 fragment, so named because it contains 10 β-strands, with a small GFP11 fragment that provides the eleventh β-strand (21). Fluorescence occurs upon the association of the large and small GFP fragments. We previously employed the bipartite system to determine the topology of some VACV membrane proteins using a recombinant VACV that expressed GFP1-10 (22). The tripartite system employs a GFP detector that contains nine β-folds (GFP1-9) and requires an interaction with two small GFP fragments (GFP10 and GFP11) each containing one β-strand. Importantly, the GFP1-9 fragment cannot complement the GFP10 and GFP11 fragments unless interacting partners bring the latter two together, as depicted in Fig. 1. Once brought together, the assembled GFP is stabilized. Advantages of the tripartite system include the small size of the tags that are appended to the interacting proteins and absence of significant background fluorescence. The tripartite system has been used to detect protein interactions in yeast, plants and mammalian cells (23–25). Here, we extend the tripartite system to study interactions of viral proteins by constructing a novel recombinant VACV that expresses GFP1-9 and plasmids with EFC proteins tagged with GFP10 or GFP11. With this approach, we confirmed previous EFC protein interactions and found new ones. Most striking is evidence for proximity of the smallest EFC protein, O3, with itself and each of the other EFC components.
FIG 1.
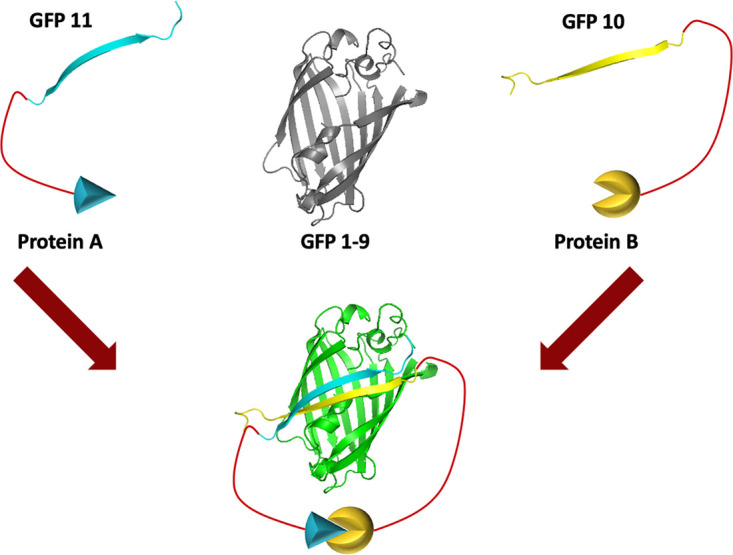
Model of the tripartite GFP complementation system. The system consists of one protein tagged with GFP10, another protein tagged with GFP11, and the GFP1-9 sensor. When proteins A and B interact, GFP10 and GFP11 are brought together, allowing complementation with GFP1-9 and green fluorescence.
RESULTS
Construction of recombinant VACV expressing GFP1-9 and plasmids expressing EFC proteins fused to GFP10 and GFP11.
We adapted the tripartite split GFP system to investigate the proximity of proteins comprising the EFC in infected cells. Recombinant VACV-GFP1-9 and VACV-GFP1-10, in which expression of the detector GFP fragment was regulated by the VACV strong early/late promoter, were constructed as depicted in Fig. 2A. These recombinant viruses are derived from the Western Reserve (WR) strain and are replication competent and unmodified except for their expression of the GFP constructs. Accordingly, VACV GFP1-9 and VACV GFP1-10 still express each of the native EFC proteins, which localize in the viral membrane. In addition, VACV expression plasmids were made in which the 20-amino-acid GFP10 or GFP11 sequence was fused via a linker to a hemagglutinin (HA) or V5 epitope tag at the ectodomain of individual EFC proteins (Fig. 2B). Each of the plasmids contained the same late promoter regulating transcription of the EFC open reading frames, which is recognized by the poxvirus cytoplasmic transcription system but not by that of the host. Synthesis of each of the EFC V5 (Fig. 2C)- and HA (not shown)-tagged proteins was demonstrated in Western blots of lysates from VACV-infected cells that had been transfected with the expression plasmids. Synthesis of GFP1-9 by several newly isolated recombinant viruses and the longer GFP1-10 by a previously isolated recombinant virus (22) in rabbit kidney 13 (RK13) cells were demonstrated by Western blotting (Fig. 3A).
FIG 2.

Adaptation of the tripartite GFP assay for analysis of EFC interactions. (A) Construction of VACV-GFP1-9 and VACV-GFP1-10. Infusion PCR was used to insert DNA encoding GFP1-9 or GFP1-10 into the transfer plasmid pRB21 so as to be regulated by the VACV early-late promoter. The resulting GFP1-9 and GFP1-10 plasmids were transfected into BS-C-1 cells that had been infected with vRB12 to allow homologous recombination and large plaque formation. Virus from large plaques was clonally purified by repeat plaque formation to obtain VACV-GFP1-9 and VACV-GFP1-10. (B) Construction of plasmids expressing tagged EFC proteins. GFP10 or GFP11 sequences were fused to DNA encoding individual EFC proteins with an HA or V5 tag at the ectodomain that are regulated by the late p11 VACV promoter. (C) Expression of EFC proteins with V5 tags in VACV-infected cells was demonstrated by Western blotting. Similar results were obtained with HA-tagged EFC proteins (not shown).
FIG 3.
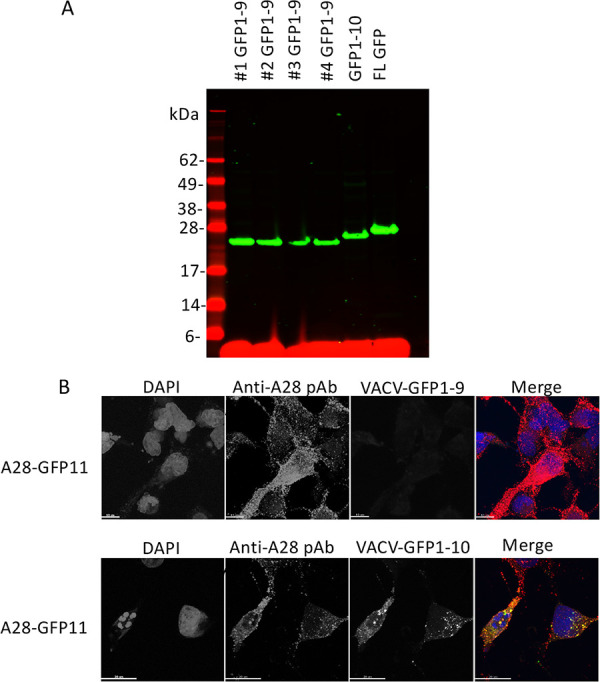
Expression of recombinant GFP1-9 and GFP1-10 by recombinant VACV. (A) RK13 cells were infected with four individual clones of VACV-GFP1-9, VACV-GFP1-10, and a VACV expressing full-length (FL) GFP. Lysates were analyzed by SDS-PAGE, followed by Western blotting with an anti-GFP antibody and a secondary fluorescent antibody. Molecular weight markers are shown on the left. (B) RK13 cells on coverslips were infected with VACV-GFP1-9 or VACV-GFP1-10 and transfected with a plasmid expressing A28-V5-GFP11. After overnight incubation at 37°C, the cells were fixed, permeabilized, and stained with polyclonal antibody (pAb) to A28, followed by fluorescent secondary antibody and DAPI. Green fluorescence was detected in cells infected with VACV-GFP1-10 but not with VACV-GFP1-9. The merge shows DAPI (blue), anti-A28 (red), GFP (green), and overlap of anti-A28 and GFP (yellow).
Interaction of EFC proteins within cytoplasmic factories of infected cells.
The experimental protocol consisted of infecting cells with VACV-GFP1-9 and then transfecting one plasmid expressing an EFC protein with an HA-GFP-10 tag and another with a V5-GFP11 tag simultaneously. All EFC proteins are expressed by VACV-GFP1-9, although the interaction of only the two expressed by the plasmids would be detected by fluorescence. Initial experiments were carried out with cells infected by VACV-GFP1-9 and transfected with plasmids expressing GFP10- and GFP11-tagged EFC proteins known to interact with each other, i.e., A28-H2 (26) and G3-L5 (15). We demonstrated that expression of A28-V5-GFP11 and H2-HA-GFP-10 induced green fluorescence in cells infected with VACV-GFP1-9 (Fig. 4A), whereas expression of A28-V5-GFP-11 alone was only sufficient to induce fluorescence in cells infected with VACV-GFP1-10 (Fig. 3B). Poxviral DNA replication, late gene transcription, and virion assembly all occur in cytoplasmic factory areas. As anticipated, the A28-V5-GFP11 and H2-HA-GFP10, detected with antibodies to the HA and V5 tags, localized in cytoplasmic viral factories that stained with DAPI (4′,6′-diamidino-2-phenylindole) (Fig. 4A). The detection of green fluorescence where GFP11 and GFP10 overlapped indicated their proximity, presumably within the viral membrane. Similar results were obtained when the tags on A28 and H2 were reversed (Fig. 4B) and when G3 and L5 interactions were analyzed (Fig. 4C and D). In contrast there was no significant GFP fluorescence when G3 and L5 had the same HA-GFP10 tag even though the proteins localized in virus factories (Fig. 4E) indicating that the proteins with GFP11 and GFP10 must interact. Furthermore, there was no detectable fluorescence when G3-V5-GFP11 and G3-HA-GFP10 were transfected, indicating that there was low or no self-interaction of G3 with itself that could be discerned by confocal microscopy (Fig. 4F).
FIG 4.

Interaction of EFC proteins and complementation of GFP1-9 occur in cytoplasmic virus factories. RK13 cells on coverslips were infected with VACV-GFP1-9 and transfected with plasmids expressing A28-V5-GFP11 and H2-HA-GFP10 (A), H2-V5-GFP11 and A28-HA-GFP10 (B), G3-V5-GFP11 and L5-HA-GFP10 (C), L5-V5-GFP11 and G3-HA-GFP10 (D), G3-HA-GFP10 and L5-HA-GFP10 (E), and G3-V5-GFP11 and G3-HA-GFP10 (F). Cells were stained with DAPI, mouse antibody to V5, rabbit antibody to HA, and secondary fluorescent antibodies and analyzed by confocal microscopy. GFP fluorescence resulting from tripartite interactions that fluoresce due to DAPI and secondary antibodies was determined. Arrows point to cytoplasmic factories in the DAPI images.
Quantitation of EFC protein interactions by flow cytometry.
Having demonstrated the specificity of the fluorescence for interacting GFP10- and GFP11-tagged proteins and cytoplasmic factory localization, we used flow cytometry to investigate the EFC interactions more sensitively and quantitatively. Cells were gated on fluorescence due to anti-V5 or both anti-V5 plus anti-HA conjugated antibodies and the mean GFP fluorescence intensity was determined. During optimization of the system, we compared the GFP fluorescence with flexible 2 and 12-amino-acid spacers between the epitope tag and GFP-10 or GFP-11. Although the fluorescence was less intense with the short spacer, the assay appeared more stringent, and it was therefore used routinely. Scatterplots obtained with VACV-GFP1-9 alone (Fig. 5A) and with plasmids expressing noninteracting A16 and A28 (Fig. 5B) and interacting H2 and A28 (Fig. 5C) illustrate the gating.
FIG 5.

Flow cytometry gating strategy. (A) Mock: cells were infected with VACV-GFP1-9 and mock transfected. From left to right are the gated cell population, V5-stained cells from the gated cell population, and GFP fluorescence. (B) Negative interaction. Cells were infected with VACV-GFP1-9 and transfected with A16-HA-GFP10 and A28-V5-GFP11. From left to right: gated cell population, V5-stained cells from gated cell population, GFP expression in V5-stained population. (C) Positive interaction. Cells were infected with VACV-GFP1-9 and transfected with H2-HA-GFP10 and A28-V5-GFP11. The images as described for panel B.
We made 11 GFP10/GFP11 sets; each set was comprised of a plasmid expressing one EFC protein with an HA-GFP10 tag as a probe and 11 plasmids expressing the individual EFC proteins with V5-GFP11 tags as baits. The cells were infected with VACV-GFP1-9 and plasmid cotransfections were carried out in triplicate. Negative-control plasmids expressing A28-HA-GFP-10 + H2-HA-GFP-10 and A28-V5-GFP11 + H2-V5-GFP11 were also cotransfected and yielded negligible GFP fluorescence, confirming that both GFP-10 and GFP-11 were required. Each set also contained plasmids expressing interacting A28-HA-GFP-10 and H2-V5-GFP-11 with 12-amino-acid spacers, which gave approximately twice the GFP fluorescence of the shorter spacer. The strong GFP fluorescence of the positive control was set as 100% and used for normalization of each set of interactions since they could not all be carried out at the same time.
We calculated Z-scores based on the GFP fluorescence intensities for probe and bait protein pairs in each set of transfections. The filled bars in Fig. 6 and the “+” symbols in Table 2 indicate positive Z-scores, and “*” and “**” indicate scores >1 and >2 standard deviations above the mean. Note, however, that interaction of the probe with several baits will elevate the mean value of the population and that values below the mean may still be biologically significant. Each EFC protein had one or more interacting partners that provided strong GFP fluorescence. The specificity of the assay was confirmed by the previously determined interaction of A28 with H2, which was found when either A28 or H2 was the probe. The previously known interaction of G3 with L5 was also found when either was the probe, and the GFP fluorescence was similar in intensity to that of the A28-H2 interaction. An additional interaction of G3 with J5 was suggested by fluorescence intensities with positive Z-scores. Most remarkable was the strong GFP fluorescence due to interaction of each EFC probe with O3, suggesting multiple copies of O3 in the complex. The latter idea was consistent with the finding that the highest GFP fluorescence occurred when O3 was the probe and the bait. Because each of the EFC proteins interact with O3, only those exhibiting the highest fluorescence rose above the mean when O3 was the probe. Positive Z-scores suggested self-interactions of A28, F9, G3, and J5. The A16 and G9 paralogs are the largest EFC proteins each with multiple disulfide bonds (Table 1), which might negatively affect their folding and insertion into the viral membrane when expressed by transfection. The latter may explain the relatively low GFP fluorescence intensities when A16 was the probe and the failure to confirm the association of A16 and G9 previously determined in detergent-treated lysates (14) or the interactions of G9 with H2, previously found by chemical cross-linking of intact virions (16),
FIG 6.

Flow cytometry analysis of EFC interactions. RK13 cells in 48-well plates were infected with VACV-GFP1-9, and triplicate wells of each set were transfected with an EFC-GFP10 plasmid and individual EFC-GFP11 plasmids. The cells were suspended with EDTA and transferred to a 96-well plate where they were fixed, permeabilized, and stained with mouse MAb to V5 and secondary fluorescent antibody. Flow cytometry was carried out by gating on V5-positive cells and determining the GFP mean fluorescence. Each set also contained cells transfected with H2-HA-GFP10 and A28-V5-GFP11, which served as a positive control used for normalization of other values. Mock-transfected cells and cells transfected with A28HA-GFP10 and H2V5-GFP10 and with A28V5-GFP11 and H2V5-GFP served as negative controls. The EFC protein attached to GFP10 is indicated near the top of each panel, and the individual EFC proteins attached to GFP11 are indicated below the x axis. The standard errors of the mean are shown. Z-scores were based on the fluorescence intensities for probe and bait protein pairs in each set of transfections. Filled bars indicate positive Z-scores; “*” and “**” indicate >1 and >2 standard deviations above the mean.
TABLE 2.
EFC interactions
| Probe | Baita |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A16 | A21 | A28 | F9 | G3 | G9 | H2 | J5 | L1 | L5 | O3 | |
| A16 | + | + | ** | ||||||||
| A21 | + | ** | |||||||||
| A28 | + | * | ** | ||||||||
| F9 | + | + | ** | ||||||||
| G3 | + | + | * | ** | |||||||
| G9 | + | ** | |||||||||
| H2 | ** | * | |||||||||
| J5 | + | + | ** | ||||||||
| L1 | + | ** | |||||||||
| L5 | ** | + | |||||||||
| O3 | + | + | + | + | ** | ||||||
Z-scores: +, positive; *, >1 log above mean; **, >2 logs above mean.
Confirmation of J5, G3, and L5 interactions.
The proximity assay suggested an interaction of J5 with G3 and possibly with L5. Since interactions of J5 with other EFC proteins had not previously been reported, we used an independent method to confirm these results. Uninfected cells were transfected with combinations of plasmids expressing HA-J5, G3-V5, and L5-myc, and the tags were used for affinity purification with anti-epitope antibody bound to beads. When HA-J5 and G3-V5 were coexpressed, J5 was isolated in association with purified G3, and G3 was isolated in association with purified J5 (Fig. 7A). As a specificity control, neither G3 nor J5 was recovered when the extracts were applied to beads with bound antibody to myc. When HA-J5 and L5-myc were coexpressed, J5 was associated with purified L5 and L5 was recovered with purified J5, whereas neither was obtained from beads with antibody to V5 (Fig. 7B). Finally, when G3-V5 and L5-myc were coexpressed, G3 was associated with purified L5, and L5 was associated with purified G3, whereas neither was captured with anti-HA beads (Fig. 7C). The results are summarized in Fig. 7D.
FIG 7.

J5, G3, and L5 interactions. RK13 cells were transfected with plasmids expressing HA-J5 and G3-V5 (A), HA-J5 and L5-myc (B), and G3-V5 and L5-myc (C). Lysates were prepared and immunopurified by incubation with beads to which anti-V5, anti-HA, or anti-myc antibody had been bound. Input lysates and proteins following immunoprecipitation (IP) were analyzed by SDS-PAGE and Western blotting with antibodies shown on left of each panel. A table summarizing the data is presented in panel D. Plus signs indicate interaction.
When a similar approach in which uninfected cells were transfected with plasmids expressing O3-myc, little or no association with HA-J5 or G3-V5 was detected, suggesting that viral membranes may be needed for such associations (data not shown).
DISCUSSION
Although the EFC proteins are embedded in the viral membrane, the approaches used to determine EFC component interactions have mostly involved a detergent lysis step. The soluble epitope-tagged EFC proteins in the detergent lysate were captured with affinity beads, and the associated proteins were identified by Western blotting or mass spectrometry. Under conditions in which formation of the holocomplex was prevented, three bimolecular interactions—A28:H2, A16:G9, and G3:L5—were discovered (7, 14, 15). The G3:L5 interaction and a close association of J5:F9 and H2:G9 were revealed by chemical cross-linking of proteins in intact virions (16). Nevertheless, additional interactions are required to link together all 11 subunits into a complex.
Proximity assays provide a way to identify neighboring proteins in living cells. We considered that the tripartite system of Cabantous and coworkers (19) would be particularly applicable for our studies since short epitope tags the size of GFP10 and GFP11 were previously attached to the ectodomains of the EFC proteins without compromising their function and the VACV genome was shown to accommodate the large GFP1-10 fragment. An important aspect of the approach used here was expression of all EFC proteins by VACV-GFP1-9, so that the interactions between the GFP-tagged proteins occurred within this setting. Using confocal microscopy, we showed that synthesis of the VACV GFP1-9 and the EFC-linked GFP-10 and GFP-11 was coordinated in time and location and that fluorescence occurred within the virus factory where VACV membranes form and particles assemble. Flow cytometry was used to determine the EFC protein interactions with increased sensitivity and quantitation. Some previous interactions, notably A28:H2 and G3:L5, were confirmed in the proximity assay, and new ones, including a triple interaction of J5, L5, and G3, were suggested. Most remarkable, however, was the proximity of O3 to each of the other EFC proteins. It seems likely that these interactions occur within the membrane, since the N-terminal hydrophobic domain of O3 is sufficient for function (27). Promiscuous binding of O3 to hydrophobic domains is likely since the EFC proteins have diverse sequences. Although the hydrophobic domain of O3 is tolerant of multiple mutations, a segment of similar length and hydrophobicity from another EFC protein could not substitute for O3 (28). A recent finding in an experimental evolution study was that deletion of O3 can be partially compensated by spontaneous mutations in the hydrophobic domains of F9 and L5 (29). Directed mutagenesis studies could extend this analysis to additional EFC proteins.
A diagram illustrating the interactions of the EFC components determined by a variety of methods is shown in Fig. 8. The dashed lines in the diagram illustrate the proximity interactions represented with Z-scores >1 or 2 logs above the mean and illustrated by one or two asterisks in Fig. 6. The interactions between each of the EFC proteins with O3 and the very strong interaction that occurred when O3 was both probe and bait suggest that there may be multiple copies of O3 within the complex, as depicted in the model. Interactions based on copurification and chemical cross-linking are also illustrated (Fig. 8). Since each of the EFC proteins has a transmembrane domain, the EFC may have a flat two-dimensional structure in the viral membrane, which is stabilized by copies of O3. Whether the EFC is a discrete assemblage with a defined number of subunits or exists as an extended interacting protein mat in the viral membrane remains to be determined.
FIG 8.

EFC interaction model. Individual EFC proteins are indicated by spheres. Diameters are relative to molecular weight of individual proteins. Paralogs are in identical colors. Dashed lines connect proteins that together complement GFP1-9 in proximity analysis with Z-scores > 1 log above the mean. Blue lines connect protein that copurify on antibody beads. Red lines connect proteins that were chemically cross-linked.
MATERIALS AND METHODS
Cells.
RK13 cells (ATCC CCL-37) were propagated in minimum essential medium with Earle’s salts supplemented with 10% fetal bovine serum, 100 U of penicillin, and 100 μg of streptomycin per ml (Quality Biologicals, Gaithersburg, MD).
Construction of recombinant VACV.
DNA encoding GFP1-9 and GFP1-10 (19) was inserted into plasmid pRB21 (30) under the control of the early/late synthetic promoter. The plasmids were transfected into cells infected with vRB12, which has a disrupted F13L open reading frame, to allow recombination and formation of large plaques. Virus from large plaques was clonally purified by repeated plaque assays. Expression of GFP1-9 and GFP1-10 were demonstrated by SDS-PAGE and Western blotting with antibody to GFP.
Confocal microscopy.
RK13 cells were grown on glass coverslips and infected with 5 PFU/cell of VACV-GFP1-9 or VACV-GFP1-10. After 1 h at 37°C, the cells were transfected with plasmids mixed with Lipofectamine 3000 (Thermo Fisher), and the incubation continued overnight. The coverslips were then blocked with 3% bovine serum albumin and stained with fluorescence-conjugated rabbit anti-V5 and mouse anti-HA antibodies from Thermo Fisher. Nuclei and virus factories were stained with DAPI. Coverslips were mounted on slides using ProLong Diamond antifade reagent (Thermo Fisher). Images were collected on a Leica SP5 confocal microscope with a 63× oil immersion objective and processed using ImageJ software to adjust the brightness
Affinity purification of proteins and Western blotting.
Cells were washed with cold phosphate-buffered saline and lysed in 20 mM Tris (pH 7.4), 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100 containing protease inhibitor on ice for 30 min. Lysates were centrifuged for 10 min at 20,000 × g at 4°C, and the supernatant was incubated with anti-HA, anti-V5, or anti-myc agarose beads (Thermo Fisher). After extensive washing with lysis buffer, the bound proteins were eluted with lithium dodecyl sulfate (LDS) buffer supplemented with 0.05 M dithiothreitol and resolved by SDS-PAGE. The proteins were transferred to a nitrocellulose membrane and probed with antibodies to HA and V5 from Thermo Fisher and myc from Covance. The membranes were washed three times with Tris-buffered saline, followed by incubation with secondary antibodies conjugated with horseradish peroxidase in blocking buffer for 1 h at room temperature. Protein was detected with SuperSignal West Dura substrate (Thermo Fisher).
Flow cytometry.
RK13 cells were infected with 4 PFU/cell of VACV-GFP1-9 in 48-well plates. After 1 h at 37°C, the cells were transfected with plasmids mixed with Lipofectamine 3000, and incubation was continued for 16 h. The cells were dislodged with EDTA and transferred to a 96-well plate, where they were fixed with 2% paraformaldehyde and permeabilized with Triton X-100. The cells were stained with rabbit anti-HA antibody (Covance) and mouse anti-V5 antibody (Life Technologies), followed by anti-rabbit IgG conjugated to phycoerythrin and anti-mouse IgG conjugated to Alexa Fluor 647. From 50,000 to 150,000 cells were acquired on a FACSCalibur cytometer using CellQuest software (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (TreeStar, Cupertino, CA). Cells were gated on expression of both HA and V5 and green fluorescence determined. Fluorescence was normalized to values obtained in cells infected with VACV-GFP1-9 and transfected with A28-HA-GFP-10 + H2-HA-GFP-11.
ACKNOWLEDGMENTS
We thank Catherine Cotter for providing cells, Andrea Weisberg for adjusting one of the figures, and members of the Genetic Engineering Section for discussions.
A.M.S. and U.S.D. were supported by an NIH Baccalaureate Intramural Research Training Award and the Visiting Fellow Program, respectively. Research was funded by the Division of Intramural Research, NIAID.
Contributor Information
Bernard Moss, Email: bmoss@NIH.gov.
Joanna L. Shisler, University of Illinois at Urbana-Champaign
REFERENCES
- 1.Moss B. 2013. Poxviridae, p 2129–2159. In Knipe DM, Howley PM (ed), Fields virology, vol 2. Lippincott/Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Townsley AC, Weisberg AS, Wagenaar TR, Moss B. 2006. Vaccinia virus entry into cells via a low pH-dependent-endosomal pathway. J Virol 80:8899–8908. 10.1128/JVI.01053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt FI, Bleck CK, Helenius A, Mercer J. 2011. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J 30:3647–3661. 10.1038/emboj.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bengali Z, Satheshkumar PS, Moss B. 2012. Orthopoxvirus species and strain differences in cell entry. Virology 433:506–512. 10.1016/j.virol.2012.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang SJ, Shih AC, Tang YL, Chang W. 2012. Vaccinia mature virus fusion regulator A26 protein binds to A16 and G9 proteins of the viral entry fusion complex and dissociates from mature virions at low pH. J Virol 86:3809–3818. 10.1128/JVI.06081-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laliberte JP, Weisberg AS, Moss B. 2011. The membrane fusion step of vaccinia virus entry is cooperatively mediated by multiple viral proteins and host cell components. PLoS Pathog 7:e1002446. 10.1371/journal.ppat.1002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senkevich TG, Ojeda S, Townsley A, Nelson GE, Moss B. 2005. Poxvirus multiprotein entry-fusion complex. Proc Natl Acad Sci U S A 102:18572–18577. 10.1073/pnas.0509239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moss B. 2012. Poxvirus cell entry: how many proteins does it take? Viruses 4:688–707. 10.3390/v4050688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maruri-Avidal L, Weisberg AS, Moss B. 2011. Vaccinia virus L2 protein associates with the endoplasmic reticulum near the growing edge of crescent precursors of immature virions and stabilizes a subset of viral membrane proteins. J Virol 85:12431–12441. 10.1128/JVI.05573-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maruri-Avidal L, Weisberg AS, Bisht H, Moss B. 2013. Analysis of viral membranes formed in cells infected by a vaccinia virus L2-deletion mutant suggests their origin from the endoplasmic reticulum. J Virol 87:1861–1871. 10.1128/JVI.02779-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maruri-Avidal L, Weisberg AS, Moss B. 2013. Direct formation of vaccinia virus membranes from the endoplasmic reticulum in the absence of the newly characterized L2-interacting protein A30.5. J Virol 87:12313–12326. 10.1128/JVI.02137-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gray RDM, Albrecht D, Beerli C, Huttunen M, Cohen GH, White IJ, Burden JJ, Henriques R, Mercer J. 2019. Nanoscale polarization of the entry fusion complex of vaccinia virus drives efficient fusion. Nat Microbiol 4:1636–1644. 10.1038/s41564-019-0488-4. [DOI] [PubMed] [Google Scholar]
- 13.Nelson GE, Wagenaar TR, Moss B. 2008. A conserved sequence within the H2 subunit of the vaccinia virus entry/fusion complex is important for interaction with the A28 subunit and infectivity. J Virol 82:6244–6250. 10.1128/JVI.00434-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagenaar TR, Ojeda S, Moss B. 2008. Vaccinia virus A56/K2 fusion regulatory protein interacts with the A16 and G9 subunits of the entry fusion complex. J Virol 82:5153–5160. 10.1128/JVI.00162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolfe CL, Moss B. 2011. Interaction between the G3 and L5 proteins of the vaccinia virus entry-fusion complex. Virology 412:278–283. 10.1016/j.virol.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirzakhanyan Y, Gershon P. 2019. The vaccinia virion: filling the gap between atomic and ultrastructure. PLoS Pathog 15:e1007508. 10.1371/journal.ppat.1007508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su HP, Garman SC, Allison TJ, Fogg C, Moss B, Garboczi DN. 2005. The 1.51-Å structure of the poxvirus L1 protein, a target of potent neutralizing antibodies. Proc Natl Acad Sci U S A 102:4240–4245. 10.1073/pnas.0501103102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diesterbeck US, Gittis AG, Garboczi DN, Moss B. 2018. The 2.1 angstrom structure of protein F9 and its comparison to L1, two components of the conserved poxvirus entry-fusion complex. Sci Rep 8:16807. 10.1038/s41598-018-34244-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cabantous S, Nguyen HB, Pedelacq JD, Koraichi F, Chaudhary A, Ganguly K, Lockard MA, Favre G, Terwilliger TC, Waldo GS. 2013. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci Rep 3:2854. 10.1038/srep02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pedelacq JD, Waldo GS, Cabantous S. 2019. High-throughput protein-protein interaction assays using tripartite split-GFP complementation. Methods Mol Biol 2025:423–437. 10.1007/978-1-4939-9624-7_20. [DOI] [PubMed] [Google Scholar]
- 21.Kaddoum L, Magdeleine E, Waldo G, Joly E, Cabantous S. 2010. One-step split GFP staining for sensitive protein detection and localization in mammalian cells. Biotechniques 49:727–736. 10.2144/000113512. [DOI] [PubMed] [Google Scholar]
- 22.Hyun SI, Maruri-Avidal L, Moss B. 2015. Topology of endoplasmic reticulum-associated cellular and viral proteins determined with split-GFP. Traffic 16:787–795. 10.1111/tra.12281. [DOI] [PubMed] [Google Scholar]
- 23.Finnigan GC, Duvalyan A, Liao EN, Sargsyan A, Thorner J. 2016. Detection of protein-protein interactions at the septin collar in Saccharomyces cerevisiae using a tripartite split-GFP system. Mol Biol Cell 27:2708–2725. 10.1091/mbc.E16-05-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu TY, Chou WC, Chen WY, Chu CY, Dai CY, Wu PY. 2018. Detection of membrane protein-protein interaction in planta based on dual-intein-coupled tripartite split-GFP association. Plant J 94:426–438. 10.1111/tpj.13874. [DOI] [PubMed] [Google Scholar]
- 25.Koraichi F, Gence R, Bouchenot C, Grosjean S, Lajoie-Mazenc I, Favre G, Cabantous S. 2018. High-content tripartite split-GFP cell-based assays to screen for modulators of small GTPase activation. J Cell Sci 131:jcs21419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senkevich TG, Moss B. 2005. Vaccinia virus H2 protein is an essential component of a complex involved in virus entry and cell-cell fusion. J Virol 79:4744–4754. 10.1128/JVI.79.8.4744-4754.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satheshkumar PS, Chavre J, Moss B. 2013. Role of the vaccinia virus O3 protein in cell entry can be fulfilled by its sequence flexible transmembrane domain. Virology 444:148–157. 10.1016/j.virol.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satheshkumar PS, Moss B. 2012. Sequence-divergent chordopoxvirus homologs of the O3 protein maintain functional interactions with components of the vaccinia virus entry-fusion complex. J Virol 86:1696–1705. 10.1128/JVI.06069-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tak AI, Americo JL, Diesterbeck US, Moss B. 2021. Loss of the vaccinia virus 35-amino-acid hydrophobic O3 protein is partially compensated by mutations in the transmembrane domains of other entry proteins. J Virol 95. 10.1128/JVI.02228-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blasco R, Moss B. 1995. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 158:157–162. 10.1016/0378-1119(95)00149-z. [DOI] [PubMed] [Google Scholar]
- 31.Ojeda S, Senkevich TG, Moss B. 2006. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J Virol 80:51–61. 10.1128/JVI.80.1.51-61.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Townsley A, Senkevich TG, Moss B. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J Virol 79:9458–9469. 10.1128/JVI.79.15.9458-9469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Senkevich TG, Ward BM, Moss B. 2004. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J Virol 78:2357–2366. 10.1128/jvi.78.5.2357-2366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner PC, Dilling BP, Prins C, Cresawn SG, Moyer RW, Condit RC. 2007. Vaccinia virus temperature-sensitive mutants in the A28 gene produce non-infectious virions that bind to cells but are defective in entry. Virology 366:62–72. 10.1016/j.virol.2007.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown E, Senkevich TG, Moss B. 2006. Vaccinia virus F9 virion membrane protein is required for entry but not virus assembly, in contrast to the related l1 protein. J Virol 80:9455–9464. 10.1128/JVI.01149-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Izmailyan RA, Huang CY, Mohammad S, Isaacs SN, Chang W. 2006. The envelope G3L protein is essential for entry of vaccinia virus into host cells. J Virol 80:8402–8410. 10.1128/JVI.00624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ojeda S, Domi A, Moss B. 2006. Vaccinia virus G9 protein is an essential component of the poxvirus entry-fusion complex. J Virol 80:9822–9830. 10.1128/JVI.00987-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bisht H, Weisberg AS, Moss B. 2008. Vaccinia virus L1 protein is required for cell entry and membrane fusion. J Virol 82:8687–8694. 10.1128/JVI.00852-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Townsley A, Senkevich TG, Moss B. 2005. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is required for cell entry and cell-cell fusion. JVI 79:10988–10998. 10.1128/JVI.79.17.10988-10998.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satheshkumar PS, Moss B. 2009. Characterization of a newly Identified 35 amino acid component of the vaccinia virus entry/fusion complex conserved in all chordopoxviruses. J Virol 83:12822–12832. 10.1128/JVI.01744-09. [DOI] [PMC free article] [PubMed] [Google Scholar]