

Abstract
Autoimmunity plays a significant role in the pathogenesis of demyelination. Multiple sclerosis (MS), neuromyelitis optica spectrum disorders (NMOSD) and myelin oligodendrocyte glycoprotein antibody‐associated disease (MOGAD) are now recognised as separate disease entities under the amalgam of human central nervous system demyelinating disorders. While these disorders share inherent similarities, investigations into their distinct clinical presentations and lesion pathologies have aided in differential diagnoses and understanding of disease pathogenesis. An interplay of various genetic and environmental factors contributes to each disease, many of which implicate an autoimmune response. The pivotal role of the adaptive immune system has been highlighted by the diagnostic autoantibodies in NMOSD and MOGAD, and the presence of autoreactive lymphocytes in MS lesions. While a number of autoantigens have been proposed in MS, recent emphasis on the contribution of B cells has shed new light on the well‐established understanding of T cell involvement in pathogenesis. This review aims to synthesise the clinical characteristics and pathological findings, discuss existing and emerging hypotheses regarding the aetiology of demyelination and evaluate recent pathogenicity studies involving T cells, B cells, and autoantibodies and their implications in human demyelination.
Keywords: AQP4 antibody, autoimmune demyelination, MOG antibody, multiple sclerosis, neuromyelitis optica spectrum disorders, pathology
This review explores the aetiology and pathogenic mechanisms involved in multiple sclerosis, neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein antibody‐associated disease. We explore the overlapping clinical features and compare lesion pathology across these diseases. Various genetic and environmental factors associated with demyelination are discussed and the pathogenic role and potential mechanisms of T cells, B cells and autoantibodies are considered.

Introduction
Central nervous system (CNS) demyelination occurs when the myelin responsible for the insulation of axons is damaged, resulting in poor conduction of action potentials, impaired neuronal signalling and, in some cases, partial or complete neuronal loss. Adaptive immunity enables a rapid and intensive response against subsequent exposures to previously encountered antigens. B and T lymphocytes are the key mediators of this branch of the immune system and are responsible for the humoral and cell‐mediated adaptive immune response, respectively. Antibodies or immunoglobulins (Igs) are specialised proteins produced by B cells with a precise affinity and specificity for their target antigen. Historically, the brain and spinal cord were perceived as immune‐privileged sites, lacking the conventional lymphatic system accessible to the remainder of the body. 1 Recent decades have shed light upon the complexity of ongoing immune trafficking across the blood–brain and blood–cerebrospinal fluid (CSF) barriers. 2 This intricate interplay between the CNS and the immune system has highlighted the notion that certain neurological demyelinating disorders are attributable to an inflammatory autoimmune pathophysiology.
In this review, we explore the complex role of autoimmunity in CNS demyelinating disorders, namely multiple sclerosis (MS), neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody‐associated disease (MOGAD). Firstly, we investigate the similarities and differences in the clinical characteristics and pathologies between the three disease entities. We then consider hypotheses regarding the autoimmune aetiology of these disorders and the mechanisms involved in disease pathogenesis. Lastly, we discuss trends in the diagnosis and therapy of these disorders as well as future directions in the field of autoimmune demyelination.
Clinical and pathological features of autoimmune demyelinating disorders
Multiple sclerosis
Multiple sclerosis is a chronic inflammatory neurological disorder characterised by numerous white matter lesions or plaques throughout the CNS. The prevalence of MS is rising globally with an estimated 35.9 people per 100 000 affected by MS which varies significantly depending on geography and ethnicity. 3 Of those diagnosed with MS, there is a clear female preponderance with females 2–4 times as likely to be affected than males. 3 Relapsing–remitting MS (RRMS) is the most common form of the disease and involves the onset of symptoms that are alleviated during periods of recovery until an eventual subsequent attack. While some patients experience recovery from symptoms during remission phases, others persist with residual disability following attacks. 4 Relapse rates can vary, with reported annualised relapse rates of 0.27–1.66 relapses per year in treatment‐naïve MS patients. 5 Between 50 and 80% of patients with RRMS develop debilitating symptoms which worsen in a progressive manner. 6 At this stage, relapsing–remitting patients are recognised as having transitioned to secondary progressive MS (SPMS). There are limited clinical and pathological indicators that identify the transition of RRMS to SPMS. 4 Approximately 15% of MS patients are diagnosed with primary progressive MS and experience persistent accumulation of neurological deficits from disease onset. 7 Disease‐modifying therapeutics are often inefficacious at alleviating symptoms of the progressive forms of MS. 8
Diagnosis of MS has been achieved by expert consensus as defined by 2010 McDonald criteria and its subsequent revision in 2017 and particularly emphasises the clinical presentation of demyelinating episodes or attacks that are disseminated in space and time. 9 These episodes are complemented by paraclinical findings of typical white matter lesions observed using magnetic resonance imaging (MRI), abnormalities in visually evoked potentials (VEPs) which evaluate the function of visual pathways, and laboratory CSF testing including the presence of oligoclonal bands (OCBs). 10 There are currently no clinical biomarkers that can predict or distinguish between different MS disease courses. 4 , 11
The pathological features of MS lesions strongly support the notion that inflammation‐driven mechanisms contribute to the disease. Most demyelinating sites form confluent, perivenous focal lesions observed throughout the CNS in both white and grey matter regions with variable axonal loss and reactive gliosis. 12 Spatial distribution of MS lesions is partial to periventricular, juxtacortical white matter, infratentorial areas such as the cerebellum and the pons, and short lesions of the cervical and thoracic spinal cord. 13 Lesion pathologies are heterogeneous and vary across MS patients, and are composed of mainly activated macrophages and microglia and CD8+ T lymphocytes, with smaller populations of CD4+ T cells, B lymphocytes and plasma cells 12 , 14 (Figure 1). Although distinct neuropathological patterns have been classically defined, 14 recent recommendations have been made to classify lesions based on the distribution and contents of activated macrophages and microglia. 15 Active lesions can be temporally divided into early or late demyelinating lesions, with the latter characterised by macrophages and microglia containing the more abundant, and thus less readily cleared myelin proteins such as myelin basic protein and myelin proteolipid protein (PLP), while the former also contains these in addition to less abundant, more readily cleared proteins such as myelin oligodendrocyte glycoprotein (MOG) and myelin‐associated glycoprotein. 15 Contrastingly, inactive lesions have significantly reduced numbers of macrophages and microglia, are mostly devoid of oligodendrocytes and predominate in MS patients with extended disease durations and progressive forms of MS. 15 , 16
Figure 1.
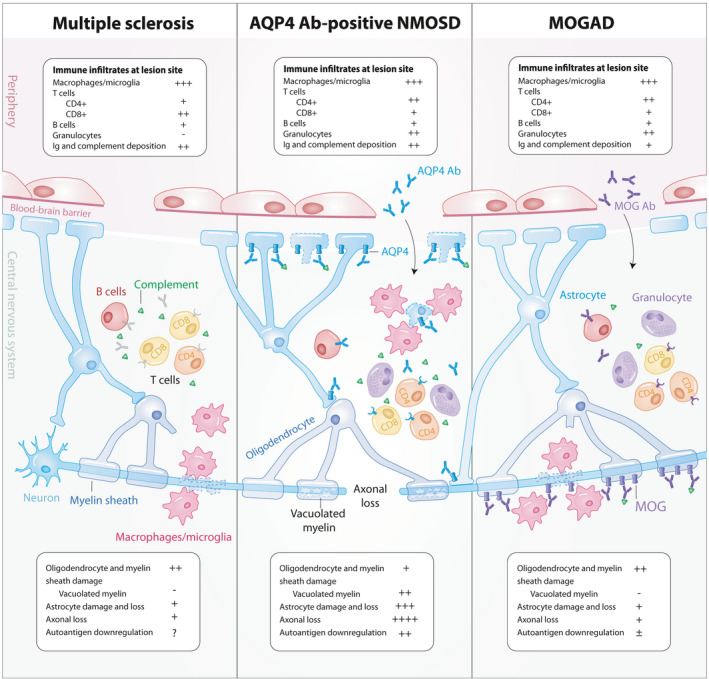
Pathological features of lesions in autoimmune demyelination. Demyelinating lesions commonly consist of immune cell infiltrates predominated by activated macrophages and microglia, lymphocytes, and varying degrees of immunoglobulin and complement deposition. CD4+ T cells outnumber CD8+ T cells in MOGAD and NMOSD while CD8+ T cells predominate in MS. Granulocytic infiltration is seen in MOGAD and NMOSD while not frequently observed in MS lesions. Axon and astrocyte loss is profound in NMOSD while astrocytes and axons are largely preserved in MS and MOGAD. AQP4 downregulation is observed in NMOSD while conflicting reports of MOG internalisation have been seen in MOGAD. Ab, antibody; AQP4, aquaporin‐4 water channel; Ig, immunoglobulin; MOG, myelin oligodendrocyte glycoprotein; MOGAD, MOG Ab‐associated disease; MS, multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorders.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders, previously known as Devic's disease or neuromyelitis optica (NMO), refer to a class of demyelinating syndromes characterised by the co‐occurrence of inflammation in the optic nerves and spinal cord. The prevalence of NMOSD ranges between 0.5 and 4 people affected per 100 000 17 and has a significantly greater female preponderance compared to MS with a female:male ratio of up to 9:1, particularly in Afro‐Caribbean populations. 18 , 19 Although a subtype of MS with optic nerve and spinal cord lesions was historically identified in the Asian population, termed ‘opticospinal MS’, this has since been included under the diagnosis of NMOSD. 20 The age of onset of NMOSD is higher compared to MS and is rarely seen in paediatric patients. 21 NMOSD patients relapse more frequently compared to MS and accumulate greater disability as measured by median expanded disability status scale (EDSS) scores. 21 Furthermore, longitudinally extensive spinal cord lesions were more frequent in NMOSD than in MS. 21
The discovery that the large majority of NMOSD patients harboured IgG antibodies targeting the aquaporin‐4 (AQP4) water channel on astrocytes has since become pivotal in NMOSD diagnosis. 22 , 23 Consensus diagnostic criteria for NMOSD have been defined by Wingerchuk and colleagues in 2007 24 and refined in 2015 20 subdividing patients into AQP4 antibody (Ab)‐positive or AQP4 antibody‐negative NMOSD, with the former making up between 60 and 70% of NMOSD cases. 25 Core clinical characteristics defined for NMOSD include optic neuritis (ON), acute myelitis, area postrema syndrome, acute brainstem syndrome, and observation of NMOSD‐typical brain lesions with diencephalic clinical syndrome and symptomatic cerebral syndrome. 20 Diagnosis of NMOSD requires the observation of at least one of the aforementioned clinical characteristics upon discovery of AQP4 Ab while more stringent diagnostic requirements need to be met if AQP4 Ab serostatus is negative or unknown. 20 The co‐occurrence of other autoantibody‐mediated diseases such as systemic lupus erythematosus, Sjogren's syndrome and myasthenia gravis is observed more frequently in NMOSD compared to MS and can strengthen the diagnosis of NMOSD. 20
Pathology of NMOSD lesions is driven by the binding of pathogenic AQP4 Ab on astrocytic endfeet surrounding endothelial cells and has been long identified as a demyelinating disease that is secondary to a primary astrocytopathy. 26 In contrast to the CD8+ T cell predominance in MS lesions, activated CD4+ T cells infiltrating the CNS are central mediators in NMOSD lesion formation 27 (Figure 1). Preferential localisation of NMOSD lesions in the spinal cord and optic nerves can be accounted for by the higher expression of AQP4 in these regions relative to the brain. 28 Lesions in NMOSD can be present with relative preservation of myelin or as classically demyelinated lesions. Demyelinated lesions are characterised by infiltration of macrophages containing myelin and astrocyte debris, marked axonal loss, astrocytic damage, decreased AQP4 expression, granulocytic inflammation, immunoglobulin and complement deposition, and vacuolated myelin. 26 , 29 , 30
MOG antibody‐associated disease
Myelin oligodendrocyte glycoprotein (MOG) has been extensively studied as a candidate autoantigen in demyelination initially because of its involvement in animal studies of experimental autoimmune encephalitis (EAE), a leading in vivo rodent model of MS. The development of assays which presented MOG in its native conformation was the first to uncover the presence of high titres of MOG Ab in paediatric demyelination cohorts, particularly prominent in children with acute disseminated encephalomyelitis (ADEM). 31 , 32 , 33 , 34 In particular, the live cell‐based assay which involves the recombinant expression of conformational MOG on the cell surface of eukaryotic cell lines has become the gold standard detection method for MOG Ab. Further investigations using these methods revealed the presence of MOG Ab in 20–50% of adults with AQP4 Ab‐negative NMOSD. 25 , 35 , 36 , 37 This has sparked the current consensus to distinguish patients with MOG Ab as a separate disease entity from NMOSD, termed MOGAD.
Despite numerous international recommendations, determination of consensus diagnostic criteria for MOGAD is still ongoing. The typical clinical characteristics of MOGAD include ON, myelitis, and encephalitis, and these phenotypes can be monophasic or relapsing in disease course. 38 The clinical spectrum of MOGAD is continually expanding with emerging reports of MOG Ab associated with seizures and cortical encephalitis. 39 Interestingly, despite the lack of MOG expression in the peripheral nervous system (PNS), MOG Ab has been described in patients with co‐existing inflammatory PNS syndromes. 40 The co‐incidence of these PNS symptoms and their direct association with MOGAD warrants further investigation.
Prevalence studies involving MOGAD cohorts are scarce. In a recent UK study, the prevalence of MOGAD was approximately two per 100 000 with the female:male ratio estimated at 1.8:1, proportionally affecting more men compared to NMOSD and MS. 41 Patients with MOGAD have been reported as having less residual functional impairment compared to AQP4 Ab‐positive NMOSD, 25 , 42 , 43 although this is not universal with some patients, particularly those with a relapsing disease course, suffering permanent visual loss or paraplegia. 44 The co‐existence of MOG Ab and AQP4 Ab in the same individual is seldom reported and occurs in as low as 0.06–8% of patients with demyelination. 45 , 46 A small subset of MS patients are seropositive for MOG Ab. 47 , 48 , 49 In contrast to MS, CSF findings in adult and paediatric MOGAD cohorts show that OCBs are less commonly observed in MOGAD compared to MS, and are seen in 10–30% of patients. 50 , 51 Radiological differences highlight that MOGAD and NMOSD patients with ON are more likely to have bilateral and longitudinally extensive optic nerve lesions compared to MS patients with ON. 52 Comparatively, MOGAD patients with ON more frequently experienced lesions in the anterior visual pathway relative to NMOSD patients with ON in which lesions were observed posteriorly including the optic chiasm and optic tract. 52 Area postrema involvement is less common in MOGAD and more predominant in NMOSD. 53 Similar to NMOSD, longitudinally extensive transverse myelitis (TM) is observed in MOGAD and rarely observed in MS. 54 Short lesions of TM similar to those seen in MS also occur in MOGAD. 55 While there is currently no clinical predictor of relapse in MOGAD patients, persistent MOG Ab seropositivity has been associated with a relapsing disease course while those with transient seropositivity were more likely to be monophasic. 56 , 57
MOGAD lesions are characterised by perivenous, confluent demyelination, axonal preservation, reactive astrocyte gliosis, prominent intracortical demyelination, with immune cell infiltrates, complement deposition and evidence of oligodendrogliopathy. 58 , 59 Granulocytes and CD4+ T cell infiltrates are observed in high frequencies in MOGAD lesions with astrocyte populations preserved and AQP4 expression sustained in contrast to NMOSD 58 (Figure 1). Intracortical lesions were more frequent in MOGAD than in MS and NMOSD. 58 Similar to MS, subpial demyelination was also observed in MOGAD. 58 , 59 Complement and Ig deposition were less frequently observed in MOGAD lesions compared to NMOSD. 59 Although AQP4 internalisation is an established pathogenic mechanism in NMOSD, some reports have observed MOG‐dominant myelin loss at MOGAD lesions, 59 while others have shown evidence of complement deposition without selective loss of MOG expression. 58
Autoimmune aetiology and triggers of CNS demyelination
While clinical presentations are similar between MS, NMOSD and MOGAD, differences in the origin of the autoimmune response in each disease remain to be explored and these have distinct implications to our understanding of each disease entity. Currently, the location of the initiating pathogenic event in demyelination remains contentious. In the ‘outside‐in’ model, disease pathogenesis begins peripherally, in which autoreactive immune cells traffic into the CNS and elicit an autoimmune response. EAE rodent models are a classical demonstration of this, whereby demyelination is initiated through immunisation of adjuvant‐administered myelin proteins resulting in expansion and activation of myelin‐reactive CD4+ T cells. 60 , 61 , 62 , 63 , 64 The likelihood of peripheral immune‐mediated pathogenesis is strengthened by large genome‐wide association studies which have shown MS susceptibility to be enriched in genes associated with B and T lymphocytes, natural killer cells and microglia. 65 , 66 Conversely, an ‘inside‐out’ model posits that disease is resultant of pre‐existing damage to oligodendrocytes and myelin, prompting immune cell recruitment to sites of injury, following which an inflammatory response proceeds. This is exemplified in cuprizone‐induced models of demyelination which directly elicits oligodendrocyte cytotoxicity and subsequent inflammatory demyelination. 67 Furthermore, alterations to myelin‐related genes support this ‘inside‐out’ model of disease pathogenesis, as seen in PLP1 missense mutations found in a subset of MS patients and cause oligodendrocyte apoptosis. 68 While continued investigation is warranted into the compartment of disease onset, several genetic and environmental factors have been commonly investigated across these diseases.
HLA genotypes
The presence of HLA‑DRB1*15:01 and the absence of HLA‑A*02 have been frequently associated with a significantly increased risk of developing MS. 69 In some cases, MS has been postulated to be a neurodegenerative disorder where the inflammatory response is secondary to neurodegeneration. 70 , 71 However, in a multinational genome‐wide association study of 9772 cases of MS, genes relevant to inflammation‐independent neurodegenerative pathways lacked an association to MS, while genes for CD4+ T cell differentiation were over‐represented and implicated in disease pathogenesis. 66 In a next‐generation sequencing study of 31 Japanese NMOSD patients, HLA‐DQA1*05:03 was found to be significantly associated with NMOSD. 72 More recently, in a cohort of 165 NMOSD patients, HLA‐DRB1*08:02 and HLA‐DPB1*05:01 were found to be susceptibility alleles while HLA‐DRB1*09:01 was found to be protective. 73 In contrast, HLA‐A*01, HLA‐B*08 and HLA‐DRB1*03 were associated with NMOSD in a recent Dutch study while no association between HLA and MOGAD was observed. 74 This study, however, did not separate adult and paediatric MOGAD cohorts, whereas in a recent Chinese study of 95 MOGAD patients (51 paediatric, 44 adult), HLA‐DQB1*05:02–DRB1*16:02 alleles were associated with paediatric onset of MOGAD while no association between adult‐onset MOGAD and HLA genotype was observed. 75
Infectious prodrome
Viral infection is among many environmental factors that have been commonly linked to or seemingly preceding autoimmune disease. Arguably, most renowned is the association between MS and Epstein–Barr virus (EBV) with almost all MS patients having been previously infected with EBV. 76 EBV Ab seropositivity is observed at higher rates in MS patients and is strongly correlated with disease onset. 77 , 78 Additionally, EBV‐induced infectious mononucleosis is found to be associated with a twofold risk of developing MS. 77 Autoreactive B cells latently infected with EBV are proposed to contribute to MS pathogenesis by evading elimination of CD8+ T cells and accumulating within the CNS, producing myelin‐reactive antibodies and survival signals for T cells. 78 Induction of MOGAD by a viral infection has also been reported, with the most common occurrence observed in paediatric patients who develop ADEM following viral infection. Postinfectious MOGAD cases include HSV‐1 infection followed by ADEM 79 and ON 80 and rubella infection followed by ON. 81 Up to 40% of patients experience a non‐specific viral prodrome with or without fever, prior to clinical onset. Most recently emerging, two reported cases of SARS‐CoV‐2 infection were followed by MOG Ab‐associated ON 82 and NMOSD. 83 Despite these cases, the association between viral infection and demyelination remains unclear and leading hypotheses regarding molecular mimicry of myelin proteins because of their structural overlap with viral proteins remain largely unvalidated in humans. Other than viral infection, there is also emerging evidence regarding non‐infectious clinical prodromes preceding autoimmune demyelination. Within 5 years of disease onset, patients with MS were reported to have greater fatigue, sleep disorders, anaemia and pain compared to healthy controls. 84 Neuropathic pain has been reported in MOG Ab‐ and AQP4 Ab‐positive patients, 85 with another study identifying prodromal headaches in almost 50% of patients with MOG Ab‐positive ON. 86
Vitamin D and UV exposure
Low vitamin D levels, insufficient UV exposure and higher latitudes have each been linked to an increased risk of developing MS. 87 In a recent study of two independent multicentre cohorts, lower disease severity was seen in MS patients with higher vitamin D levels. 88 Although UV exposure is correlated with the maintenance of sufficient levels of vitamin D, the role of sun exposure in the development of MS may be vitamin D‐independent. 89 For instance, while whole‐body UV irradiation of mice prevents the development of EAE, 90 UV‐induced suppression of EAE was still observed in mice lacking the vitamin D receptor. 91 Interestingly, in a recent cross‐sectional study of 29 NMOSD patients, increased sun exposure and serum calcifediol (a vitamin D metabolite) were seen in AQP4 Ab‐negative NMOSD patients compared to AQP4 Ab‐positive patients, potentially implicating a role of vitamin D in AQP4 Ab synthesis. 92 Presently, associations of vitamin D and sun exposure have yet to be explored in MOGAD cohorts. 93
Paraneoplastic syndromes
A paraneoplastic association has been described in a few cases of MOGAD and AQP4 Ab‐positive NMOSD and may be associated with the pathogenesis of demyelination. More recently, expression of oligodendrocyte markers, including MOG, and CD4+ and CD8+ T cell infiltration have been described in the teratoma tissue of a MOG Ab‐positive ON patient with ovarian teratoma. 94 This is in contrast to the more frequently associated lung and breast adenocarcinomas in a small number of patients with NMOSD 95 ; however, cases of ovarian teratomas in NMOSD have also been reported. 96 A patient positive for CSF MOG Ab and presenting with longitudinally extensive TM, BON, and brainstem encephalitis coincidentally with lung adenocarcinoma has been recently reported 97 ; however, it is clear that the paraneoplastic nature of demyelination is rare and warrants further investigation.
Gut microbiota
Intestinal microbiota influence both local and systemic immune responses through its interaction with the gut–brain axis and offer several hypotheses regarding the origin of the autoimmune response. A recent study found that RRMS patients with active disease had increased numbers of intestinal Th17 cells, and expansion of this population was reliant on the composition of gut microbiota. 98 An increased relative abundance of anti‐inflammatory Prevotella strains was seen in healthy controls and RRMS patients without active disease. 98 More recently, clonally expanded IgA‐expressing B cells have been found to actively traffic to the CNS with specificity against certain phyla of gut microbiota and were detected in the CSF and tissue of active MS‐associated lesions and could therefore serve as a systemic biomarker for active disease in MS. 99 Recent investigations into the involvement of gut microbiota in NMOSD pathogenesis have revealed the role of Clostridia perfringens in the disease. This strain was enriched in NMOSD patients and has been implicated in the regulation of Treg and Th17 cell populations. Specifically, AQP4‐specific T cells in NMOSD patients exhibited a Th17 phenotype and cross‐reacted with a C. perfringens peptide sequence. 100 A newly isolated Erysipelotrichaceae bacterium has been recently described that when co‐cultured with Lactobacillus reuteri which has homology to MOG peptides, Th17 inflammatory responses were induced from MOG‐specific Th17 cells and increased EAE severity. 101 Other instances of molecular mimicry to MOG have been demonstrated in a small proportion of MS patients harbouring antibodies targeting MOG peptides which cross‐reacted with the bovine milk protein butyrophilin. 102 Additionally, butyrophilin has been shown to induce a MOG‐specific T‐cell response in rats, while also having the potential to produce a tolerogenic response and alleviate EAE. 103 As this was investigated in the context of MS, which is now known to comprise a small minority of MOG Ab‐positive patients, these findings have yet to be investigated in a MOGAD cohort.
Pathogenic mechanisms in autoimmune demyelination
While MS has been classically considered a T cell‐driven disease, the efficacy of B‐cell‐targeted therapies has warranted recent investigation into the regulatory or antigen‐presenting role of B cells. 104 , 105 , 106 Conversely, even though NMOSD and MOGAD have a clear antibody association, the role of antigen‐specific T cells in these disorders remains largely undefined. Overall, dysregulation of both cell‐mediated and humoral compartments of the adaptive immune system likely exists across MS, NMOSD and MOGAD.
T cells
The contribution of pathogenic T cells has been considerably explored in the context of MS which has been classically described as a T cell‐mediated disease. In particular, in EAE, the role of CD4+ Th17 cells is well established as being the primary mediators of disease pathogenesis 107 with recent findings showing increased frequencies of distinct Th17 subsets in the blood and CSF of patients with RRMS compared to SPMS. 108 Regardless of disease course, CD8+ T cells in MS lesions far outweigh the frequency of other infiltrating lymphocytes such as B cells and CD4+ T cells. 109 CD8+ T cells are enriched in the CSF of RRMS patients early in disease, 110 and these CD8+ T cells found in the CSF shared the same repertoire as CNS‐resident CD8+ T cells. 111 However, the role these CD8+ T cells play in disease is not well understood. For instance, myelin‐specific CD8+ T cells from MS patients have demonstrated a proinflammatory profile when expanded in vitro and expressed CD20 in greater proportions compared to control subjects. 105 These myelin‐specific CD20+CD8+ T cells were preferentially depleted by anti‐CD20 therapy which has been shown to be effective in MS treatment. 105 On the contrary, a subset of clonally expanded regulatory CD8+ T cells have been recently described to suppress the pathogenicity of CD4+ T cells in EAE, 112 and thus, CD8+ T cells may play a multifaceted role as either a regulatory or pathogenic mediator of human demyelination.
Antigen‐specific T cells in NMOSD and MOGAD have been seldom detected despite central dogma for the necessity of these cells in their role for autoantibody production and pathogenicity. Interestingly, AQP4‐specific CD4+ T cells were detected in AQP4 Ab‐positive patients, with T cell reactivity observed in a peptide sequence at amino acids 156–170 which overlaps with the epitope recognised by AQP4 Ab. 113 , 114 Contrastingly, the same study could not detect MOG‐specific T cells in MOG Ab‐positive patients and this was postulated to be because of the failure of synthetic peptides in mimicking antigen processing and MHC presentation. 113 Indeed, a previous study using Escherichia coli‐expressed MOG coupled to fluorescent beads was able to detect MOG‐specific CD4+ T cells producing IFN‐gamma, IL‐22 and IL‐17A in 16 out of 29 MS patients. 115 However, MOG Ab was detected in only one of these patients reinforcing the notion that MOG Ab seropositivity in MS patients is rare and that the role of MOG‐specific T cells in MS patients remains difficult to explore. 115 Although antigen‐specific T cells in demyelination have been challenging to isolate, the cytokine profile of demyelination patients implicates the role of specific T cell subpopulations. The proinflammatory Th17‐related cytokines IL‐6 and G‐CSF were elevated in paediatric MOGAD patients compared to MOG Ab‐negative patients with demyelination. 116 In a cross‐sectional study, adult and paediatric MOGAD patients were found to have an upregulation of Th17, Th1‐related and Treg‐related cytokines. 117 These cytokines were similar to those upregulated in the CSF of AQP4 Ab‐positive NMOSD patients; however, they differed significantly from MS patients. 117
B cells and antibodies
The initiation of an autoimmune response against CNS antigens can lead to the breach of the blood–brain barrier (BBB) to allow leukocyte infiltration, antibody deposition and inflammation in the local CNS region. 118 , 119 , 120 Autoantibodies are implicated in the exacerbation of autoantigen destruction through executing effector functions such as the activation of complement‐dependent cytotoxicity (CDC) or antibody‐dependent cellular cytotoxicity (ADCC). 121
Even though a central autoantigen in MS remains unidentified, evidence exists for the contribution of humoral mechanisms in disease pathogenesis in addition to the T cell‐driven response. 109 , 122 Intrathecal OCBs are a hallmark feature of MS, in which the presence and quantity are strongly associated with disease severity and disability in patients. 123 , 124 , 125 , 126 , 127 Several studies have evidenced the role of B cells in the production of OCBs, 128 , 129 , 130 thus implicating the importance of the humoral response in promoting characteristic MS demyelination. This is further supported by the observation that B cells are responsible for mediating brain homing Th1 cell autoproliferation, and B‐cell depletion by anti‐CD20 therapies reduces T cells and inflammation both in vitro and in patients with MS. 104 MS patients commonly display elevated intrathecal Ig, specifically IgG1 and IgG3. 131 These findings have been supported by recent studies observing higher serum IgG3 may be predictive of the progression of CIS towards MS. 132 , 133 Subsequent functional assays on antibody pathogenicity in animal and human models have confirmed MS lesion observations. Injection of CSF from MS patients was observed to induce demyelination and axonal damage in mice. 134 Evidencing the specific role of antibodies in this process, recombinant IgG1 from MS patient CSF was observed to initiate CDC, rapid demyelination and microglia activation on mouse organotypic cerebellar slices. 135 In addition to CSF antibodies, serum autoantibodies have the capacity to disrupt the BBB, target brain microvessels and initiate destruction of myelin. 136
Defining a putative autoantibody responsible for MS demyelination is encumbered by the potential heterogeneity of target autoantigens in MS. Various autoantibody targets have been investigated (Table 1). For instance, the presence of isotype‐switched antibodies against myelin PLP is significantly elevated in patients with RRMS and SPMS who have specific HLA types, and PLP Ab titres correlated with disease severity. 137 Additionally, a subgroup of MS patients with brainstem and cerebellar lesions have been identified with PLP antibodies. 138 In other cases, the pathogenic potential of these autoantibodies has been evidenced through the initiation of CNS inflammation following the passive transfer to rodent models 139 ; however, validation of their pathogenic roles in humans remains unprecedented. Distinct subgroupings of MS patients with precise disease course, clinical and radiological features may facilitate discovery of new autoantigenic targets and discrete clinical entities, as has been the case for the discovery of AQP4 Ab and MOG Ab.
Table 1.
Summary of evidence for the pathogenicity of demyelinating autoantibodies
| Pathogenicity criteria | MS | NMOSD | MOGAD |
|---|---|---|---|
| Autoantibodies present in affected patients |
MOG Ab 0–5% of adult MS patients 47 , 48 , 49 (dependent on selection criteria and cohort size) ADEM patients with persistent MOG Ab titres over 5 years were eventually diagnosed with MS 166 |
AQP4 Ab Inflammatory demyelinating CNS diseases: 18.1% 164 |
MOG Ab Optic neuritis: 1.7–4% 194 , 195 Paediatric ADEM: 40% 31 AQP4 Ab‐negative NMOSD: 39% 35 |
|
PLP Ab Isotype‐switched PLP Ab present in 84% of MS patients 137 |
ANA Ab 36% (52/143) in AQP4‐positive NMOSD cohort 196 |
||
|
NFL Ab Progressive and relapsing–remitting MS associated with higher levels of NFL and NFL Ab 197 | |||
|
NFM Ab Increased NFM Ab in MS patients 198 | |||
|
HERV‐W Ab 19/22 (86%) patients with relapsing–remitting MS (RRMS) harboured HERV‐W Ab; compared to 7/22 (32%) AQP4 Ab‐positive and 20/22 (91%) MOG Ab‐positive patients 199 | |||
|
Kir4.1 Ab 46.9% MS patients harboured Kir4.1 Ab, 139 but varied prevalence in subsequent studies; from 8 to 28%, 200 , 201 , 202 , 203 and a number of studies detected no significant level of Kir4.1 Ab in MS patient serum 204 , 205 , 206 | |||
|
Talin1 Ab Talin1 Ab levels increased in MS compared to controls, but negatively correlate to demyelination activity 207 | |||
|
Recombinant IgG1 from MS patient CSF target myelin and astrocyte‐specific antigens on mouse organotypic cerebellar slices and cause oligodendrocyte loss and demyelination through CDC and microglia activation 135 Autoreactive antibodies found in phagocytic macrophages within active demyelinating lesions; may be contributing to lesion formation or initiation 208 Astrocytes in active MS lesions are immunoreactive to and express Kir4.1 25 Antibody and complement deposition on CNS lesions in patients with MS 209 , 210 Deposition of Ig and complement (C9neo) in areas of active demyelination, alongside activated macrophages with immunoreactivity to myelin antigens, Ig and C9neo 211 | |||
| Antibody interacts with target antigen |
AQP4 Ab induces demyelination through complement‐dependent cytotoxicity, antibody‐dependent cell‐mediated cytotoxicity and inflammation initiated by granulocytes and macrophages 141 Necrotic CNS lesions display AQP4 loss and deposits of Ig and complement 30 , 209 , 212 Serum AQP4 IgG1 from NMO patients binds to AQP4‐expressing cells to activate complement and initiate endocytosis 213 , 214 ADCC‐mediated cell death when incubated with AQP4 Ab‐positive patient serum and NK cells 140 |
Human MOG Ab induces complement‐mediated myelin loss in murine organotypic brain slices 215 Human 37 and murine 171 MOG Ab causes rearrangement and destabilisation of oligodendrocyte cytoskeleton Children with MOG Ab harbour elevated IL‐6, G‐CSF and deposits of IgG, C1q and activated microglia around early brain lesions 169 |
|
| Passive antibody transfer reproduces disease features | Injection of Kir4.1 Ab in mice increased astrocyte expression of glial fibrillary acidic protein, activated complement cascade surrounding Kir4.1 and decreased Kir4.1 expression. 139 However, importance of this autoantigen in MS patients is debated |
Monoclonal AQP4 Ab transferred to rats induces CNS lesion formation and AQP4 loss without assisted antibody entry into CNS 161 Passive transfer of human AQP4 Ab to rats causes infiltration of inflammatory cells to CNS and exacerbation of lesion formation 216 Recombinant AQP4 Ab derived from NMOSD patients initiates perivascular astrocyte depletion, myelinolysis and deposition of complement and Ig in rats with EAE 151 |
Affinity‐purified MOG Ab induces complement‐dependent demyelination when co‐transferred with MOG‐specific T cells in rat models of EAE 173 |
| Immunisation with antigen produces model disease |
MOG Mice sensitised with MOG35–55 peptide induces an acute and reproducible EAE phenotype 61 Immunisation with non‐inflammatory MOG mRNA suppresses EAE in mice 191 EAE exacerbated after passive transfer of MOG monoclonal IgG in mice, causing extensive demyelinating plaques and fatal relapses 217 |
Injection of AQP4 peptide to mice with experimental autoimmune myasthenia gravis aggravates disease symptoms 158 Rats immunised with mimotopes of conformational AQP4 epitopes produce AQP4 Ab detectable in sensitive cell‐based assays; however, no concurrent pathology was observed 218 Encephalomyelitic syndrome was initiated when Rag1−/− mice reconstituted with mature T cells of AQP4−/− mice were immunised with AQP4. NMO‐specific lesions within CNS occur only with the additional presence of AQP4 Ab 163 |
Macaques immunised with rhMOG/IFA develop EAE comparable to MOGAD in children; increased IL‐6 and G‐CSF cytokines and early brain lesions with deposits of IgG, C1q and activated microglia 169 |
|
PLP Mice immunised with PLP139–151 produce EAE which models clinical relapses and RRMS 62 | |||
|
MBP Mice immunised with bovine 63 or rat 64 MBP produce a monophasic EAE model | |||
| Reduction of antibody levels ameliorates disease |
Therapeutics reducing inflammation and immune response ameliorate MS symptoms and treat relapse, including prednisone, methylprednisone, plasmapheresis and ocrelizumab 104 Relapse rate higher in MOG Ab‐seropositive patients 131 , 219 |
AQP4 Ab seropositivity is predictive of relapse and titres increase during relapse 220 , 221 , 222 High titres associated with increased disease activity (complete blindness or extensive CNS involvement) 223 Treatment with rituximab causes significant improvement of disease activity most patients, 224 and AQP4 Ab concentration decreases in serum after immunosuppressive therapy 222 Chimeric, high avidity AQP4 Ab blocks patient IgG binding to AQP4, thus preventing CDC in vitro 225 Coexistence of AQP4 and ANA Ab associated with more severe disease phenotypes 196 |
Disease ameliorated upon treatment with steroids and rituximab 44 High MOG Ab titres are predictive of a more severe, relapsing course and increase during active disease 56 , 168 |
Ab, antibody; ADCC, antibody‐dependent cellular cytotoxicity; ADEM, acute disseminated encephalomyelitis; ANA, anti‐nuclear antibody; AQP4, aquaporin‐4; CDC, complement‐dependent cytotoxicity; CSF, cerebrospinal fluid; EAE, experimental autoimmune encephalitis; EAMG, experimental autoimmune myasthenia gravis; G‐CSF, granulocyte colony‐stimulating factor; HERV‐W, human endogenous retrovirus‐w; Ig, immunoglobulin; IL‐6, interleukin 6; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; MOGAD, MOG antibody‐associated disorder; mRNA, messenger ribonucleic acid; MS, multiple sclerosis; NFL, neurofilament light; NFM, neurofilament medium; NK cell, natural killer cell; NMOSD, neuromyelitis optica spectrum disorder; PLP, myelin proteolipid protein; rAb, recombinant antibody; rhMOG/IFA, recombinant human MOG/incomplete Freund's adjuvant; RRMS, relapsing–remitting multiple sclerosis; SLE, systemic lupus erythematosus.
The mechanisms of AQP4 Ab pathogenesis have been investigated extensively through functional assays in animals and human tissue. An early in vitro study revealed that serum‐derived IgG from NMOSD patients binds to astrocytes and increases BBB permeability to permit autoimmune reactivity within the CNS. 140 Further, human AQP4 Ab binding to astrocytes initiates injury, secondary oligodendrocytopathy and demyelination through CDC, ADCC or induction of inflammation through granulocyte activation. 141 , 142 , 143 The process by which astrocytopathy leads to demyelination in NMOSD remains unclear. Bystander formation of membrane attack complexes in the process of CDC has been shown in neurons and oligodendrocytes co‐cultured with astrocytes. 144 , 145 Other studies demonstrate internalisation and downregulation of the astrocytic glutamate receptor, EAAT2, alongside AQP4 upon Ab binding, 146 and have therefore implicated glutamate excitotoxicity to oligodendrocytes as a contributing factor in disease pathogenesis. 147 The production of recombinant antibodies from NMOSD patient periphery‐ 148 , 149 , 150 and CSF‐derived 149 , 151 , 152 B cells revealed that AQP4 Ab initiates pathogenic effects both in vitro and in vivo because of defective central and peripheral B cell tolerance mechanisms. Recombinant AQP4 Ab was observed to specifically bind to the H101/L104 epitope and initiate CDC. 152 Supporting these findings, recent clinical trials indicate that eculizumab, a complement inhibitor, reduces relapse rates in patients. 153 Additionally, the IL‐6 cytokine pathway has been strongly implicated in NMOSD pathogenesis 154 , 155 , 156 and using antibodies against the IL‐6 receptor has demonstrated a strong potential to block this pathway through reducing the survival of AQP4 Ab‐secreting plasmablasts. 157
Evidence of disease initiation and exacerbation following passive transfer of AQP4 Ab further outlines the pathogenic role of these autoantibodies. Immunisation with AQP4 peptides exacerbated experimental autoimmune myasthenia gravis symptoms in mice, including fatigue, weakness and decreased nerve responsiveness. 158 The administration of recombinant AQP4 Ab to rodents has provided strong evidence to support in vivo studies. Recombinant AQP4 Ab generated from NMOSD patient CSF‐derived plasma cells was observed to initiate perivascular astrocyte depletion, myelinolysis, and Ig and complement deposition in EAE rats. 151 In line with functional assays on pathogenicity, the observation that seropositivity is associated with increased chances of opticospinal involvement, longitudinally extensive spinal cord lesions, relapse rate and lower EDSS scores further evidences the role of the autoantibodies in driving demyelinating pathogenesis. 159 Although the mechanism of pathogenesis initiation within the CNS is still debated, current evidence points towards a peripherally initiated response in NMOSD. For instance, intrathecal production of AQP4 Ab is rarely observed in NMOSD patients. 160 Additionally, a recent report demonstrated that systemic injection of AQP4 Ab has a number of entry sites into the CNS and produced NMOSD‐like lesions. 161 Although some studies report the necessity for T cells in AQP4 Ab lesion formation, 162 others suggest that AQP4 Ab alone can induce NMOSD pathology without T cell help. 161 It is likely that both pathways are required in NMOSD pathogenesis, as indicated in a study which observed that while immunisation with AQP4 initiates encephalomyelitis in Rag1−/− mice with the presence of T cells alone, lesions are only produced in the presence of both antibodies and T cells against AQP4. 163
MOG Ab has been shown to contribute to CNS demyelination through CDC or ADCC‐mediated lysis of MOG‐expressing cells. 31 , 164 , 165 These findings are supported by the observation that the majority of MOG Ab are IgG1, 33 , 166 , 167 , 168 , 169 a subclass efficient in fixing complement and binding to Fc receptors for ADCC initiation although the potency of these effector mechanisms may be dependent on autoantibody affinity. 170 Further studies suggest murine and human MOG Ab induce cytoskeleton disruption and microtubule destabilisation. 37 , 171
Several studies have further observed CNS inflammation and complement‐dependent lysis of MOG‐expressing cells following passive transfer of high‐affinity patient MOG Ab to rodents. 172 , 173 A recent report demonstrated the pathogenicity of an antibody derived from the RNA of post‐mortem human brain tissue and showed similar demyelinating activity to a prototypical murine‐derived MOG Ab. 174 However, these studies use rodent‐reactive antibodies which are only present in a minor proportion of MOGAD patients, with between 70 and 80% of human MOG Ab recognising a conformational epitope at position 42 of the extracellular domain of MOG where proline exists in humans as opposed to serine in rodents. 168 , 175 Autoantibody pathogenicity in a primate model has since been demonstrated in an in vivo macaque model in which complement‐dependent vacuolisation of myelin, lesion pathology, EAE and MOG Ab were observed after injection of recombinant human MOG. 169
Current trends and future directions
Standardised serological testing
With ongoing attempts to make the delineation between MS, NMOSD and MOGAD progressively clearer, discernment of the correct disease entity remains reliant on evaluation of clinical presentations and accurate paraclinical testing. Increasingly evident is the need to standardise assay methodologies for the accurate detection of demyelination‐associated autoantibodies. 109 , 176 Several multicentre studies over recent years have evaluated the accuracy, sensitivity and specificity of antibody testing in NMOSD and MOGAD. A multicentre study of 21 AQP4 Ab assays across 15 European centres found that cell‐based assays using microscopy produced the highest sensitivities and specificities. 177 It also further reinforced the use of the M23 isoform of AQP4, with improved sensitivity in contrast to the M1 isoform, 177 attributed to findings that formation of orthogonal array of particles (OAPs) exclusive to the M23 isoform, is important for pathogenic human AQP4 Ab binding. 141 Recent validation studies have demonstrated lower concordance in MOG Ab testing when determining the presence of MOG Ab in patients with borderline and low MOG Ab titres tested across international centres. 178 Differences in assay methodologies, data analyses and cut‐offs for MOG Ab positivity largely account for these discrepancies. 179 , 180 Moreover, studies on the isoform of MOG have shown that C‐terminal truncation of the full‐length protein decreases binding of human MOG Ab 181 and is likely because of the effect of truncation on the second hydrophobic domain of MOG which has recently been revealed to be important in the bivalent binding of human MOG Ab. 170 Although major human MOG Ab epitopes have been described in the extracellular domain of the protein, 168 , 175 it has been postulated that the transmembrane domains and cytoplasmic tail may play an important role in the surface expression and oligomerisation of the antigen. Further, the use of fixatives in cell‐based assays has been shown to alter the native conformation of MOG and significantly decrease the sensitivity of the MOG Ab test. 168 , 178 Hence, cell‐based assays using live cells are the current gold standard in the detection of these antibodies, 38 , 178 , 182 and therefore, translation and standardisation of these assays for routine diagnostic testing are highly warranted.
Recent studies are also shedding light on the co‐occurrence of disease‐associated autoantibodies in CNS neurological disorders. For instance, the co‐existence of NMDAR Ab and other autoantibodies associated with autoimmune encephalitis in MOGAD and AQP4 Ab‐positive NMOSD patients has been observed. 183 , 184 AQP4 Ab‐positive NMOSD has been found to occur in patients with AChR Ab‐positive myasthenia gravis with NMOSD onset most commonly occurring after diagnosis of MG. 185 This was further reflected in antibody titres, with AChR Ab decreasing as AQP4 Ab increased over the disease course.
Predictors of disease
Predicting disease course remains a significant challenge as residual disability and worsening of clinical outcomes often accumulates with further relapses in MS, NMOSD and MOGAD. Intriguingly, a recent study found an association between epitope binding patterns and likelihood to relapse, with 75% of adult MOGAD patients that did not recognise the Proline42 epitope, the major immunodominant binding region of human MOG Ab, presented with a relapsing disease course. 168 This provides evidence that characterising the epitope of autoantibodies in demyelination may prove useful in predicting disease course, thereby enabling refinement of therapeutic pathways. Characterisation of binding patterns in AQP4 Ab‐positive NMOSD showed a change in AQP4 Ab epitope in only a minority of patients over the disease course. 186 Similarly, the immunodominant epitope in MOGAD patients also remains stable over time. 168 Additionally, while MOGAD patients exhibited the same Ab characteristics in serum and CSF, paired CSF‐serum samples containing AQP4 Ab taken at the same time exhibited different binding patterns potentially suggesting the contribution of two independent sources of antibody production in NMOSD. 186
Given the exemplars of MOG Ab and AQP4 Ab, the discovery and validation of new biomarkers may have significant implications in predicting disease features. Axonal damage is variable across demyelinating diseases and can be measured by neurofilament levels. Recent findings have shown elevated blood and CSF neurofilament levels in RRMS patients associated with disease activity, disability score and more frequent relapse. 187 Additionally, in 18 MOGAD patients followed longitudinally, elevated neurofilament light‐chain levels at baseline remained stable or decreased over time, signifying peak axonal damage occurring at disease onset. 188 Interestingly, similar associations have been reported between elevated serum levels of glial fibrillary acidic protein (GFAP) 189 and NMOSD, in which higher GFAP levels were correlated with greater disability and shorter time to relapse. 190
Preventative treatment
Evolution of new technologies such as single‐cell RNA sequencing has shed light on impairments to immune tolerance checkpoints that result in CNS autoreactivity (reviewed in Zou et al. 150 ). Delivery of mRNA to lymphoid tissues in order to mimic and correct peripheral tolerance mechanisms has recently been demonstrated in an EAE mouse model. 191 In this study, vaccination using MOG peptide‐encoding mRNA enabled the expansion of MOG‐specific Foxp3+ Treg cells and prevented the development and worsening of symptoms in MOG‐induced EAE mice. 191 Single‐cell RNA sequencing revealed that the expansion of effector Tregs played a significant role in suppressing proinflammatory Th1 and Th17 cells that would otherwise cause disease. Further, the importance of PD‐1 and CTLA‐4, which are inhibitory receptors on activated T cells, were emphasised when blockade of these receptors abolished the protective effect of MOG mRNA vaccination. Overall, the study demonstrated the first instance of a protective mRNA vaccine in an autoimmune disease, and showed that pathogenic T cells are inhibited rather than deleted and were reliant on PD‐1 and CTLA‐4 signalling.
Conclusion
In summary, MS, NMOSD and MOGAD can present with similar and often overlapping clinical and radiological characteristics that have made the differential diagnoses of these disorders challenging. The discovery of AQP4 Ab and MOG Ab has become essential in the diagnoses of NMOSD and MOGAD, respectively. Remarkably, despite expression of these autoantigens on distinct cell types of the CNS with heterogeneous pathologies, their immunopathogenic mechanisms lead to similar clinical features. Although the pathogenic role of AQP4 Ab has been more thoroughly investigated, evidence of MOG Ab pathogenicity is emerging. While current focus lies on the autoantibodies in NMOSD and MOGAD, there remains a need to clarify the contribution of T cells in pathogenesis. Although extensive research in MS disease models has demonstrated a primary pathogenic role of CD4+ T cells, B cell and antibody involvement in MS is being investigated, with a present demand to define patient subgroups in MS with regard to specific autoantigens. Along with associations to environmental factors such as infection, vitamin D and UV exposure, advancing research in the contribution of the gut microbiome provides a potential avenue for novel therapeutic interventions and for modifiable factors involved in brain health. As immunosuppressive and immunomodulatory therapies differ in efficacy across these disorders, a deeper understanding of the aetiology and pathogenic mediators of disease is essential to determine targeted therapeutic approaches to improve treatment decision‐making and patient outcomes.
Conflict of Interest
JAL reports funding from the Research Training Program Scholarship (Australia). SR has received competitive research funding from the National Health and Medical Research Council (Australia), the Petre Foundation (Australia), the Brain Foundation (Australia), the Royal Australasian College of Physicians and the University of Sydney; and is currently supported by an NHMRC Early Career Fellowship (APP1141169). SR is a consultant on an advisory board for UCB and Limbic Neurology and has received honoraria from Biogen and Limbic Neurology as an invited speaker. RCD and FB have received research funding from The Trish Multiple Sclerosis Research Foundation, Multiple Sclerosis Research Australia, the Petre Foundation and the National Health Medical Research Council (Australia). They have received honoraria from Biogen Idec and Merck as invited speakers. MD declares no competing interests.
Author Contributions
Joseph A Lopez: Conceptualization; Writing‐original draft; Writing‐review & editing. Martina Denkova: Writing‐original draft; Writing‐review & editing. Sudarshini Ramanathan: Writing‐review & editing. Russell C Dale: Writing‐review & editing. Fabienne Brilot: Conceptualization; Funding acquisition; Writing‐original draft; Writing‐review & editing.
Acknowledgments
This work was supported by the Australian National Health and Medical Research Council [APP1078643 and APP1183968] (NHRMC, Australia), Multiple Sclerosis Research Australia and a Sydney Research Excellence Initiative grant (University of Sydney, Australia).
References
- 1. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol 2015; 36: 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marchetti L, Engelhardt B. Immune cell trafficking across the blood‐brain barrier in the absence and presence of neuroinflammation. Vasc Biol 2020; 2: H1–H18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walton C, King R, Rechtman L et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Multi Scler J 2020; 26: 1816–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lublin FD, Reingold SC, Cohen JA et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014; 83: 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inusah S, Sormani MP, Cofield SS et al. Assessing changes in relapse rates in multiple sclerosis. Mult Scler J 2010; 16: 1414–1421. [DOI] [PubMed] [Google Scholar]
- 6. Burtchell J, Fetty K, Miller K, Minden K, Kantor D. Two sides to every story: perspectives from four patients and a healthcare professional on multiple sclerosis disease progression. Neurol Ther 2019; 8: 185–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Antel J, Antel S, Caramanos Z, Arnold DL, Kuhlmann T. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol 2012; 123: 627–638. [DOI] [PubMed] [Google Scholar]
- 8. Ontaneda D, Fox RJ. Progressive multiple sclerosis. Curr Opin Neurol 2015; 28: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thompson AJ, Banwell BL, Barkhof F et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018; 17: 162–173. [DOI] [PubMed] [Google Scholar]
- 10. Polman CH, Reingold SC, Banwell B et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011; 69: 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harris V, Tuddenham J, Sadiq S. Biomarkers of multiple sclerosis: current findings. Degener Neurol Neuromuscul Dis 2017; 7: 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lassmann H. Multiple sclerosis pathology. Cold Spring Harbor Perspect Med 2018; 8: a028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Filippi M, Preziosa P, Banwell BL et al. Assessment of lesions on magnetic resonance imaging in multiple sclerosis: practical guidelines. Brain 2019; 142: 1858–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000; 47: 707–717. [DOI] [PubMed] [Google Scholar]
- 15. Kuhlmann T, Ludwin S, Prat A, Antel J, Brück W, Lassmann H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol 2017; 133: 13–24. [DOI] [PubMed] [Google Scholar]
- 16. Frischer JM, Weigand SD, Guo Y et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 2015; 78: 710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Etemadifar M, Nasr Z, Khalili B, Taherioun M, Vosoughi R. Epidemiology of neuromyelitis optica in the world: a systematic review and meta‐analysis. Mult Scler Int 2015; 2015: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gold SM, Willing A, Leypoldt F, Paul F, Friese MA. Sex differences in autoimmune disorders of the central nervous system. Semin Immunopathol 2019; 41: 177–188. [DOI] [PubMed] [Google Scholar]
- 19. Pandit L, Asgari N, Apiwattanakul M et al. Demographic and clinical features of neuromyelitis optica: a review. Mult Scler J 2015; 21: 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wingerchuk DM, Banwell B, Bennett JL et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bukhari W, Clarke L, O’Gorman C et al. The clinical profile of NMOSD in Australia and New Zealand. J Neurol 2020; 267: 1431–1443. [DOI] [PubMed] [Google Scholar]
- 22. Lennon VA, Wingerchuk DM, Kryzer TJ et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364: 2106–2112. [DOI] [PubMed] [Google Scholar]
- 23. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic‐spinal multiple sclerosis binds to the aquaporin‐4 water channel. J Exp Med 2005; 202: 473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007; 6: 805–815. [DOI] [PubMed] [Google Scholar]
- 25. Sato DK, Callegaro D, Lana‐Peixoto MA et al. Distinction between MOG antibody‐positive and AQP4 antibody‐positive NMO spectrum disorders. Neurology 2014; 82: 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lucchinetti CF, Guo Y, Popescu BFG, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol 2014; 24: 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bradl M, Reindl M, Lassmann H. Mechanisms for lesion localization in neuromyelitis optica spectrum disorders. Curr Opin Neurol 2018; 31: 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matiello M, Schaefer‐Klein J, Sun D, Weinshenker BG. Aquaporin 4 expression and tissue susceptibility to neuromyelitis optica. JAMA Neurol 2013; 70: 1118. [DOI] [PubMed] [Google Scholar]
- 29. Popescu BFG, Guo Y, Jentoft ME et al. Diagnostic utility of aquaporin‐4 in the analysis of active demyelinating lesions. Neurology 2015; 84: 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Misu T, Fujihara K, Kakita A et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 2007; 130: 1224–1234. [DOI] [PubMed] [Google Scholar]
- 31. Brilot F, Dale RC, Selter RC et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol 2009; 66: 833–842. [DOI] [PubMed] [Google Scholar]
- 32. O'Connor KC, McLaughlin KA, De Jager PL et al. Self‐antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med 2007; 13: 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McLaughlin KA, Chitnis T, Newcombe J et al. Age‐dependent B cell autoimmunity to a myelin surface antigen in pediatric multiple sclerosis. J Immunol 2009; 183: 4067–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hennes E‐M, Baumann M, Schanda K et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology 2017; 89: 900–908. [DOI] [PubMed] [Google Scholar]
- 35. Ramanathan S, Reddel SW, Henderson A et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm 2014; 1: e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kitley J, Woodhall M, Waters P et al. Myelin‐oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012; 79: 1273–1277. [DOI] [PubMed] [Google Scholar]
- 37. Dale RC, Tantsis EM, Merheb V et al. Antibodies to MOG have a demyelination phenotype and affect oligodendrocyte cytoskeleton. Neurol Neuroimmunol Neuroinflamm 2014; 1: e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jarius S, Paul F, Aktas O et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation 2018; 15: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Armangue T, Olive‐Cirera G, Martinez‐Hernandez E et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol 2020; 19: 234–246. [DOI] [PubMed] [Google Scholar]
- 40. Rinaldi S, Davies A, Fehmi J et al. Overlapping central and peripheral nervous system syndromes in MOG antibody–associated disorders. Neurol Neuroimmunol Neuroinflamm 2021; 8: e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Connell K, Hamilton‐Shield A, Woodhall M et al. Prevalence and incidence of neuromyelitis optica spectrum disorder, aquaporin‐4 antibody‐positive NMOSD and MOG antibody‐positive disease in Oxfordshire, UK. J Neurol Neurosurg Psychiatry 2020; 91: 1126–1128. [DOI] [PubMed] [Google Scholar]
- 42. Kitley J, Waters P, Woodhall M et al. Neuromyelitis optica spectrum disorders with aquaporin‐4 and myelin‐oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 2014; 71: 276–283. [DOI] [PubMed] [Google Scholar]
- 43. Cobo‐Calvo A, Ruiz A, Maillart E et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology 2018; 90: e1858–e1869. [DOI] [PubMed] [Google Scholar]
- 44. Ramanathan S, Mohammad S, Tantsis E et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody‐associated demyelination. J Neurol Neurosurg Psychiatry 2018; 89: 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kunchok A, Chen JJ, McKeon A, Mills JR, Flanagan EP, Pittock SJ. Coexistence of myelin oligodendrocyte glycoprotein and aquaporin‐4 antibodies in adult and pediatric patients. JAMA Neurol 2020; 77: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yan Y, Li Y, Fu Y et al. Autoantibody to MOG suggests two distinct clinical subtypes of NMOSD. Sci China Life Sci 2016; 59: 1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cobo‐Calvo A, d'Indy H, Ruiz A et al. Frequency of myelin oligodendrocyte glycoprotein antibody in multiple sclerosis: a multicenter cross‐sectional study. Neurol Neuroimmunol Neuroinflamm 2020; 7: e649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Spadaro M, Gerdes LA, Krumbholz M et al. Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 2016; 3: e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Waters PJ, Komorowski L, Woodhall M et al. A multicenter comparison of MOG‐IgG cell‐based assays. Neurology 2019; 92: e1250–e1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jarius S, Pellkofer H, Siebert N et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: results from 163 lumbar punctures in 100 adult patients. J Neuroinflammation 2020; 17(261). 10.1186/s12974-12020-01824-12972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jarius S, Lechner C, Wendel EM et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 2: results from 108 lumbar punctures in 80 pediatric patients. J Neuroinflammation 2020; 17(262). 10.1186/s12974-12020-01825-12971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ramanathan S, Prelog K, Barnes EH et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin‐4 antibodies, and multiple sclerosis. Mult Scler J 2016; 22: 470–482. [DOI] [PubMed] [Google Scholar]
- 53. Salama S, Khan M, Shanechi A, Levy M, Izbudak I. MRI differences between MOG antibody disease and AQP4 NMOSD. Mult Scler J 2020; 26: 1854–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dubey D, Pittock SJ, Krecke KN et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol 2019; 76: 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ciron J, Cobo‐Calvo A, Audoin B et al. Frequency and characteristics of short versus longitudinally extensive myelitis in adults with MOG antibodies: a retrospective multicentric study. Mult Scler J 2020; 26: 936–944. [DOI] [PubMed] [Google Scholar]
- 56. Lopez‐Chiriboga AS, Majed M, Fryer J et al. Association of MOG‐IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG‐IgG‐associated disorders. JAMA Neurol 2018; 75: 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Waters P, Fadda G, Woodhall M et al. Serial anti‐myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol 2020; 77: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hoftberger R, Guo Y, Flanagan EP et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol 2020; 139: 875–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Takai Y, Misu T, Kaneko K et al. Myelin oligodendrocyte glycoprotein antibody‐associated disease: an immunopathological study. Brain 2020; 143: 1431–1446. [DOI] [PubMed] [Google Scholar]
- 60. Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol 2017; 133: 223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mendel I, De Rosbo NK, Ben‐Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H‐2b mice: Fine specificity and T cell receptor Vβ expression of encephalitogenic T cells. Eur J Immunol 1995; 25: 1951–1959. [DOI] [PubMed] [Google Scholar]
- 62. Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J Immunol 1989; 142: 1523–1527. [PubMed] [Google Scholar]
- 63. Faunce DE, Terajewicz A, Stein‐Streilein J. Cutting edge: in vitro‐generated tolerogenic APC induce CD8+ T regulatory cells that can suppress ongoing experimental autoimmune encephalomyelitis. J Immunol 2004; 172: 1991–1995. [DOI] [PubMed] [Google Scholar]
- 64. Linker RA, Gold R. MBP‐induced experimental autoimmune encephalomyelitis in C57BL/6 mice. J Immunol 2004; 173: 2896. [DOI] [PubMed] [Google Scholar]
- 65. International Multiple Sclerosis Genetics C . Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019; 365: eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. International Multiple Sclerosis Genetics C , Wellcome Trust Case Control C , Sawcer S et al. Genetic risk and a primary role for cell‐mediated immune mechanisms in multiple sclerosis. Nature 2011; 476: 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Caprariello AV, Rogers JA, Morgan ML et al. Biochemically altered myelin triggers autoimmune demyelination. Proc Natl Acad Sci USA 2018; 115: 5528–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cloake N, Yan J, Aminian A, Pender M, Greer J. PLP1 mutations in patients with multiple sclerosis: identification of a new mutation and potential pathogenicity of the mutations. J Clin Med 2018; 7: 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol 2017; 13: 25–36. [DOI] [PubMed] [Google Scholar]
- 70. Chaudhuri A. Multiple sclerosis is primarily a neurodegenerative disease. J Neural Transm (Vienna) 2013; 120: 1463–1466. [DOI] [PubMed] [Google Scholar]
- 71. Correale J, Gaitán MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 2016; 140: 527–546. [DOI] [PubMed] [Google Scholar]
- 72. Ogawa K, Okuno T, Hosomichi K et al. Next‐generation sequencing identifies contribution of both class I and II HLA genes on susceptibility of multiple sclerosis in Japanese. J Neuroinflammation 2019; 16: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Watanabe M, Nakamura Y, Sato S et al. HLA genotype‐clinical phenotype correlations in multiple sclerosis and neuromyelitis optica spectrum disorders based on Japan MS/NMOSD Biobank data. Sci Rep 2021; 11: 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bruijstens AL, Wong YYM, Van Pelt DE et al. HLA association in MOG‐IgG– and AQP4‐IgG–related disorders of the CNS in the Dutch population. Neurol Neuroimmunol Neuroinflamm 2020; 7: e702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sun X, Qiu W, Wang J et al. Myelin oligodendrocyte glycoprotein‐associated disorders are associated with HLA subtypes in a Chinese paediatric‐onset cohort. J Neurol Neurosurg Psychiatry 2020; 91: 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pender MP, Burrows SR. Epstein‐Barr virus and multiple sclerosis: potential opportunities for immunotherapy. Clin Transl Immunol 2014; 3: e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Houen G, Trier NH, Frederiksen JL. Epstein‐Barr Virus and multiple sclerosis. Front Immunol 2020; 11: 587078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bar‐Or A, Pender MP, Khanna R et al. Epstein‐Barr Virus in multiple sclerosis: theory and emerging immunotherapies. Trends Mol Med 2020; 26: 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sato R, Okanari K, Maeda T, Kaneko K, Takahashi T, Kenji I. Postinfectious acute disseminated encephalomyelitis associated with antimyelin oligodendrocyte glycoprotein antibody. Child Neurol Open 2020; 7: 2329048X2094244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nakamura M, Iwasaki Y, Takahashi T et al. A case of MOG antibody‐positive bilateral optic neuritis and meningoganglionitis following a genital herpes simplex virus infection. Mult Scler Relat Disord 2017; 17: 148–150. [DOI] [PubMed] [Google Scholar]
- 81. Choi S‐J, Oh DA, Chun W, Kim S‐M. The relationship between anti‐myelin oligodendrocyte glycoprotein antibody‐associated disease and the Rubella Virus. J Clin Neurol 2018; 14: 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhou S, Jones‐Lopez EC, Soneji DJ, Azevedo CJ, Patel VR. Myelin oligodendrocyte glycoprotein antibody‐associated optic neuritis and myelitis in COVID‐19. J Neuroophthalmol 2020; 40: 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. De Ruijter NS, Kramer G, Gons RAR, Hengstman GJD. Neuromyelitis optica spectrum disorder after presumed coronavirus (COVID‐19) infection: a case report. Mult Scler Relat Disord 2020; 46: 102474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yusuf FL, Wijnands JM, Kingwell E et al. Fatigue, sleep disorders, anaemia and pain in the multiple sclerosis prodrome. Mult Scler J 2020; 27: 290–302. [DOI] [PubMed] [Google Scholar]
- 85. Asseyer S, Schmidt F, Chien C et al. Pain in AQP4‐IgG‐positive and MOG‐IgG‐positive neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin 2018; 4: 205521731879668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Asseyer S, Hamblin J, Messina S et al. Prodromal headache in MOG‐antibody positive optic neuritis. Mult Scler Relat Disord 2020; 40: 101965. [DOI] [PubMed] [Google Scholar]
- 87. Jelinek GA, Marck CH, Weiland TJ, Pereira N, Van Der Meer DM, Hadgkiss EJ. Latitude, sun exposure and vitamin D supplementation: associations with quality of life and disease outcomes in a large international cohort of people with multiple sclerosis. BMC Neurol 2015; 15: eabc7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ostkamp P, Salmen A, Pignolet B et al. Sunlight exposure exerts immunomodulatory effects to reduce multiple sclerosis severity. Proc Natl Acad Sci USA 2021; 118: e2018457118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lucas RM, Ponsonby AL, Dear K et al. Sun exposure and vitamin D are independent risk factors for CNS demyelination. Neurology 2011; 76: 540–548. [DOI] [PubMed] [Google Scholar]
- 90. Hauser SL, Weiner HL, Che M, Shapiro ME, Gilles F, Letvin NL. Prevention of experimental allergic encephalomyelitis (EAE) in the SJL/J mouse by whole body ultraviolet irradiation. J Immunol 1984; 132: 1276–1281. [PubMed] [Google Scholar]
- 91. Irving AA, Marling SJ, Seeman J, Plum LA, Deluca HF. UV light suppression of EAE (a mouse model of multiple sclerosis) is independent of vitamin D and its receptor. Proc Natl Acad Sci USA 2019; 116: 22552–22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shaygannejad V, Maljaei MB, Bank SS, Mirmosayyeb O, Maracy MR, Askari G. Association between sun exposure, vitamin D intake, serum vitamin D level, and immunoglobulin G level in patients with neuromyelitis optica spectrum disorder. Int J Prev Med 2018; 9: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Koduah P, Paul F, Dörr J‐M. Vitamin D in the prevention, prediction and treatment of neurodegenerative and neuroinflammatory diseases. EPMA J 2017; 8: 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wildemann B, Jarius S, Franz J, Ruprecht K, Reindl M, Stadelmann C. MOG‐expressing teratoma followed by MOG‐IgG‐positive optic neuritis. Acta Neuropathol 2021; 141: 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sepúlveda M, Sola‐Valls N, Escudero D et al. Clinical profile of patients with paraneoplastic neuromyelitis optica spectrum disorder and aquaporin‐4 antibodies. Mult Scler J 2018; 24: 1753–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bernard‐Valnet R, Cobo‐Calvo A, Siegfried A et al. Paraneoplastic neuromyelitis optica and ovarian teratoma: a case series. Mult Scler Relat Disord 2019; 31: 97–100. [DOI] [PubMed] [Google Scholar]
- 97. Li K, Zhan Y, Shen X. Multiple intracranial lesions with lung adenocarcinoma: A rare case of MOG‐IgG‐associated encephalomyelitis. Mult Scler Relat Disord 2020; 42: 102064. [DOI] [PubMed] [Google Scholar]
- 98. Cosorich I, Dalla‐Costa G, Sorini C et al. High frequency of intestinal T H 17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci Adv 2017; 3: e1700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Probstel AK, Zhou X, Baumann R et al. Gut microbiota‐specific IgA+ B cells traffic to the CNS in active multiple sclerosis. Sci Immunol 2020; 5: eabc7191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zamvil SS, Spencer CM, Baranzini SE, Cree BAC. The gut microbiome in neuromyelitis optica. Neurotherapeutics 2018; 15: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Miyauchi E, Kim S‐W, Suda W et al. Gut microorganisms act together to exacerbate inflammation in spinal cords. Nature 2020; 585: 102–106. [DOI] [PubMed] [Google Scholar]
- 102. Guggenmos J, Schubart AS, Ogg S et al. Antibody cross‐reactivity between myelin oligodendrocyte glycoprotein and the milk protein butyrophilin in multiple sclerosis. J Immunol 2004; 172: 661–668. [DOI] [PubMed] [Google Scholar]
- 103. Stefferl A, Schubart A, Storch M et al. Butyrophilin, a milk protein, modulates the encephalitogenic T cell response to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis. J Immunol 2000; 165: 2859–2865. [DOI] [PubMed] [Google Scholar]
- 104. Jelcic I, Al Nimer F, Wang J et al. Memory B cells activate brain‐homing, autoreactive CD4+ T cells in multiple sclerosis. Cell 2018; 175: 85–100.e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sabatino JJ, Wilson MR, Calabresi PA, Hauser SL, Schneck JP, Zamvil SS. Anti‐CD20 therapy depletes activated myelin‐specific CD8+ T cells in multiple sclerosis. Proc Natl Acad Sci USA 2019; 116: 25800–25807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sabatino JJ, Zamvil SS, Hauser SL. B‐cell therapies in multiple sclerosis. Cold Spring Harb Perspect Med 2019; 9: a032037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KHG. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol 2010; 162: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kalra S, Lowndes C, Durant L et al. Th17 cells increase in RRMS as well as in SPMS, whereas various other phenotypes of Th17 increase in RRMS only. Mult Scler J Exp Transl Clin 2020; 6: 205521731989969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Machado‐Santos J, Saji E, Troscher AR et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue‐resident CD8+ T lymphocytes and B cells. Brain 2018; 141: 2066–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jilek S, Schluep M, Rossetti AO et al. CSF enrichment of highly differentiated CD8+ T cells in early multiple sclerosis. Clin Immunol 2007; 123: 105–113. [DOI] [PubMed] [Google Scholar]
- 111. Salou M, Garcia A, Michel L et al. Expanded CD 8 T cell sharing between periphery and CNS in multiple sclerosis. Ann Clin Transl Neurol 2015; 2: 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Saligrama N, Zhao F, Sikora MJ et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature 2019; 572: 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hofer LS, Ramberger M, Gredler V et al. Comparative analysis of T‐cell responses to aquaporin‐4 and myelin oligodendrocyte glycoprotein in inflammatory demyelinating central nervous system diseases. Front Immunol 2020; 11: 1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Owens GP, Ritchie A, Rossi A et al. Mutagenesis of the aquaporin 4 extracellular domains defines restricted binding patterns of pathogenic neuromyelitis optica IgG. J Biol Chem 2015; 290: 12123–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bronge M, Ruhrmann S, Carvalho‐Queiroz C et al. Myelin oligodendrocyte glycoprotein revisited—sensitive detection of MOG‐specific T‐cells in multiple sclerosis. J Autoimmun 2019; 102: 38–49. [DOI] [PubMed] [Google Scholar]
- 116. Kothur K, Wienholt L, Tantsis EM et al. B Cell, Th17, and neutrophil related cerebrospinal fluid cytokine/chemokines are elevated in MOG antibody associated demyelination. PLoS One 2016; 11: e0149411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Kaneko K, Sato DK, Nakashima I et al. CSF cytokine profile in MOG‐IgG+ neurological disease is similar to AQP4‐IgG+ NMOSD but distinct from MS: a cross‐sectional study and potential therapeutic implications. J Neurol Neurosurg Psychiatry 2018; 89: 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Filiano AJ, Xu Y, Tustison NJ et al. Unexpected role of interferon‐γ in regulating neuronal connectivity and social behaviour. Nature 2016; 535: 425–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014; 505: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rustenhoven J, Kipnis J. Bypassing the blood‐brain barrier. Science 2019; 366: 1448–1449. [DOI] [PubMed] [Google Scholar]
- 121. Sinmaz N, Amatoury M, Merheb V, Ramanathan S, Dale RC, Brilot F. Autoantibodies in movement and psychiatric disorders: updated concepts in detection methods, pathogenicity, and CNS entry. Ann NY Acad Sci 2015; 1351: 22–38. [DOI] [PubMed] [Google Scholar]
- 122. Li R, Patterson KR, Bar‐Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol 2018; 19: 696–707. [DOI] [PubMed] [Google Scholar]
- 123. Joseph FG, Hirst CL, Pickersgill TP, Ben‐Shlomo Y, Robertson NP, Scolding NJ. CSF oligoclonal band status informs prognosis in multiple sclerosis: a case control study of 100 patients. J Neurol Neurosurg Psychiatry 2009; 80: 292–296. [DOI] [PubMed] [Google Scholar]
- 124. Calabrese M, Agosta F, Rinaldi F et al. Cortical lesions and atrophy associated with cognitive impairment in relapsing‐remitting multiple sclerosis. Arch Neurol 2009; 66: 1144–1150. [DOI] [PubMed] [Google Scholar]
- 125. Ferreira D, Voevodskaya O, Imrell K et al. Multiple sclerosis patients lacking oligoclonal bands in the cerebrospinal fluid have less global and regional brain atrophy. J Neuroimmunol 2014; 274: 149–154. [DOI] [PubMed] [Google Scholar]
- 126. Heussinger N, Kontopantelis E, Gburek‐Augustat J et al. Oligoclonal bands predict multiple sclerosis in children with optic neuritis. Ann Neurol 2015; 77: 1076–1082. [DOI] [PubMed] [Google Scholar]
- 127. Farina G, Magliozzi R, Pitteri M et al. Increased cortical lesion load and intrathecal inflammation is associated with oligoclonal bands in multiple sclerosis patients: a combined CSF and MRI study. J Neuroinflammation 2017; 14: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wootla B, Denic A, Keegan BM et al. Evidence for the role of B cells and immunoglobulins in the pathogenesis of multiple sclerosis. Neurol Res Int 2011; 2011: 780712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bankoti J, Apeltsin L, Hauser SL et al. In multiple sclerosis, oligoclonal bands connect to peripheral B‐cell responses. Ann Neurol 2014; 75: 266–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Eggers EL, Michel BA, Wu H et al. Clonal relationships of CSF B cells in treatment‐naive multiple sclerosis patients. JCI Insight 2017; 2: e92724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Greve B, Magnusson CG, Melms A, Weissert R. Immunoglobulin isotypes reveal a predominant role of type 1 immunity in multiple sclerosis. J Neuroimmunol 2001; 121: 120–125. [DOI] [PubMed] [Google Scholar]
- 132. Trend S, Jones AP, Cha L et al. Higher serum immunoglobulin G3 levels may predict the development of multiple sclerosis in individuals with clinically isolated syndrome. Front Immunol 2018; 9: 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Marsh‐Wakefield F, Ashhurst T, Trend S et al. IgG3+ B cells are associated with the development of multiple sclerosis. Clin Transl Immunol 2020; 9: e01133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Cristofanilli M, Gratch D, Pagano B et al. Transglutaminase‐6 is an autoantigen in progressive multiple sclerosis and is upregulated in reactive astrocytes. Mult Scler 2017; 23: 1707–1715. [DOI] [PubMed] [Google Scholar]
- 135. Liu Y, Given KS, Harlow DE et al. Myelin‐specific multiple sclerosis antibodies cause complement‐dependent oligodendrocyte loss and demyelination. Acta Neuropathol Commun 2017; 5: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Long Y, Gao C, Qiu W, Hu X, Peng F, Lu Z. Antibodies target microvessels in neuromyelitis optica and multiple sclerosis patients. Neurol Res 2013; 35: 922–929. [DOI] [PubMed] [Google Scholar]
- 137. Greer JM, Trifilieff E, Pender MP. Correlation between anti‐myelin proteolipid protein (PLP) antibodies and disease severity in multiple sclerosis patients with PLP response‐permissive HLA types. Front Immunol 2020; 11: 1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Greer JM, Csurhes PA, Muller DM, Pender MP. Correlation of blood T cell and antibody reactivity to myelin proteins with HLA type and lesion localization in multiple sclerosis. J Immunol 2008; 180: 6402–6410. [DOI] [PubMed] [Google Scholar]
- 139. Srivastava R, Aslam M, Kalluri SR et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med 2012; 367: 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Vincent T, Saikali P, Cayrol R et al. Functional consequences of neuromyelitis optica‐IgG astrocyte interactions on blood‐brain barrier permeability and granulocyte recruitment. J Immunol 2008; 181: 5730–5737. [DOI] [PubMed] [Google Scholar]
- 141. Phuan PW, Ratelade J, Rossi A, Tradtrantip L, Verkman AS. Complement‐dependent cytotoxicity in neuromyelitis optica requires aquaporin‐4 protein assembly in orthogonal arrays. J Biol Chem 2012; 287: 13829–13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Alexopoulos H, Kampylafka EI, Fouka P et al. Anti‐aquaporin‐4 autoantibodies in systemic lupus erythematosus persist for years and induce astrocytic cytotoxicity but not CNS disease. J Neuroimmunol 2015; 289: 8–11. [DOI] [PubMed] [Google Scholar]
- 143. Bennett JL, O'Connor KC, Bar‐Or A et al. B lymphocytes in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 2015; 2: e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Tradtrantip L, Yao X, Su T, Smith AJ, Verkman AS. Bystander mechanism for complement‐initiated early oligodendrocyte injury in neuromyelitis optica. Acta Neuropathol 2017; 134: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Duan T, Smith AJ, Verkman AS. Complement‐dependent bystander injury to neurons in AQP4‐IgG seropositive neuromyelitis optica. J Neuroinflammation 2018; 15: 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hinson SR, Clift IC, Luo N, Kryzer TJ, Lennon VA. Autoantibody‐induced internalization of CNS AQP4 water channel and EAAT2 glutamate transporter requires astrocytic Fc receptor. Proc Natl Acad Sci USA 2017; 114: 5491–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Marignier R, Nicolle A, Watrin C et al. Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain 2010; 133: 2578–2591. [DOI] [PubMed] [Google Scholar]
- 148. Cotzomi E, Stathopoulos P, Lee CS et al. Early B cell tolerance defects in neuromyelitis optica favour anti‐AQP4 autoantibody production. Brain 2019; 142: 1598–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kowarik MC, Astling D, Gasperi C et al. CNS Aquaporin‐4‐specific B cells connect with multiple B‐cell compartments in neuromyelitis optica spectrum disorder. Ann Clin Transl Neurol 2017; 4: 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zou A, Ramanathan S, Dale RC, Brilot F. Single‐cell approaches to investigate B cells and antibodies in autoimmune neurological disorders. Cell Mol Immunol 2021; 18: 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Bennett JL, Lam C, Kalluri SR et al. Intrathecal pathogenic anti‐aquaporin‐4 antibodies in early neuromyelitis optica. Ann Neurol 2009; 66: 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Soltys J, Liu Y, Ritchie A et al. Membrane assembly of aquaporin‐4 autoantibodies regulates classical complement activation in neuromyelitis optica. J Clin Invest 2019; 129: 2000–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Pittock SJ, Berthele A, Fujihara K et al. Eculizumab in aquaporin‐4–positive neuromyelitis optica spectrum disorder. N Engl J Med 2019; 381: 614–625. [DOI] [PubMed] [Google Scholar]
- 154. Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti‐IL‐6 receptor monoclonal antibody tocilizumab. Mod Rheumatol 2013; 23: 827–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ayzenberg I, Kleiter I, Schröder A et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti‐CD20 therapy. JAMA Neurol 2013; 70: 394. [DOI] [PubMed] [Google Scholar]
- 156. Jarius S, Ruprecht K, Kleiter I et al. MOG‐IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long‐term outcome. J Neuroinflamm 2016; 13: 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Chihara N, Aranami T, Sato W et al. Interleukin 6 signaling promotes anti‐aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci USA 2011; 108: 3701–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Mizrachi T, Brill L, Rabie M et al. NMO‐IgG and AQP4 peptide can induce aggravation of EAMG and immune‐mediated muscle weakness. J Immunol Res 2018; 2018: 5389282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Abdullah S, Wong WF, Tan CT. The prevalence of anti‐aquaporin 4 antibody in patients with idiopathic inflammatory demyelinating diseases presented to a tertiary hospital in Malaysia: presentation and prognosis. Mult Scler Int 2017; 2017: 1359761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Jarius S, Franciotta D, Paul F et al. Cerebrospinal fluid antibodies to aquaporin‐4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation 2010; 7: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Hillebrand S, Schanda K, Nigritinou M et al. Circulating AQP4‐specific auto‐antibodies alone can induce neuromyelitis optica spectrum disorder in the rat. Acta Neuropathol 2019; 137: 467–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Akaza M, Tanaka K, Tanaka M et al. Can anti‐AQP4 antibody damage the blood‐brain barrier? Eur Neurol 2014; 72: 273–277. [DOI] [PubMed] [Google Scholar]
- 163. Vogel A‐L, Knier B, Lammens K et al. Deletional tolerance prevents AQP4‐directed autoimmunity in mice. Eur J Immunol 2017; 47: 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kim SM, Woodhall MR, Kim JS et al. Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol Neuroimmunol Neuroinflamm 2015; 2: e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Mader S, Gredler V, Schanda K et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation 2011; 8: 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Probstel AK, Dornmair K, Bittner R et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology 2011; 77: 580–588. [DOI] [PubMed] [Google Scholar]
- 167. Mariotto S, Ferrari S, Monaco S et al. Clinical spectrum and IgG subclass analysis of anti‐myelin oligodendrocyte glycoprotein antibody‐associated syndromes: a multicenter study. J Neurol 2017; 264: 2420–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Tea F, Lopez JA, Ramanathan S et al. Characterization of the human myelin oligodendrocyte glycoprotein antibody response in demyelination. Acta Neuropathol Commun 2019; 7: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Serguera C, Stimmer L, Fovet CM et al. Anti‐MOG autoantibodies pathogenicity in children and macaques demyelinating diseases. J Neuroinflammation 2019; 16: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Macrini C, Gerhards R, Winklmeier S et al. Features of MOG required for recognition by patients with MOG antibody‐associated disorders. Brain 2021: awab105. 10.1093/brain/awab1105 [DOI] [PubMed] [Google Scholar]
- 171. Dyer CA, Matthieu J‐M. Antibodies to myelin/oligodendrocyte‐specific protein and myelin/oligodendrocyte glycoprotein signal distinct changes in the organization of cultured oligodendroglial membrane sheets. J Neurochem 1994; 62: 777–787. [DOI] [PubMed] [Google Scholar]
- 172. Kinzel S, Lehmann‐Horn K, Torke S et al. Myelin‐reactive antibodies initiate T cell‐mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol 2016; 132: 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Spadaro M, Winklmeier S, Beltran E et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann Neurol 2018; 84: 315–328. [DOI] [PubMed] [Google Scholar]
- 174. Beltrán E, Paunovic M, Gebert D et al. Archeological neuroimmunology: resurrection of a pathogenic immune response from a historical case sheds light on human autoimmune encephalomyelitis and multiple sclerosis. Acta Neuropathol 2021; 141: 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Mayer MC, Breithaupt C, Reindl M et al. Distinction and temporal stability of conformational epitopes on myelin oligodendrocyte glycoprotein recognized by patients with different inflammatory central nervous system diseases. J Immunol 2013; 191: 3594–3604. [DOI] [PubMed] [Google Scholar]
- 176. Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol 2019; 15: 89–102. [DOI] [PubMed] [Google Scholar]
- 177. Waters P, Reindl M, Saiz A et al. Multicentre comparison of a diagnostic assay: aquaporin‐4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry 2016; 87: 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Reindl M, Schanda K, Woodhall M et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm 2020; 7: e674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Tea F, Pilli D, Ramanathan S et al. Effects of the positive threshold and data analysis on human MOG antibody detection by live flow cytometry. Front Immunol 2020; 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Marchionatti A, Woodhall M, Waters PJ, Sato DK. Detection of MOG‐IgG by cell‐based assay: moving from discovery to clinical practice. Neurol Sci 2021; 42: 73–80. [DOI] [PubMed] [Google Scholar]
- 181. Waters P, Woodhall M, O'Connor KC et al. MOG cell‐based assay detects non‐MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2015; 2: e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Yeh EA, Nakashima I. Live‐cell based assays are the gold standard for anti‐MOG‐Ab testing. Neurology 2019; 92: 501–502. [DOI] [PubMed] [Google Scholar]
- 183. Tao S, Zhang Y, Ye H, Guo D. AQP4‐IgG‐seropositive neuromyelitis optica spectrum disorder (NMOSD) coexisting with anti‐N‐methyl‐D‐aspartate receptor (NMDAR) encephalitis: A case report and literature review. Mult Scler Relat Disord 2019; 35: 185–192. [DOI] [PubMed] [Google Scholar]
- 184. Titulaer MJ, Höftberger R, Iizuka T et al. Overlapping demyelinating syndromes and anti‐N‐methyl‐D‐aspartate receptor encephalitis. Ann Neurol 2014; 75: 411–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Leite MI, Coutinho E, Lana‐Peixoto M et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: A multicenter study of 16 patients. Neurology 2012; 78: 1601–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Tuller F, Holzer H, Schanda K et al. Characterization of the binding pattern of human aquaporin‐4 autoantibodies in patients with neuromyelitis optica spectrum disorders. J Neuroinflammation 2016; 13: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Khalil M, Teunissen CE, Otto M et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018; 14: 577–589. [DOI] [PubMed] [Google Scholar]
- 188. Mariotto S, Gastaldi M, Grazian L et al. NfL levels predominantly increase at disease onset in MOG‐Abs‐associated disorders. Mult Scler Relat Disord 2021; 50: 102833. [DOI] [PubMed] [Google Scholar]
- 189. Watanabe M, Nakamura Y, Michalak Z et al. Serum GFAP and neurofilament light as biomarkers of disease activity and disability in NMOSD. Neurology 2019; 93: e1299–e1311. [DOI] [PubMed] [Google Scholar]
- 190. Schindler P, Grittner U, Oechtering J et al. Serum GFAP and NfL as disease severity and prognostic biomarkers in patients with aquaporin‐4 antibody‐positive neuromyelitis optica spectrum disorder. J Neuroinflammation 2021; 18: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Krienke C, Kolb L, Diken E et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021; 371: 145–153. [DOI] [PubMed] [Google Scholar]
- 192. Jarius S, Jacobi C, de Seze J et al. Frequency and syndrome specificity of antibodies to aquaporin‐4 in neurological patients with rheumatic disorders. Mult Scler 2011; 17: 1067–1073. [DOI] [PubMed] [Google Scholar]
- 193. Asgari N, Jarius S, Laustrup H et al. Aquaporin‐4‐autoimmunity in patients with systemic lupus erythematosus: a predominantly population‐based study. Mult Scler 2018; 24: 331–339. [DOI] [PubMed] [Google Scholar]
- 194. Chen JJ, Flanagan EP, Jitprapaikulsan J et al. Myelin oligodendrocyte glycoprotein antibody‐positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol 2018; 195: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Soelberg K, Jarius S, Skejoe H et al. A population‐based prospective study of optic neuritis. Mult Scler 2017; 23: 1893–1901. [DOI] [PubMed] [Google Scholar]
- 196. Fan R, Zhang Y, Xu Y et al. Serum antinuclear antibodies associate with worse prognosis in AQP4‐positive neuromyelitis optica spectrum disorder. Brain Behavior 2021; 11: e01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Novakova L, Zetterberg H, Sundström P et al. Monitoring disease activity in multiple sclerosis using serum neurofilament light protein. Neurology 2017; 89: 2230–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Iwanowski P, Losy J. Immunological differences between classical phenotypes of multiple sclerosis. J Neurol Sci 2015; 349: 10–14. [DOI] [PubMed] [Google Scholar]
- 199. Arru G, Sechi E, Mariotto S et al. Antibody response against HERV‐W in patients with MOG‐IgG associated disorders, multiple sclerosis and NMOSD. J Neuroimmunol 2020; 338: 577110. [DOI] [PubMed] [Google Scholar]
- 200. Marnetto F, Valentino P, Caldano M, Bertolotto A. Detection of potassium channel KIR4.1 antibodies in multiple sclerosis patients. J Immunol Methods 2017; 445: 53–58. [DOI] [PubMed] [Google Scholar]
- 201. Malyavantham K, Weinstock‐Guttman B, Suresh L et al. Humoral responses to diverse autoimmune disease‐associated antigens in multiple sclerosis. PLoS One 2015; 10: e0129503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Brill L, Goldberg L, Karni A et al. Increased anti‐KIR4.1 antibodies in multiple sclerosis: could it be a marker of disease relapse? Mult Scler 2015; 21: 572–579. [DOI] [PubMed] [Google Scholar]
- 203. Nerrant E, Salsac C, Charif M et al. Lack of confirmation of anti‐inward rectifying potassium channel 4.1 antibodies as reliable markers of multiple sclerosis. Mult Scler J 2014; 20: 1699–1703. [DOI] [PubMed] [Google Scholar]
- 204. Higuchi O, Nakane S, Sakai W et al. Lack of KIR4.1 autoantibodies in Japanese patients with MS and NMO. Neurol Neuroimmunol Neuroinflamm 2016; 3: e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Navas‐Madroñal M, Valero‐Mut A, Martínez‐Zapata MJ et al. Absence of antibodies against KIR4.1 in multiple sclerosis: A three‐technique approach and systematic review. PLoS One 2017; 12: e0175538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Pröbstel A‐K, Kuhle J, Lecourt A‐C et al. Multiple Sclerosis and Antibodies against KIR4.1. N Engl J Med 2016; 374: 1496–1498. [DOI] [PubMed] [Google Scholar]
- 207. Muto M, Mori M, Hiwasa T et al. Novel serum autoantibodies against talin1 in multiple sclerosis: possible pathogenetic roles of the antibodies. J Neuroimmunol 2015; 284: 30–36. [DOI] [PubMed] [Google Scholar]
- 208. Breij EC, Brink BP, Veerhuis R et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol 2008; 63: 16–25. [DOI] [PubMed] [Google Scholar]
- 209. Lucchinetti CF, Bruck W, Lassmann H. Evidence for pathogenic heterogeneity in multiple sclerosis. Ann Neurol 2004; 56: 308. [DOI] [PubMed] [Google Scholar]
- 210. Metz I, Weigand SD, Popescu BFG et al. Pathologic heterogeneity persists in early active multiple sclerosis lesions. Ann Neurol 2014; 75: 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: in situ evidence for antibody‐ and complement‐mediated demyelination. Ann Neurol 1998; 43: 465–471. [DOI] [PubMed] [Google Scholar]
- 212. Roemer SF, Parisi JE, Lennon VA et al. Pattern‐specific loss of aquaporin‐4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007; 130: 1194–1205. [DOI] [PubMed] [Google Scholar]
- 213. Hinson SR, Pittock SJ, Lucchinetti CF et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007; 69: 2221–2231. [DOI] [PubMed] [Google Scholar]
- 214. Waters P, Jarius S, Littleton E et al. Aquaporin‐4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol 2008; 65: 913–919. [DOI] [PubMed] [Google Scholar]
- 215. Peschl P, Schanda K, Zeka B et al. Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement‐dependent demyelination. J Neuroinflammation 2017; 14: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Kinoshita M, Nakatsuji Y, Moriya M et al. Astrocytic necrosis is induced by anti‐aquaporin‐4 antibody‐positive serum. NeuroReport 2009; 20: 508–512. [DOI] [PubMed] [Google Scholar]
- 217. Linnington C, Webb M, Woodhams PL. A novel myelin‐associated glycoprotein defined by a mouse monoclonal antibody. J Neuroimmunol 1984; 6: 387–396. [DOI] [PubMed] [Google Scholar]
- 218. Tsymala I, Nigritinou M, Zeka B et al. Induction of aquaporin 4‐reactive antibodies in Lewis rats immunized with aquaporin 4 mimotopes. Acta Neuropathol Commun 2020; 8: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Tomassini V, De Giglio L, Reindl M et al. Anti‐myelin antibodies predict the clinical outcome after a first episode suggestive of MS. Mult Scler J 2007; 13: 1086–1094. [DOI] [PubMed] [Google Scholar]
- 220. Weinshenker BG, Wingerchuk DM, Vukusic S et al. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 2006; 59: 566–569. [DOI] [PubMed] [Google Scholar]
- 221. Matiello M, Lennon VA, Jacob A et al. NMO‐IgG predicts the outcome of recurrent optic neuritis. Neurology 2008; 70: 2197. [DOI] [PubMed] [Google Scholar]
- 222. Jarius S, Aboul‐Enein F, Waters P et al. Antibody to aquaporin‐4 in the long‐term course of neuromyelitis optica. Brain 2008; 131: 3072–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Takahashi T, Fujihara K, Nakashima I et al. Anti‐aquaporin‐4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 2007; 130: 1235–1243. [DOI] [PubMed] [Google Scholar]
- 224. Damato V, Evoli A, Iorio R. Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders. JAMA Neurol 2016; 73: 1342. [DOI] [PubMed] [Google Scholar]
- 225. Miyazaki‐Komine K, Takai Y, Huang P et al. High avidity chimeric monoclonal antibodies against the extracellular domains of human aquaporin‐4 competing with the neuromyelitis optica autoantibody, NMO‐IgG. Br J Pharmacol 2016; 173: 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]