

Abstract
A series of novel purine-modified 2′,3′-dideoxy-2′,3′-difluro-D-arabinonucleosides, including fluorinated analogs of fludarabine and nelarabine, have been prepared via anion glycosylation reactions of salts of 2-fluoropurine derivatives with the glycosyl bromide. A short and efficient synthetic route to the carbohydrate precursor 5-O-benzoyl-2,3-difluoro-α-d-arabinofuranosyl bromide was developed in five steps from d-xylose. Improved synthesis of methyl 5-O-benzoyl-2,3-difluoro-α-d-arabinofuranoside based upon the study of diethylaminosulfur trifluoride (DAST)-reactions with 5-O-protected methyl D-xylosides was explored using mild reaction conditions on the key step. New peculiarities for selective fluorinations of 5-O-benzoylated α- and β-D-pentofuranosides with DAST leading to the formation of mono and difluoro-furanoside derivatives are reported.
Keywords: d-xylose, Regioselective benzoylation, Fluorination, Fluorodeoxy sugars, Glycosylation, Fluoronucleosides
1. Introduction
Modified nucleosides analogs are an important class of compounds possessing antiviral and anticancer activities. Fluorinated by the sugar moiety or in the heterocyclic base nucleosides have drawn a lot of attention because of their interesting biological properties and several of them are essential chemotherapeutics [1,2]. Introduction of fluorine atom(s) to synthetic nucleoside analog can influence conformational properties, metabolism, enhance bioavailability, binding affinity and selectivity of fluorinated derivative, and plays a prominent role in the drug design due to improvement in the pharmacokinetic properties and a change in the bioactivity [3,4].
A number of purine-based nucleosides bearing a halogen atom at the 2-position in the nucleobase have been prepared and investigated as potential anticancer gents. Among these derivatives purine analogs with 2-fluorine or 2-chlorine atom have been shown to possess activity against human tumor cells. Fludarabine (1a) and its prodrug, fludarabine phosphate (9-β-d-arabinofuranosyl-2-fluroadenine 5′-O-phosphate, 1b) (Fig. 1), [5–7] were explored as anticancer agents, and, after the clinical development, fludarabine phosphate is currently used for the treatment of chronic lymphocytic leukemia [8]. 2-Fluoro-2′-deoxyadenosine (2), a deoxyanalog of fludarabine (1a) with excellent substrate properties for E. coli purine nucleoside phorphorylase demonstrated better activity against tumors that express E. coli than fludarabine phosphate (1b) and is considered as a valuable asset in the suicide gene therapy strategy [9]. 2-Fluoro-3′-deoxyadenosine (3), the fluorinated analog of cordycepin was also synthesized using chemical or chemoenzymatic approaches [10,11] and identified as a selective and potent compound with trypanocidal activity in vitro and in vivo. Synthetic routes to 2′-deoxy-2′-fluoro-arabino and ribo 2-fluoroadenine nucleosides 4–5 (Fig. 1) were developed, and these compounds were evaluated for their cytotoxicity and in vitro anti-HIV activity [7,12,13].
Fig. 1.
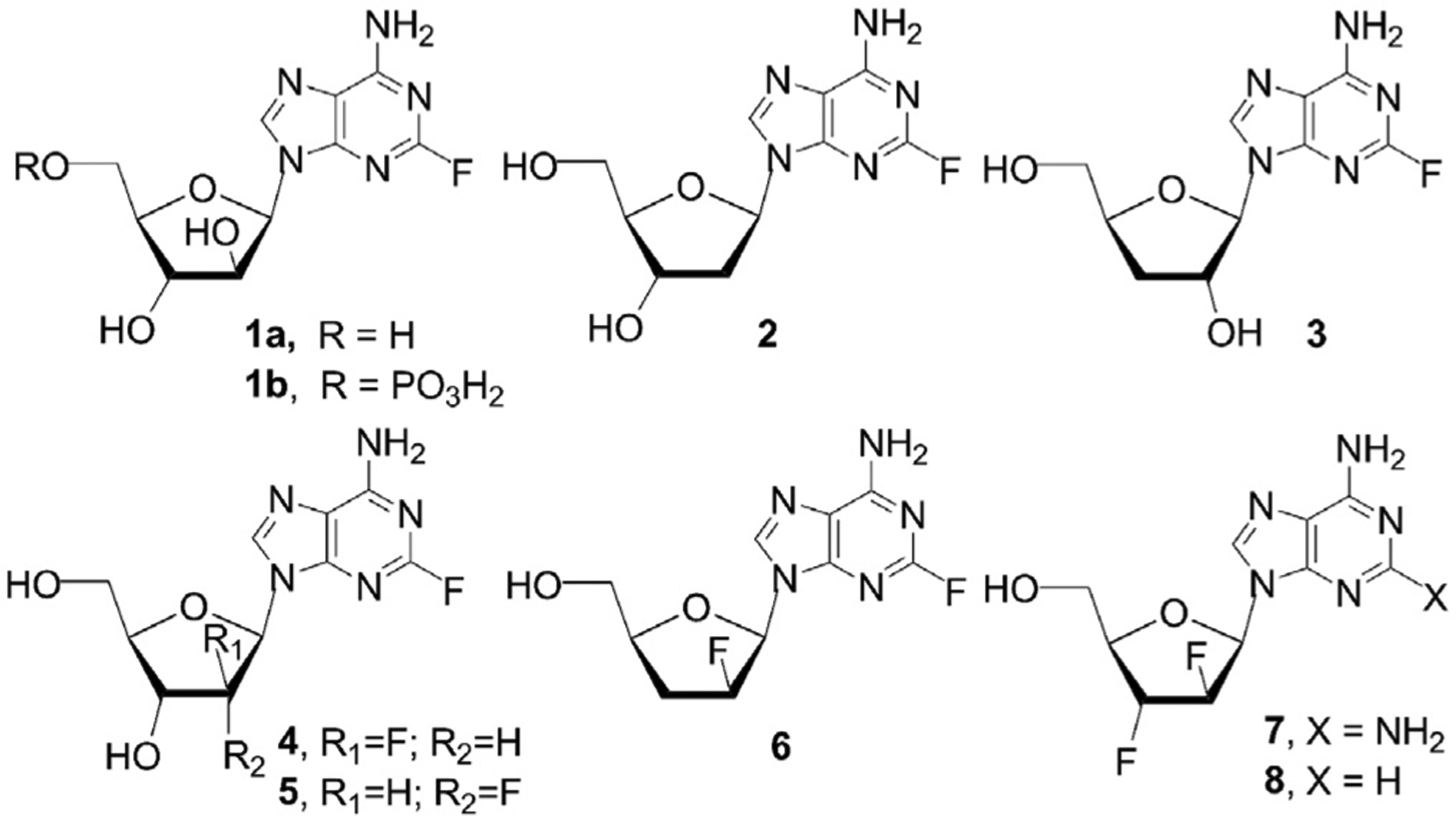
Fluorinated purine nucleosides with anticancer, antiviral and antibacterial activities.
2′-Fluoro-analog of fludarabine 4 showed cytotoxicities to a series of human tumor cell lines [7,12]. Besides, 2-fluorolodenosine (6) [7] as well as a number of other purine-modified 2′-fluoro-2′,3′-dideoxyarabinofuranosyl nucleosides were prepared and evaluated in vitro as anti-HIV agents [14,15]. 2′,3′-Dideoxy-2′,3′-difluoro D-arabino purine nucleosides were also prepared and the studies of in vitro antiviral activities led to the discovery of 2,6-diaminopurine (7) [16] and adenine (8) [17] 2′,3′-difluoro-β-D-arabinosides which display selective anti-HIV-1 activities. In order to extend this series of bioactive fluoronucleosides and evaluate their anticancer and antiviral potential, the further development of the synthetic methodology to 2′,3′-difluoroarabinofuranosyl nucleosides with structural modifications in the base was continued from the carbohydrate precursor and herein we report the synthesis of novel 2-fluorine and 2,6-disubstituted derivatives of purine 2′,3′-difluorinated nucleosides.
2. Results and discussions
2.1. Syntheses of 5-O-benzoylated mono and difluoro-d-pentofuranoside derivatives from d-xylose
Our synthetic strategy for designing purine-modified difluoro nucleosides starts from d-xylose and involves study of synthesis of the key intermediates, methyl 5-O-benzoyl-2,3-difluoro-α-d-arabinoside and the 1-α-bromosugar, for which several approaches were earlier investigated [16,17]. In order to improve the methods developed for the preparation of the difluoro sugar 15 [17] from methyl 5-O-benzoyl-α-d-xylofuranoside (11a), different synthetic routes to the latter were explored in detail on the first step (Scheme 1).
Scheme 1. Reagents and conditions.
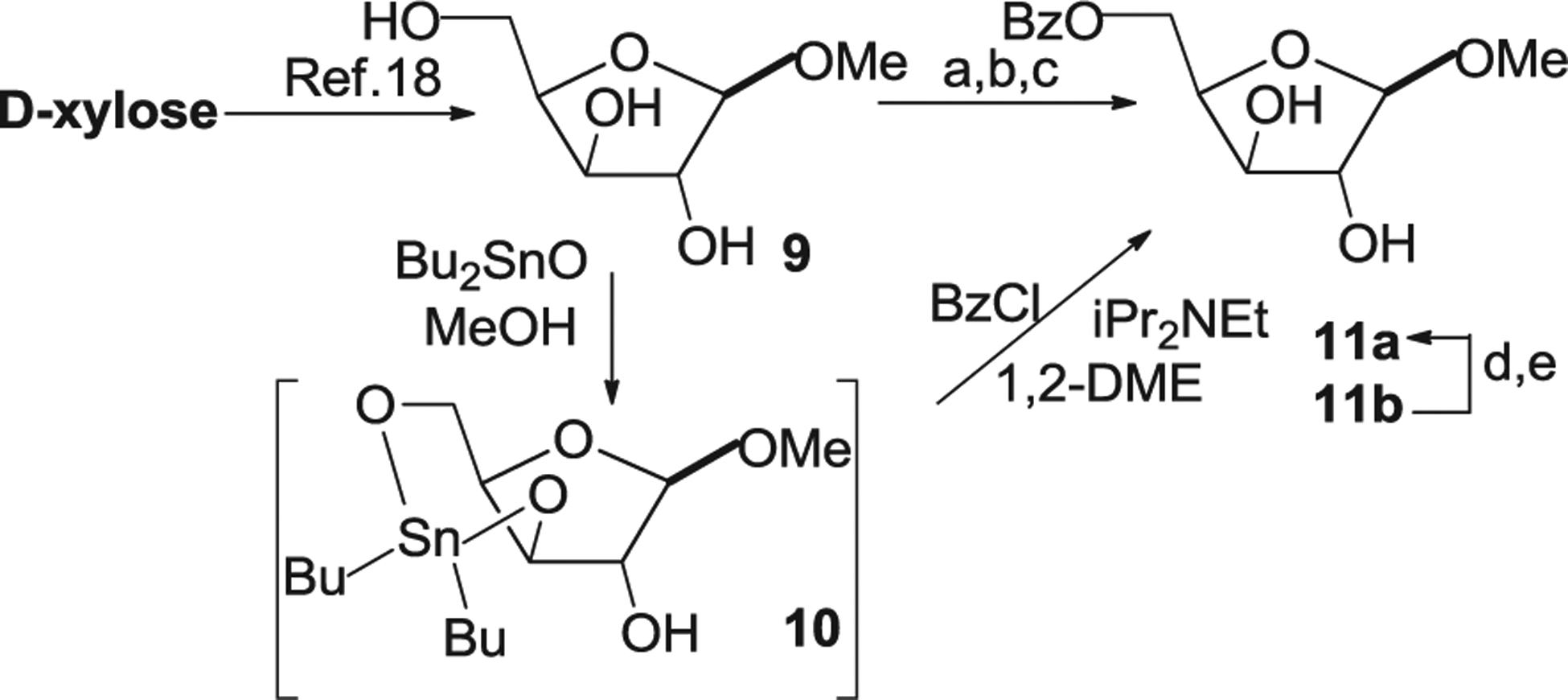
a) 9, BzCl/CH2Cl2, Et3N/iPr2NEt, 0 °C→rt, 18 h, 46% overall to 11a,b, 39% to 11a; 45% to 11b; b) i) 9, Bu2SnO/MeOH, reflux, 1 h; ii) BzCl/iPr2NEt, toluene, rt, 5 h, 46% overall to 11a,b, 35% to 11a; 41% to 11b; c) i) 9, Bu2SnO/MeOH, reflux, 1 h; ii) BzCl/iPr2NEt,1,2-DME, rt, 5 h, 97% overall to 11a,b, 82% to 11a; 89% to 11b; d) 11b, MeOH/CH3SO3H, rt, 18 h, 32% to 11a; e) 11b, MeOH/HCl, rt, 3 h, 47% to 11a.
Direct and selective acylation of primary hydroxyl groups in methyl α/β-xylofuranosides (9), prepared quantitatively from readily available from d-xylose [18], using an excess of benzoyl chloride as the acylating agent was tested under various conditions (Table 1, entries 1–3). First, benzoylation of 9 in CH2Cl2/Py in the presence of Et3N as a base catalyst gave 5-O-benzoates 11a,b with a low yield (Table 1, entry 1). Second, the use of a mixture of Et3N/iPr2NEt in the acylation reaction led to 11a,b in 39–46% (entries 2–3), but the target α-xyloside 11a was isolated in 39% yield after chromatography on silica gel (entry 3). Next, the Bu2SnO method [19] widely used in carbohydrate chemistry for acylation and alkylation of D-hexopyranosides [20–22], and benzylation of 1,2-O-isopropylidene-d-xylofuranose [23] was investigated for selective 5-O-monobenzoylation of xylofuranosides 9 with benzoyl chloride (Table 1, entries 4,5).
Table 1.
Selective 5-O-benzoylation of methyl α/β-xylofuranosides (9) with PhCOCl
| Entry | Regents and conditions | Base (equiv.) | BzCl (equiv.) | Time (h) | Ratios of 11b/11aa | Yields (%)b |
|---|---|---|---|---|---|---|
| 1 | BzCl/CH2Cl2/Py 0 °C→rt | Et3N (1.0) | 1.2 | 18 | 1.24:1 | 29 |
| 2 | BzCl/CH2Cl2 0 °C→rt | Et3N/iPr2NEt(1.7/0.7) | 1.3 | 18 | 1.29:1 | 39 |
| 3 | BzCl/CH2Cl2 0 °C→rt | Et3N/iPr2NEt(1.7/0.8) | 1.3 | 18 | 1.32:1 | 46 (39) |
| 4 | Bu2SnO/MeOH reflux; BzCl/toluene, rt | iPr2NEt (1.9) | 2.0 | 5 | 1:0.72 | 46 (35) |
| 5 | Bu2SnO/MeOH reflux; BzCl/1,2-DME, rt | iPr2NEt (2.3) | 3.0 | 5 | 1:0.80 | 97 (82) |
Ratio of 5-O-benzoyl α- and β-xylofuranosides determined from 1H NMR spectrum of the anomeric mixture after chromatography.
Combined isolated yields of 5-O-benzoates 11a,b after chromatography on silica gel. Figures in parentheses refer to isolated yield of methyl 5-O-benzoyl α-xylofuranoside 11a (calculated from α:β ratio of 1:1.25/1.14 in the starting methyl xyloside 9).
Different reaction conditions have been reported [20–22] for regioselective and efficient acylations of sugars via activation of OH groups by Bu2SnO followed by treatment of the cyclic tin intermediate with excess acyl chloride and triethylamine. The reaction of methylα/β-xylofuranosides (9) with an equimolar amount of dibutyltin oxide in methanol under reflux produced intermediate 3,5-O-di-n-butylstannylene derivative 10 which was converted to 5-O-benzoates 11a in 46% overall yield (Table 1, entry 4) by reaction with benzoyl chloride and iPr2NEt in toluene (Table 1, entry 4). The yield of the target products was improved to 97% via the dibutylstannylene derivatives in anhydrous 1,2-dimethoxyethane (entry 5). It should be noted that under the above reaction conditions (Table 1) benzoylation of a mixture of methyl α/β-xylosides 9 derived from d-xylose yielded the higher yields for 5-O-benzoyl derivative of β-xylofuranoside compared to that of the α-xyloside. Further, to improve yield of the target pentofuranoside, conversion of 5-O-benzoylated methyl β-xylofuranoside 11b to isomeric α-xyloside 11a was studied under mild acidic conditions (Table 2). Isomerization of 11b to 11a was tested in anhydrous MeOH in the presence of CH3SO2OH at room temperature (entry 1) and the yield of 11a made up 32% after the methanolysis and treatment of the reaction mixture with aqueous NaHCO3. Methanolysis of 11b in MeOH/HCl generated by adding acetyl chloride to methanol at 0 °C afforded only a mixture of two anomers 11b and 11b with a ratio of 1:0.90 (from 1H NMR data) and 47% yield of α-xyloside 11a (entry 2). These findings indicate that acidic methanolysis resulted in the anomerization of 11b with the formation of α-anomer 11a and this simple procedure can be used for the increase of overall yield of the target 5-O-benzoylated α-xyloside (Scheme 1) after separation of the anomeric mixture with column chromatography on silica gel. Thus, novel synthetic approach to methyl 5-O-benzoyl-α,β-xylofuranoside (11a,b) via the 3,5-O-dibutylstannylene intermediates 10 was developed from d-xylose. This method gave 5-O-acylated xylosides in higher combined yields compared to the method reported earlier via 5-O-protected 1,2-O-isopropylidene-α-d-xylofuranose [24].
Table 2.
Study of isomerization of β-xylofuranoside 11b to α-xylofuranoside 11a under acidic conditions.
| Entry | Regents and conditions | Acid (equiv.) | 11b:11aa | Yields (%)b |
|---|---|---|---|---|
| 1 | MeOH/CH3SO2OH, rt, 18 h | 5.5 | 1:0.66 | 32 |
| 2 | MeOH/HCl, rt, 3 h | 0.87 | 1:0.90 | 47 |
Ratio of 5-O-benzoyl β/α -xylofuranosides 11b and 11a determined from the 1H NMR spectrum of the anomeric mixture after treatment.
Yields of methyl 5-O-benzoyl α-xylofuranoside 11a refer to NMR yields.
Fluorination of individual xylosides 11a and 11b with DAST, the widely used fluorinating agent [25,26] in carbohydrate chemistry [27,28], was reported for the synthesis of deoxyfluoro D-pentofuranosides [23]. In addition, fluorination of mixtures of methyl 5-O-benzyl or 5-O-p-toluoyl-α/β-D-xylofuranosides with DAST and Deoxo-Fluor was studied for preparation of 5-O-protected methyl 3-fluoro-3-deoxy-α/β-D-ribofuranosides [29,30].
Several pathways for the preparation of the 2,3-difluoride 15 were also explored from 2-O-sulfonates of methyl 3-fluoro-3-deoxy-α-d-ribofuranoside and selectively protected α-d-xylofuranoside 11a [15]. As an extension of this work, new investigations on the DAST-reaction with 11a were undertaken to develop a shorter and more efficient synthetic route to methyl 5-O-benzoyl-2,3-difluoroarabinoside (15) (Scheme 2). Treatment of 11a with 5.6 eqiuv. of DAST in methylene chloride under 25–29 °C for 10 h resulted in the difluoride 15 (18%) and methyl 5-O-benzoyl-3-deoxy-3-fluoro-α-d-ribofuranoside (14) (51%) after workup of reaction mixture and chromatography on silica gel. In this case, decrease of the reaction time and an excess of the fluorinated agent compared to the previous data described for the double fluorination of the xyloside 11a [15] gave rise to 15 with a lower yield and increased the formation of the α-riboside 14. The results obtained when studying introduction of fluorine at C-2 of O-protected pyranoside and furanoside derivatives with DAST [24,31–34] indicate that the nature of the C1-substituent and its stereochemistry, the protecting groups in the pentofuranose ring, conformational peculiarities of the C-2-O-SF2NEt2 intermediates prepared by DAST-activation of hydroxyl groups, the temperature, and solvent should be considered as important factors affecting the SN2-displacement reaction by fluoride in selectively protected pentofuranoside derivatives. From the above reasoning fluorination of the α-riboside 14 with an excess of DAST was carried out in a mixture of methylene chloride and pyridine under mild heating 35–37 °C for 18 h to give the difluoride 15 in 66% yield after chromatographic isolation (Scheme 2). A short and optimized synthesis of the key intermediate 15 was studied from d-xylose using mild reaction conditions for the fluorination reaction.
Scheme 2. Reagents and conditions.
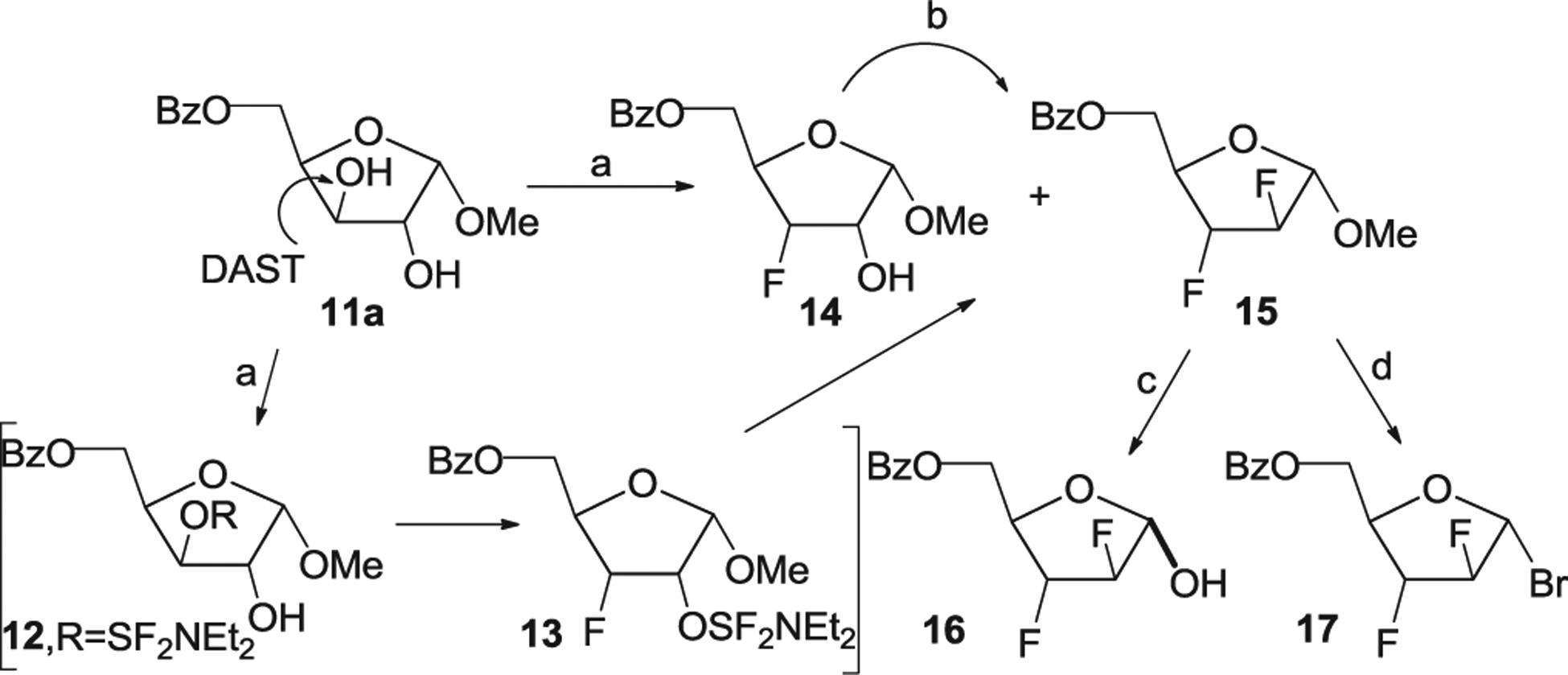
a) 11a, DAST/CH2Cl2, 25–29 °C, 10 h, 18% to 15, 51% to 14; b) 14, DAST/CH2Cl2/Py, 35–37 °C, 18 h, 66% to 15; c) CH3COOH/Ac2O, 34%-HBr in AcOH, 18 h, rt, 16, 50%; d) i) 15, CH3COOH/Ac2O/H2SO4, 0 °C→24–25 °C, 3 h; ii) TMSBr/CH2Cl2/ZnBr2, 0 °C→rt, 16 h, 84%.
Next, synthesis of the 1-α-bromide 17 from 15 was explored taking into account the known two-step procedures [16,17]. Two methods were tested to directly prepare the bromide 17 from the α-arabinoside 15 without preparation of intermediate 1-O-acetates. Bromination of 15 with 34% HBr in CH3COOH in the presence of TMSBr in CH2Cl2 for 18 h failed to prepare the target bromide and only the starting sugar was detected in the reaction mixture. Reaction of 15 with 34% HBr in CH3COOH in a mixture of CH3COOH/Ac2O at room temperature did not also resulted in the bromide. 1H NMR spectrum of the crude reaction mixture after removal of the reagents in vacuum under mild heating showed the absence of the signals corresponding to the bromo sugar. 5-O-Benzoyl-2,3-dideoxy-2,3-difluoro-α,β-d-arabinofuranose (16) derived likely from hydrolysis of the intermediate reaction products was isolated in 50% yield by chromatography on silica gel. Nevertheless, optimized reaction conditions for preparation of the 1-α-bromide 17 were found which include the acetolysis reaction of 15 in a mixture of CH3COOH/Ac2O/H2SO4 at 24–25 °C with a slight increase of sulfuric acid content and the reaction time followed by bromination of the intermediate 1-O-acetates with TMSBr in the presence of the catalyst (Scheme 2).
In continuation, fluorination of the β-xyloside 11b with an excess of DAST in methylene chloride was carried out under 25–27 °C for 18 h (Scheme 3). Four products were prepared after the workup of the reaction mixture and the 3-fluoro-3-deoxy-β-D-riboside 21, 2,3-difluoride 15, 1,3-difluoride 22, and methyl 5-O-benzoyl-2,3-anhydro-β-D-riboside (not presented in scheme) were isolated in 55%, 3%, 3% and 14% yields, respectively, after column chromatography on silica gel. A thorough analysis of prepared reaction products made it possible to recognize new interesting peculiarities for the fluorination reaction of 11b. In present study the formation of the 3-fluoro-β-riboside 21 was observed as the main product along with two difluoro derivatives of α-d-arabinofuranose 15 and 22 as side-products unlike a previous work [24].
Scheme 3. Reagents and conditions.
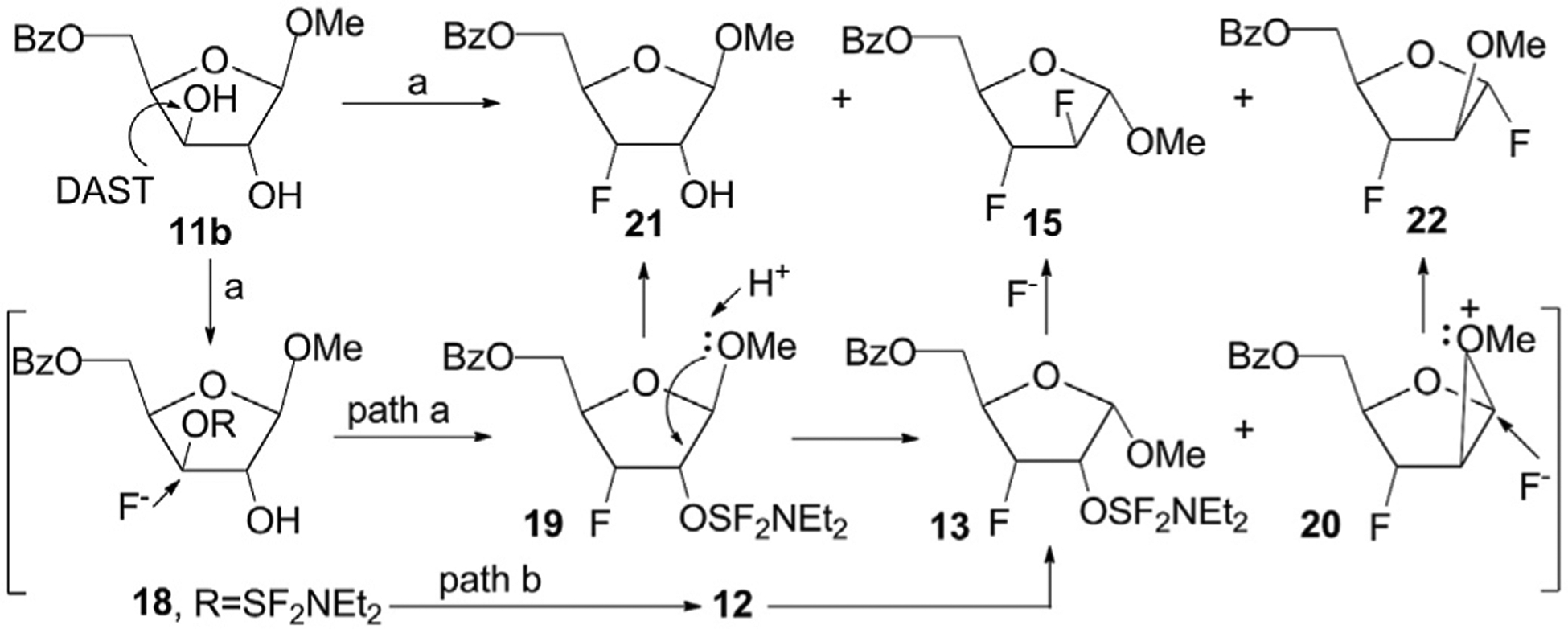
a) 11b, DAST/CH2Cl2, 25–27 °C, 18 h, 21, 55%, 15, 3%, 22, 3%.
The structures of fluorinated sugars were confirmed by 1H, 13C and 19F NMR, mass and IR-spectroscopy. 1H and 13C NMR spectral data (CDCl3) of isomeric difluorides 15 and 22 are distinguished essentially for H-1 and H-2 protones, C-1 and C-2 carbons (experimental part). Signals of the H-1 and H-2 protons of the 1,3-difluoride 22 displayed as a doublet at 5.90 ppm (J1,F-1 = 60.4 and J1,2 < 1.0 Hz) and a multiplet at 4.17 ppm in the 1H NMR spectrum unlike those of isomeric 2,3-difluoride 15. H-1 and H-2 resonances of the latter are revealed as a broad doublet at 5.19 ppm (J1,F-2 = 10.0 and J1,2 < 1.0 Hz) and a complex multiplet at 5.08 ppm, respectively. Small magnitudes of 3JH-1,H-2 coupling constants are characteristic of the 1H NMR data for the both difluorinated α-d-arabinofuranoside derivatives 15 and 22. The large value of 3JC-1,F-1 = 225.7 Hz inherent in the glycosyl fluorides [35] was displayed in the 13C NMR spectrum of the 1,3-difluoride 22. Very similar IR-spectroscopic data obtained in film were observed for isomeric difluorides 15 and 22 which differ from that of the 2,3-difluoride 16 with a hydroxy group at C-1. 19F NMR data of 5-O-benzoylated fluorodeoxy monosaccharides 14–16, 21–22 are indicative of the assigned structures of synthesized compounds. Fluorine resonance signals of 3-deoxy fluoro ribosides, 1,3 and 2,3-difluoro-containing arabinofuranose derivatives are revealed as double triplets or two complex multiplets, respectively, in their 19F NMR spectra.
The proposed mechanism of the DAST-reaction with 11b resulting to the fluoride 21, difluorides 15 and 22 is outlined in Scheme 3. The results of fluorination of xyloside 11b point to the interaction of DAST with hydroxyl groups of the β-xyloside as well as the α-xyloside 11a gives rise to the predominant generation of classical C-3-O-SF2NEt2 derivative 18 on the first step. In addition, the formation of 5-O-benzoylated 2,3-anhydro-β-D-riboside via transient intermediate 18 took also place in this reaction. Conversions of the key intermediate 19 are likely to proceed with participation of β-methoxy group and production of the 2-O-activated intermediate 13 (via isomerization at C-1), and a methyloxiranium ion 20 (via 1,2-alkoxy migration) [32] followed by attacks with fluoride on C-2 or C-1 atoms, respectively, leading to difluorides 15 and 22 (path a). The formation of methyl 2,3-difluoro α-d-arabinofuranoside derivative 15, but not the β-counterpart, from the β-xyloside 11b is unexpected fact which may also be explained by another reaction pathway b via anomerization of the intermediate 18 forming on the first step to an isomeric intermediate 12 with a α-methoxy group under acidic reaction conditions (HF/CH2Cl2) followed by consecutive transformations of the latter with DAST into the C-2-O-SF2NEt2 derivative of the α-riboside 13 and the 2,3-difluoride 15 as in the case of the fluorination of the α-xyloside 11a (Scheme 2).
Furthermore, the DAST-reaction of a mixture of isomeric xylosides 11a and 11b (a ratio - 3:2) was carried out in methylene chloride under 24–27 °C for 18 h to give to the 3-fluoro-α-D-riboside 14 (41%) and its β-anomer 21 (40%) in 40% overall yield as the main reaction products along with difluoro derivatives 15 (24%) and 22 (4%), and 5-O-benzoylated the 2,3-ribo-epoxide (14%). It should be noted that the fluorination reaction of a mixture of xylosides 11a,b with predominant content of the α-anomer afforded 3-fluoro-3-deoxy-D-ribosides 13 and 20 in a similar yield, and the target 2,3-difluoro sugar 15 in moderate yield.
Based on the above findings, it may be concluded that the DAST-reactions with 5-O-protected α-11a and β-11b xylofuranosides resulted in fluorinations on several positions (C-3; C-3 and C-2; or C-3 and C-1) of the pentofuranose rings via the DAST-activation of free hydroxyl groups with formation of the C-3-O-SF2NEt2, and then C-2-O-SF2NEt2 derivatives, 1,2-alkoxy-migration or anomerization for the β-xyloside intermediate, and intermolecular attacks by fluoride-anion to afford mono-, difluorinated pentofuranoside derivatives.
2.2. Synthesis of novel purine-modified 2′,3′-difluoro-d-arabino nucleosides from the 1-α-bromide 17
As an extension of our previous studies, we prepared 2′,3′-dideoxy-2′,3′-difluoro-d-arabinofuranosyl nucleosides featuring fluorine substituted purine nucleobases from the universal carbohydrate precursor. A series of purine-modified 2′,3′-difluoro arabinonucleosides were synthesized via glycosylation reactions of 2-fluoroadenine and 6-chloro-2-fluoropurine with the bromide 17 prepared from d-xylose. Nucleobase anion glycosylation reaction of potassium salt of the 2-fluoroadenine, generated in the presence of potassium t-butoxide in anhydrous 1,2-dimethoxyethane, with the bromo sugar 17 in a mixture of anhydrous acetonitrile/methylene chloride in the presence of calcium hydride at room temperature afforded a mixture of protected N9-β-D- and N9-α-D nucleosides 23 and 24 which were separated by column chromatography on silica gel in 28% yield (Scheme 4). Debenzoylation of individual blocked nucleosides 23 and 24 with LiOH in aqueous acetonitrile [7] at room temperature gave pure β-2′,3′-difluoroarabino nucleoside 25 and its α-anomer 26 in 73% and 87% yield, respectively.
Scheme 4. Reagents and conditions.
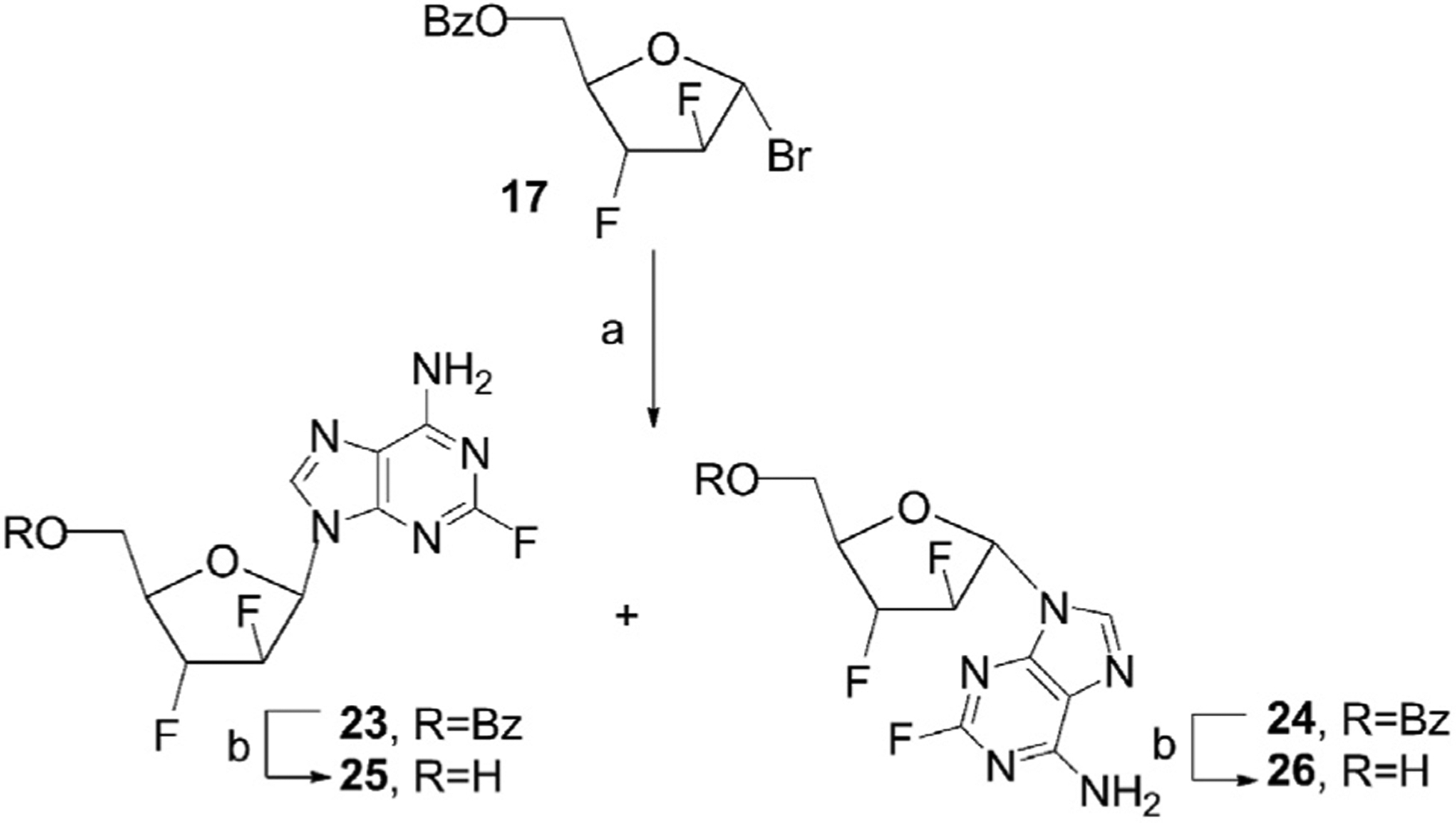
a) 17, K-salt of 2-fluoroadenine, CH3CN/CH2Cl2, CaH2, rt, 23, 28%; 24, 28%; b) 23, LiOH·H2O, CH3CN/H2O, rt, 25, 73%; 24, LiOH·H2O, CH3CN/H2O, rt, 26, 87%.
An alternative approach was investigated to prepare the target 2-fluorosubstituted nucleoside 25 through the coupling reaction of 6-chloro-2-fluoropurine instead of 2-fluoroadenine with the carbohydrate precursor 17 followed by transformations of intermediate protected N9-β-d-arabinoside 27 (Scheme 5). Reaction of the potassium salt of 6-chloro-2-fluoropurine, prepared in the presence of potassium t-butoxide in anhydrous 1,2-dimethoxyethane, with the bromo sugar 17 at room temperature gave a mixture of N9-β- and N9-α-D-2′,3′-difluoroarabinonucleosides (a β/α-ratio = 8.5:1 according to 1H NMR data) from which protected β-6-chloro-2-fluoropurine nucleoside 27 was isolated in 66% yield after column chromatography (Scheme 5). Standard deacylation of 27 with LiOH at room temperature gave β-6-chloro-2-hydroxypurine nucleoside 28 as the main product of the reaction in 61% yield after column chromatography on silica gel. The treatment of 27 with a saturated solution of ammonia in anhydrous 1,2-dimethoxyethane at room temperature for 18 h gave 5-O-benzoylated β-nucleosides of 6-chloro-2-aminopurine 29 and 2-fluoroadenine 23 which were successively isolated by chromatography on silica gel in 55% and 23% yield, respectively.
Scheme 5. Reagents and conditions.
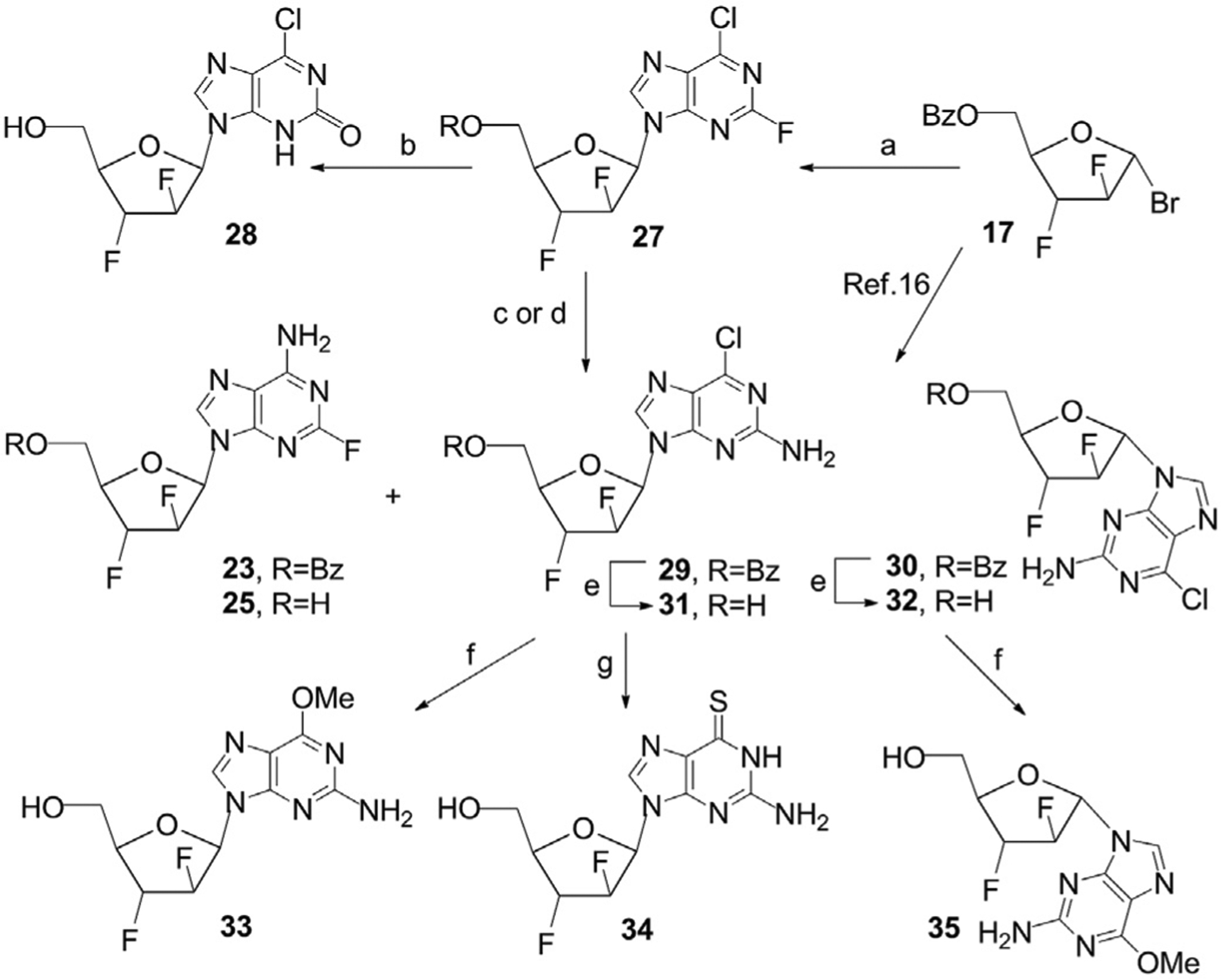
a) 17, K-salt of 2-fluoro-6-chloropurine, CH3CN, rt, 27, 66%; b) LiOH·H2O, CH3CN/H2O, rt, 87%; c) THF/NH3 in MeOH, 31, 54%; 25, 15%; d) 1,2-DME/NH3, 0 °C, 1 h, then 24 °C, 18 h, 29, 55%, 23, 23%; e) NH3, MeOH, rt; f) 31, MeOH, K2CO3, 80–82 °C, 33, 72%; 32, MeOH, K2CO3, 80–82 °C, 35, 75%; g) 31, EtOH, CS(NH2)2, reflux, 76%.
In this case, substitution reactions of halogen atoms in the purine base of β-D-nucleoside 27 with ammonia gave 5-O-protected 6-chloro-2-aminopurine β-nucleoside 29 as predominant product in contrast to the results prepared earlier under the same treatment of the protected N9-β/α-D-2′,3′-difluoroarabinoside of 2,6-dicloropurine [17]. Deacylation of protected intermediate N9-β-D-arabinonucleoside 27 with saturated at 0 °C methanolic ammonia in anhydrous tetrahydrofuran resulted in 6-chloro-2-aminopurine β-D-nucleoside 31 (54%) along with β-2-fluoroadenine derivative 25 (15%) which were separated by column chromatography on silica gel (Scheme 5). Deprotection of N9-β-D-arabinonucleoside 27 with ammonia in a mixture of methanol/THF unexpectedly afforded also 2-amino-6-chloropurine derivative 31 as the main product after selective displacement of a fluorine atom at C-2 and the removal of benzoyl protecting group, and the target 2-fluorine-substituted nucleoside was prepared in low yield. The structures of nucleosides 29 and 31 were confirmed by comparison of their NMR data with those of the β-nucleosides synthesized via the glycosylation reaction of 6-chloro-2-aminopurine with the bromo sugar 17 followed by deacylation of intermediate nucleosides 28 and 29, following a procedure we established earlier [16]. The above selective transformations in 27 demonstrate that this protected halogenated nucleoside as well as the 2,6-dichloropurine analog [16,17] is a valuable intermediate for the synthesis of different 2,6-substituted purine nucleoside derivatives of this class.
The treatment of β-anomer 31 with potassium carbonate in methanol at 80–82 °C gave 2-amino-6-methoxypurine β-arabinoside 33, a close analog of the antileukemic nucleoside nelarabine (9-β-d-arabinofuranosyl-2-amino-6-methoxypurine) which is used for treatment of T-cell acute lymphatic leukemia [36]. 2′,3′-Difluoronelarabine 33 was isolated by chromatography on silica gel in 72% yield, while the α-isomer 35 was prepared from 2-amino-6-chloropurine derivative 32 in 75% yield through the nucleophilic displacement of the chlorine atom by the methoxide anion (Scheme 5). Thiation of 6-chloro-2-aminopurine β-arabinonucleoside 31 with thiourea in EtOH under refluxing gave 9-(2′,3′-difluoro-β-d-arabinofuranosyl)-6-thioguanine (34) (76%), a difluoro analog of 9-(β-d-arabinofuranosyl)-6-thioguanine [37] which displayed cytotoxicity against L1210 mouse leukemia.
The structures of fluorinated nucleosides were confirmed by 1H, 13C, 19F NMR, and mass spectroscopy. The assignments of the configurations of synthesized purine nucleosides at the anomeric centres were based upon 1H and 13C NMR data. The diagnostic for the β-anomeric configuration of difluoronucleosides 23,25, 27–31, 33 and 34 is the five-bond couplings of 1.3–2.4 Hz between the H-8 protons of modified purine bases (doublets in their 1H NMR spectra) and 2′-β-fluorine atoms [7,14–17,38]. The absence of the same long-range couplings is characteristic of the proton NMR spectra of α-anomers 24, 26, 35 and signals of the H-8 protons are displayed as singlets. The magnitudes of 2JC-1′,F-2′ coupling constants for β- and α-anomers of 2′,3′-difluoronucleosides 23 and 25 (16.9 and 17.1 Hz), 24 and 26 (30.9 and 35.9 Hz) in their 13C NMR spectra also confirmed the stereochemistry of the purine analogs at the anomeric centres [16,17,39]. 19F NMR data of nucleosides provided evidence in favor of the assigned structures of purine derivatives with 2′,3′-difluoro-d-arabinofuranosyl moiety. F-2′ and F-3′ resonance signals of the nucleosides are revealed as complex multiplets for all series and F-2 resonances of 2-fluorine-substituted purine derivatives 23–26, 27 appeared as singlets at a range of −49 to −53 ppm [40] in the 19F NMR spectra.
3. Conclusions
In summary, synthetic routes to a series of new purine-modified 2′,3′-dideoxy-2′,3′-difluoro-D-arabino nucleosides were developed from readily available d-xylose using selective anion glycosylation reactions of 2-fluoropurine derivatives by the 1-α-bromide. A short and improved on key steps synthesis of 5-O-benzoyl 2,3-dideoxy-2,3-difluoro-α-d-arabinofuranosyl bromide was devised which includes a new and selective 5-O-monobenzoylation of methyl xylosides via their stannylene derivatives with benzoyl chloride, the efficient method for synthesis of 2,3-dideoxy-2,3-difluoro-α-d-arabinofuranoside derivative via fluorination reaction of 5-O-benzoylated methyl 3-fluoro-3-deoxy-α-d-ribofuranoside with DAST under mild heating, and bromination of the intermediate 1-O-acetates prepared under optimized conditions for the acetolysis reaction. New insights into the selective fluorination procedures of 5-O-benzoylated D-pentofuranosides with DAST have been obtained and these findings are of interest for the development of practical synthetic methods for different deoxyfluoro sugars. Novel 2-fluoro-substituted purine 2′,3′-difluoro D-arabino nucleosides have been prepared via selective coupling reactions of potassium salts of 2-fluoroadenine and 2-fluoro-6-chloropurine with the bromide. We showed that the protected 2-fluoro-6-chloropurine N9-β-d-arabinoside can be used as a versatile and valuable intermediate to prepare various 2′,3′-difluoro-β-d-arabinofuranosyl 2,6-disubstituted purine nucleosides. Using the multi-step approach, we have successfully prepared 9-(2′,3′-difluoro-β-d-arabinofuranosyl)-2-fluoroadenine and 9-(2′,3′-difluoro-β-d-arabinofuranosyl)-2-amino-6-methoxypurine as sugar-difluorinated nucleoside analogs of fludrabine and nelarabine.
4. Experimental
4.1. General information
Column chromatography was performed on silica gel 60H (70–230 mesh; Merck, Darmstadt, Germany), and thin-layer chromatography (TLC) on Merck silica gel aluminum 60 F254 pre-coated plates. The anhydrous solvents were distilled over CaH2, P2O5 or magnesium prior to the use. All commercially available reagents were used without further purification. 1H, 13C, and 19F NMR spectra were recorded in CDCl3, CD3OD and DMSO-d6 with a Bruker Avance-500-DRX spectrometer at 500.13, 126.76 and 470.59 MHz, respectively. 1H and 13C NMR chemical shifts (δ, ppm) are relative to internal chloroform peak (7.26 ppm for 1H and 77.0 for 13C NMR). Chemical shifts are also reported downfield from internal SiMe4 (1H) or external CFCl3 (19F). J values are reported in Hz. Melting points were determined on a Boetius apparatus and were uncorrected. IR spectra were measured on Perkin-Elmer Spectrum 100FT-IR spectrometer. High resolution mass spectra (HRMS) were recorded on an Agilent Q-TOF 6550 Instrument (USA) using ESI (electrospray ionization).
4.2. Syntheses of 5-O-benzoylated mono and di-fluorinated d-pentofuranoside derivatives from d-xylose
4.2.1. Selective 5-O-benzoylation of methyl α,β-d-xylofuranoside (9)
Method A.
To methyl α,β-d-xylofuranoside (9) (1.09 g, 6.64 mmol), prepared in quantitative yield after methanolysis of d-xylose [18] and purification on silica gel, in anhydrous CH2Cl2 (15 mL) was added triethylamine (1.57 mL, 11.26 mmol) and Pr2NEt (1.0 mL, 5.6 mmol) and then to prepared solution was added slowly dropwise benzoyl chloride (1.0 mL, 8.62 mmоl) in anhydrous CH2Cl2 (6 mL) at 0 °С (ice and sodium chloride). The reaction mixture was stirred for 15 min under cooling and then 18 h at room temperature. The solution was diluted with CH2Cl2 (60 mL), washed with water, the aqueous phase was extracted with CH2Cl2 (30 mL). The combined organic extracts were washed cooled 5% NaHCO3, dried over anh. Na2SO4 and evaporated to dryness. The residue was chromatographed on silica gel using a mixtures of EtOAc-petroleum ether to give a mixture of 5-O-benzoates α/β-d-xylofuranosides of 11a,b (819 mg, 46% overall yield). Chromatography of the prepared mixture, using a linear gradient of EtOAc (14 → 66%, v/v; 500 mL) in petroleum ether, afforded benzoate β-xyloside 11b (428 mg, 45%). Mp.107–108 °C. 1H NMR (CDCl3): 7.42–8.07 (m, 5H, Ar-H), 4.89 (s, 1H, H-1), 4.67 (dd, 1H, J5,4 = 4.5, J5,5′ = 11.2, H-5), 4.64 (m, 1H, H-4), 4.50 (dd, 1H, J5,4 = 6.6, H-5′), 4.25 (br.s, 1H, H-2), 4.16 (br.d, 1H, H-3), 3.40 (s, 3H, OCH3). 13C NMR (CDCl3): 166.7 (C=O, Bz), 133.2, 129.8, 129.4 (C6H5CO-), 108.7 (C-1), 80.8 (C-4), 79.6 (C-2), 76.4 (C-3), 64.4 (C-5), 55.4 (OCH3). HRMS (ESI+): m/z calcd for [C13H16O6+Na]+: 291.0840, found 291.0839.
And benzoate α-xyloside 11a (324 mg, 39%) as a syrup. 1H NMR (CDCl3): 7.43–8.05 (m, 5H, Ar-H), 5.05 (d, 1H, J1,2 = 4.4, H-1), 4.71 (dd, 1H, J5,4 = 5.5, J5,5′ =11.4, H-5), 4.43 (m, 1H, H-4), 4.38 (dd, 1H, J5,4 = 5.0, H-5′), 4.26 (t, 1H, J3,2 = 3.8, J3,4 = 3.8, H-3), 4.18 (t, 1H, H-2), 3.51 (s, 3H, OCH3). 13C NMR (CDCl3): 166.9 (C=O, Bz), 133.3, 129.8, 129.7, 128.5 (C6H5CO-), 102.0 (C-1), 77.9 (C-4),76.9 (C-2), 76.6 (C-3), 62.6 (C-5), 56.2 (OCH3). HRMS (ESI+): m/z calcd for [C13H16O6+Na]+: 291.0840, found 291.0841.
Method B.
To methyl α,β-d-xylofuranoside 9 (256 mg, 1.56 mmol) in anhydrous MeOH (10 mL) was added Bu2SnO (388 mg, 1.56 mmol) and then the reaction mixture was refluxed for 1 h and stirred for 15 min at rt. Prepared solution was evaporated dryness, to the residue was added anhydrous toluene (6 mL), then dropwise iPr2NEt (0.53 mL, 2.96 mmol) and benzoyl chloride (0.36 mL, 3.10 mmоl). The reaction mixture was stirred for 5 h at room temperature, and then evaporated. The residue was chromatographed on silica gel using a mixtures of EtOAc-petroleum ether to give a mixture of 5-O-benzoylated α/β-d-xylofuranosides 11a,b (194 mg, 46% overall yield). Chromatography of the prepared mixture, using a linear gradient of EtOAc (14 → 66%, v/v; 500 mL) in petroleum ether, afforded β-xyloside 11b (96 mg, 41%) and α-xyloside 11a (65 mg, 35%).
Method C.
To methyl α,β-d-xylofuranoside 9 (256 mg, 1.56 mmol) in anhydrous MeOH (10 mL) was added Bu2SnO (388 mg, 1.56 mmol) and then the reaction mixture was refluxed for 1 h and stirred for 15 min at rt. Prepared solution was evaporated dryness, to the residue was added 1,2-dimethoxyethane (4 mL) and then dropwise iPr2NEt (0.65 mL, 3.63 mmol) and benzoyl chloride (0.54 mL, 4.65 mmоl). The reaction mixture was stirred for 5 h at room temperature, and then evaporated. The residue was chromatographed on silica gel using a mixtures of EtOAc-petroleum ether to give a mixture of 5-O-benzoylated α/β-d-xylofuranosides 11 a,b (404 mg, 97% overall yield). Chromatography of the prepared mixture using a linear gradient of EtOAc (14 → 66%, v/v; 500 mL) in petroleum ether afforded crystalline β-xyloside 10b (206 mg, 89%) and α-xyloside 11a (153 mg, 82%) as a syrup.
4.2.2. Isomerization of β-d-xylofuranoside 11b to α-d-xylofuranoside 11a
Method A.
To methyl β-d-xylofuranoside 11b (108 mg, 0.64 mmol) in anhydrous MeOH (2.0 mL) was added CH3SO2OH (0.04 mL, 2.48 mmol) and then the reaction mixture was stirred for 18 h at rt. Prepared solution was treated by cooled 5%-aqueous NaHCO3, the aqueous phase was extracted with CH2Cl2 (3 × 35 mL). The combined organic extracts were dried over anh. Na2SO4 and evaporated to dryness. A mixture of anomers was prepared as oil product (115 mg). Ratio of 5-O-benzoyl α/β-xylofuranosides 11b and 11a (1:0.66) was determined from the 1H NMR spectrum of the mixture in CDCl3.
Method B.
To methyl 5-O-benzoyl-β-d-xylofuranoside 11b (155 mg, 0.58 mmol) was added MeOH/HCl (3.0 mL, 0.5 mmol HCl) from a solution prepared by adding 0.18 mL acetyl chloride to anhydrous 15 mL MeOH at 0 °C and then the reaction mixture was stirred for 3 h at rt. Prepared solution was evaporated under diminished pressure at 30–35 °C, coevaporated with anhydrous toluene. A mixture of anomers was prepared as oil product (155 mg). Ratio of 5-O-benzoyl α/β-xylofuranosides 11b and 11a (1:0.90) was determined from the 1H NMR spectrum of the mixture in CDCl3.
4.2.3. DAST-reaction with xyloside 11a
To a solution of methyl 5-O-benzoyl-α-xylofuranoside 11a (272 mg, 1.01 mmol) in anhydrous CH2Cl2 (5.9 mL) was added dropwise 0.75 mL (5.66 mmоl) DAST at room temperature. The reaction mixture was stirred for 30 min at rt and then for 10 h at 25–29 °C. The solution was diluted CH2Cl2 (10 mL), poured gradually into cooled 5%-aqueous NaHCO3 with stirring, then after stirring for 30 min the aqueous phase was extracted with CH2Cl2 (3 × 60 mL). The combined organic extracts was washed with water (20 mL), and dried over anhydrous Na2SO4 and evaporated to dryness. The residue was chromatographed on a silica gel, using a mixture of 12:1, 9:1, and 1:1 hexane-EtOAc to afford the difluoride 15 (49 mg, 18%) as a syrup. IR (film, CHCl3): v 2931, 2854, 1726, 1275, 1116, 1076, 712 cm−1. 1H NMR (CDCl3): 7.44–8.06 (m, 5H, Ar-H), 5.19 (br.d, 1H, J1,2 < 1.0, J1,F-2 = 10.3, H-1), 5.10 (dm, 1H, H-3), 5.08 (ddd, 1H, H-2), 4.51–4.61 (m, 3H, H-4 an 2H-5), 3.44 (s, 3H, OCH3). 13C NMR (CDCl3): 166.1 (s, C=O, Bz), 133.3, 129.8, 129.4, 128.4 (4s, C6H5CO-), 105.9 (dd, JC-1,F-3 = 4.0, JC-1,F-2 = 35.2, C-1), 97.4 (dd, JC-2,F-2 = 181.5, JC-2,F-3 = 28.0, C-2), 94.9 (dd, JC-3,F-2 = 30.5, JC-3,F-3 = 186.5, C-3), 80.2 (d, JC-4, F-3 = 28.9, C-4), 62.9 (d, JC-5,F-3 = 5.4, C-5), 55.0 (OCH3). 19F NMR (CDCl3): −195.35 (m, F-2), −192.93 (m, F-3). HRMS (ESI+): m/z calcd for [C13H14F2O4+Na]+: 295.0753, found 295.0751.
And methyl 5-O-benzoyl-3-deoxy-3-fluoro-α-d-ribofuranoside (14) (140 mg, 51%) as a syrup. IR (film, CHCl3): v 3487, 2937, 1722, 1275, 1076, 712 cm−1. 1H NMR (CDCl3): 7.44–7.99 (m, 5H, Ar-H), 4.98 (d, 1H, J1,2 = 4.8, H-1), 4.91 (ddd, 1H, J3,2 = 5.6, J3,4 = 1.8, J3,F = 55.7, H-3), 4.59 (ddt, 1H, J4,F = 25.9, H-4), 4.51 (dd, 1H, J5,4 = 3.7, J5,5′ = 12.0, H-5), 4.45 (dd, 1H, J5,4 = 3.5, H-5′), 4.21 (dt, 1H, J2,F = 22.1, H-2), 3.50 (s, 3H, OCH3). 13C NMR (CDCl3): 166.0 (C=O, Bz), 133.4, 129.5, 129.4, 129.3, 128.5 (4s, C6H5CO-), 102.2 (C-1), 90.4 (d, JC-3,F-3 =186.5, C-3), 80.5 (d, JC-4, F-3 = 25.2, C-4), 72.3 (d, JC-2,F-3 = 16.6, C-2), 63.7 (d, JC-5,F-3 = 10.3, C-5), 55.7 (OCH3). 19F NMR (CDCl3): −195.3 (dt, F-3). HRMS (ESI+): m/z calcd for [C13H15O5F+Na]+: 293.0796, found 293.0796.
4.2.4. Methyl 5-O-benzoyl-2,3-dideoxy-2,3-difluoro-α-d-arabinofuranoside (15)
To a solution of methyl 5-O-benzoyl-3-deoxy-3-fluoro-α-ribofuranoside 14 (190 mg, 0.7 mmol) in anhydrous CH2Cl2 (6.6 mL) and pyridine (0.1 mL, 1.23 mmol) was added dropwise 0.34 mL (2.57 mmоl) DAST at room temperature. The reaction mixture was stirred for 30 min and then for 18 h at 35–37 °C. The solution was diluted CH2Cl2 (10 mL), poured gradually into cooled 5%-aqueous NaHCO3, the aqueous phase was extracted with CH2Cl2 (2 × 25 mL). The combined organic extracts was washed with water (10 mL), and dried over anhydrous Na2SO4 and evaporated to dryness. The residue was chromatographed on a silica gel, using a mixture of 12:1, 9:1 and 6:1 hexane-EtOAc to afford the difluoride 15 (127 mg, 66%) as a syrup.
4.2.5. Methyl 5-O-benzoyl-2,3-dideoxy-2,3-difluoro-α,β-d-arabinofuranose (16)
To methyl arabinoside 15 (35 mg, 0.12 mmol) in CH3COOH (0.27 mL), Ac2O (0.064 mL) was added 34% HBr in acetic acid (0.3 mL). The resulting mixture was stirred for 18 h at room temperature, and then evaporated at 35–40 °C, coevaporated with anhydrous toluene. The residue was dissolved in CH2Cl2 (10 mL), and washed with water (3 mL), the organic layer was dried over anhydrous Na2SO4 and evaporated to dryness. The yellowish oil was chromatographed on a silica gel, using a mixture of 3:1, 1:1 hexane-EtOAc to afford 16 mg (50%) the difluoride 16 as a syrup. IR (film, CHCl3): v 3451, 2957, 2927, 1726, 1278, 1073, 715 cm−1. 1H NMR (CDCl3): 7.49–8.11 (3m, 7.08H, Bz), 5.68 (d, 1H, J1,F-2 = 10.4, H-1α), 5.55 (m, 0.3H, H-1β), 5.34 (ddt, 0.3H, H-2β or H-3β), 5.12–5.27 (dm, 2H, H-2α and H-3α), 4.78 (dm, 1H, J4,F = 22.76, H-4α), 4.62 (dd, 1H, J5,4 = 4.6, J5,5′ = 12.0, H-5α), 4.56 (dd, 1H, J5,4 = 4.9, H-5′α), 4.53–4.65 (m, H-5β and H-5″β), 4.5 (dm, 0.3H, J4,F = 23.08, H-4β). 13C NMR (CDCl3): 166.2 (s, C=O, Bz), 133.4, 129.8, 129.4, 128.5 (4s, C6H5CO-), 100.3 (dd, JC-1,F-2 = 35.1, JC-1,F-3 = 3.1, C-1), 97.4 (d, JC-2,F-2 = 181.5, JC-2,F-3 = 27.8, C-2), 94.8 (dd, JC-3,F-3 = 182.5, JC-3,F-2 = 30.7, C-3), 80.6 (d, JC-4, F-3 = 38.0, C-4), 63.1 (d, JC-5,F-3 = 6.9, C-5). 19F NMR (CDCl3): −192.5 (m, F-2 or F-3), −195.4 (m, F-3 or F-2). HRMS (ESI+): m/z calcd for [C12H12F2O4−OH]+: 241.0676, found 241.0675; calcd for [C12H12F2O4+Na]+: 281.0596, found 281.0593; calcd for [C12H12F2O4+MeOH+Na]+: 313.0858, found 313.0858.
4.2.6. 5-O-benzoyl-2,3-dideoxy-2,3-difluoro-α-d-arabinofuranosyl bromide (17)
Concentrated H2SO4 (0.03 mL) was added to a solution of difluoride 15 (60 mg, 0.22 mmol) in acetic acid (0.38 mL) and acetic anhydride (0.12 mL) at 0 °С. The reaction mixture was stirred at this temperature for 20 min and then 3 h at 24–25 °С. To solution was added ice. After the ice melted, the aqueous phase was extracted with CH2Cl2 (3 × 20 mL). Cooled aqueous NaHCO3 was added to the aqueous layer and then it was extracted with CH2Cl2 (20 mL), the combined organic extracts dried over anhydrous Na2SO4 and evaporated to dryness. The residue was used on the next step after coevaporation with anhydrous toluene (2 × 6 mL). To a suspension of intermediate 1-O-acetates and anhydrous ZnBr2 (10 mg) in anhydrous CH2Cl2 (1.7 mL) was added TMSBr (0.06 mL, 0.46 mmol) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h, then 18 h at room temperature. The reaction mixture was poured into saturated cooled aqueous NaHCO3, extracted with CH2Cl2 (3 × 30 mL). The combined organic extracts were dried over anhydrous Na2SO4 and evaporated to dryness, and coevaporated with anhydrous toluene to give 59 mg (combined yield 84%) of the bromide 17 as a yellowish oil which was used in the next step without an additional purification.
4.2.7. DAST-reaction with xyloside 11b
To a solution of β-d-xylofuranoside 11b (86 mg, 0.32 mmol) in anhydrous CH2Cl2 (2.0 mL) was added dropwise 0.25 mL (1.92 mmоl) DAST at room temperature. The reaction mixture was stirred for 18 h at 24–27 °C. The solution was diluted CH2Cl2 (10 mL), poured into cooled 5%-aqueous NaHCO3, the aqueous phase was extracted with CH2Cl2 (3 × 30 mL). The combined organic extracts was washed with water (15 mL), and dried over anhydrous Na2SO4 and evaporated to dryness. The residue was chromatographed on a silica gel, using a mixture of 12:1, 9:1, 4:1 and 1:1 hexane-EtOAc to afford the difluoride 15 (3 mg, 3%) as a syrup.
5-O-benzoyl-2-O-methyl-3-deoxy-3-fluoro-α-d-arabinofuranosyl fluoride (22) (3 mg, 3%) as a syrup. IR (film, CHCl3): v 2943, 2851, 1725, 1276, 1112, 712 cm−1. 1H NMR (CDCl3): 7.46–8.13 (m, 5H, Ar-H), 5.87 (d, 1H, J1,F-1 = 60.4, J1,2 < 1.0 Hz, H-1), 5.03 (dd, 1H, J3,F-3 = 52.1, J3,4 = 2.8, H-3), 4.83 (dm, 1H, J4,F-3 = 25.64, H-4), 4.55 (dd, 1H, J5,4 = 4.8, J5,5′ =11.9, H-5), 4.49 (dd, 1H, J5,4 = 5.1, H-5′), 3.51 (s, 3H, OMe), 4.17 (ddd, 1H, J2,3 = 0.9 Hz, H-2). 13C NMR (CDCl3): 166.2 (s, C=O, Bz), 133.4,129.9, 129.5, 128.5 (4s, C6H5CO-),112.7 (dd, JC-1,F-1 = 225.7, C-1), 94.1 (d, JC-3,F-3 = 186.5, C-3), 87.7 (dd, J = 34.7, J = 25.1, C-2), 83.4 (d, JC-4, F-3 = 28.3, C-4), 63.0 (d, JC-5,F-3 = 7.1, C-5), 56.1 (OCH3). 19F NMR (CDCl3): −123.59 (dt, F-1), −188.77 (m, F-3). HRMS (ESI+): m/z calcd for [C13H14F2O4+Na]+: 295.0753, found 295.0754.
Methyl 5-O-benzoyl-2,3-anhydro-β-d-ribofuranoside (11 mg, 14%) as a syrup. 1H NMR (CDCl3): 7.49–8.13 (m, 5H, Ar-H), 5.05 (s, 1H, H-1), 4.44–4.52 (m, 3H, H-4, H-5 and H-5′), 3.90 (d, 1H, H-2), 3.78 (d, 1H, H-3), 3.43 (s, 3H, OCH3). 13C NMR (CDCl3): 166.1 (s, C=O, Bz), 133.3, 129.8, 128.5 (C6H5CO-), 102.5 (C-1), 76.1 (C-4), 64.3 (C-5), 56.4, 55.5, 55.2 (C-2, C-3, OCH3). HRMS (ESI+): m/z calcd for [C13H14O5+Na]+: 273.0734, found 273.0737.
And methyl 5-O-benzoyl-3-deoxy-3-fluoro-β-d-ribofuranoside (21) (48 mg, 55%) as a syrup. IR (film, CHCl3): v 3454, 2940, 2937, 1722, 1275, 1123, 712 cm−1. 1H NMR (CDCl3): 7.44–8.07 (m, 5H, ArH), 5.2 (dt, 1H, J3,2 = 4.7, J3,4 = 4.7, J3,F =54.18, H-3), 4.91 (br.s, 1H, H-1), 4.52–4.59 (m, 1H, H-4 and H-5), 4.43 (dd, 1H, J5,4 = 3.7, J5,5′ = 12.0, H-5′), 4.25 (dt, 1H, J2,F = 22.1, H-2), 3.45 (s, 3H, OCH3). 13C NMR (CDCl3): 165.2 (s, C=O, Bz), 133.2, 129.7, 129.4 (3s, C6H5CO-), 107.9 (d, JC-1, F-2 = 2.7, C-1), 92.9 (d, JC-3,F-3 = 185.5, C-3), 78.4 (d, JC-4, F-3 = 25.5, C-4), 74.4 (d, JC-2,F-3 = 15.2, C-2), 64.1 (d, JC-5,F-3 = 4.5, C-5), 55.5 (OCH3). 19F NMR (CDCl3): −195.3 (dt, F-3). HRMS (ESI+):m/z calcd for [C13H15O5F+Na]+: 293.0796, found 293.0796
4.2.8. DAST-reaction with a mixture of xylosides 11a,b
To a solution of methyl 5-O-benzoyl-α/β-xylofuranoside 11a,b (300 mg, 1.12 mmol, 3:2, ratio of 11a/11b) in anhydrous CH2Cl2 (6.2 mL) was added dropwise 0.88 mL (6.75 mmоl) DAST at 24–26 °C. The reaction mixture was stirred for 18 h and then diluted CH2Cl2, poured into cooled 5%-aqueous NaHCO3, the aqueous phase was extracted with CH2Cl2 (3 × 60 mL). The combined organic extracts was washed with water (20 mL), and dried over anhydrous Na2SO4 and evaporated to dryness. The residue was chromatographed on a silica gel, using a mixture of 12:1, 9:1, 6:1 and 3:1 hexane-EtOAc to afford difluoride 15 (44 mg, 24%) as a syrup, 1,3-difluoride 22 (5 mg, 4%) as syrup, methyl 5-O-benzoyl-3-deoxy-3-fluoro-α-ribofuranoside (14) (74 mg, 41%) as a syrup, and methyl 5-O-benzoyl-3-deoxy-3-fluoro-β-ribofuranoside (22) (48 mg, 40%) as a syrup.
4.3. Synthesis of novel purine-modified 2′,3′-difluoro-d-arabino nucleosides from the 1-α-bromide
4.3.1. 2-Fluoro-9-(5-O-benzoyl-2,3-dideoxy-2,3-difluoro-β-d-arabinofuranosyl)adenine (23) and its α-anomer 24
Potassium t-butoxide (32 mg, 0.28 mmol) was added to 2-fluoroadenine (42 mg, 0.25 mmol) in anhydrous 1,2-dimethoxyethane (10 mL) at 0 °C and then the resulting solution was stirred for 9 min under cooling and then for 40 min at room temperature and evaporated to dryness coevaporated with anhydrous acetonitrile. Anhydrous acetonitrile (8 mL) was added to the residue and the suspension was stirred under argon at room temperature for 10 min, then a solution of the bromide 17 (80 mg, 0.25 mmol) in anhydrous methylene chloride (4 mL) and CaH2 (12 mg, 0.28 mmol) were added sequentially to the suspension of prepared potassium salt of the purine. The reaction mixture was stirred under argon at room temperature for 20 h. Insoluble materials were removed by filtration and the solids were washed with MeCN (20 mL). The solvent was removed under reduced pressure and the residue was chromatographed on silica gel, eluting with EtOAc/petroleum ether 1:5, 3:2 and 2:1 to afford β-nucleoside 23 (28 mg, 28%). Mp.174–176 °С (EtOH). 1H NMR (CDCl3): 8.06 (d, 1H, JH-8, F-2′ = 1.4, H-8), 7.46–8.07 (3m, 5H, Bz), 6.48 (dt, 1H, J1′,2′ = 2.7, J1′,F-3′ = 2.7, J1′,F-2′ = 21.79, H-1′), 5.43 (ddm, 1H, J3′,F-2′ = 12.18, J3′,F′ = 49.69, H-3′), 5.33 (ddm, 1H, J2′,1′ = 2.7, J2′,F-2′ = 49.38, J2′,F-3′ = 9.2, H-2′), 4.71 (dd, 1H, H-5′), 4.66 (dd, 1H-5″), 4.63 (dm, 1H, H-4′). 13C NMR (CDCl3): 166.3 (C=O, Bz), 159.3 (d, JC-2,F-2 = 212.6, C-2), 158.4 (d, J C6,F-2 = 21.9, C-6), 150.5 (d, JC-6,F-2 = 20.3, C-4),139.6 (d, JC-8,F-20′~ 4.0, C-8), 133.6, 129.6, 129.0, 129.0, 128.6 (C6H5CO-), 116.3 (s, C-5), 93.6 (dd, JC-2′,F-2′ = 192.5, JC-2′,F-3′ = 30.9, C-2′), 91.5 (dd, JC-3′,F-2′ = 29.9, JC-3′,F-3′ = 191.5, C-3′), 83.3 (d, JC-1′, F-2′ = 16.9, C-1′), 80.5 (d, JC-4′,F-3′ = 27.1, C-4′), 62.6 (d, JC-5′,F-3′ = 9.2, C-5′). 19F NMR (CDCl3): −49.86 (s, F-2), −188.71 (m, F-2′ or F-3′), −203.72 (m, F-3′ or F-2′). HRMS (ESI+): m/z calcd for [C17H14N5F3O3 + H]+: 394.1122, found 394.1123.
And α-nucleoside 24 (28 mg, 28%). Mp. 238–240 °С (EtOH). 1H NMR (DMSO-d6): 8.32 (s, 1H, H-8), 7.56–8.05 (3m, 5H, Bz), 6.49 (dd, 1H, J1′,2′ = 2.3, J1′,F-2′ = 15.0, H-1′), 6.17 (ddm, 1H, H-2′), 5.75 (ddm, 1H, H-3′), 5.10 (dm, 1H, H-4′), 4.63 (dd, 1H, H-5′), 4.57 (dd, 1H, H-5″). 13C NMR (CDCl3): 165.9 (C=O, Bz),159.1 (d, JC-2,F-2 = 206.5, C-2), 158.2 (d, JC6,F-2 = 21.7, C-6), 150.7 (d, JC-6,F-2 = 20.2, C-4), 139.8 (C-8), 134.1, 129.8, 129.7, 129.3 (C6H5CO-), 118.0 (d, JC-5,F-2 = 3.4, C-5), 96.6 (dd, JC-2′,F-2′ = 186.5, JC-2′,F-3′ = 28.9, C-2′), 94.5 (dd, JC-3′,F-2′ = 28.9, JC-3′,F-3′ = 193.5, C-3′), 86.8 (dd, JC-1′, F-2′ = 30.9, JC-1′,F-3′ = 9.5, C-1′), 81.3 (d, JC-4′,F-3′ = 25.9, JC-4′,F-2′ = 4.1, C-4′), 63.7 (d, JC-5′,F-3′ = 5.2, C-50). 19F NMR (CDCl3+CD3OD): −50.14 (s, F-2), −192.25 (m, F-2′ an F-3′). HRMS (ESI+): m/z calcd for [C17H14N5F3O3+H]+: 394.1122, found 394.1121.
4.3.2. 2-Fluoro-9-(2,3-dideoxy-2,3-difluoro-β-d-arabinofuranosyl) adenine (25)
To solution of β-nucleoside 23 (13 mg, 0.03 mmol) in a mixture of acetonitrile (1.5 mL) and water (0.5 mL) was added lithium hydroxide monohydrate (4.7 mg, 0.11 mmol). The reaction mixture was stirred at room temperature for 6 h, then neutralized with acetic acid, evaporated, and coevaporated with toluene to dryness The residue was chromatographed on silica gel using for elution EtOAc, EtOAc:MeOH-9:1 and 6:1 to afford nucleoside 25 (7 mg, 73%). Mp. 221–224 °С (CH2Cl2/EtOH). 1H NMR (CD3OD): 8.25 (d, 1H, JH-8, F-2′ = 2.3, H-8), 6.44 (ddd, 1H, J1′,2′ = 4.0, J1′,F-3′ = 1.74, J1′,F-2′ = 17.3, H-1′), 5.52 (dddd, 1H, J2′,1′ = 4.1, J3′,2′ = 2.37, J2′,F-2′ = 50.34, J2′,F-3′ = 15.06, H-2′), 5.50 (dm, 1H, J3′,F-3′ = 51.37, J3′,4′ = 3.9, J3′,F-2′ = 17.3, H-3′), 4.31 (dm, 1H, J4′,F-3′ = 20.77, H-4′), 3.90 (ddd, 1H, H-5′), 3.87 (dd, 1H, H-5″). 13C NMR (CD3OD): 159.4 (d, JC-2,F-2 = 209.8, C-2), 157.8 (d, JC6,F-2 = 20.1, C-6), 150.6 (d, JC4,F-2 =18.8), 140.1 (br.s), 116.5 (d, J C-5,F-2 = 3.0) (C-4, C-8, C-5), 93.4 (dd, JC-2′,F-2′ = 182.33, JC-2′,F-3′ = 28.5, C-2′), 92.6 (dd, JC-3′,F-2′ = 28.9, JC-3′,F-3′ = 191.8, C-3′), 83.7 (dd, JC-1′,F-2′ = 17.1, JC-1′,F-3′ = 3.1, C-1′), 80.7 (dd, JC-4′, F-3′ = 25.2, JC-4′, F-2′ = 2.2, C-4′), 60.1 (d, JC-5′,F-3′ = 6.2, C-5′). 19F NMR (CD3OD): −53.17 (s, F-2), −195.92 (m, F-2′ or F-3′), −204.4 (m, F-3′ or F-2′). HRMS (ESI+): m/z calcd for [C10H10N5F3O2+H]+: 290.0860, found 290.0861.
4.3.3. 2-Fluoro-9-(2,3-dideoxy-2,3-difluoro-α-d-arabinofuranosyl) adenine (26)
To a solution of protected α-nucleoside 24 (14 mg, 0.03 mmol) in a mixture of acetonitrile (1.5 mL) and water (0.5 mL) was added lithium hydroxide monohydrate (4 mg, 0.09 mmol). The reaction mixture was stirred at room temperature for 7 h, then neutralized with acetic acid, evaporated, and coevaporated with toluene to dryness. The residue was chromatographed on silica gel using for elution EtOAc, EtOAc:MeOH - 9:1 and 6:1 to afford nucleoside 26 (9 mg, 87%). Mp. 187–191 °С (CH2Cl2/EtOH). 1H NMR (CD3OD): 8.17 (s, 1H, H-8), 6.36 (dd, 1H, J1′,F-2′ = 15.1, J1′,2′ = 2.56, H-1′), 6.01 (ddt, 1H, J2′,3′ = 2.81, J2′,F-2′ = 14.1, J2′,F′ = 50.3, H-2′), 5.31 (dddd, 1H, J3′,2′ = 2.9, J3′,F-3′ = 51.93, J3′,4′ = 4.1, J3′,F-2′ = 16.35, H-3′), 4.76 (dm, 1H, J4′,F-3′ = 20.77, H-4′), 3.79 (d, 2H, 2H-5′). 13C NMR (CD3OD): 155.6 (d, J C-2,F-2 = 206.5, C-2), 154.1 (d, J C6,F-2 = 20.83, C-6), 146.5 (C-4), 135.6 (C-8), 113.6 (C-5), 92.8 (dd, JC-2′,F-2′ = 187.0, JC-2′,F-3′ = 19.2, C-2′), 89.9 (dd, JC-3′,F-2′ = 27.7, JC-3′,F-3′ = 183.5, C-3′), 83.7 (dd, JC-1′,F-2′ = 35.9, JC-1′,F-3′ = 5.4, C-1′), 80.7 (dd, JC-4′, F-3′ = 24.8, JC-4′,F-2′ = 2.6, C-4′), 56.5 (d, JC-5′,F-3′ = 5.4, C-5′). 19F NMR (CD3OD): −52.78 (s, F-2), −196.13 (m, F-2′ or F-3′), −197.43 (m, F-3′ or F-2′). HRMS (ESI+): m/z calcd for [C10H10N5F3O2+H]+: 290.0860, found 290.0861.
4.3.4. 2-Fluoro-6-chloro-9-(5-O-benzoyl-2,3-dideoxy-2,3-difluoro-β-d-arabinofuranosyl)-purine (27)
Potassium t-butoxide (19 mg, 0.17 mmol) was added to 2-fluoro-6-chloropurine (30 mg, 0.17 mmol) in anhydrous 1,2-dimethoxyethane (3.0 mL) at 0 °C and then the resulting solution was stirred for 7 min under cooling and 30 min at room temperature and then evaporated to dryness, coevaporated with anhydrous acetonitrile. Acetonitrile (1.5 mL) was added to residue and the suspension was stirred under argon at room temperature for 10 min, then a solution of bromide 17 (51 mg, 0.16 mmol) in an anhydrous acetonitrile (2.0 mL) was added to prepared potassium salt of the purine. The reaction mixture was stirred under argon at room temperature for 18 h. Insoluble materials were removed by filtration and the solids were washed with acetonitrile (20 mL). The solvent was removed under reduced pressure and the residue was chromatographed on silica gel, eluting with EtOAc/petroleum ether 1:5, 1:3 and 1:2.5 to afford β-nucleoside 27 (43 mg, 66%). Mp. 150–152 °С (EtOAc/petroleum ether). 1H NMR (CDCl3): 8.36 (d, 1H, JH-8, F-2′ = 1.97, H-8), 7.52–8.36 (3m, 5H, Bz), 6.59 (dt, 1H, J1′,2′ = 2.7, J1′,F-3′ = 2.7, J1′,F-2′ = 21.79, H-1′), 5.52 (ddt, 1H, J3′,F-2′ = 12.21, J3′,F′ = 49.37, H-3′), 5.40 (dddd, 1H, J1′,2′ = 2.76, J2′,F-2′ = 49.37, J2′,F-3′ = 8.97, H-2′), 4.70–4.80 (m, 3H, H-4′, H-5′ and H-5″). 13C NMR (CDCl3): 166.1 (C=O, Bz), 157.4 (d, JC-2,F-2 = 222.4, C-2), 153.3 (d, JC-6,F-2 = 7.4, C-6), 144.7 (d, J C-8,F-2’ = 2.99, C-8), 133.8, 129.8, 129.8, 129.0, 128.7 (C6H5CO- and C-5), 93.5 (dd, JC-2′,F-2′ = 184.5, JC-2′,F-3′ = 29.92, C-2′), 91.4 (dd, JC-3′,F-2′ = 30.9, JC-3′,F-3′ = 192.5, C-3′), 83.8 (d, JC-1′, F-2′ = 16.7, C-1′), 81.1 (d, JC-4′,F-3′ = 29.9, C-4′), 62.3 (d, JC-5′,F-3′ = 9.1, C-5′). 19F NMR (CDCl3): −48.68 (s, F-2), −188.60 (m, F-2′ or F-3′), −203.41 (m, F-3′ or F-2′). HRMS (ESI+): m/z calcd for [C17H12N4F3O3Cl+H]+: 413.0623, found 413.0618.
4.3.5. 2-Hydroxy-6-chloro-9-(2,3-dideoxy-2,3-difluoro-β-d-arabinofuranosyl)purine (28)
To solution of β-nucleoside 27 (11 mg, 0.02 mmol) in a mixture of acetonitrile (1.1 mL) and water (0.36 mL) was added lithium hydroxide monohydrate (4.7 mg, 0.11 mmol). The reaction mixture was stirred at room temperature for 280 min, then neutralized with acetic acid, evaporated, and coevaporated with toluene to dryness. The residue was chromatographed on silica gel using for elution EtOAc:hexane 2:1, EtOAc:MeOH-7:1 and 6:1 to afford nucleoside 28 (5 mg, 61%) as oil. 1H NMR (CD3OD): 8.41 (d, 1H, JH-8, F-2′ = 1.3, H-8), 6.53 (ddd, 1H, J1′,2′ = 3.2, J1′,F-3′ = 1.2, J1′,F-2′ = 17.4, H-1′),5.45–5.62 (dm, H-2′ and H-3′), 4.32 (dm, 1H, J4′,F-3′ = 24.8, H-4′), 3.91 (ddd, 1H, H-5′), 3.88 (dd, 1H, H-5″). 13C NMR (CD3OD): 163.5, 163.4, 152.1, 111.5 (C-6, C-2, C-4, C-5), 144.4 (br.d, C-8), 94.8 (dd, JC-2′,F-2′ = 180.5, JC-2′,F-3′ = 28.6, C-2′), 93.9 (dd, JC-3′,F-2′ = 28.6, JC-3′,F-3′ = 192.5, C-3′), 84.2 (br.d, JC-1′,F-2′ = 20.9,C-1′), 83.5 (d, JC-4′,F-3′ = 23.9, C-4′), 61.5 (d, JC-5′,F-3′ = 6.2, C-5′). 19F NMR (CD3OD): −185.38 (m, F-2′ or F-3′), −193.35 (m, F-3′ or F-2′). HRMS (ESI+): m/z calcd for [C10H9N4F2O3Cl+H]+: 307.0404, found 307.0400.
4.3.6. 2-Amino-6-chloro-9-(5-O-benzoyl-2,3-dideoxy-2,3-difluoro-β-d-arabinofuranosyl)purine (29)
A solution of β-nucleoside 27 (20 mg, 0.05 mmol) in anhydrous 1,2-dimehoxyethane (10.0 mL) was saturated by dry ammonia for 1 h at 0 °C, and then the reaction mixture was left for 18 h at room temperature and evaporated. The residue was chromatographed on silica gel using for elution EtOAc:hexane 1:3, 1:1 and 2:1 to afford nucleoside 29 (11 mg, 55%) as an amorphous powder, and nucleoside 23 (4.4 mg, 23%). NMR spectroscopic data for nucleosides 29 and 23 were identical to those prepared by glycosylation reactions of 6-chloro-2-aminopurine [16] and 2-fluoroadenine with the bromide 16.
4.3.7. 2-Amino-6-chloro-9-(2,3-dideoxy-2,3-difluoro-β-d-arabinofuranosyl)purine (31)
To solution of β-nucleoside 27 (25 mg, 0.06 mmol) in anhydrous THF (2.0 mL) was added in 4.8 mL methanol saturated at 0 °C with ammonia, the reaction mixture was stirring for 4 h under 0 °C, then for 18 h at room temperature and evaporated. The residue was chromatographed on silica gel using for elution EtOAc:hexane 2:1 and 3:1 and EtOAc:EtOH to afford nucleoside 31 (10 mg, 54%) as a syrup, and nucleoside 25 (2.6 mg, 15%).
4.3.8. 2-Amino-6-methoxy-9-(2,3-dideoxy-2,3-difluoro-β-d-arabinofuranosyl)purine (33)
To solution of β-nucleoside 31 (7 mg, 0.02 mmol) in MeOH (1.0 mL) was added anhydrous potassium carbonate (12 mg, 0.087 mmol). The reaction mixture was stirred at 80–82 °C for 30 min, then cooled to room temperature and evaporated. The residue was chromatographed on silica gel using for elution CHCl3, CHCl3:MeOH-10:1 and 5:1 to afford nucleoside 33 (5 mg, 72%) as a syrup. 1H NMR (CD3OD): 8.03 (d, 1H, JH-8, F-2′ = 2.4, H-8), 6.43 (ddd, 1H, J1′,2′ = 3.9, J1′,F-3′ = 1.92, J1′,F-2′ = 18.27, H-1′), 5.49 (dddd, 1H, J2′,1′ = 4.0, J3′,2′ = 2.2, J2′,F-2′ = 51.29, J2′,F-3′ = 14.42, H-2′), 5.48 (dm, 1H, J3′,F-3′ = 50.65, J3′,4′ = 3.8, J3′,F-2′ = 11.22, H-3′), 4.28 (dm, 1H, J4′,F-3′ = 24.68, H-4′), 4.09 (s, 3H, OCH3), 3.88 (d, 2H, 2H-5). 13C NMR (CD3OD): 161.3 (C-6), 160.7, 153.3, 113.3 (C-2, C-4, C-5), 138.4 (d, JC-8,F-2′ = 4.4, C-8), 93.6 (dd, JC-2′,F-2′ = 182.33, JC-2′,F-3′ = 28.5, C-2′), 92.5 (dd, JC-3′,F-2′ = 28.9, JC-3′,F-3′ = 191.85, C-3′), 82.6 (dd, JC-1′, F-2′ = 17.2, JC-1′, F-3′ = 2.5, C-1′), 81.9 (dd, JC-4′,F-3′ = 25.6, JC-4′,F-2′ = 1.7, C-4′), 60.2 (d, JC-5′,F-3′ = 6.7, C-5′), 52.9 (OCH3). 19F NMR (CD3OD): −195.07 (m, F-2′ or F-3′), −204.61 (m, F-3′ or F-2′). HRMS (ESI+):m/z calcd for [C11H13N5F2O3+H]+: 302.1060, found 302.1061, calcd for [C11H13N5F2O3+Na]+: 324.0879, found 324.0879.
4.3.9. 9-(2,3-Dideoxy-2,3-difluoro-β-d-arabinofuranosyl)-6-thioguanine (34)
A mixture of nucleoside 31 (8 mg, 0.03 mmol), thiourea (14 mg, 0.18 mmol) in 5.0 mL ethanol was heated at reflux for 2 h. After cooling, the mixture was evaporated in vacuum. The residue was chromatographed on silica gel using for elution CHCl3, CHCl3:MeOH-10:1 to afford nucleoside 34 (6 mg, 76%) as an amorphous powder. 1H NMR (CD3OD): 8.04 (d, 1H, JH-8, F-2′ = 2.46, H-8), 6.35 (ddd, 1H, J1′,2′ = 3.6, J1′,F-3′ = 1.7, J1′,F-2′ = 17.6, H-1′), 5.40–5.55 (dm, H-2′ and H-3′), 4.27 (dm, 1H, J4′,F-3′ = 25.0, H-4′), 3.87 (d, 2H, 2H-5′). 13C NMR (CD3OD): 177.5 (C-6), 176.1 (C-2), 155.1 (C-4), 140.6 (d, JC-8,F-2′ = 4.4, C-8), 129.2 (C-5), 95.0 (dd, JC-2′,F-2′ = 181.5, JC-2′,F-3′ = 28.9, C-2′), 93.9 (dd, JC-3′,F-2′ = 28.9, JC-3′,F-3′ = 192.48, C-3′), 83.8 (dd, JC-1′, F-2′ = 17.9, JC-1′, F-3′ = 3.3, C-1′), 83.3 (br.d, JC-4′,F-3′ = 27.9, C-4′), 61.6 (d, JC-5′,F-3′ = 6.4, C-5′). 19F NMR (CD3OD): −195.27 (m, F-2′ or F-3′), −204.65 (m, F-3′ or F-2′). HRMS (ESI+): m/z calcd for [C10H11N5O2F2S+H]+: 304.0680, found 304.0685.
4.3.10. 2-Amino-6-methoxy-9-(2,3-dideoxy-2,3-difluoro-α-d-arabinofuranosyl)purine (35)
In a similar way, starting from (6 mg, 0.02 mmol) of α-nucleoside 32 was prepared nucleoside 35 (4.4 mg, 75%) as an amorphous powder. 1H NMR (CD3OD): 7.97 (s,1H, H-8), 6.31 (dd,1H, J1′,2′ = 2.73, J1′,F-2′ = 15.3, H-1′), 6.07 (ddt, 1H, J2′,3′ = 2.83, J2′,F-2′ = 50.33, J2′,F-3′ = 14.43, H-2′), 5.42 (dddd, 1H, J3′,F-3′ = 52.5, J3′,4′ = 4.1, J3′,F-2′ = 16.67, H-3′), 4.76 (dm, 1H, J4′,F-3′ = 20.52, H-4′), 4.08 (s, 3H, OCH3), 3.79 (d, 2H, 2H-5′). 13C NMR (CD3OD): 161.3 (C-6), 160.7, 153.2, 137.5, 115.6 (C-2, C-4, C-8, C-5), 96.4 (dd, JC-2′,F-2′ = 185.5, JC-2′,F-3′ = 28.9, C-2′), 93.8 (dd, JC-3′,F-2′ = 26.9, JC-3′,F-3′ = 184.5, C-3′), 87.2 (dd, JC-1′, F-2′ = 35.9, JC-1′, F-3′ = 5.3, C-1′), 84.2 (d, JC-4′,F-3′ = 25.2, C-4′), 60.2 (d, JC-5′,F-3′ = 6.7, C-5′), 52.8 (OCH3). 19F NMR (CD3OD): −196.42 (m, F-2′ or F-3′), −197.79 (m, F-3′ or F-2′). HRMS (ESI+): m/z calcd for [C11H13N5F2O3+H]+: 302.1060, found 302.1063.
Supplementary Material
Acknowledgments
This work was supported from State Program of “Biologically active compounds” (Grant 2.18), National Academy of Sciences of Belarus, and in part by NIH grant 5P30-AI-50409(CFAR, Unated States).
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version, at https://doi.org/10.1016/j.tet.2019.02.027. These data include MOL files and InChiKeys of the most important compounds described in this article.
Conflicts of interest
The authors declared no potential conflicts of interest in this work.
References
- [1].Qiu X-L, Xu X-H, Qing F-L, Tetrahedron 66 (2010) 789–843. [Google Scholar]
- [2].Liu P, Sharon A, Chu Ch.-K., J. Fluorine Chem 129 (2008) 743–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Meanwell NA, J. Med. Chem 61 (2018) 5822–5880. [DOI] [PubMed] [Google Scholar]
- [4].Muller K, Faech C, Diederich F, Science 317 (2007) 1881–1886. [DOI] [PubMed] [Google Scholar]
- [5].Mongometry JA, Hewson K, J. Med. Chem 12 (1969) 498–504. [DOI] [PubMed] [Google Scholar]
- [6].Secrist JA, Shortancy AT, Mongometry JA, J. Med. Chem 31 (1988) 405–410. [DOI] [PubMed] [Google Scholar]
- [7].Mongometry JA, Shortancy-Fowler AT, Clayton SD, Riodan JM, Secrist III JA, J. Med. Chem 35 (1992) 397–401. [DOI] [PubMed] [Google Scholar]
- [8].Shelton JJ, Lu X, Hollenbaugh JA, Cho JH, Amblard F, Schinazi RF, Chem. Rev 116 (2016) 14379–14455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Parker WP, Allan PW, Hassan AEA, Secrist III JA, Sorscher EJ, Wand WR, Cancer Gene Ther. 10 (2003) 23–29. [DOI] [PubMed] [Google Scholar]
- [10].Vodnala SK, Lundback T, Yehenskieli E, et al. , J. Med. Chem 56 (2013) 9861–9873. [DOI] [PubMed] [Google Scholar]
- [11].Denisov AO, Tokunova YA, Fateev IV, et al. , Synthesis 49 (2017) 4853–4860. [Google Scholar]
- [12].Thomas HJ, Tiwari KN, Clayton SJ, et al. , Nucleosides Nucleotides 13 (1994) 309–323. [Google Scholar]
- [13].Maruyama T, Utsumi K, Sato Y, Richman DD, Nucleosides Nucleotides 13 (1994) 1219–1230. [Google Scholar]
- [14].Barchi JJ, Marquez VE, Driscoll JS, Ford H, Mitsuya H, Shirasaka T, Aoki S, Kelley JA, J. Med. Chem 34 (1991) 1647–1655. [DOI] [PubMed] [Google Scholar]
- [15].Ford H Jr., Driscoll JS, Siddiqui M, Kelley JA, Mitsuya H, Shirasaka T, Johns DG, Marquez VE, Nucleosides Nucleotides 13 (1994) 213–234. [Google Scholar]
- [16].Schinazi RF, Sivets GG, Detorio MA, McBrayerT R, Whitaker T, Coats SJ, Amblard F, Heterocycl. Commun 21 (2015) 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sivets GG, Kalinichenko EN, Mikhailopulo IA, Detorio MA, McBrayer TR, Whitaker T, Schinazi RF, Nucleos Nucleot. Nucleic Acids 28 (2009) 519–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Thome MA, Giudicelli MB, Picq D, Anker D, J. Carbohydr. Chem 10 (1991) 923–926. [Google Scholar]
- [19].Lawandi J, Rocheleau S, Moitessier N, Tetrahedron 72 (2016) 6283–6319. [Google Scholar]
- [20].Munavu RM, Szmant HH, J. Org. Chem 41 (1976) 1832–1836. [Google Scholar]
- [21].Nashed MA, Anderson L, Tetrahedron Lett. 39 (1976) 3503–3506. [Google Scholar]
- [22].Tsuda Y, Haque E, Yoshimoto K, Chem. Pharm. Bull 31 (1983) 1612–1624. [Google Scholar]
- [23].Alper PB, Hendrix M, Sears P, Wong C-H, J. Am. Chem. Soc 120 (1998) 1965–1978. [Google Scholar]
- [24].Mikahilopulo IA, Sivets GG, Helv. Chim. Acta 82 (1999) 2052–2065. [Google Scholar]
- [25].Middelton WJ, J. Org. Chem 40 (1975) 574–578. [Google Scholar]
- [26].Middelton WJ, Bingham EM, Org. Synth 57 (1977) 50–52. [Google Scholar]
- [27].Card PJ, J. Org. Chem 48 (1983) 393–395. [Google Scholar]
- [28].Tewson TJ, Welch MJ, J. Org. Chem 43 (1978) 1090–1092. [Google Scholar]
- [29].Gopishetty B, Zhu J, Rajan R, Sobczak AJ, Wnuk SF, Bell CE, Pei D, J. Am. Chem. Soc 131 (2009) 1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ren H, An H, Hatala PJ, Stevens WC, Tao J Jr., He B, Beilstein J. Org. Chem 11 (2015) 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mikahilopulo IA, Sivets GG, Synlett 2 (1996) 173–174. [Google Scholar]
- [32].Dax KIA, Albert M, Ortner J, Paul B, J. Carbohydr. Res 327 (2000) 47–86. [DOI] [PubMed] [Google Scholar]
- [33].Scharer OD, Verdine GL, J. Am. Chem. Soc 117 (1995) 10781–10782. [Google Scholar]
- [34].Wilds CJ, Damha MJ, Nucleic Acids Res 28 (2000) 3625–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Michalik M, Hein M, Frank M, Carbohydr. Res 327 (2000) 185–218. [DOI] [PubMed] [Google Scholar]
- [36].Cohen MH, Johnson JR, Massie T, et al. , Clin. Cancer Res 12 (2006) 5329–5335. [DOI] [PubMed] [Google Scholar]
- [37].Lee WW, Martinez AP, Blackfound RW, Bartuska VJ, Reist EJ, Goodman L, J. Med. Chem 14 (1971) 819–823. [DOI] [PubMed] [Google Scholar]
- [38].Herdewijn P, Van Aerschot A, Kerremans L, Nucleosides Nucleotides 8 (1989) 65–96. [Google Scholar]
- [39].Mansuri M, Krishnan B, Martin JC, Tetrahedron Lett. 32 (1991) 1287–1290. [Google Scholar]
- [40].Braenvang M, Gundersen L-L, Synthesis 18 (2006) 2993–2995. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.