

Abstract
Necrotizing enterocolitis (NEC) is a life-threatening intestinal disease that primarily affects preterm infants during their first weeks after birth. Mortality rates associated with NEC are 15-30%, and surviving infants are susceptible to multiple serious, long-term complications. The disease is sporadic and, with currently available tools, unpredictable. We are creating an early warning system that uses stool microbiome features, combined with clinical and demographic information, to identify infants at high risk of developing NEC. Our approach uses a multiple instance learning, neural network-based system that could be used to generate daily or weekly NEC predictions for premature infants. The approach was selected to effectively utilize sparse and weakly annotated datasets characteristic of stool microbiome analysis. Here we describe initial validation of our system, using clinical and microbiome data from a nested case-control study of 161 preterm infants. We show receiver-operator curve areas above 0.9, with 75% of dominant predictive samples for NEC-affected infants identified at least 24 hours prior to disease onset. Our results pave the way for development of a real-time early warning system for NEC using a limited set of basic clinical and demographic details combined with stool microbiome data.
Keywords: Necrotizing enterocolitis, attention-based neural networks, premature infants, prediction, multiple instance learning
1. BACKGROUND
Necrotizing enterocolitis (NEC) is the most common intestinal emergency among preterm infants. It is characterized by rapidly progressive intestinal necrosis, bacteremia, acidosis, and high rates of morbidity and mortality (Figure 1). Up to 11,000 premature infants are affected in the U.S. annually. Mortality rates are 15-30%, and survivors are at increased risk for intestinal strictures, nutritional malabsorption, and neurocognitive impairments [12, 21].
Figure 1:
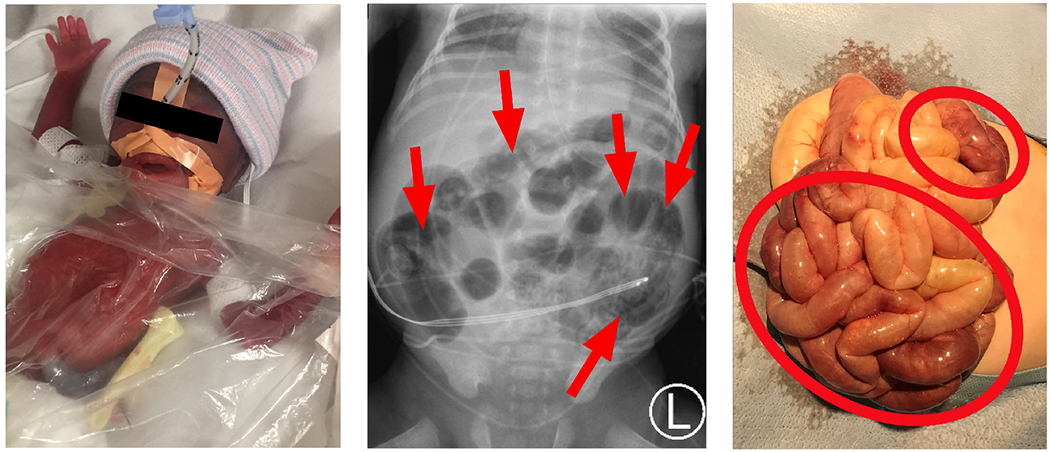
Necrotizing enterocolitis (NEC) manifests as feeding intolerance and abdominal swelling in preterm infants (left). Current diagnosis relies on clinical signs and characteristic X-ray features (middle). The result is irreversible intestinal necrosis (right).
Currently there is no effective way to predict NEC before disease onset, when it is often too late to successfully intervene. Causes of NEC are not well-understood, but several studies have focused on shifts in the intestinal microbiome, the bacteria in the intestine whose composition can be determined from DNA sequencing from small stool samples [19]. Certain microbiome patterns have been found to precede NEC, although these patterns are variable and incompletely reliable [6, 11, 24, 36]. We hypothesized that a machine learning approach to modeling clinical, demographic, and microbiome data from preterm patients might allow discrimination of patients at high risk for NEC long before clinical disease onset, which would permit early intervention and mitigation of serious complications.
As a clinical implementation, we envision an early warning system built around rapid stool DNA extraction and sequencing technology, such as the Nanopore sequencing system [20], combined with efficient bioinformatic tools to rapidly and directly map sequence reads to a comprehensive bacterial sequence database (e.g. Kraken2, used in this paper and described below). Longitudinal microbiome compositional data for each patient would then be analyzed using a machine learning early prediction system such as described here. The process would be fast enough that it could be completed for all at-risk newborns in a neonatal ICU on a daily basis by a single technician. Daily NEC risk assessments from the system would be returned to the medical team so that infants at high risk for imminent NEC could receive enhanced observation or preventative measures such as restricted enteral intake and/or prophylactic antibiotic therapy.
Real life neonatal data are imperfect, however. Clinical data are noisy (from imprecise measurement, input errors, and clinician-to-clinician variability) and microbiome data from stool samples are sparse (most stool samples have similar microbial composition, representing only a tiny fraction of the bacterial kingdom). The suboptimal nature of the data presents a significant challenge to a machine learning approach to predicting NEC in an individual patient.
In order to contend with these limitations, we adapted an attention-based, multiple instance learning (MIL) approach that has been successfully deployed in the context of medical image interpretation [17]. We chose this strategy after recognizing similarities between microbiome and medical image data–both feature mostly extraneous information, minimal labeling, and both are usually accompanied by limited clinical information. We also used several pre-processing steps to normalize our input data and reduce the dimensionality of the dataset, both through rational handling of zero counts and hierarchical feature selection. These pre-processing steps proved crucial to generating highly accurate and early NEC predictions in a computationally tractable algorithm that could be performed in real-time in a clinical setting.
Other groups have used computational strategies to analyze various aspects of NEC or suggested hypothetical approaches to do so [35]. One group applied linear discriminant analysis to clinical findings associated with the disease [18]. Clinical findings consistent with NEC often occur too late to effectively intervene, however, while combining clinical data with microbiome analysis may permit earlier detection. Machine learning has also been used in an exploratory capacity to discern bacterial metabolic pathways that may contribute to pathogenesis [22]. Ours is the first described system for a clinically applicable machine learning model that combines microbiome, demographic, and clinical data that could be collected and monitored in real-time in a neonatal ICU. We employ recently developed microbiome pre-processing and machine learning methods, extending their applicability to a new area of predictive monitoring.
This paper makes the following contributions:
Provides a framework for microbiome data pre-processing that recognizes and appropriately attends to the compositional and hierarchical aspects of the data.
Investigates suitable machine learning methodologies in the case study of predicting NEC in preterm infants that could translate to other medical problems with similar types of data.
Shows successful results that present a pathway toward a clinically useful tool to predict individual patient risk of developing imminent NEC.
2. COHORT
We tested machine learning algorithms on a dataset from a large, prospective cohort study with a nested case-control design by Warner et al.—performed between 2009 and 2013—in which 2,895 stool samples were studied from 161 preterm infants, 45 of whom developed NEC [36] (Figure 2). Infants were enrolled at three neonatal ICU sites: St. Louis Children’s Hospital, ChildrenâĂŹs Hospital at Oklahoma University Medical Center, and Kosair Children’s Hospital.
Figure 2:
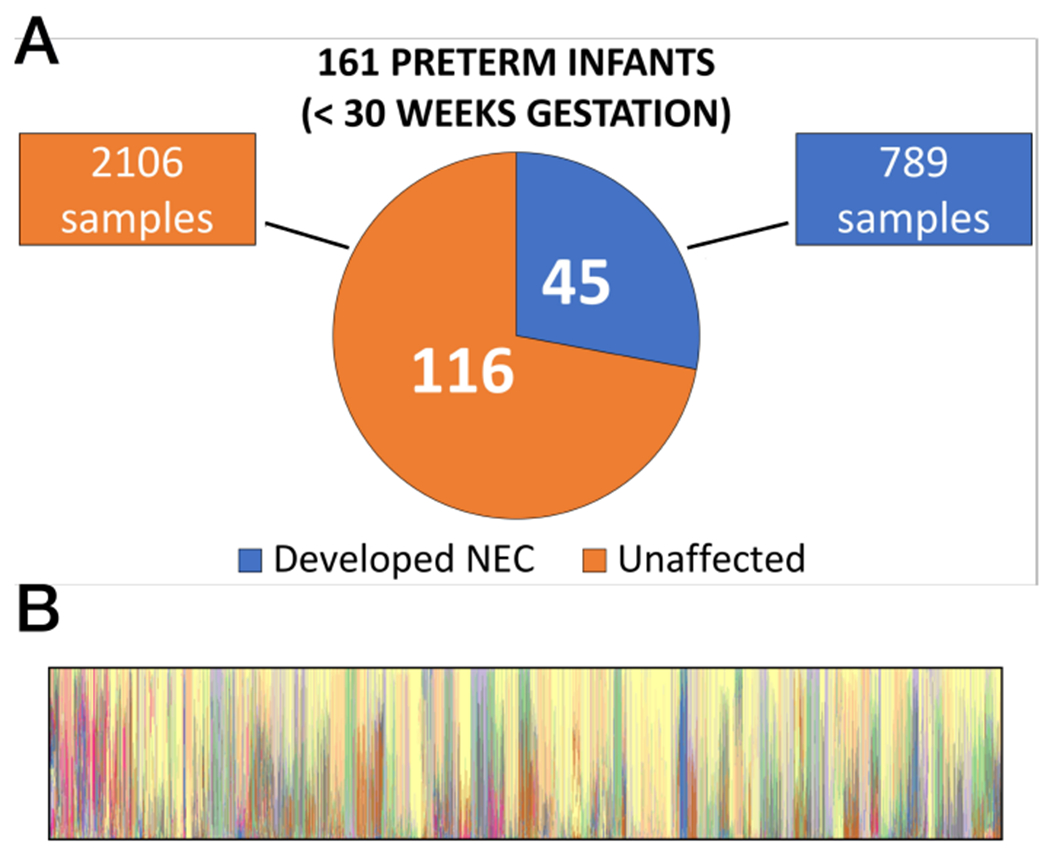
Summary images show disease characteristics of the nested case-control cohorts from [36] used to develop the NEC early warning system (A). Panel B illustrates stool microbiome composition characteristics for patients in the study. Each vertical strip represents the complete microbiome of one stool sample; colored portions indicate genus level microbiome community members detected in that sample.
2.1. Cohort Selection
Inclusion criteria included birthweight under 1,500 g and an expectation of survival beyond seven days from birth. All stools from enrolled infants were sampled up to 60 days of life, hospital discharge, development of NEC, or death (whichever came first). Stool samples were stored frozen until thawed for DNA extraction. NEC was evaluated using the Bell staging system, and subjects were diagnosed with NEC only if they met criteria for Bell stage 2 (clinical and radiographic signs of intestinal pathology) [27]. Infants who were classified as having spontaneous intestinal perforation (a clinical entity distinct from NEC) were excluded from analysis, as were any subjects found to have major cardiovascular abnormalities. Clinical and demographic data were collected and stored for all enrolled infants.
At the end of the enrollment period, subjects who had developed NEC were demographically matched to 1-4 unaffected control subjects. These two groups made up the case (n=45) and control (n=116) cohorts. DNA was extracted from all 2,895 stool samples from both cohorts. Following DNA extraction, bacterial 16S ribosomal protein DNA sequences were PCR amplified using primers flanking the V3 and V5 regions. Amplicons were sequenced on the Roche 454 GS FLX Titanium platform and uploaded to the National Center for Biotechnology Information.
2.2. Features
After obtaining authorized access to clinical and demographic data, we downloaded the raw sequence reads for 2,895 clinical samples from the Warner et al. study. We used Kraken2 [38] to directly align the sequence reads to a database of microbiome DNA sequences (the miniKraken2 database), after which viral, fungal, and archaeal alignments were removed, leaving only bacterial matches.
We also included 27 clinical metadata features collected and reported in the Warner et al. dataset, eliminating from our analyses those metadata features (such as whether the patient survived) that would not be available to a neonatologist caring for a patient.
We replicated key findings from the initial study, including DNA sequence-based characterization of the taxonomic distribution of bacteria present in the samples and demonstration that infants who developed NEC showed trends toward Firmicutes-depleted and Proteobacteria-enriched intestinal microbiota, relative to unaffected control infants. Both of the latter findings were variable, however, and existed within a background of complex and noisy microbiome data, as visualized in Figure 2B.
3. PRE-PROCESSING
3.1. Feature normalization
Using microbiome raw data in downstream applications without careful pre-processing can lead to spurious results for several reasons. At a basic level, pre-processing is essential because the microbial community in each biological sample may be represented by datasets that differ significantly from each other simply as a result of differential sequencing efficiency, even if the underlying populations are actually very similar [15, 16]. Put differently, two microbiome sequencing experiments from the exact same sampling site might yield results that–at least on the surface–appear distinct from one another. However, with proper pre-processing, many of the artifacts introduced by methodological or technical variation can be minimized.
One challenge that pre-processing must overcome is the fact that most microbiome tables are sparse, meaning that they contain a high proportion of zero counts (often around 90%). On the other hand, in most intestinal microbiome samples, a handful of bacteria account for most of the sequence reads. This imbalance between read counts for abundant species and rare ones ensures that the counts of rare species are uncertain, since they are at the limit of detection for the sequencing instrument. The exact number of read counts for these rare components will depend on the sequencing depth: some (though perhaps not all) will be detected if sequencing is performed very deeply, while rare components will not be detected at all with more shallow sequencing. Again, this fact can be largely accounted for through careful consideration of pre-processing techniques.
The next challenge for which pre-processing must account is the fact that microbiome data are fundamentally compositional. The term compositional, in this context, means that the total number of reads obtained for a sample does not reflect the absolute number of microbes present, since the sample is just a fraction of the original environment [15, 16]. Compositional data are constrained by the simplex (sum to 1) and are not free floating in the Euclidean space [34]. As a result, standard statistical approaches must be adjusted for the underlying compositionality, otherwise surprising but misleading patterns will be identified. For example, without accounting for compositionality, an increase in abundance of one prevalent bacterial taxon can lead to spurious negative correlations for the abundance of other taxa.
A cornerstone of microbiome data pre-processessing is normalization, which adjusts for the sample-to-sample differences most likely to result from purely methodological or technical features of the experiment. There are several possible approaches to normalization [37]. Ideally, the strategy selected should effectively deal with the distinct but related issues of sparsity and compositionality. The normalization strategy may be (and usually is) an algorithm built from several sub-processes.
A typical first step is to eliminate the many zeros present in a sparse microbiome dataset. There are two main strategies for doing so. The first is to apply a pseudocount where zeros are replaced by a uniform small number, such as two-thirds of the sequencer’s limit of detection. Alternatively, Bayesian approaches can be employed to individually compare taxon counts across all samples and substitute zeros with individually calculated estimates. Again, these estimates tend to be small numbers. A drawback of the Bayesian approach is that it is computationally demanding, especially when the study consists of a large number of biological samples. Furthermore, it is not clear that the computational demands of the Bayesian approach yield superior downstream results compared to the pseudocount approach [37].
For our dataset, we elected to use the non-Bayesian approach. Zeros were replaced with a uniform value: 0.66, which was 2/3 of the smallest possible read count of 1.
The next step is to perform some form of logarithmic transformation to the data to restore data shape features that make them more tractable to traditional statistical analyses. Several log transformations have been described, and interested readers are directed to several papers on the topic [1, 14–16, 37]. Among the most widely used compositional data transformation, however, is Aitchison’s log-ratio approach [2]. The log-ratio approach rests on the recognition that the abundance values are not informative by themselves and that the relevant information is contained in the ratios of abundances between the different taxa. A simple log-ratio approach would involve taking the logarithm of ratios between each count in a sample and the count of an invariant reference taxa. However, since selecting a reference value can introduce its own set of artifacts, a less biased alternative was developed: the centered log-ratio approach. Here each taxon within a sample is transformed by taking the log-ratio counts for that taxon within a sample divided by the geometric mean of the counts of all taxa.
Figure 4 shows the effects of our pre-processing. Here, the top row shows population distributions for major microbiome taxa before pre-processing. The histograms are highly skewed and not amenable to further statistical analysis. After pre-processing (bottom row), previously obscured population characteristics emerge, making downstream analyses feasible and informative.
Figure 4:
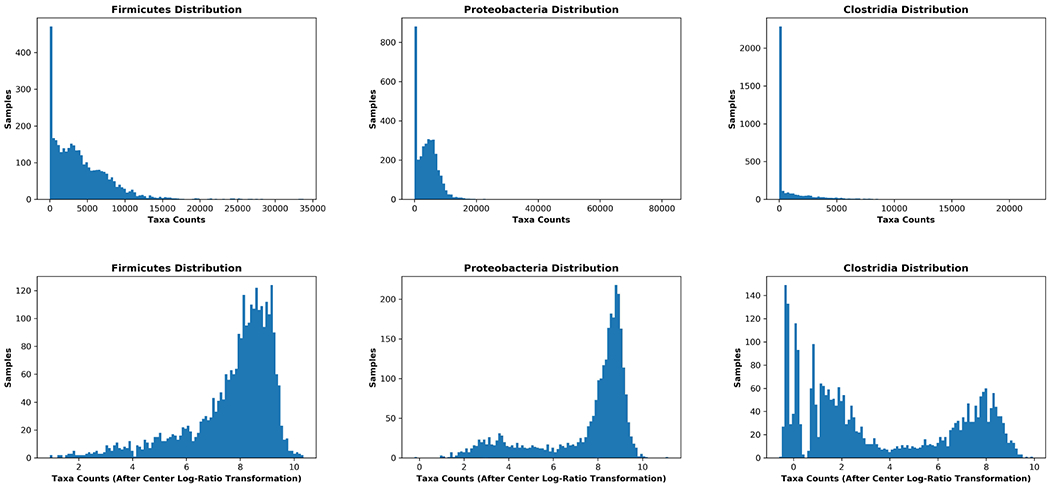
Pre-processing of microbiome data allows population characteristics to emerge, enabling downstream analyses. The top row shows raw-count taxonomic population data prior to pre-processing. The curves are skewed and compressed. The bottom row shows the same populations after pre-processing as described in the text. Following pre-processing, population distributions are normalized, less skewed, and demonstrate patterns that were obscured in the unprocessed raw data.
3.2. Hierarchical Feature Selection
Another special characteristic of microbiome data is its hierarchically structured feature space, which can be exploited for more efficient and accurate modeling through taxonomy dimensionality reduction (e.g., [23, 26]).
16S sequencing allows the generation of thousands of features. While this facilitates the categorization of microbiota to different groups of organisms (from the highest to the lowest level: kingdom, phylum, class, order, family, genus and species) based on their shared characteristics, it creates challenges when it comes to applying machine learning algorithms. Indeed, a high number of features compared to the sample size is linked to the curse of dimensionality [4]: a phenomenon where the data points are so sparse in the high-dimensional space that it becomes extremely hard for any sophisticated machine learning algorithm to generalize well without overfitting the training sample. Overfitting leads ultimately to unreliable models that will do poorly on new unseen samples. Concretely, in the Warner et al. dataset, the data is a matrix n × m where n represents the total number of samples, with n = 2895 for 161 patients, and m = 3702, the number of non-zero taxa derived by 16S sequencing. This low samples-to-features ratio will subvert efforts at machine learning. Therefore, some form of taxonomy dimensionality reduction is required to improve model performance. Taxonomy dimensionality reduction has been successfully applied to colorectal cancer prediction [25].
Feature selection in hierarchical feature space [28], adopted by Oudah and Henschel in [23], has been shown to be an effective approach for using microbiome taxonomy for feature engineering and dimensionality reduction. Their proposed methodology, Hierarchical Feature Engineering (HFE), allows identification of a reduced set of features that can then be fed to machine learning algorithms. An additional advantage of this reduction is that it provides insights by identifying a small set of informative features that are linked to the outcome of interest.
HFE uses a series of filtering steps and heuristics to decrease the number of dimensions. In brief, given a microbiome taxonomy, HFE proceeds as follows: (1) a feature engineering phase where higher taxonomic features abundance is obtained by summing the weighted abundance of their children, (2) a heuristic-based phase where Pearson correlation coefficients are calculated for each pair (parent, child) in the taxonomy. Any child node with a correlation greater than a given threshold is pruned, (3) a supervised information gain-based filtering is performed on all possible paths from the root to the leaves of the taxonomy. The outcome label of interest is then used to calculate the information gain for each node. Those with a low score (zero, or smaller than the average information gain along the path, are discarded). Incomplete paths where the full taxonomic information was not calculable will be handled by comparing the information gain of the leaves to the global information gain across the taxonomy.
Similarly to the HFE method [23], we reduce the dimensionality in the Warner data by removing any node that is redundant with the parent, using a Pearson correlation threshold of 0.7. This reduced the the numbers of taxa from 3,702 to 2,282.
Information gains were also calculated for each node of the taxonomy tree using the NEC target label. Any node with an information gain of zero was discarded. This process allowed us to prune the number of features further to 362 features.
4. METHODOLOGY
We evaluated several machine learning methods in order to determine the best strategy for predicting NEC from microbiome data. We found optimal performance from a gated attention-based multiple instance learning (MIL) approach based on a multi-layer neural network architecture [17]. This approach was successfully applied to medical imaging.
We adopt a MIL approach to predicting NEC. We model each example (patient) as a bag of instances (samples). MIL is a form of weakly supervised learning [7, 10], where the training instances are arranged in sets, called bags, and the label is provided for the entire bag. More formally, we have a set of labeled examples:
where each bag Xi is a set of instances of the stool microbiome of the form Xi = {xi1, xi2, …, xik} along with clinical metadata features. Note that k corresponding to the number of samples can vary from one patient (bag) to another. The overall label yi ∈ {0, 1} denotes the bag class label–that is, whether the baby developed NEC or not.
There is no access to the instance labels themselves. Therefore, the whole bag is labeled with yi = 1, the NEC outcome, if it includes at least one positive instance. Therefore the example is considered weakly labeled, because only a subset of those instances are the drivers of that outcome.
The bag label yi is given by:
| (1) |
Where yik denotes the latent label of the kth instance of bag i. These yik labels are not available during training while the bag label yi is observed.
There are two main approaches to solve the MIL problem. In instance level approaches, predictions are made for each instance and aggregated to obtain the bag level label Y. In embedding level approaches, instances X are mapped to a vectorial embedded space and fed to a final classifier. We used a recently developed embedding level strategy, known as attention-based multiple instance learning [17] that was applied to medical imaging.
In [17], initial embedding of features is performed by a neural network, which passes an embedded value to an attention-based MIL pooling algorithm that delivers the bag label X. The embeddings are aggregated using the attention weights, which are then fed to the fully connected layer with a sigmoid activation function that produces a bag probability. This model assumes that a bag label is distributed as a Bernoulli distribution θ(X) ∈ [0, 1] and trains it by optimizing the log likelihood function.
The attention-based pooling function is constructed as follows. Obtaining the embeddings H = {h1, …, hk} through a neural network fψ(∗), where hk = fψ(xk), the embeddings are aggregated by the weighted mean operator [17]:
| (2) |
where:
| (3) |
where and are parameters. Attention-based MIL can be further enhanced by adding a gated mechanism [9, 17], a feature included in our prototype:
| (4) |
Where are parameters and ⊙ is an element-wise multiplication. The sigm(·) is a sigmoid function that serves to introduce non-linearity to the attention-based MIL pooling, improving efficiency in learning complex relationships that may be significant in determining the bag label. An overview of the computational architecture of our system is shown in Figure 5.
Figure 5:
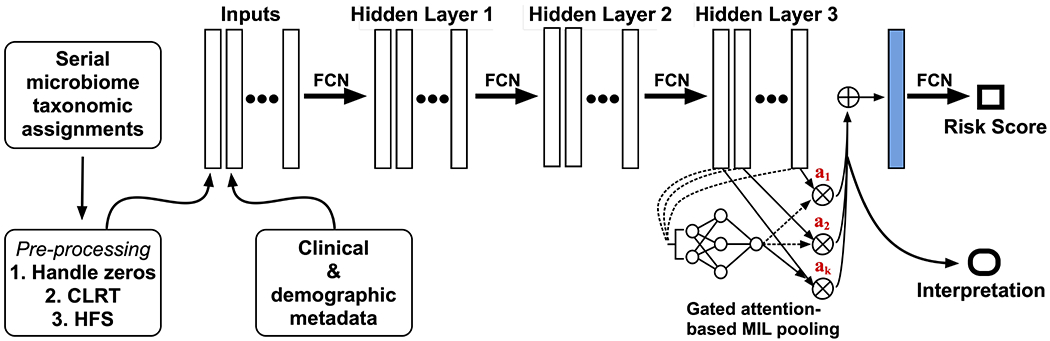
The architecture of our prototype NEC early warning system, which consists of multiple layers of fully convolutional networks (FCN) culminating in an interpretable gated attention mechanism and passage of embedded data to a sigmoid function that generates a risk prediction. CLRT=centered-log ratio transformation; HFS=hierarchical feature selection. Image derived, with modifications, from [17].
In NEC risk assessment, the instances consist of microbiome taxonomy data and demographic metadata. Each patient is defined as a collection of instances, and the task is to assign a NEC risk classification to the patient.
5. EMPIRICAL RESULTS
We implemented in Python for the general pipeline and used Py-Torch as a deep learning framework. Our implementation is available on Github 1. We tested our MIL-based NEC prediction system on the dataset described in Section 2 [36].
In NEC risk assessment with our system, the instances consist of microbiome taxonomy data after normalization (3.1) and hierarchical feature selection (3.2), combined with commonly collected demographic and clinical metadata (e.g. gestational age at birth, mode of delivery, birth weight, gender). Each patient is defined as a collection of instances, and the task is to assign a NEC risk classification to the patient.
We used stratified sampling to partition the Warner et al. dataset into a training and testing sets. We conducted five trials of this partitioning to avoid any sampling bias. We used a cross-validation modeling strategy whereby we trained our system on a portion of the dataset, then repeatedly tested the resultant model on the data subset that was withheld during training. The final model obtained was then applied to the test set. We averaged the results across all trials.
We evaluated our system using two main metrics:
How well can it determine, based on stool microbiome and clinical data, whether or not a given patient developed NEC?
How long before disease onset did the dominant instance occur?
Alternative Methods.
Two of the instance level approaches that we have included as comparison are mi-SVM and MISVM [3]. Both of these algorithms try to identify the maximum margin hyperplane that separates negative bag instances from a selected representative instance from a positive bag. These two algorithms assume that all instances are independently distributed and therefore structure information—the potential interrelationships between instances, such as important temporal variation in microbiome features—is not taken into account. MILboost is a MIL variant of AdaBoost that has been mostly used for object detection in images. In this model, weights are assigned to each instance to maximize the likelihood of bags [39]. As with the two instance-based approaches, the MILboost algorithm does not take account of relationships between the instances. We also include a logistic regression (LR) comparitor. For SVM-based methods, we used linear and radial basis function (RBF) kernels. For all alternative methods, we did hyperparameter tuning accordingly (for example, C for linear, C and Gamma for RBF in SVM), using cross-validation. This is to ensure a fair comparison to the attention method proposed and other baselines used. Finally, we show the improvement in model performance that stems from center-log ratio transformation pre-processing.
Performance of these alternative approaches, shown in Figure 6, was generally poor. Predictive accuracy was barely above chance, with receiver-operator characteristics demonstrating limited ability to differentiate NEC from non-NEC cases.
Figure 6:
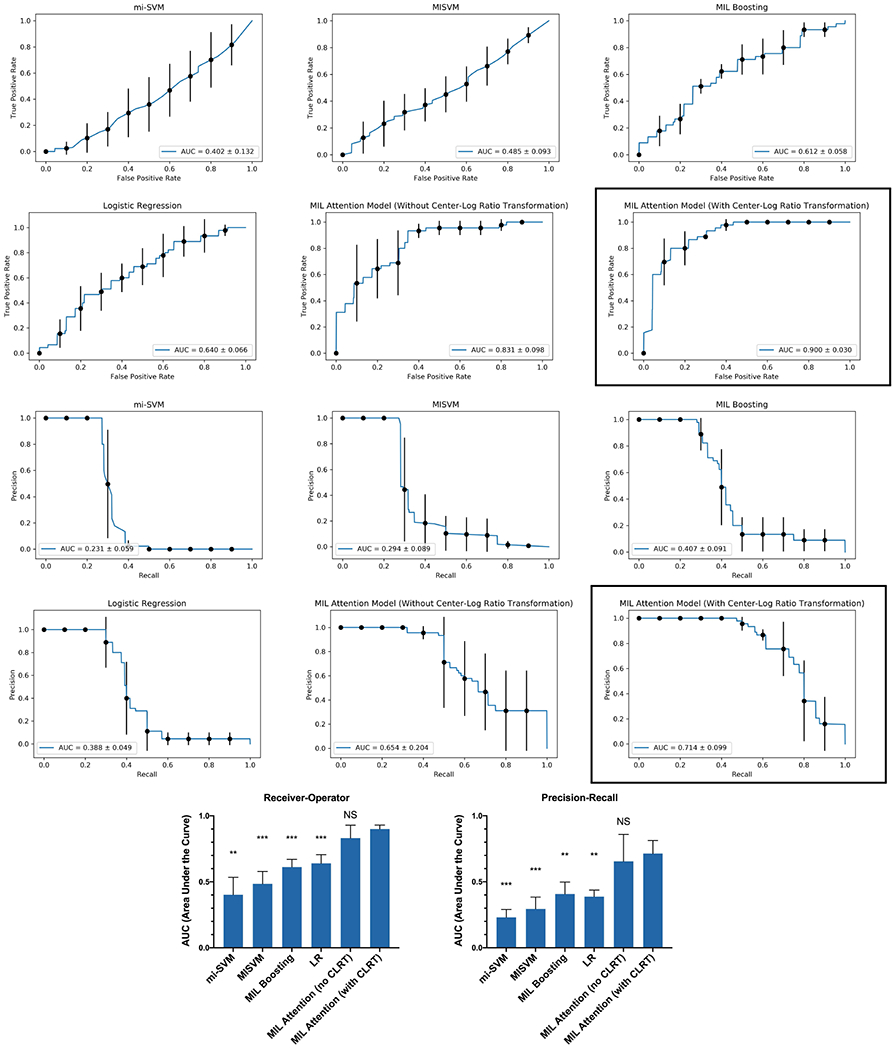
Receiver-operator and precision-recall curves generated by different models. The MIL Attention Model with CLRT applied (black boxes) was the best performer, and was used to determine the prediction lead-times for affected patients. For the histograms (bottom), ANOVA with Dunnett’s correction for multiple comparisons was performed comparing all models to the final MIL attention model with CLRT. ** p<0.01; *** p<0.001; LR=logistic regression; CLRT=center-log ratio transformation
Experimental System Predictive Performance.
In repeated trials using the approach described above, we demonstrated ROC AUCs around 0.9, suggesting a good balance of sensitivity and specificity (Figure 6). Precision-recall characteristics of the experimental system also exceeded any of the alternative methods, with precision-recall AUC values around 0.7.
We note that the models trained on all the features from microbiome taxonomic classification did not converge, due to the high dimentionality of the data. This highlights the importance of the hierarchical feature selection.
Model interpretability.
Most neural network-based machine learning systems cannot provide interpretability [13], leaving the basis of prediction unexplained. An attractive feature of our system is the interpretability potential of its models, permitting identification of key samples, over 75% of which are identified at least 24 hours prior to disease onset (Figure 7). These key samples will help direct further studies toward interpretability in the model. Interpretability will enhance our understanding of the etiology of NEC, increase our confidence in using the models in a clinical setting, and help refine our data collection to build even more accurate NEC scoring models.
Figure 7:
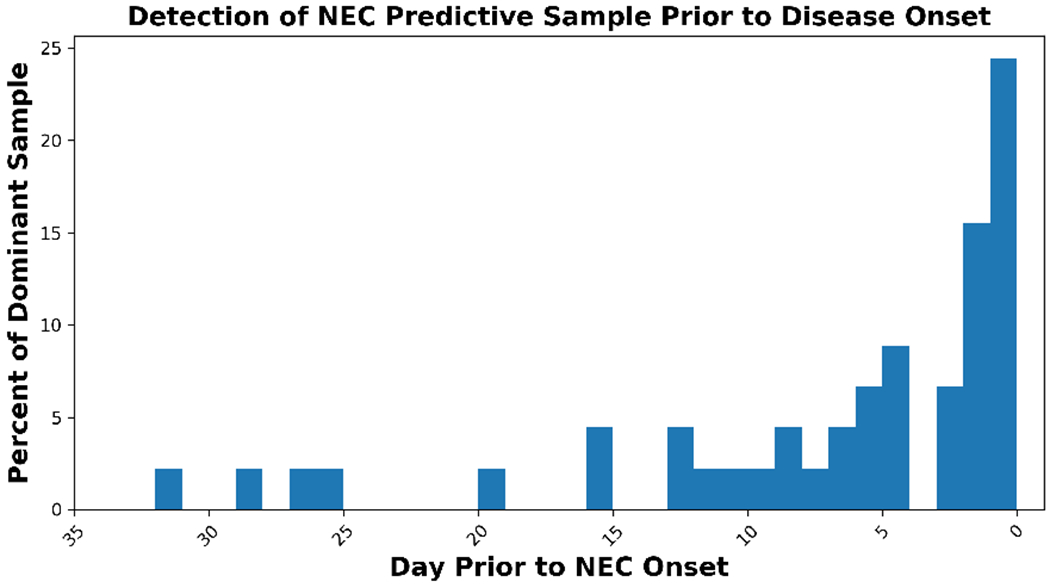
As a key step toward model interpretability, identification of dominant samples that inform NEC predictions well before disease onset.
6. CONCLUSION AND FUTURE WORK
We have developed a prototype of a machine learning system for predicting NEC in preterm infants, using stool microbiome and clinical metadata features. NEC is a serious illness for which there is currently no effective method to predict individual patient risk and no single predictive biomarker. This makes NEC an appealing target for a semi-supervised machine learning system that can detect subtle associations from complex datasets with minimal operator instruction.
To handle the sparseness, compositionality, and high dimensionality of raw microbiome sequencing data, we have employed pre-processing steps that include normalization, centered-log ratio transformation, and hierarchical feature selection. The hierarchical feature selection was performed to maintain only features that are essential for prediction. The cut-off point of 0.7 is a conservative threshold for Pearson correlation (where in fact any correlation exceeding 0.5 could be deemed as strong). In addition, any node in the taxonomy tree with an information gain of zero was discarded. Our results demonstrate the importance of addressing these two problems with this type of data. We used a MIL system that has been effectively deployed in medical image interpretation—another realm in which weak signals exist in a complex and mostly irrelevant data space. We show empirically that the MIL attention model outperforms other MIL and non MIL methods.
Since there is no meaningful clinical benchmark against which to compare our system, we have instead assessed its performance relative to closely related multiple instance learning systems and logistic regression analysis, demonstrating superior predictive accuracy and discrimination.
To pursue this research further, we envision multiple potential paths from our current instantiation to a clinically useful tool. Our future work will be to integrate our prediction model into a clinical research protocol whereby at-risk neonates under 1,500 g will have bacterial DNA from stool samples sequenced daily using rapid and inexpensive Nanopore sequencing technology [20]. Sequence data will be automatically fed into Kraken2 for microbial population characterization, followed by pre-processing as described above. Relevant clinical data for each patient will be attached to the biological data, which will then be fed into our MIL early warning system. The goal of our initial study will be to validate our predictions in a real-time neonatal ICU cohort. Once this validation phase is complete, we will start planning a randomized clinical trial of using our predictive system as a driver of neonatal ICU medical decision-making.
Future work will explore the use of interpretability models, which have shown great promise in explaining the prediction of machine learning models for individual patients [8]. Specifically, given the high predictive power of our MIL approach, when it correctly predicts that a patient developed NEC, we could explore further the characteristics of what instance (composed of microbiome and clinical data) drove that prediction. Examples of interpretability approaches include using rules such as the work in [29], and generating characterizations of key features based on the ranked list of the attention weights assigned by the MIL attention model to each instance within the bag label [30–32]. Interpretability models will be deployed on those key instances and could impact clinical practice while providing explanations to the patient’s parents for any model-driven clinical decisions.
We will also explore devising a hierarchical feature selection approach more suitable for the multiple instance learning framework. The method we employed is univariate and works at the instance level, not the bag level. Given that only a few instances are driving the bag label, we would devise an iterative approach to focus on the high-attention samples.
While considerable attention has been devoted to machine learning applications in adult medicine [5, 33], there has been less emphasis on potential uses in pediatric diagnosis and prevention, and the subspecialty of neonatology has been so far almost entirely overlooked by machine learning teams. However, neonatal patients are—in some ways—ideal candidates for machine learning approaches. They suffer from complex, multifactorial illnesses driven by a host of maternal and intrinsic risk factors; neonatal ICUs generate considerable physiologic and laboratory data amenable to machine learning strategies; and because infants cannot communicate directly they are highly vulnerable and would benefit substantially from effective computation systems to identify and prevent their illnesses. We see the work presented here as an initial step in that direction.
Figure 3:
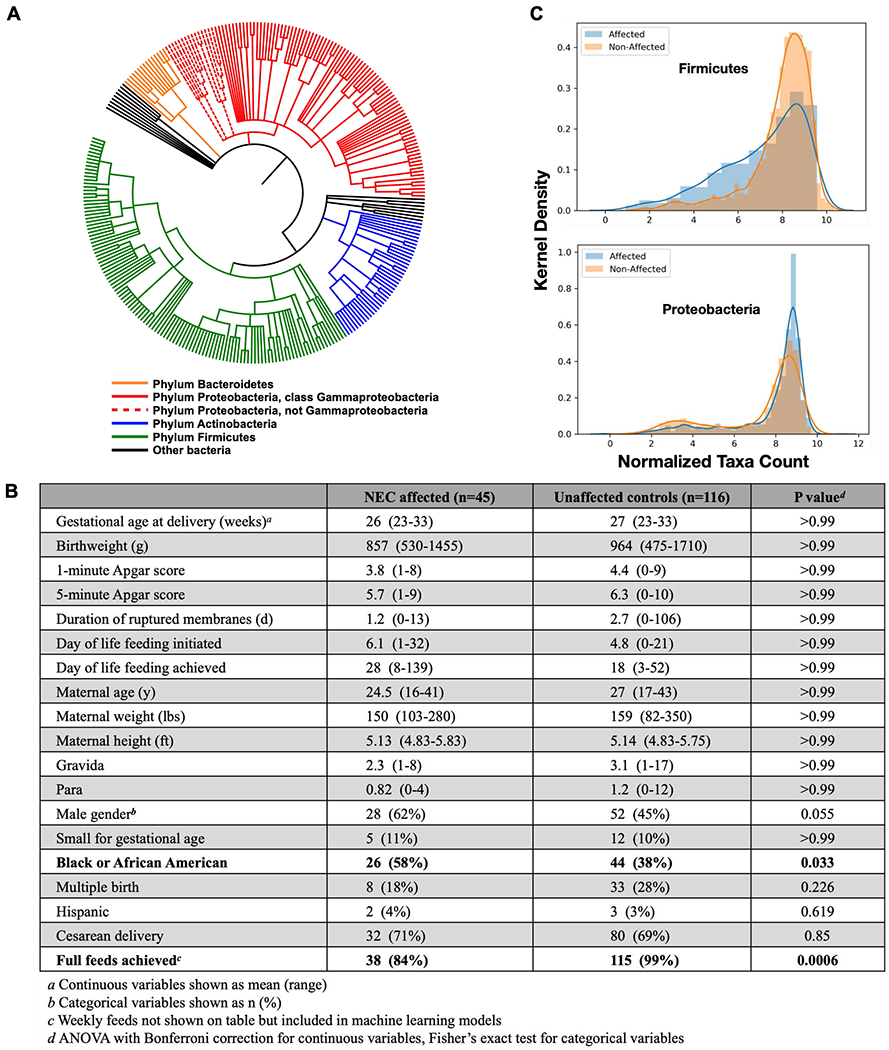
A phylogenetic tree (A) showing microbiome community members identified through 16S sequencing of DNA from study subject stools. This tree reflects Kraken2 sequence mapping followed by hierarchical feature selection. The table (B) lists clinical metadata features included in our model and statistical comparisons. We confirmed two microbiome trends reported in the initial study: NEC-affected infant microbiota contain less Firmicutes bacteria and more Proteobacteria relative to unaffected control infant microbiota (C).
CCS CONCEPTS.
• Applied computing → Health informatics; Bioinformatics.
7. ACKNOWLEDGEMENTS
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number K08AI132555 to T.A.H.
T.A.H. gratefully acknowledges fruitful conversations and helpful insights from Dr. Michael Hooven.
Footnotes
Contributor Information
Thomas A. Hooven, University of Pittsburgh, Pittsburgh, USA
Adam (Yun Chao) Lin, Columbia University, New York, USA.
Ansaf Salleb-Aouissi, Columbia University, New York, USA.
REFERENCES
- [1].Aitchison J. Reducing the dimensionality of compositional data sets. Journal of the International Association for Mathematical Geology, 16(6):617–635, 1984. doi: 10.1007/bf01029321. [DOI] [Google Scholar]
- [2].Aitchison J. The Statistical Analysis of Compositional Data. Chapman & Hall, Ltd., GBR, 1986. [Google Scholar]
- [3].Andrews Stuart, Tsochantaridis Ioannis, and Hofmann Thomas. Support vector machines for multiple-instance learning. In Advances in neural information processing systems, pages 577–584, 2003. [Google Scholar]
- [4].Beyer Kevin, Goldstein Jonathan, Ramakrishnan Raghu, and Shaft Uri. When is “nearest neighbor” meaningful? In Beeri Catriel and Buneman Peter, editors, Database Theory — ICDT’ 99, pages 217–235, Berlin, Heidelberg, 1999. Springer Berlin Heidelberg. [Google Scholar]
- [5].Bhattacharyya Malay. From Machine Learning to Learning Machines âĂŞ A Perspective toward Personalized Medicine. Nature Precedings, 2012. doi: 10.1038/npre.2012.7118.1. [DOI] [Google Scholar]
- [6].Brown Christopher T., Xiong Weili, Olm Matthew R., Thomas Brian C., Baker Robyn, Firek Brian, Morowitz Michael J., Hettich Robert L., and Banfield Jillian F.. Hospitalized premature infants are colonized by related bacterial strains with distinct proteomic profiles. mBio, 9(2), 2018. doi: 10.1128/mBio.00441-18. URL https://mbio.asm.org/content/9/2/e00441-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Carbonneau Marc-André, Cheplygina Veronika, Granger Eric, and Gagnon Ghyslain. Multiple instance learning: A survey of problem characteristics and applications. CoRR, abs/1612.03365, 2016. URL http://arxiv.org/abs/1612.03365. [Google Scholar]
- [8].Caruana Rich, Lou Yin, Gehrke Johannes, Koch Paul, Sturm Marc, and Elhadad Noemie. Intelligible models for healthcare: Predicting pneumonia risk and hospital 30-day readmission. In Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ’15, pages 1721–1730, New York, NY, USA, 2015. ACM. doi: 10.1145/2783258. URL http://doi.acm.org/10.1145/2783258.2788613. [DOI] [Google Scholar]
- [9].Dauphin Yann N., Fan Angela, Auli Michael, and Grangier David. Language modeling with gated convolutional networks. CoRR, abs/1612.08083, 2016. URL http://arxiv.org/abs/1612.08083. [Google Scholar]
- [10].Dietterich Thomas G., Lathrop Richard H., and Lozano-Perez Tomas. Solving the multiple instance problem with axis-parallel rectangles. Artificial Intelligence, 89 (1-2):31–71, 1997. URL http://lis.csail.mit.edu/pubs/tlp/multiple-instance-aij.pdf. [Google Scholar]
- [11].Dobbler Priscila T., Procianoy Renato S., Mai Volker, Silveira Rita C., Corso Andréa L., Rojas Bruna S., and Roesch Luiz F. W.. Low microbial diversity and abnormal microbial succession is associated with necrotizing enterocolitis in preterm infants. Frontiers in Microbiology, 8:2243, 2017. doi: 10.3389/fmicb.2017.02243. URL https://www.frontiersin.org/article/10.3389/fmicb.2017.02243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dominguez Kathleen M. and Lawrence Moss R. Necrotizing enterocolitis. Clinics in Perinatology, 39(2):387–401, 2012. doi: 10.1016/j.clp.2012.04.011. URL http://www.sciencedirect.com/science/article/pii/S0095510812000279. [DOI] [PubMed] [Google Scholar]
- [13].Doshi-Velez Finale and Kim Been. A roadmap for a rigorous science of interpretability. CoRR, abs/1702.08608, 2017. URL http://arxiv.org/abs/1702.08608. [Google Scholar]
- [14].Egozcue Juan JosâĹŽÂl’ and Pawlowsky-Glahn Vera. Compositional data: the sample space and its structure. TEST, 28(3):599–638, 2019. doi: 10.1007/s11749-019-00670-6. [DOI] [Google Scholar]
- [15].Gloor Gregory B, Wu Jia Rong, Pawlowsky-Glahn Vera, and Egozcue Juan JosâĹŽÂl’. It’s all relative: analyzing microbiome data as compositions. Annals of epidemiology, 26(5):322–9, 2016. doi: 10.1016/j.annepidem.2016.03.003. [DOI] [PubMed] [Google Scholar]
- [16].Gloor Gregory B., Macklaim Jean M., Pawlowsky-Glahn Vera, and Egozcue Juan J.. Microbiome Datasets Are Compositional: And This Is Not Optional. Frontiers in Microbiology, 8:2224, 2017. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ilse Maximilian, Tomczak Jakub M., and Welling Max. Attention-based deep multiple instance learning. In Proceedings of the 35th International Conference on Machine Learning, ICML 2018, Stockholmsmässan, Stockholm, Sweden, July 10-15, 2018, pages 2132–2141, 2018. URL http://proceedings.mlr.press/v80/ilse18a.html. [Google Scholar]
- [18].Ji Jun, Bruce Ling Xuefeng, Zhao Yingzhen, Hu Zhongkai, Zheng Xiaolin, Xu Zhening, Wen Qiaojun, Kastenberg Zachary J., Li Ping Chung, Abdullah FSC, Brandt Mary L., Ehrenkranz Richard A., Harris Mary Catherine, Lee Timothy Charles Philip, Simpson Barry J., Bowers Corinna, Moss Richard L, and Sylvester Karl G.. A data-driven algorithm integrating clinical and laboratory features for the diagnosis and prognosis of necrotizing enterocolitis. In PloS one, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Johnson Coreen L. and Versalovic James. The human microbiome and its potential importance to pediatrics. Pediatrics, 129(5):950–960, 2012. doi: 10.1542/peds.2011-2736. URL https://pediatrics.aappublications.org/content/129/5/950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kai Shinichi, Matsuo Yoshiyuki, Nakagawa So, Kryukov Kirill, Matsukawa Shino, Tanaka Hiromasa, Iwai Teppei, Imanishi Tadashi, and Hirota Kiichi. Rapid bacterial identification by direct PCR amplification of 16S rRNA genes using the MinIONâĂŽÃŚÂć nanopore sequencer. FEBS Open Bio, 9(3):548–557, 2019. doi: 10.1002/2211-5463.12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Martin Joyce A., Hamilton Brady E., Osterman Michelle J.K., Driscoll Anne K., and Drake Patrick. Births: Final data for 2017. National Vital Statistics Reports Centers Dis Control Prev National Cent Heal Statistics National Vital Statistics Syst, 67(8):1–50, 2018. [PubMed] [Google Scholar]
- [22].Olm Matthew R., Bhattacharya Nicholas, Crits-Christoph Alexander, Firek Brian A., Baker Robyn, Song Yun S., Morowitz Michael J., and Banfield Jillian F.. Necrotizing enterocolitis is preceded by increased gut bacterial replication, klebsiella, and fimbriae-encoding bacteria that may stimulate tlr4 receptors. bioRxiv, 2019, doi: 10.1101/558676. URL https://www.biorxiv.org/content/early/2019/02/22/558676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Oudah Mai and Henschel Andreas. Taxonomy-aware feature engineering for microbiome classification. BMC bioinformatics, 19(1):227, June 2018. doi: 10.1186/s12859-018-2205-3. URL http://europepmc.org/articles/PMC6003080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pammi Mohan, Cope Julia, Tarr Phillip I., Warner Barbara B., Morrow Ardythe L., Mai Volker, Gregory Katherine E., Simon Kroll J, McMurtry Valerie, Fer-ris Michael J., Engstrand Lars, Lilja Helene Engstrand, Hollister Emily B., Versalovic James, and Neu Josef. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: a systematic review and meta-analysis. Microbiome, 5(1):31, 2017. doi: 10.1186/s40168-017-0248-8. URL 10.1186/s40168-017-0248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Qu Kaiyang, Gao Feng, Guo Fei, and Zou Quan. Taxonomy dimension reduction for colorectal cancer prediction. Computational Biology and Chemistry, 83:107160, 2019. doi: 10.1016/j.compbiolchem.2019.107160. URL http://www.sciencedirect.com/science/article/pii/S1476927119305626. [DOI] [PubMed] [Google Scholar]
- [26].Qu Kaiyang, Gao Feng, Guo Fei, and Zou Quan. Taxonomy dimension reduction for colorectal cancer prediction. Computational biology and chemistry, 83:107160, December 2019. doi: 10.1016/j.compbiolchem.2019.107160. URL 10.1016/j.compbiolchem.2019.107160. [DOI] [PubMed] [Google Scholar]
- [27].Rich Barrie S and Dolgin Stephen E. Necrotizing Enterocolitis. Pediatrics in review, 38(12):552–559, 2017. doi: 10.1542/pir.2017-0002. [DOI] [PubMed] [Google Scholar]
- [28].Ristoski Petar and Paulheim Heiko. Feature selection in hierarchical feature spaces. In Džeroski Sašo, Panov Panče, Kocev Dragi, and Todorovski Ljupčo, editors, Discovery Science, pages 288–300, Cham, 2014. Springer International Publishing. [Google Scholar]
- [29].Salleb Ansaf, Turmeaux Teddy, Vrain Christel, and Nortet Cyril. Mining quantitative association rules in a atherosclerosis dataset. In Proceedings of the PKDD Discovery Challenge 2004 (co-located with the 6th European Conference on Principles and Practice of Knowledge Discovery in Databases), pages 98–103, Pisa, Italy, 2004. [Google Scholar]
- [30].Salleb-Aouissi Ansaf, Vrain Christel, and Nortet Cyril. Quantminer: A genetic algorithm for mining quantitative association rules. In Veloso Manuela M., editor, IJCAI, pages 1035–1040, 2007. [Google Scholar]
- [31].Salleb-Aouissi Ansaf, Huang Bert, and Waltz David. Discovering characterization rules from rankings. In 2009 International Conference on Machine Learning and Applications, pages 154–161, December 2009. doi: 10.1109/ICMLA.2009.67. [DOI] [Google Scholar]
- [32].Salleb-Aouissi Ansaf, Vrain Christel, Nortet Cyril, Kong Xiangrong, Rathod Vivek, and Cassard Daniel. Quantminer for mining quantitative association rules. Journal of Machine Learning Research, 14:3153–3157, 2013. URL http://jmlr.org/papers/v14/salleb-aouissi13a.html. [Google Scholar]
- [33].Shameer Khader, Johnson Kipp W, Glicksberg Benjamin S, Dudley Joel T, and Sengupta Partho P. Machine learning in cardiovascular medicine: are we there yet? Heart, 104:1156, 2018. doi: 10.1136/heartjnl-2017-311198. [DOI] [PubMed] [Google Scholar]
- [34].Tsilimigras Matthew C B and Fodor Anthony A. Compositional data analysis of the microbiome: fundamentals, tools, and challenges. Annals of epidemiology, 26 (5):330ÃćÂĂÂŤ335, May 2016. doi: 10.1016/j.annepidem.2016.03.002. URL 10.1016/j.annepidem.2016.03.002. [DOI] [PubMed] [Google Scholar]
- [35].van Druten J, Sharif MS, Khashu M, and Abdalla H. A proposed machine learning based collective disease model to enable predictive diagnostics in necrotising enterocolitis. In 2018 International Conference on Computing, Electronics Communications Engineering (iCCECE), pages 101–106, August 2018. doi: 10.1109/iCCECOME.2018.8658948. [DOI] [Google Scholar]
- [36].Warner Barbara B, Deych Elena, Zhou Yanjiao, Hall-Moore Carla, Weinstock George M, Sodergren Erica, Shaikh Nurmohammad, Hoffmann Julie A, Linneman Laura A, Hamvas Aaron, Khanna Geetika, Rouggly-Nickless Lucina C, Malick Ndao I, Shands Berkley A, Escobedo Marilyn, Sullivan Janice E, Radmacher Paula G, Shannon William D, and Tarr Phillip I. Gut bacteria dysbiosis and necrotising enterocolitis in very low birthweight infants: a prospective case-control study. The Lancet, 387(10031):1928–1936, 2016. doi: 10.1016/S0140-6736(16)00081-7. URL http://www.sciencedirect.com/science/article/pii/S0140673616000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Weiss Sophie, Xu Zhenjiang Zech, Peddada Shyamal, Amir Amnon, Bittinger Kyle, Gonzalez Antonio, Lozupone Catherine, Zaneveld Jesse R, VâĹŽÂřzquez-Baeza Yoshiki, Birmingham Amanda, Hyde Embriette R, and Knight Rob. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome, 5(1):27, 2017. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wood Derrick E, Lu Jennifer, and Langmead Ben. Improved metagenomic analysis with Kraken 2. Genome biology, 20(1):257, 2019. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang Cha, Platt John C, and Viola Paul A. Multiple instance boosting for object detection. In Advances in neural information processing systems, pages 1417–1424, 2006. [Google Scholar]