

Abstract
Nonsteroidal anti‐inflammatory drugs (NSAIDs) are widely available drugs with anti‐inflammatory and analgesic properties. Their mechanism of action is associated with the enzymes of the arachidonic acid cycle (cyclooxygenases: COX‐1 and COX‐2). The cyclooxygenase pathway results in the formation of prostanoids (prostaglandins [PGs], prostacyclins, and thromboxanes). It affects various structures of the human body, including the kidneys. Medical literature associates the usage of NSAIDs with acute kidney injury (AKI), tubulointerstitial nephritis (TIN), as well as nephrotic syndrome and chronic kidney disease (CKD). AKI associated with the chronic consumption of NSAIDs is mainly attributed to pharmacological polytherapy and the presence of cardiovascular or hepatic comorbidities. The pathomechanism of AKI and CKD is associated with inhibition of the biosynthesis of prostanoids involved in the maintenance of renal blood flow, especially PGE2 and PGI2. It is suggested that both COX isoforms play opposing roles in renal function, with natriuresis increased by COX‐1 inhibition followed by a drop in a blood pressure, whereas COX‐2 inhibition increases blood pressure and promotes sodium retention. TIN after NSAID use is potentially associated with glomerular basement membrane damage, reduction in pore size, and podocyte density. Therefore, nephrotic proteinuria and impairment of renal function may occur. The following article analyzes the association of NSAIDs with kidney disease based on available medical literature.
Keywords: AKI, CKD, nephrotoxicity, NSAIDs
The NSAIDs are one of the most frequently used drugs worldwide available over the counter. Medical literature associates the usage of NSAIDs with acute kidney injury (AKI), tubulointerstitial nephritis, as well as nephrotic syndrome and chronic kidney disease.
Abbreviations
- ACE
angiotensin‐converting enzyme
- AKI
acute kidney injury
- CKD
chronic kidney disease
- LOX
lipoxygenase
- NKCC2
Na+‐K+‐2Cl− cotransporter type 2
- NSAID
nonsteroidal anti‐inflammatory drug
- PG
prostaglandin
- RAA
renin–angiotensin–aldosterone
- RAAS
renin–angiotensin–aldosterone system
- TIN
tubulointerstitial nephritis
1. INTRODUCTION
Nonsteroidal anti‐inflammatory drugs (NSAIDs) are among the most commonly prescribed drugs in the world, including over the counter availability. 1 , 2 This is due to their wide range of action as agents with anti‐inflammatory and analgesic properties. The classification of NSAIDs can be based on their chemical structure, as well as their selectivity toward cyclooxygenase. Due to structural differences, one can distinguish salicylates, indole acetic acid derivatives (e.g., indomethacin), phenylacetic acid derivatives (e.g., diclofenac, aceclofenac), phenylpropionic acid derivatives (e.g., ibuprofen, naproxen, ketoprofen), and enolic acid derivatives (e.g., oxicams). 3 , 4 The mechanism of action of NSAIDs is related to the arachidonic acid cycle and depends on the release of prostaglandins (PGs) and their effects on the tissues. 5 The production of the aforementioned substances depends on the activity of phospholipase A2 toward membrane phospholipids and the release of arachidonic acid. In the next stage, arachidonic acid undergoes a series of reactions known as arachidonic acid cascade. Those reactions can be divided into two pathways dependent on cyclooxygenase (COX‐1, COX‐2) or lipoxygenase (LOX). 6 The cyclooxygenase pathway results in the formation of prostanoids (PGs, prostacyclins, and thromboxanes), whereas LOX catalyzes the production of leukotrienes. 4 , 6 The arachidonic acid cascade is presented in Figure 1.
FIGURE 1.
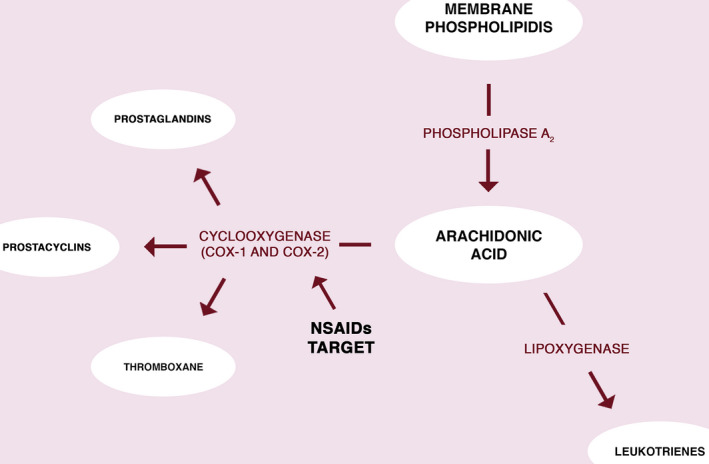
The arachidonic acid cascade and NSAIDs biological target. The arachidonic acid undergoes series of chemical reaction depended on cyclooxygenases (COX‐1 or COX‐2) or lipoxygenase. NSAIDs are inhibitors of the COX enzymes, blocking that pathway of the arachidonic acid cascade. NSAID, nonsteroidal anti‐inflammatory drugs
As mentioned earlier, NSAIDs can be classified according to their activity against cyclooxygenase. At least two isoforms of this enzyme have been identified: COX‐1 and COX‐2. The COX‐1 enzyme, considered a "constitutive" enzyme, is dominant in the human body. It is commonly involved in various homeostatic functions such as the regulation of the gastrointestinal mucosa, especially of the stomach, renal epithelial cells, platelets, and vascular endothelium. COX‐2 has been defined as the "inducible" isoform because its basal expression is more restricted and its activity is upregulated mainly by inflammatory mediators. 5 In addition, it was found that COX‐2 is induced by hormones secreted by the ovaries, uterine muscle, and fetal membranes. COX‐2 regulates the course of the ovulation cycle and the implantation of a fertilized egg. COX‐2 has a constitutional expression in the CNS, testis, kidneys, and bronchial epithelial cells. 7 The discovery of the second cyclooxygenase isoform led to the emergence of selective inhibitors affecting this enzyme, which could reduce the adverse effects associated with the use of COX‐1 inhibitors. It was believed that COX‐2 isoform was only induced at sites of inflammation, which could prevent the negative effects associated with the inhibition of COX‐1. 8
Both isoforms of COX are located in the kidneys, however, their influences on renal function are contrary and so the selectivity of chosen NSAID could alter the renal function in different ways. The inhibition of COX‐1 was suggested to lower blood pressure by increasing natriuresis, whereas blockade of COX‐2 could result in sodium and water retention, elevating blood pressure. 9
The best‐known PGs involved in the regulation of kidney function are PGE2 and prostacyclin (PGI2). PGs increase renal blood flow and glomerular filtration rate in conditions of reduced circulation. 10 This results in a greater tubular flow as well as the release of potassium ions. Under conditions of reduced renal perfusion, renal PG production is an important compensatory mechanism. PGI2, and possibly PGE2, increases potassium secretion mainly by stimulating renin secretion and activating the renin–angiotensin system, which leads to increased aldosterone secretion. Additionally, PGE2 participates in the regulation of sodium and water reabsorption and acts as a counter‐regulatory factor in conditions of increased sodium reabsorption. 11 PGE2 reduces sodium reabsorption on the thick ascending limb of the Henle loop, possibly by inhibiting the Na+‐K+‐2Cl− cotransporter type 2 (NKCC2). Cyclooxygenase inhibitors can increase urine concentrating capacity, in part by affecting NKCC2 regulation in the thick ascending limb of the Henle loop and aquaporin‐2 in the collection tube. 12 Therefore, they may be useful in the treatment of Bartter's syndrome and nephrogenic diabetes insipidus. Moreover, arginine vasopressin releases kinins to the distal nephron and interstitial space of the kidneys, where PGE2 synthesis in the collecting ducts is enhanced. The increase in PGE2 inhibits the hydroosmotic effect of vasopressin and increases spinal blood flow. PGs also buffer the renal vasoconstrictor and antidiuretic effects of angiotensin II. 13
The renal medulla is known to be the main region of sodium, chloride, and water reabsorption in the kidneys. The effects of COX‐2 inhibition, like sodium and water retention, could be assigned to a decreased level of medullary PGE2. It was proposed that COX‐2 induced PGs are vasodilators that allow to maintain renal blood flow impaired by the vasoconstrictors such as angiotensin II or noradrenaline. 8 Furthermore, a study involving mice showed that inhibition of COX‐1 reduced the amount of PGs in the renal cortex, medulla, and aorta, and inhibition of COX‐2 reduced PGs only in the renal medulla. 14 The pressure exerted by NSAIDs was presumed to have an unfavorable effect on kidney function. The mechanisms are summarized in Figure 2.
FIGURE 2.
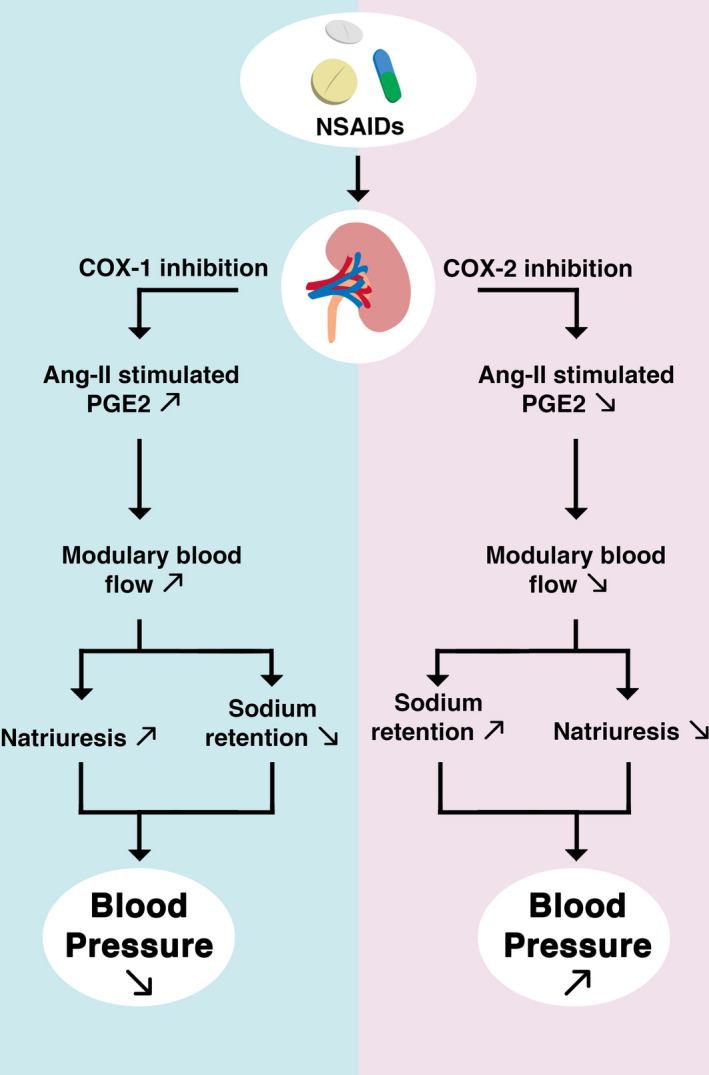
Renal effects of COX‐1 and COX‐2 inhibition. Ang‐II, angiotensin II; NSAID, nonsteroidal anti‐inflammatory drugs; PGE2, prostaglandin E2
The aforementioned effects of NSAIDs on their biological target, that is, COX explain the potentially toxic effects of these drugs on the kidneys. The medical literature describes NSAIDs induced kidney injury from acute injury, tubulointerstitial injury, as well as nephrotic syndrome and chronic kidney disease (CKD). 15 In the presented work, the available medical literature has been analyzed in order to explain potential pathomechanisms that may contribute to the occurrence of these pathologies.
2. ACUTE KIDNEY INJURY AND NSAIDS
Acute kidney injury (AKI) is an abrupt reduction in kidney function: oliguria of 0.5 ml/kg/h for >6 h, increase in serum creatinine of either ≥0.3 mg/dl, or 1.5‐fold from baseline. 16 , 17 Stages of AKI defined by urine output and serum creatine are presented in Table 1. 18 Almost all NSAIDs may be associated with AKI, 17 , 19 however, kidney damage caused by the use of NSAIDs is not common. Certain factors such as advanced age, comorbidities, and medications (e.g., co‐trimoxazole, aminoglycosides, or cimetidine) taken on a regular basis already lead to a reduction in glomerular filtration rate (GFR), increasing the risk of nephrotoxicity associated with NSAIDs use, contributing to the development of side effects. One of the important risk factors that should be considered is arterial hypertension. Patients who suffer from it develop an increased activity of two systems: renin–angiotensin–aldosterone (RAA) and the sympathetic nervous system. Increased activity of these systems contributes to vasoconstriction and thus inhibition of PG synthesis. Adding the mechanism that blocks COX by NSAIDs, the compensatory mechanism is lost in the form of dilation of the renal vessels and their damage. 20 PGs are one of the factors that influence blood pressure through their impact on vascular tone and the transport of fluids and electrolytes in the renal structures. Alterations caused by NSAIDs have minimal influence on blood pressure in normotensive patients, however, they could elevate blood pressure in hypertensive patients and attenuate the control with antihypertensive drugs. 15 , 21 In rodents, deletion of COX‐1 causes natriuresis and increases sensitivity to angiotensin‐converting enzyme (ACE) inhibitors, meaning that COX‐1 deficiency lowers blood pressure despite activation of the RAA system (RAAS). Pharmacological inhibition and genetic removal of COX‐1 eliminate the hypertensive response to angiotensin II. 9 The effects of COX‐2 blockade or deletion are opposite. The vasoconstrictive response to angiotensin II and blood pressure are both elevated, due to decreased renal medullary blood flow and sodium excretion. 9 , 22 It is believed that mainly high doses of NSAIDs, have a significant impact on the formation of AKI, especially in the elderly. 11 The study by Griffin et al. reported an increased risk of AKI among patients receiving doses of ibuprofen >1200 mg/day. 23
TABLE 1.
Table summarizing selected retrospective studies determining the influence of NSAIDs on kidney functioning
| Author | Number of patients | Kidney disease | Results | References |
|---|---|---|---|---|
| Griffin et al. | 1799 AKI cases and 9899 patients in the control group | AKI | In population over 65 years old, the usage of NSAIDs was associated with a higher risk of AKI (OR = 1.58; 95% CI: 1.34–1.86) | 23 |
| Zhang et al. | 1,609,163 participants from 10 studies | AKI | In general population, NSIADs usage was reported to increase the risk of AKI (OR 1.73 95% CI: 1.44–2.07) | 24 |
| Dreischulte et al. | 78,379 | AKI | The simultaneous usage of NSAIDs and any of RASI or diuretic was associated with a higher risk of AKI development (OR = 1.66; 95% CI: 1.40–1.97) | 25 |
| Yu et al. | 23,073 | AKI | The risk of AKI was higher among patients using NSAIDs (OR = 2.39; 95% CI: 1.25–4.58) | 26 |
| Chiu et al. | 12,579 patients in the study group and 37,737 in the control group | CKD | Usage of NSAIDs was associated with a higher risk of development of CKD (OR = 2.01; 95% CI: 1.31–3.09) | 31 |
Abbreviations: AKI, acute kidney injury; CKD, chronic kidney disease; NSAID, nonsteroidal anti‐inflammatory drugs; RASI, renin–angiotensin system inhibitors.
Nevertheless, there are reports that therapeutic doses of NSAIDs in susceptible patients can also cause acute kidney damage, probably in similar pathomechanism to aforementioned. Inhibition of the biosynthesis of prostanoids involved in the maintenance of renal blood flow, especially PGE2 and PGI2, appears to be of key importance. Zhang et al. have identified a higher risk of AKI in the general population, especially in the elderly, as well as in patients with cardiovascular, hepatic, renal, or chronic diseases or with reduced circulating blood volume, such as patients using NSAIDs in combination with diuretics and RAAS inhibitors. 24 Dreischulte et al. in a follow‐up study of nearly 80,000 patients chronically using NSAIDs in combination with diuretics and/or ACE inhibitors showed a strong association with the occurrence of kidney damage. They proved that combined therapy was responsible for a large increase in the risk of AKI during the year of their use compared with antihypertensive treatment only. 25
Yu et al. conducted a retrospective study, which included 21,131 patients. The authors reported an association between NSAIDs and drug‐induced AKI—OR = 2.39 (95% CI: 1.25–4.58). 26 Furthermore, Pham et al. found that antenatal indomethacin increases the risk of AKI in neonates. A higher probability of AKI was observed when indomethacin was administrated <48 h before child delivery. 27
NSAIDs might as well present nephroprotective properties. A recent study showed that continuation of aspirin therapy is associated with a lower incidence of AKI after coronary artery bypass grafting than discontinuation between 24–48 h prior to surgery. 28 Quite contrary, in a study including more than 5000 patients, early NSAIDs administration after major gastrointestinal surgeries was not significantly associated with a lower risk of AKI. However, the result was close to statistical significance threshold—OR = 0.80 (95% CI: 0.63–1.00), p = .057. 29 Moreover, Han et al. did not find an association between NSAIDs usage for patient‐controlled analgesia after laparoscopic nephrectomy and AKI. 30
3. CHRONIC KIDNEY DISEASE AND NSAIDS
There are not many studies showing the long‐term effects of NSAIDs on the development of CKD. However, Melgaco et al. reported, that daily use of NSAIDs for over a year increases the risk of developing CKD. 20 Patients who continue to use NSAIDs may progress to the already existing weakened kidney function, contributing to their fibrosis. 20 A recent cohort study by Chiu et al. published in 2015 showed that in the elderly, chronically ill patients, regardless of the class and selectivity of the drug, NSAIDs increase the risk of CKD in a dose‐dependent matter. Among over 12 thousand patients who had comorbidities but were using NSAIDs about 10% of them developed CKD. 31 The result was statistically significant.
The negative influence of the long duration of NSAIDs treatment on kidney function was observed in a cohort of children with juvenile idiopathic arthritis. 32 The risk of CKD progression in patients using NSAIDs might be different depending on their initial GFR and comorbidities. 33 Terrill et al. found that patients with CKD might be prone to AKI after topical NSAIDs administration. 34 Moreover, CKD progression is probably more likely when NSAIDs are combined with acetaminophen. 35 Opioid use has a stronger association with adverse events like kidney failure with kidney replacement therapy, hospitalization, and death compared to NSAIDs amongst patients with CKD. 36 In stable patients with stage 1 and 2 CKD without predisposing risk factors, management may be similar to that of patients without renal disease. In patients with stage 3 CKD who have minimized predisposing risk factors, NSAID use for the short term (up to 5 days) is an acceptable pain management strategy with an acceptably low risk of nephrotoxicity. 37 Routine monitoring of laboratory parameters of kidney function over 2–3 weeks of use is sufficient to monitor for side effects. A greater risk of side effects in these patients is associated with long‐term and chronic use. This refers to a longer exposure period during which additional risk factors for NSAID toxicity may develop. Moreover, short‐acting drugs are preferable to long‐acting drugs. Additionally, it is recommended to optimize the state of cardiac volume and function before and during treatment. The dosage intervals of NSAIDs should be adjusted to take into account the reduced elimination of the drug such as mineralocorticoid inhibitors and trimethoprim in CKD. 38 Patients with stage 4 CKD require a more individual and careful approach to NSAID therapy as many of these patients struggle with severe AKI events, acid‐base disorders (especially hyperkalemia and metabolic acidosis), and hypervolemia. In patients with stable CKD in stage 4, low doses of preparations with short half‐lives with appropriate dosing intervals of 5 days or less and close monitoring during the treatment period are required to be used. 33 However, it is too soon to draw a conclusion on the best analgesic agent in patients with CKD. 39
4. TUBULOINTERSTITIAL NEPHRITIS AND NSAIDS
Tubulointerstitial nephritis (TIN) is an inflammatory process, whether acute or chronic, that develops in the extra‐glomerular structures of the kidney. One factor with proven acute risk is the use of NSAIDs. In up to 80% of cases, it occurs with nephrotic proteinuria and more frequently in patients using naproxen, phenoprofen, and ibuprofen. 40 The exact mechanism of its occurrence has not been fully elucidated, however, a delayed hypersensitivity reaction and activation of T lymphocytes are suspected. Additionally, the effect of leukotrienes (an alternative pathway in the arachidonic acid cascade), through activated T lymphocytes, cause minimal lesion disease and nephrotic syndrome for several days after the use of NSAIDs. 41 These drugs also have long‐term effects, examples of which are described below.
There have been numerous reports of TIN in patients taking NSAIDs. Henao et al. reported a case of interstitial nephritis proven in biopsy in a 73‐year‐old woman taking celecoxib for over a year. This resulted in the above‐mentioned inflammation with proteinuria and AKI requiring hemodialysis. Discontinuation of the drug for 2 weeks resulted in an improvement in renal parameters. 42 In 2018. Szponar et al. discussed a case of a 61‐year‐old patient who developed acute TIN after using diclofenac for a few days for gout symptoms. After discontinuation of the drug, an improvement in renal parameters was observed after a few days and the patient was discharged on day 11 of hospitalization. 43 In 2018 by a team led by Hsiu‐Wen Chang described a case of a 28‐year‐old woman taking diclofenac 25 mg or mefenamic acid 500 mg two to three times a day for migraine headaches. Due to severe pain that persisted after taking the drug, she was taken to the emergency department where 30 mg of ketorolac was administered intravenously. The renal parameters were normal at the time (eGFR = 113.3 ml/min). After 2 days, she was admitted to the hospital due to bilateral lumbar pain caused by acute interstitial nephritis. Symptoms of inflammation resolved after drug discontinuation, whereas the use of bilateral double‐J and Foley catheter to reduce bladder pressure and facilitate its outflow resulted in the resolution of clinical symptoms. 44
One of the mechanisms, through which NSAIDs cause kidney damage, is basement membrane injury. Nasrallah et al. evaluated the kidney effects of NSAIDs in diabetic rats. Studies have shown that chronic PG inhibition through the use of NSAIDs (celecoxib, 20 weeks) is associated with thinning of the glomerular basement membrane, a reduction in the slit pore diameter, a decrease in the density of podocytes, and an increase in the mesangium. Those alterations lead to exacerbation of the patient's condition and acceleration of disease progression. Ibuprofen induced more severe symptoms than celecoxib, resulting in severe necrotizing pyelonephritis. 45
Bakhriansyah et al. conducted a cohort analysis of patients from the British primary care. The effect of NSAID exposure was compared with the development of nephrotic syndrome. It has been proven that the use of these drugs for more than 14 days is associated with a significantly higher risk of nephrotic syndrome (OR = 1.34). 46
5. SUMMARY
NSAIDs have an inhibitory effect on both COX‐1 and COX‐2, that is, enzymes that inhibit PG synthesis. Both forms of this isoenzyme are found in the kidney. Blocking one or both of these enzymes can affect different kidney functions. 8 , 10 , 11 COX‐2‐derived PGs have profound effects on renal homeostasis, suggesting that selective COX‐2 inhibitors such as celecoxib may have the same potential for renal adverse effects as traditional nonselective NSAIDs, especially in clinical situations associated with a renal impairment such as sodium depletion, hypovolemia, cirrhosis, congestive heart failure, nephrotic syndrome, and CKD. 15 NSAIDs can cause sodium and fluid retention (especially in the elderly) and increase blood pressure or worsen pre‐existing high blood pressure. 47 These compounds may, in a dose‐dependent manner, increase the risk of AKI, especially in elderly people with underlying disease, as well as the use of a combination of ACE inhibitors or angiotensin II blockers, diuretics, and NSAIDs. 24 Whether regular use of NSAIDs is a direct risk factor for end‐stage renal disease is debated in the literature. Figure 3 summarizes mechanisms of NSAIDs induced AKI, CKD, and TIN. The associations between NSAIDs treatment and various kidney diseases highlight that this group of agents should be used cautiously, for a short duration as it is possible. Selected large retrospective studies are summarized in Table 1. Pain management in patients with end‐stage kidney disease requires experience in using NSAIDs and these opioids, which seem to be the safest for the kidney function, including buprenorphine, fentanyl, hydromorphone methadone, and oxycodone. 48
FIGURE 3.
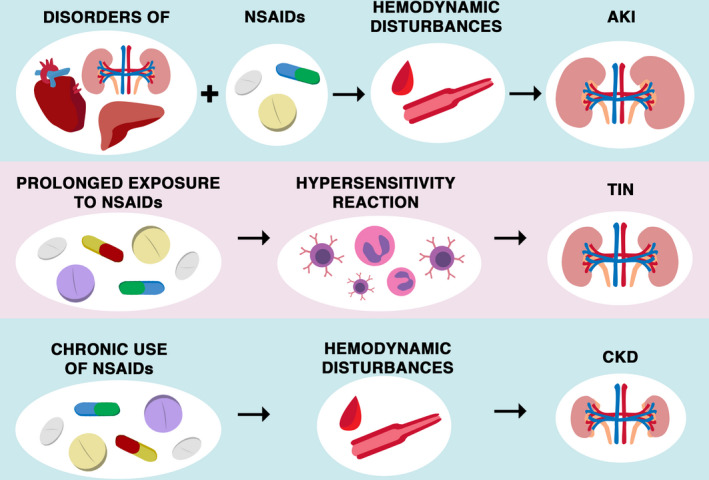
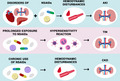
Summarization of main renal pathomechanisms associated with NSAIDs usage. The usage of NSAIDs could disturb kidney function in multiple pathways. The chronic usage of NSAIDs could lead to CKD as the effect of hemodynamic disturbances. The TIN could be the effect of the consequence of prolonged exposure to NSAIDs. A possible mechanism is assigned to a delayed hypersensitivity reaction, with interstitial infiltration of eosinophils and T cells. NSAIDs could also lead to AKI, especially in patients with comorbidities and polypragmasia. AKI, acute kidney injury; CKD, chronic kidney disease; NSAID, nonsteroidal anti‐inflammatory drugs; TIN, tubulointerstitial nephritis
DISCLOSURE
The authors declare no conflict of interest.
AUTHOR CONTRIBUTION
Sylwester Drożdżal, Kacper Lechowicz: Conceptualization, methodology, writing—original draft. Bartosz Szostak, Jakub Rosik: Writing‐ review and editing, conceptualization of figures. Katarzyna Kotfis, Anna Machoy‐Mokrzyńska, Monika Białecka, Kazimierz Ciechanowski, Barbara Gawrońska‐Szklarz: Conceptualization, methodology, investigation, supervision.
ACKNOWLEDGMENT
The authors thank Piotr Michalski from the Academy of Art in Szczecin for his assistance in composing the figures.
Drożdżal S, Lechowicz K, Szostak B, et al. Kidney damage from nonsteroidal anti‐inflammatory drugs—Myth or truth? Review of selected literature. Pharmacol Res Perspect. 2021;9:e00817. 10.1002/prp2.817
Funding information
This research received no external funding.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials. The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Han Y, Balkrishnan R, Hirth RA, et al. Assessment of prescription analgesic use in older adults with and without chronic kidney disease and outcomes. JAMA Netw Open. 2020;3:e2016839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barbieri MA, Rottura M, Cicala G, et al. Chronic kidney disease management in general practice: a focus on inappropriate drugs prescriptions. J Clin Med. 2020;9:1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Friedewald VE, Bennett JS, Christo JP, et al. AJC Editor’s consensus: selective and nonselective nonsteroidal anti‐inflammatory drugs and cardiovascular risk. Am J Cardiol. 2010;106:873‐884. [DOI] [PubMed] [Google Scholar]
- 4. Samborski W, Filipiak KJ, Kaczmarczyk J, Tykarski A. Niesteroidowe leki przeciwzapalne a powikłania sercowo‐naczyniowe i gastroenterologiczne—algorytm wyboru. Chor Serca i Naczyń. 2016;13:257‐264. [Google Scholar]
- 5. Antman EM, Bennett JS, Daugherty A, et al. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634‐1642. [DOI] [PubMed] [Google Scholar]
- 6. Shimizu T, Wolfe LS. Arachidonic acid cascade and signal transduction. J Neurochem. 1990;55:1‐15. [DOI] [PubMed] [Google Scholar]
- 7. Zidar N, Odar K, Glavač D, Jerše M, Zupanc T, Štajerc D. Cyclooxygenase in normal human tissues—is COX‐1 really a constitutive isoform, and COX‐2 an inducible isoform? J Cell Mol Med. 2009;13:3753‐3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khan S, Andrews KL, Chin‐Dusting JPF. Cyclo‐oxygenase (COX) inhibitors and cardiovascular risk: are non‐steroidal anti‐inflammatory drugs really anti‐inflammatory? Int J Mol Sci. 2019;20:4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qi Z, Hao C‐M, Langenbach RI, et al. Opposite effects of cyclooxygenase‐1 and ‐2 activity on the pressor response to angiotensin II. J Clin Invest. 2002;110:61‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Navar LG, Inscho EW, Majid SA, Imig JD, Harrison‐Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiol Rev. 1996;76:425‐536. [DOI] [PubMed] [Google Scholar]
- 11. Villa E, Garcia‐Robles R, Haas J, Romero JC. Comparative effect of PGE2 and PGI2 on renal function. Hypertens. 1997;30:664‐666. [DOI] [PubMed] [Google Scholar]
- 12. Shankar SS, Brater DC. Loop diuretics: from the Na‐K‐2Cl transporter to clinical use. Am J Physiol Renal Physiol. 2003;284:F11‐F21. [DOI] [PubMed] [Google Scholar]
- 13. Arima S, Ren Y, Juncos LA, Carretero OA, Ito S. Glomerular prostaglandins modulate vascular reactivity of the downstream efferent arterioles. Kidney Int. 1994;45:650‐658. [DOI] [PubMed] [Google Scholar]
- 14. Qi Z, Cai H, Morrow JD, Breyer MD. Differentiation of cyclooxygenase 1‐ and 2‐derived prostanoids in mouse kidney and aorta. Hypertens. 2006;48:323‐328. [DOI] [PubMed] [Google Scholar]
- 15. Hörl WH. Nonsteroidal anti‐inflammatory drugs and the kidney. Pharmaceuticals. 2010;3:2291‐2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kellum JA, Lameire N. Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1). Crit Care. 2013;17:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. 2012;120:c179‐c184. [DOI] [PubMed] [Google Scholar]
- 18. Kellum JA, Sileanu FE, Murugan R, Lucko N, Shaw AD, Clermont G. Classifying AKI by urine output versus serum creatinine level. J Am Soc Nephrol. 2015;26:2231‐2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380:756‐766. [DOI] [PubMed] [Google Scholar]
- 20. Melgaço SSC, Saraiva MIR, Lima TTC, Silva Junior GB, Daher EF. Nonsteroidal antiinflammatory drugs nephrotoxicity. Med. 2010;43:382‐390. [Google Scholar]
- 21. Armstrong EP, Malone DC. The impact of nonsteroidal anti‐inflammatory drugs on blood pressure, with an emphasis on newer agents. Clin Ther. 2003;25:1‐18. [DOI] [PubMed] [Google Scholar]
- 22. Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase‐1, and cardiovascular function. J Clin Invest. 2006;116:1391‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Griffin MR, Yared A, Ray WA. Nonsteroidal antiinflammatory drugs and acute renal failure in elderly persons. Am J Epidemiol. 2000;151:488‐496. [DOI] [PubMed] [Google Scholar]
- 24. Zhang X, Donnan PT, Bell S, Guthrie B. Non‐steroidal anti‐inflammatory drug induced acute kidney injury in the community dwelling general population and people with chronic kidney disease: systematic review and meta‐analysis. BMC Nephrol. 2017;18:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dreischulte T, Morales DR, Bell S, Guthrie B. Combined use of nonsteroidal anti‐inflammatory drugs with diuretics and/or renin‐angiotensin system inhibitors in the community increases the risk of acute kidney injury. Kidney Int. 2015;88:396‐403. [DOI] [PubMed] [Google Scholar]
- 26. Yu C, Guo D, Yao C, et al. Clinical characteristics of hospitalized patients with drug‐induced acute kidney injury and associated risk factors: a case‐control study. Biomed Res Int. 2020;2020:9742754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pham JT, Jacobson JL, Ohler KH, Kraus DM, Calip GS. Evaluation of the risk factors for acute kidney injury in neonates exposed to antenatal indomethacin. J Pediatr Pharmacol Ther. 2020;25:606‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aboul‐Hassan SS, Marczak J, Stankowski T, et al. Association between preoperative aspirin and acute kidney injury following coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2020;160:712‐719. [DOI] [PubMed] [Google Scholar]
- 29. Collaborative S. Perioperative Nonsteroidal Anti‐inflammatory Drugs (NSAID) Administration and acute kidney injury (AKI) in major gastrointestinal surgery: a prospective, multicenter, propensity matched cohort study. Ann Surg. 2020. 10.1097/sla.0000000000004314. [DOI] [PubMed] [Google Scholar]
- 30. Han J, Jeon YT, Oh AY, Koo CH, Bae YK, Ryu JH. Comparison of postoperative renal function between non‐steroidal anti‐inflammatory drug and opioids for patient‐controlled analgesia after laparoscopic nephrectomy: a retrospective cohort study. J Clin Med. 2020;9:2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chiu H‐Y, Huang H‐L, Li C‐H, et al. Increased risk of chronic kidney disease in rheumatoid arthritis associated with cardiovascular complications—a national population‐based cohort study. PLoS One. 2015;10:e0136508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gicchino MF, Di Sessa A, Guarino S, Miraglia Del Giudice E, Olivieri AN, Marzuillo P. Prevalence of and factors associated to chronic kidney disease and hypertension in a cohort of children with juvenile idiopathic arthritis. Eur J Pediatr. 2020. 10.1007/s00431-020-03792-4. [DOI] [PubMed] [Google Scholar]
- 33. Baker M, Perazella MA. NSAIDs in CKD: are they safe? Am J Kidney Dis. 2020;76:546‐557. [DOI] [PubMed] [Google Scholar]
- 34. Terrill M, Soden M, Srivastava V. Survey of Australasian Renal and Rheumatology Specialists investigating topical NSAID use and adverse renal outcomes. Musculoskelet Care. 2020;18:134‐139. [DOI] [PubMed] [Google Scholar]
- 35. Mitsuboshi S, Yamada H, Yamazaki S, Kobayashi M, Ueno K, Nagai K. Is concomitant therapy with acetaminophen and low‐dose aspirin a risk factor for CKD progression? A 6‐year cohort study. Yakugaku Zasshi. 2020;140:943‐947. [DOI] [PubMed] [Google Scholar]
- 36. Zhan M, Doerfler RM, Xie D, et al. Association of opioids and nonsteroidal anti‐inflammatory drugs with outcomes in CKD: findings from the CRIC (chronic renal insufficiency cohort) study. Am J Kidney Dis. 2020;76:184‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pham PC, Khaing K, Sievers TM, et al. 2017 update on pain management in patients with chronic kidney disease. Clin Kidney J. 2017;10:688‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perazella MA, Buller GK. NSAID nephrotoxicity revisited: acute renal failure due to parenteral ketorolac. South Med J. 1993;86:1421‐1424. [DOI] [PubMed] [Google Scholar]
- 39. Zhan M, Doerfler RM, Fink JC. In reply to ‘no obvious impact of nsaids on risk of kidney failure: causal or another selection bias’. Am J Kidney Dis. 2020;76:742‐743. [DOI] [PubMed] [Google Scholar]
- 40. Burdmann EA, Vieira Junior JM, Vidal EC. Nefropatia tóxica e tubulointersticial: nefrotoxicidade do meio de contraste radiológico. In: Koogan G, ed. Princípios de nefrologia e distúrbios hidroeletrolíticos. Rio de Janeiro: Editora Guanabara Koogan; 1996:325‐332. [Google Scholar]
- 41. Harirforoosh S, Asghar W, Jamali F. Adverse effects of nonsteroidal antiinflammatory drugs: an update of gastrointestinal, cardiovascular and renal complications. J Pharm Pharm Sci. 2013;16:821‐847. [DOI] [PubMed] [Google Scholar]
- 42. Henao J, Hisamuddin I, Nzerue CM, Vasandani G, Hewan‐Lowe K. Celecoxib‐induced acute interstitial nephritis. Am J Kidney Dis. 2002;39:1313‐1317. [DOI] [PubMed] [Google Scholar]
- 43. Szponar J, Krajewska A, Jakubowicz‐Klecha A, Tchórz M, Kwiecien‐Obara E. Ostre śródmiąższowe zapalenie nerek w przebiegu nadużywania diklofenaku—opis przypadku. Przegl Lek. 2018;75:32‐33. [Google Scholar]
- 44. Chang H‐W, Kuei C‐H, Tseng C‐F, Hou Y‐C, Tseng Y‐L. Spontaneous perirenal urinoma induced by NSAID‐associated acute interstitial nephritis. Ther Clin Risk Manag. 2018;14:595‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nasrallah R, Robertson SJ, Karsh J, Hébert RL. Celecoxib modifies glomerular basement membrane, mesangium and podocytes in OVE26 mice, but ibuprofen is more detrimental. Clin Sci. 2013;124:685‐694. [DOI] [PubMed] [Google Scholar]
- 46. Bakhriansyah M, Souverein PC, van den Hoogen MWF , de Boer A , Klungel OH. Risk of nephrotic syndrome for non‐steroidal anti‐inflammatory drug users. Clin J Am Soc Nephrol. 2019;14:1355‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. White WB. Cardiovascular effects of the cyclooxygenase inhibitors. Hypertens. 2007;49:408‐418. [DOI] [PubMed] [Google Scholar]
- 48. Roy PJ, Weltman M, Dember LM, Liebschutz J, Jhamb M. Pain management in patients with chronic kidney disease and end‐stage kidney disease. Curr Opin Nephrol Hypertens. 2020;29:671‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials. The data that support the findings of this study are available from the corresponding author upon reasonable request.