

Abstract
Equilibrativenucleoside transporters (ENTs) participate in the pharmacokinetics and disposition of nucleoside analog drugs. Understanding drug interactions with the ENTs may inform and facilitate the development of new drugs, including chemotherapeutics and antivirals that require access to sanctuary sites such as the male genital tract. This study created three-dimensional pharmacophores for ENT1 and ENT2 substrates and inhibitors using Kt and IC50 data curated from the literature. Substrate pharmacophores for ENT1 and ENT2 are distinct, with partial overlap of hydrogen bond donors, whereas the inhibitor pharmacophores predominantly feature hydrogen bond acceptors. Mizoribine and ribavirin mapped to the ENT1 substrate pharmacophore and proved to be substrates of the ENTs. The presence of the ENT-specific inhibitor 6-S-[(4-nitrophenyl)methyl]-6-thioinosine (NBMPR) decreased mizoribine accumulation in ENT1 and ENT2 cells (ENT1, ∼70% decrease, P = 0.0046; ENT2, ∼50% decrease, P = 0.0012). NBMPR also decreased ribavirin accumulation in ENT1 and ENT2 cells (ENT1: ∼50% decrease, P = 0.0498; ENT2: ∼30% decrease, P = 0.0125). Darunavir mapped to the ENT1 inhibitor pharmacophore and NBMPR did not significantly influence darunavir accumulation in either ENT1 or ENT2 cells (ENT1: P = 0.28; ENT2: P = 0.53), indicating that darunavir’s interaction with the ENTs is limited to inhibition. These computational and in vitro models can inform compound selection in the drug discovery and development process, thereby reducing time and expense of identification and optimization of ENT-interacting compounds.
SIGNIFICANCE STATEMENT
This study developed computational models of human equilibrative nucleoside transporters (ENTs) to predict drug interactions and validated these models with two compounds in vitro. Identification and prediction of ENT1 and ENT2 substrates allows for the determination of drugs that can penetrate tissues expressing these transporters.
Introduction
The equilibrative nucleoside transporters (ENTs) 1 and 2 are bidirectional, sodium-independent transporters that move nucleosides in and out of cells (Plagemann et al., 1988; Griffith and Jarvis, 1996; Baldwin et al., 2004; Young et al., 2008; Molina-Arcas et al., 2009; Yao et al., 2011; Boswell-Casteel and Hays, 2017). ENT1 and ENT2 are widely expressed proteins with ENT1 expression being highest in erythrocytes, placenta, brain, heart, liver, colon, and vascular endothelium (Pennycooke et al., 2001; Molina-Arcas et al., 2009), whereas ENT2 expression is highest in skeletal muscle, prostate, kidney, placenta, brain, heart, thymus, and pancreas (Pennycooke et al., 2001; Molina-Arcas et al., 2009). Additionally, these transporters are both expressed in the epithelial cells of the blood-testis barrier (BTB), with ENT1 on the basal membrane and ENT2 present at the apical membrane of Sertoli cells (Klein et al., 2013), providing nucleosides essential for spermatogenesis (Kato et al., 2009). Although these widely expressed ENTs could play an important role in the pharmacokinetics and disposition of nucleoside analogs, this remains a relatively understudied area (King et al., 2006; Young et al., 2008; Fukuda and Schuetz, 2012; Shimada et al., 2015; Hioki et al., 2018).
The kinetics of endogenous substrates for nucleoside transporters has been extensively studied, predominately with ENT1 (Boleti et al., 1997; Ward et al., 2000; Bone and Hammond, 2007; Boswell-Casteel and Hays, 2017; Miller et al., 2020, 2021), and recent advancements include the determination of the ENT1 crystal structure bound to two nontransported ENT1 inhibitors, 6-nitrobenzylthioinosine and dilazep (Wright and Lee, 2019), but further research is necessary to understand the roles that human ENT1 and ENT2 play in the pharmacokinetics of currently prescribed therapeutics and therapeutics currently in development. Identifying, predicting, and validating drug interactions with these transporters may inform and facilitate the drug development process for new chemotherapeutics, antivirals, and male contraceptives and fertility agents (Mruk et al., 2011; Ekins et al., 2012, 2019; Pastor-Anglada and Pérez-Torras, 2018; Rehan et al., 2019).
Computational models of drug-transporter interactions are useful for identifying potential substrates and/or inhibitors of clinically relevant transporters and can provide insight on transporter substrate selectivity (Chang and Swaan, 2006; Astorga et al., 2012; Ekins et al., 2012; Lin et al., 2015; Zdrazil et al., 2016; Jain and Ecker, 2019). There have been numerous efforts to use computational approaches to predict drug interactions with transporters, such as pharmacophores, quantitative structure-activity relationships, machine learning models, and docking in crystal structures or homology models (Ekins et al., 2012, 2015). For example, we have published several recent examples using in vitro data to generate Bayesian machine learning models that can in turn be used to score libraries of compounds and predict additional compounds (Martinez-Guerrero et al., 2016; Sandoval et al., 2018; Miller et al., 2021). These Bayesian models have been useful in identifying important molecular features in the training sets, and such models can be applied to the drug discovery and development process to identify valuable information on favorable and unfavorable drug-transporter interactions before additional in vitro and in vivo studies are conducted. Drug-drug interactions that involve transporters can increase exposure and the risk of toxicity or facilitate drug disposition (Cheng et al., 2016). Computational modeling that is implemented early in the drug discovery and development process can refine the library of screening compounds and reduce the amount of time and effort required for lead identification and optimization.
Although the ENTs are widely expressed transporters that play a role in the pharmacokinetics of nucleoside analogs, there is no comprehensive list of known ENT substrates and inhibitors. This study has curated data from the literature on human ENT1 and ENT2 substrates and inhibitors and recorded IC50 and Kt values when provided. These data, in conjunction with experimental data previously generated by our research group, were then used to generate pharmacophores for ENT1 and ENT2, develop Bayesian models to identify interactions with these transporters, and determine substrate or inhibitory interactions with compounds the models identified (Miller et al., 2021). ENT substrates identified using these models may cross the BTB through the ENT transepithelial transport pathway and enter other tissues expressing these transporters.
Materials and Methods
Reagents
Darunavir was purchased from Cayman Chemical (Ann Arbor, MI). Mizoribine and ribavirin were purchased from Sigma Aldrich (St. Louis, MO). 6-S-[(4-Nitrophenyl)methyl]-6-thioinosine (NBMPR) was purchased from Tocris Bioscience (Bristol, UK). [3H]Uridine (specific activity 35.8 Ci/mmol) and MicroScint-20 scintillation cocktail were purchased from Perkin-Elmer (Waltham, MA). Additional reagents were purchased form Thermo Fisher Scientific (Waltham, MA) unless otherwise noted.
Identification of ENT1 and ENT2 Substrates and Inhibitors
Substrates and inhibitors of human ENT1 and ENT2 were manually curated from literature searches from PubMed and included in Supplemental Table 1. When not provided in the original report, simplified molecular-input line-entry system and structure images were generated using PubChem Sketcher V2.4 and RDKit. The table includes Kt and IC50 values when these values were reported.
Ligand-Based Pharmacophores
Generation of three-dimensional quantitative structure-activity relationship pharmacophores was completed using Discovery Studio (Biovia, San Diego, CA) (Ekins et al., 2002). For the ENT1 substrate pharmacophore, 21 known substrate molecules were used, with literature reported Kt values used as a measure of biologic activity (Supplemental Table 2). For the ENT2 substrate pharmacophore, nine molecules with literature reported Kt values used as a measure of biologic activity were used (Supplemental Table 3). For inhibitor pharmacophores, IC50 values were used as a measure of biologic activity. Ten pharmacophores were generated for ENT1 substrates, ENT2 substrates, ENT1 inhibitors and ENT2 inhibitors. The following features were selected for pharmacophore generation, including hydrogen bond acceptor, hydrogen bond donor, hydrophobic, positive ionizable, and negative ionizable. Best conformation generation was used, and a maximum of 10 pharmacophores were selected. The ENT1 and the ENT2 substrate pharmacophores were also used to score the compounds we recently characterized and previously reported (Miller et al., 2021).
Receptor-Ligand Pharmacophores
The ENT1 crystal structure with dilazep bound (Protein Data Bank 60B7, 2.3 Å) had multiple crystallographic waters directly coordinated with dilazep and ENT1. These waters appear to be important for dilazep binding to ENT1, as they form hydrogen bonds with three independent oxygens within dilazep. Each of these waters also form hydrogen bonding interactions with ENT1, enhancing the likelihood that these waters are not crystallographic artifacts. Although waters may also have been important in the binding of NBMPR, the resolution was 2.9 Å, which likely precluded the ability to resolve even stable water molecules. Pharmacophore features were assigned to dilazep using the Receptor-Ligand pharmacophore generation protocol within Discovery Studio. This protocol assigns pharmacophores that correspond to those found within ENT1 only. Water molecules that directly interacted with dilazep were retained during this process. The types of pharmacophores considered were hydrogen bond acceptor, hydrogen bond donor, hydrophobic, negative ionizable, positive ionizable, and ring aromatic. The crystal structure of dilazep and ENT1 show a complex series of contacts involving multiple types of interactions including: an aromatic interaction with a neighboring phenylalanine, hydrogen bonding with glutamines, hydrophobic interactions with methionine, hydrogen bonding with a tryptophan, as well as hydrogen bonding with the previously mentioned waters.
Assay Central Bayesian Models
We used a training set curated from ChEMBL, described in a previous publication of IC50 values for human ENT1 (Miller et al., 2021), to generate Assay Central machine learning models to predict ENT1 activity from chemical structures. The data used to generate the training set is from ChEMBL (Target ID 1997: https://www.ebi.ac.uk/chembl/target_report_card/CHEMBL1997). The threshold for actives/inactives was calculated (as described previously) automatically within Assay Central at 1.9 µM (Miller et al., 2021). This model was used to predict the ENT1 literature test set of 165 compounds (after removal of compounds overlapping between the two datasets) curated for this study.
Cell Culture
ENT1 and ENT2 cells were previously generated using CRISPR/Cas9 to functionally eliminate either ENT2 or ENT1, respectively, in wild-type HeLa S3 CCL-2.2 cells (Miller et al., 2021). Cells were grown in Ham’s F-12K medium supplemented with 1.5 g/l sodium bicarbonate, 10% v/v fetal bovine serum, and 1% v/v penicillin and streptomycin. Cells were propagated using the AmericanType Culture Collection protocol for HeLa S3 CCL-2.2 cells and kept at 37°C in a humidified 5% CO2 incubator.
Transport Experiments
Cells (200,000 cells/ml) were seeded into Nunc MicroWell 96-well optical bottom plates (ThermoFisher Scientific, Waltham, MA) for experiments the following day. Transport buffers were made with Waymouth’s buffer (WB) (2.5 mM CaCl•2H2O, 28 mM d-glucose, 13 mM HEPES, 135 mM NaCl, 1.2 mM MgCl2, 0.8 mM MgSO4•7H2O, pH 7.4). For IC50 experiments, transport buffer included 1 μCi/ml (∼30 nM) of [3H]uridine and increasing concentrations of the test compound. The transport buffers for LC-MS/MS included 50 μM of either darunavir, mizoribine, or ribavirin with or without 100 μM NBMPR. Culture media was aspirated, and cells were washed once with 300 μl of room temperature WB using a Biotek 405 LS Microplate Washer (BioTek, Winooski, VT) before initiating transport by adding 50 μl of transport buffer using an Integra VIAFLO 96-well multichannel pipette (Integra Lifesciences, Plainsboro, NJ). Transport was terminated after 5 minutes by rinsing twice with ice-cold WB. 200 μl of MicroScint-20 scintillation cocktail was added to samples for IC50 experiments before analyzing data on a liquid scintillation counter.
Sample preparation for LC-MS/MS was carried out as previously described (Miller et al., 2020, 2021). After the experiment was completed, 50 μl of 1:1 methanol:acetonitrile (ACN) was added to cells containing 62.5 ng/ml of internal standard and incubated overnight at 4°C. The internal standard was cladribine for darunavir and ribavirin, and stavudine for mizoribine. Calibration curves for each compound were prepared identically to samples in MeOH:ACN + 0.1% formic acid. Samples were dried and resuspended in 50 μl of 90:10 H2O:ACN + 0.1% formic acid for darunavir and ribavirin, and in 50 μl of H2O + 0.1% formic acid for mizoribine. Cellular debris was removed by centrifugation for 10 minutes at 15,000g. The supernatant was collected for LC-MS/MS analysis.
A Shimadzu Prominence high performance liquid chromatography system (Shimadzu, Kyoto, Japan) coupled to a SCIEX QTRAP 4500 mass spectrometer (SCIEX, Framingham, MA) was used to detect intracellular accumulation of selected compounds. 10 μl of each sample was injected onto a 3.0 × 50 mm, 2.7-µm particle InfinityLab Poroshell 120 EC-C18 column (Agilent, Santa Clara, CA). Analyte intensity was determined by multiple reaction monitoring and is reported in Supplemental Table 4. Darunavir and ribavirin were detected by positive electrospray ionization, and mizoribine was detected by negative electrospray ionization. Mobile phase A was 0.1% formic acid in H2O, and mobile phase B was 0.1% formic acid in acetonitrile.
For darunavir, analytes were separated with a flow rate of 0.3 ml/min using the following gradient over 6 minutes: 10% B (0 to 1 minute), 10%–90% B (1–3 minutes), 90% B (3 to 4 minutes), 90%–10% B (4–4.5 minutes), and 10% B (4.5–6 minutes). The column was equilibrated with 10% B for 0.5 minutes between samples. For mizoribine, analytes were separated with a flow rate of 0.25 ml/min using the following gradient over 7.5 minutes: 0% B (0 to 1 minute), 0%–80% B (1–5 minutes), hold at 80% B for 1 minute, followed by a decrease from 80%–0% B (6–7.5 minutes). The column was equilibrated with 0% B for 2 minutes between samples. For ribavirin, analytes were separated with a flow rate of 0.3 ml/min using the following gradient over 6 minutes: 10% B (0 to 1 minute), 10%–15% B (1 to 2 minutes), 15%–90% B (3 to 4 minutes), 90% B (4–5.5 minutes), and 90%–10% B (5.5–6 minutes). The column was equilibrated with 10% B for 2 minutes between samples. All LC-MS/MS data were analyzed using MultiQuant MD version 3.0.2 before performing statistical analysis.
Data Analysis
Transport experiments were completed with cells from multiple passages and in triplicate per passage. Data are presented as mean ± S.D. unless otherwise noted, and analyzed using GraphPad Prism 8 (San Diego, CA). An unpaired two-tailed t test was used to calculate the statistical difference between experimental conditions in substrate and inhibitor determination studies (P ≤ 0.05 indicating a difference in the presence of NBMPR, P > 0.05 indicating no difference). Inhibition of [3H]uridine uptake by darunavir, mizoribine, and ribavirin was calculated using the equation previously described in (Miller et al., 2021):
J = [(Japp-max* T)/(IC50 + [S])] + (Kd * T)
J is total uridine transport, Japp-max is a constant (maximal rate of transport times the ratio of the IC50 for the drug and the Kt for uridine), T is [3H]uridine concentration, and S is drug concentration. Calculated IC50 values are reported as mean and 95% confidence intervals. A comparison of fits extra sum of squares F test (P ≤ 0.05) was used to compare ENT1 and ENT2 IC50 values for darunavir, mizoribine, and ribavirin.
Results
Identification of ENT1 and ENT2 Substrates and Inhibitors
A table containing data on ENT1 and ENT2 substrates and inhibitors was created (Supplemental Table 1). We identified 33 ENT1 substrates, 16 ENT2 substrates, 220 ENT1 inhibitors, and 92 ENT2 inhibitors from the literature. For data presented as percent inhibition at a specific test concentration, the IC50 value is reported as greater than (>) or less than (<) the test concentration, if inhibition was less than or more than 50%, respectively.
Ligand-Based Pharmacophores
Ligand-based pharmacophores are generated from multiple conformations of the ligands in a training set alone. The substrate pharmacophores for both transporters are distinct with partial overlap of hydrogen bond donor features. Both substrate pharmacophores are slightly different (predominantly containing hydrogen bond donors) compared with the inhibitor pharmacophores, which consist mostly of multiple hydrogen bond acceptor features. The ENT1 and the ENT2 substrate pharmacophore was then used to score compounds tested in Miller et al. (2021). Gemcitabine, mizoribine, ribavirin, GS-9667, and rabacfosfadine are mapped to the ENT1 substrate pharmacophore (Fig. 1; Table 1). Nine compounds mapped to the ENT2 substrate pharmacophore (Fig. 2), with the example of inosine shown mapped to the ENT2 substrate pharmacophore in Fig. 2. Only five compounds mapped to the ENT1 inhibitor pharmacophore using the ligand pharmacophore mapping protocol (Table 2). Of the 43 previously tested compounds, 25 mapped to the ENT1 inhibitor pharmacophore and NBMPR, dilazep, and rabacfosfadine are shown mapped to the ENT1 inhibitor pharmacophore in Fig. 3. 2,6-Bis(diethanolamino)-4,8-diisopropylamino-pyrimido[5,4-d]pyrimidine is also shown mapped to the ENT2 inhibitor pharmacophore in Fig. 4.
Fig. 1.

ENT1substrate pharmacophore correlation and fit (A) and mapping of gemcitabine (B), mizoribine (C), ribavirin (D), GS-9667 (E), and rabacfosfadine (F) to this pharmacophore. Purple features represent hydrogen bond donors, whereas gray features represent excluded volumes. Act, activity; Est, estimate; Err, error; HBD, hydrogen bond donor.
TABLE 1.
ENT1 substrate pharmacophore mapping with corresponding fit and estimated Kt values, and reported literature reported Kt values
| Compound | Fit Value | Estimate Kt | Reported Kt |
|---|---|---|---|
| µM | µM | ||
| Decitabine | 5.91932 | 7.27743 | 0.75–1 (Ueda et al., 2020), 1.09 (Ueda et al., 2015) |
| Mizoribine | 5.6915 | 12.297 | N/A (Ishida et al., 2009) |
| Ribavirin | 5.58548 | 15.6973 | 70 (Choi et al., 2015) |
| Fludarabine | 5.52084 | 18.2161 | 107 (King et al., 2006) |
| Adenosine | 5.51127 | 18.622 | 40 (Molina-Arcas et al., 2009), 11–40 (Boswell-Casteel and Hays, 2017) |
| Trifluridine | 5.39219 | 24.4968 | 710 (Takahashi et al., 2018) |
| Cytarabine | 5.32892 | 28.3385 | 540 (White et al., 1987), 383 (Wiley et al., 1983) |
| Cytidine | 5.32892 | 28.3385 | 580 (Molina-Arcas et al., 2009), 21–580 (Boswell-Casteel and Hays, 2017) |
| Tecadenoson | 5.31673 | 29.1452 | 24 (Lepist et al., 2013) |
| Azacitadine | 5.3162 | 29.1807 | N/A (Hummel-Eisenbeiss et al., 2013) |
| Cladribine | 5.21138 | 37.1463 | 23 (King et al., 2006) |
| Clofarabine | 5.09363 | 48.7156 | 108 (King et al., 2006) |
| Telbivudine | 4.20054 | 380.854 | N/A |
| Thymidine | 4.20054 | 380.854 | N/A |
| Guanosine | 4.05191 | 536.28 | 140 (Molina-Arcas et al., 2009), 48–140 (Boswell-Casteel and Hays, 2017) |
| GS-9667 | 3.9884 | 620.728 | N/A |
| Inosine | 3.6862 | 1244.8 | 170 (Pastor-Anglada and Casado, 2006), 29–170 (Boswell-Casteel and Hays, 2017) |
| Ticagrelor | 3.58954 | 1555.09 | N/A |
| Clevudine | 3.3123 | 2944.43 | N/A |
| Nelarabine | 2.92469 | 7188 | N/A |
| Gemcitabine | 2.7449 | 10,874.4 | 500–1500 (Hioki et al., 2018), 1.8–5.4 (Baer et al., 1992), 329 (Mackey et al., 1998), 160 (Mackey et al., 1999) |
| Aciclovir | 2.56893 | 16,306.9 | N/A |
| Capecitabine | 2.33461 | 27,969.8 | N/A |
| Dipyridamole | 0.108913 | 4.70313e+06 | N/A |
| Rabacfosadine | 0.0574308 | 5.29504e+06 | N/A |
N/A, not available.
Fig. 2.

ENT2substrate pharmacophore correlation and fit (A) and mapping of inosine (B) to this pharmacophore. Purple features represent hydrogen bond donors, whereas green features represent hydrogen bond acceptors. Act, activity; Est, estimate; Err, error; HBA, hydrogen bond acceptor; HBD, hydrogen bond donor; RMS, root mean square.
TABLE 2.
Compounds mapped to ENT1 inhibitor pharmacophore with corresponding fit and estimated IC50 values, and literature reported IC50 values
| Compound | Fit Value | Estimate IC50 | Reported IC50 |
|---|---|---|---|
| µM | µM | ||
| Dilazep | 5.69199 | 2.01238 | 0.009 (Vlachodimou et al., 2020) 0.330 0.0012 (Rehan et al., 2015) |
| Rabacfosadine | 5.13737 | 7.21657 | N/A |
| Darunavir | 4.50727 | 30.7916 | 85.98 (Miller et al., 2021) |
| GS-6620 | 3.83753 | 143.936 | 12.7 (Miller et al., 2021) |
| GS-9667 | 2.37685 | 4157.69 | 4.3 (Miller et al., 2021) |
N/A, not available.
Fig. 3.

ENT1inhibitor pharmacophore correlation and fit (A) and mapping of m-nitro-6-benzylthioinosine (B), dilazep (C), rabacfosidine (D), darunavir (E), GS-6620 (F), and GS-9667 (G) to this pharmacophore. Cyan features represent hydrogen bond donors, green features represent hydrogen bond acceptors, and gray features represent excluded volumes. HBA, hydrogen bond acceptor; RMS, root mean square.
Fig. 4.

ENT2inhibitor pharmacophore correlation (A) and mapping of 2,6-Bis(diethanolamino)-4,8-diisopropylamino-pyrimido[5,4-day]pyrimidine (B) to this pharmacophore. Cyan features represent hydrogen bond donors, green features represent hydrogen bond acceptors, and gray features represent excluded volumes. HBA, hydrogen bond acceptor; RMS, root mean square.
Receptor-Ligand Pharmacophores
Receptor-Ligand pharmacophores use small molecules in their conformation derived from a crystal structure. The nine dilazep analogs tested showed a similar degree of inhibition (10 nM–1 µM). An attempt was made to generate a pharmacophore that was common between them. All compounds were considered active when generating these pharmacophores, although the weaker inhibitors (?1 µM) were assigned as moderately active within Discovery Studio. The common pharmacophore generated was irrespective of the conformation of dilazep found within the crystal structure. The pharmacophore that was common after the generation of up to 255 different conformations per molecule (Fig. 5A) was different than the pharmacophore found by using the receptor-ligand pharmacophore generation protocol, with the most pronounced difference being the positive ionizable group (red) which, based on the crystal structure, was likely a hydrophobic interaction between these compounds and ENT1. Such differences would be expected with using a solely ligand-based pharmacophore versus a structure-based pharmacophore, and this highlights the limitations of the former proposing features and locations that may not be relevant. Attempting to use at least the position of the pharmacophores found by this common pharmacophore search, the pharmacophores found during the receptor-ligand pharmacophore generation were filtered based on these positions for steered docking (Fig. 5B). However, docking with these selected pharmacophores failed to generate any docking poses for the dilazep analogs (data not shown).
Fig. 5.
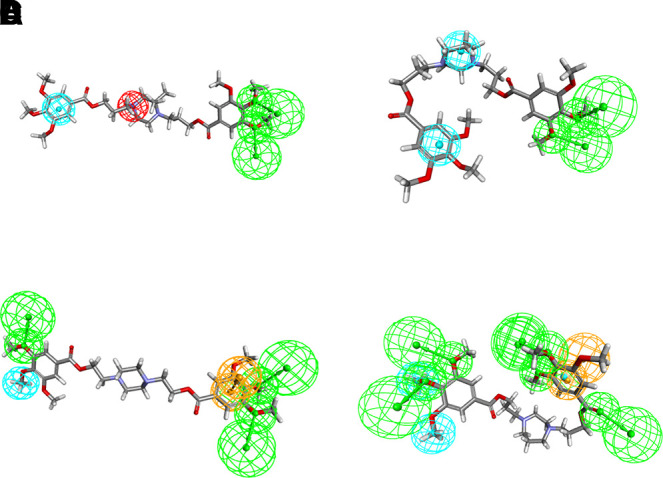
Receptor-ligand pharmacophore generation for ENT1. (A) Common Feature generation using receptor-ligand pharmacophore generation using dilazep and analogs. (B) Mimicry of common features found in crystal structure by filtering all generated pharmacophores from receptor-ligand pharmacophore generation to match actual interactions. (C) Common feature pharmacophore created using a sampling of strong, weak, and very weak inhibitors. (D) In a similar manner described above, the full pharmacophore generated from receptor-ligand pharmacophore generation was made to mimic the pharmacophores found in the common pharmacophore generation step.
To find a common pharmacophore that could describe the negative data of dilazep analogs, a smaller sampling of strong, weak, and very weak inhibitors was used. The most potent inhibitor, dilazep analog 3 (IC50 = 2.8 nM), was defined as strong (2), dilazep (15.3 nM) as well as analogs 5 (93.9 nM) and 40 (17.7 nM) were considered mildly active (1), and finally, analogs 15 (803 nM) and 20 (217 nM) were defined as inactive (0). The common feature pharmacophore shown represents the pharmacophore 2, chosen because the ligand fit correlated well with the known activity (Fig. 5C; Table 3).
TABLE 3.
Structure-based pharmacophore data using dilazep analogs with corresponding activities, fit and principal values
| Compound | Activity | Fit Value | Principal Value (Weight) |
|---|---|---|---|
| nM | |||
| Dilazep analog 3 | 2.80 | 5.00 | 2 |
| Dilazep | 15.27 | 3.03 | 1 |
| Dilazep analog 5 | 93.90 | 3.71 | 1 |
| Dilazep analog 40 | 17.70 | 3.85 | 1 |
| Dilazep analog 15 | 803.01 | 3.00 | 0 |
| Dilazep analog 20 | 217 | 3.00 | 0 |
TABLE 4.
IC50 values for select compounds on ENT1 and ENT2-mediated [3H]uridine uptake
Data are reported as mean and 95% confidence intervals. A comparison of fits extra sum of squares F test (P ≤ 0.05) was used to compare ENT1 and ENT2 IC50 values for each compound.
| Compound | ENT1 IC50 µM (95% CI) |
ENT2 IC50 µM (95% CI) |
P Value |
|---|---|---|---|
| Darunavir | 85.98 (47.94,157.50) | 8.23 (1.30, 63.03) | 0.0116 |
| Mizoribine | 2216 (934.80, 6654) | 378.2 (197.40, 754.50) | 0.0018 |
| Ribavirin | 2657 (1213, 7250) | 1253 (573.70, 3015) | 0.1887 |
The dilazep analogs were docked in the ENT1 structure using pharmacophore restraints and CDOCKER (Fig. 5). Pharmacophore-steered docking used the modified pharmacophore generated from the receptor-ligand pharmacophore generation (Fig. 5D) with the dilazep analogs (including those that were used to generate the pharmacophore). The majority of the compounds that docked within the energy parameters allowed were in the set used in the machine learning models. A notable exception was dilazep analog 15, which was one of the weakest inhibitors in this set (the poorest fit value; representative of the pharmacophore overlaps with the molecule), suggesting that this pharmacophore may represent the required features for high-affinity ENT1 inhibition. The differences between substrate and the inhibitor pharmacophores would suggest ENT1 inhibitors may be structurally quite different from substrates for the same transporter; therefore, their interactions with the protein would be different as well as possibly at different locations.
Assay Central Bayesian Models
At the calculated inhibitory threshold of 1.9 µM, the Bayesian model metrics look encouraging (i.e., receiver operating characteristic = 0.83, Fig. 6B). However, the extreme imbalance of active and inactive compounds at this threshold means that this may not be ideal to evaluate the predictive power of a training model. Alternative thresholds (Supplemental Fig. 1) did not solve the model imbalance issue, and at 50 µM, there were only 15 inactive compounds in the test set (Supplemental Fig. 1C). Figure 6A shows an updated model that integrates the original ChEMBL data with the literature data curated in this study. In comparison with the previous ENT1 model (Fig. 6B), some model metrics are diminished at the new autocalculated threshold of 1.28 µM, which is notably lower than the original ChEMBL-only dataset. At 5 µM, the metrics (i.e., receiver operating characteristic, precision, recall, and specificity) improved from the same ChEMBL-only counterpart.
Fig. 6.

(A) Updatedmodel that integrates the original models generated from ChEMBL data from (Miller et al., 2021) with the literature data curated in this study. The model on the left has an automatically calculated threshold of 1.28 µM, whereas the model on the right has a manually selected threshold of 5 µM. (B) ChEMBL training set for ENT1 in purple on the left. Calculated threshold of 1.9 µM to analyze literature inhibitor data in green on the right. MCC, Matthews correlation coefficient; ROC, receiver operating characteristic.
Inhibition of [3H] Uridine Uptake
Darunavir inhibited both ENT1 and ENT2-mediated [3H]uridine uptake with the most potency (Fig. 7A). The IC50 for darunavir was lower for ENT2 than ENT1 (ENT2: 8.23 µM 95% CI: 1.3, 63.0 versus ENT1: 85.98 µM 95% CI: 47.9, 157.5; P = 0.0116). Mizoribine inhibited ENT2-mediated [3H]uridine uptake more than ENT1-mediated [3H]uridine uptake (Fig. 7B, ENT2: 378.2 µM 95% CI: 197.4, 754.3 versus ENT1: 2216 µM 95% CI: 934.8, 6554; P = 0.0018). Ribavirin inhibited both ENT1 and ENT2-mediated [3H]uridine uptake with similar potency (Fig. 7C, ENT1: 2657 µM 95% CI: 1213, 7250 versus ENT2: 1253 µM 95% CI: 573.7, 3015).
Fig. 7.

Inhibition of ENT1 and ENT2-mediated [3H]uridine uptake in ENT1 and ENT2 cells by darunavir (A), mizoribine (B), and ribavirin (C). Data are presented as mean ± S.D. Calculated IC50 values are reported in Table 4.
Substrate and Inhibitor Determination
Mizoribine mapped to the ENT1 substrate pharmacophore (Fig. 1C) with a fit value of 5.69 (large fit values likely correspond to higher affinity) and a computationally estimated Kt of 12.3 µM. Previous data showed no inhibitory effect of 200 µM mizoribine on [3H]uridine uptake (Miller et al., 2021). Ribavirin mapped to the ENT1 substrate pharmacophore (Fig. 1D) with a fit value of 5.58 and an estimated Kt of 15.69 µM. Previous data showed no inhibitory effect of 200 µM ribavirin on ENT1-mediated [3H]uridine uptake, and a modest impact on ENT2-mediated [3H]uridine uptake (Miller et al., 2021). Darunavir mapped to the ENT1 inhibitor pharmacophore (Fig. 3E) with a fit value of 4.51 and a computationally estimated IC50 of 30.79 µM. Previous data estimated the IC50 of darunavir on [3H]uridine uptake to be 68.8 µM for ENT1 and 15.8 for ENT2 µM (Miller et al., 2021). Mizoribine, ribavirin, and darunavir accumulation in ENT1 and ENT2 cells was then measured in the presence and absence of 100 µM NBMPR (Fig. 8) to validate the findings presented by the substrate and inhibitor pharmacophores. Mizoribine accumulation in ENT1 and ENT2 cells decreased in the presence of 100 µM NBMPR (ENT1: ∼70% decrease, 38.7 pmol cm−2 versus 11.4 pmol cm−2; P = 0.0046; ENT2: ∼50% decrease 21.09 pmol cm−2 versus 10.7 pmol cm−2; P = 0.0012). Ribavirin accumulation in ENT1 and ENT2 cells decreased in the presence of 100 µM NBMPR (ENT1: ∼50% decrease 33.53 pmol cm−2 versus 17.83 pmol cm−2 P = 0.0498; ENT2: ∼30% decrease, 27.93 pmol cm−2 versus 19.14 pmol cm−2 P = 0.0125). There was no difference in the total accumulation of darunavir in the presence of NBMPR in either cell line (ENT1: 74.6 versus 66.9 pmol cm−2 P = 0.28; ENT2: 65.3 versus 61.1 pmol cm−2 P = 0.53),
Fig. 8.

Validation of pharmacophore models that identified mizoribine and darunavir as an ENT substrate and inhibitor, respectively. Mizoribine uptake in the presence of the ENT inhibitor NBMPR in ENT1 (A) and ENT2 (B) cell lines. Darunavir uptake in the presence of the ENT inhibitor NBMPR in ENT1 (C) and ENT2 (D) cell lines. Ribavirin uptake in the presence of the ENT inhibitor NBMPR in ENT1 (E) and ENT2 (F) cell lines. Data are presented as mean ± S.D. A two-tailed unpaired t test was used to determine the difference between groups with **P < 0.01.
Discussion
This study manually curated data on substrates and inhibitors of the human ENTs and used these data to develop predictive Bayesian models and pharmacophores for each transporter. Building a data base of compounds that interact with the ENTs and developing models that enable more informed decisions regarding potential ENT interactions has value for the drug discovery and development process. However, the experimental conditions and cell models chosen by different research groups to generate IC50 and Kt values varied tremendously, and probably contributed to some of the observed differences in these values for compounds investigated by multiple groups. For example, the reported ENT1 Kt values for gemcitabine range from 1.8 to 1500 µM (Baer et al., 1992; Mackey et al., 1998, 1999; Hioki et al., 2018), and this presents challenges of which value to use (whether to take the best or the worst case). There were far more substrates and inhibitors of ENT1 that were identified compared with ENT2. Several of the compounds identified as ENT1 substrates or inhibitors may also interact with ENT2, but that possibility was not explored in many published studies. For both transporters, more compounds were tested and identified as inhibitors than substrates (likely due to ease of data generation), but it is worth noting that some of the inhibitors may also be substrates, and this has yet to be experimentally determined. Published studies that determined inhibition of ENT-mediated transport of radiolabeled endogenous substrates such as uridine or adenosine only identify inhibitory and/or competitive substrate interactions, but do not define the type of interaction.
The utility of the various computational methods used here (pharmacophores and Bayesian machine learning models in particular) was supported by three predictions derived from the models. In this study, darunavir mapped to the ENT1 inhibitor ligand-based pharmacophore, but not the substrate ligand-based pharmacophore. The estimated IC50 for darunavir and ENT1 was similar to the value we determined experimentally and previously reported values (estimated: 30.8 µM; determined: 85.98 µM; reported: 68.8 µM) (Miller et al., 2021). The IC50 values for mizoribine and ribavirin were higher than estimated Kt values. These poor predictions could be due to the aforementioned limitations in the literature data used to build the models or subtle structural differences in the structure-activity relationship that are not picked up by the pharmacophores. Directly measuring the cellular accumulation of a compound of interest in the absence and presence of an ENT inhibitor (i.e., NBMPR and dilazep) or in control and knockout cell models provided information on whether a compound is substrate or inhibitor. Mizoribine and ribavirin mapped to the ENT1 substrate pharmacophore. We validated these predictions by measuring mizoribine and ribavirin accumulation in the presence of NBMPR, and the results suggest that both are substrates of ENT1 and ENT2 (Fig. 8C–F). We validated that darunavir, although clearly an inhibitor from previous data, is not a substrate of both ENT1 and ENT2 (Fig. 8, A and B) (Miller et al., 2021). Previous studies by us examined compounds in a similar manner, where lexibulin and nevirapine were determined to be ENT1 and ENT2 inhibitors, and cladribine and clofarabine were determined to be ENT1 and ENT2 competitive substrates (Miller et al., 2021). The models generated here provide furthermore opportunities to make predictions and identify ENT1 and ENT2 inhibitors and/or substrates, building on our earlier work.
The ENTs play a critical role in nucleoside homeostasis and disposition of nucleoside analogs (Baldwin et al., 2004; Young et al., 2013; Boswell-Casteel and Hays, 2017; Rehan et al., 2019). Drug disposition to the male genital tract is important for the treatment of various cancers and viral infections. The ENT1-ENT2 transepithelial transport pathway provides a mechanism for nucleoside analogs to cross the BTB where they can more effectively treat these cancers and viral infections. This can reduce the likelihood of cancer relapse or viral transmission. Clofarabine, a chemotherapeutic used to treat refractory or relapsed leukemias, mapped to the ENT1 and ENT2 substrate pharmacophores and is a known substrate of these transporters (King et al., 2006; Miller et al., 2021). Clofarabine is a substrate of ENT1 and ENT2, therefore, this may explain why it is effective in treating refractory and relapsed leukemias. This also applies to antivirals. Although there are preventative therapies that greatly reduce the risk of transmission of human immunodeficiency virus and keeps the viral load in the blood undetectable, there is still a risk of viral transmission from semen (Politch et al., 2012). Antiviral treatment regimens that are capable of crossing the BTB may eliminate a viral sanctuary site and further reduce the risk of viral transmission from semen. This may be of importance for a range of viruses reported to be sexually transmissible (e.g., Ebola, Zika, SARS-CoV-2, etc.).
Further investigation into potential inhibitors and substrates of the ENTs can improve our understanding of the structure-function relationship of these widely expressed transporters, and the computational models described herein could be useful in this regard, as they provide a starting point to select compounds to validate these hypotheses. Doubtless, there is scope for further validation and updating of the computational models, however, they may also provide useful information for predicting the ability of existing therapeutics, environmental chemicals, or natural products to interact with the ENTs, and therefore help in prioritizing compounds for testing in vitro.
Acknowledgments
We would like to thank Biovia for providing Discovery Studio used in this study and Alex Clark for software assistance.
Abbreviations
- ACN
acetonitrile
- BTB
blood-testis barrier
- ENT
equilibrative nucleoside transporter
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- NBMPR
6-S-[(4-nitrophenyl)methyl]-6-thioinosine
- WB
Waymouth’s buffer
Authorship Contributions
Participated in research design: Miller, Lane, Zorn, Ekins, Wright, Cherrington.
Conducted experiments: Miller, Lane, Zorn, Ekins.
Performed data analysis: Miller, Lane, Zorn, Ekins, Wright, Cherrington.
Wrote or contributed to the writing of the manuscript: Miller, Lane, Zorn, Ekins, Wright, Cherrington.
Footnotes
This work was supported by National Institutes of Health National Institute of General Medical Sciences [Grant 1R-41GM131433-01A1], [Grant R44-GM122196-02A1], and [Grant 5R01GM123643-04] and the National Institutes of Health National Institute of Environmental Health Sciences [Grant P30 ES006694] and [Grant 2T32ES007091-36A1].
S.E. is owner of collaborations Pharmaceuticals Inc., and K.M.Z. and T.R.L. are employees. All other authors have no conflicts of interest.
This work was previously presented as an abstract: Multiple Computational Approaches for Predicting Drug Interactions with Human Equilibrative Nucleoside Transporter 1 (Abstract), in Translation of in vitro ADMET Science to in vivo: Current Perspectives and Challenges; 2021 Mar 3. International Society for the Study of Xenobiotics (ISSX) and IQ Workshop.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Astorga B, Ekins S, Morales M, Wright SH (2012) Molecular determinants of ligand selectivity for the human multidrug and toxin extruder proteins MATE1 and MATE2-K. J Pharmacol Exp Ther 341:743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer HP, Moorji A, Ogbunude POJ, Serignese V (1992) Sodium-dependent nucleoside transport in mouse lymphocytes, human monocytes, and hamster macrophages and peritoneal exudate cells. Can J Physiol Pharmacol 70:29–35. [DOI] [PubMed] [Google Scholar]
- Baldwin SA, Beal PR, Yao SYM, King AE, Cass CE, Young JD (2004) The equilibrative nucleoside transporter family, SLC29. Pflugers Arch 447:735–743. [DOI] [PubMed] [Google Scholar]
- Boleti H, Coe IR, Baldwin SA, Young JD, Cass CE (1997) Molecular identification of the equilibrative NBMPR-sensitive (es) nucleoside transporter and demonstration of an equilibrative NBMPR-insensitive (ei) transport activity in human erythroleukemia (K562) cells. Neuropharmacology 36:1167–1179. [DOI] [PubMed] [Google Scholar]
- Bone DBJ, Hammond JR (2007) Nucleoside and nucleobase transporters of primary human cardiac microvascular endothelial cells: characterization of a novel nucleobase transporter. Am J Physiol Heart Circ Physiol 293:H3325–H3332. [DOI] [PubMed] [Google Scholar]
- Boswell-Casteel RC, Hays FA (2017) Equilibrative nucleoside transporters-A review. Nucleosides Nucleotides Nucleic Acids 36:7–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Swaan PW (2006) Computational approaches to modeling drug transporters. Eur J Pharm Sci 27:411–424. [DOI] [PubMed] [Google Scholar]
- Cheng Y, El-Kattan A, Zhang Y, Ray AS, Lai Y (2016) Involvement of drug transporters in organ toxicity: the fundamental basis of drug discovery and development. Chem Res Toxicol 29:545–563. [DOI] [PubMed] [Google Scholar]
- Choi M-K, Kim M-H, Maeng H-J, Song I-S (2015) Contribution of CNT1 and ENT1 to ribavirin uptake in human hepatocytes. Arch Pharm Res 38:904–913. [DOI] [PubMed] [Google Scholar]
- Ekins S, Clark AM, Wright SH (2015) Making transporter models for drug-drug interaction prediction mobile. Drug Metab Dispos 43:1642–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins SKim RBLeake BFDantzig AHSchuetz EGLan LBYasuda KShepard RLWinter MASchuetz JD, et al. (2002) Application of three-dimensional quantitative structure-activity relationships of P-glycoprotein inhibitors and substrates. Mol Pharmacol 61:974–981. [DOI] [PubMed] [Google Scholar]
- Ekins S, Polli JE, Swaan PW, Wright SH (2012) Computational modeling to accelerate the identification of substrates and inhibitors for transporters that affect drug disposition. Clin Pharmacol Ther 92:661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S, Puhl AC, Zorn KM, Lane TR, Russo DP, Klein JJ, Hickey AJ, Clark AM (2019) Exploiting machine learning for end-to-end drug discovery and development. Nat Mater 18:435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y, Schuetz JD (2012) ABC transporters and their role in nucleoside and nucleotide drug resistance. Biochem Pharmacol 83:1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith DA, Jarvis SM (1996) Nucleoside and nucleobase transport systems of mammalian cells. Biochim Biophys Acta 1286:153–181. [DOI] [PubMed] [Google Scholar]
- Hioki MShimada TYuan TNakanishi TTajima HYamazaki MYokono RTakabayashi MSawamoto KAkashita G, et al. (2018) Contribution of equilibrative nucleoside transporters 1 and 2 to gemcitabine uptake in pancreatic cancer cells. Biopharm Drug Dispos 39:256–264. [DOI] [PubMed] [Google Scholar]
- Hummel-Eisenbeiss J, Hascher A, Hals P-A, Sandvold ML, Müller-Tidow C, Lyko F, Rius M (2013) The role of human equilibrative nucleoside transporter 1 on the cellular transport of the DNA methyltransferase inhibitors 5-azacytidine and CP-4200 in human leukemia cells. Mol Pharmacol 84:438–450. [DOI] [PubMed] [Google Scholar]
- Ishida K, Takaai M, Yotsutani A, Taguchi M, Hashimoto Y (2009) Membrane transport mechanisms of mizoribine in the rat intestine and human epithelial LS180 cells. Biol Pharm Bull 32:741–745. [DOI] [PubMed] [Google Scholar]
- Jain S, Ecker GF (2019) In silico approaches to predict drug-transporter interaction profiles: data mining, model generation, and link to cholestasis. Methods Mol Biol 1981:383–396. [DOI] [PubMed] [Google Scholar]
- Kato R, Maeda T, Akaike T, Tamai I (2009) Characterization of nucleobase transport by mouse Sertoli cell line TM4. Biol Pharm Bull 32:450–455. [DOI] [PubMed] [Google Scholar]
- King KM, Damaraju VL, Vickers MF, Yao SY, Lang T, Tackaberry TE, Mowles DA, Ng AML, Young JD, Cass CE (2006) A comparison of the transportability, and its role in cytotoxicity, of clofarabine, cladribine, and fludarabine by recombinant human nucleoside transporters produced in three model expression systems. Mol Pharmacol 69:346–353. [DOI] [PubMed] [Google Scholar]
- Klein DM, Evans KK, Hardwick RN, Dantzler WH, Wright SH, Cherrington NJ (2013) Basolateral uptake of nucleosides by Sertoli cells is mediated primarily by equilibrative nucleoside transporter 1. J Pharmacol Exp Ther 346:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepist E-I, Damaraju VL, Zhang J, Gati WP, Yao SYM, Smith KM, Karpinski E, Young JD, Leung KH, Cass CE (2013) Transport of A1 adenosine receptor agonist tecadenoson by human and mouse nucleoside transporters: evidence for blood-brain barrier transport by murine equilibrative nucleoside transporter 1 mENT1. Drug Metab Dispos 41:916–922. [DOI] [PubMed] [Google Scholar]
- Lin L, Yee SW, Kim RB, Giacomini KM (2015) SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov 14:543–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE (1998) Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 58:4349–4357. [PubMed] [Google Scholar]
- Mackey JR, Yao SYM, Smith KM, Karpinski E, Baldwin SA, Cass CE, Young JD (1999) Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J Natl Cancer Inst 91:1876–1881. [DOI] [PubMed] [Google Scholar]
- Martínez-Guerrero LJ, Morales M, Ekins S, Wright SH (2016) Lack of influence of substrate on ligand interaction with the human multidrug and toxin extruder, MATE1. Mol Pharmacol 90:254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SR, Hau RK, Jilek JL, Morales MN, Wright SH, Cherrington NJ (2020) Nucleoside reverse transcriptase inhibitor interaction with human equilibrative nucleoside transporters 1 and 2. Drug Metab Dispos 48:603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SRZhang XHau RKJilek JLJennings EQGalligan JJFoil DHZorn KMEkins SWright SH, et al. (2021) Predicting drug interactions with human equilibrative nucleoside transporters 1 and 2 using functional knockout cell lines and bayesian modeling. Mol Pharmacol 99:147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Arcas M, Casado FJ, Pastor-Anglada M (2009) Nucleoside transporter proteins. Curr Vasc Pharmacol 7:426–434. [DOI] [PubMed] [Google Scholar]
- Mruk DD, Su L, Cheng CY (2011) Emerging role for drug transporters at the blood-testis barrier. Trends Pharmacol Sci 32:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor-Anglada M, Casado FJ (2006) Nucleoside transport into cells, in: Cancer Drug Discovery and Development: Deoxynucleoside Analogs in Cancer Therapy, pp 1–28, Humana Press. [Google Scholar]
- Pastor-Anglada M, Pérez-Torras S (2018) Emerging roles of nucleoside transporters. Front Pharmacol 9:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennycooke M, Chaudary N, Shuralyova I, Zhang Y, Coe IR (2001) Differential expression of human nucleoside transporters in normal and tumor tissue. Biochem Biophys Res Commun 280:951–959. [DOI] [PubMed] [Google Scholar]
- Plagemann PGW, Wohlhueter RM, Woffendin C (1988) Nucleoside and nucleobase transport in animal cells. Biochim Biophys Acta 947:405–443. [DOI] [PubMed] [Google Scholar]
- Politch JA, Mayer KH, Welles SL, O’Brien WX, Xu C, Bowman FP, Anderson DJ (2012) Highly active antiretroviral therapy does not completely suppress HIV in semen of sexually active HIV-infected men who have sex with men. AIDS 26:1535–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehan S, Ashok Y, Nanekar R, Jaakola V-P (2015) Thermodynamics and kinetics of inhibitor binding to human equilibrative nucleoside transporter subtype-1. Biochem Pharmacol 98:681–689. [DOI] [PubMed] [Google Scholar]
- Rehan S, Shahid S, Salminen TA, Jaakola V-P, Paavilainen VO (2019) Current progress on equilibrative nucleoside transporter function and inhibitor design. SLAS Discov 24:953–968. [DOI] [PubMed] [Google Scholar]
- Sandoval PJ, Zorn KM, Clark AM, Ekins S, Wright SH (2018) Assessment of substrate-dependent ligand interactions at the organic cation transporter OCT2 using six model substrates. Mol Pharmacol 94:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada TNakanishi TTajima HYamazaki MYokono RTakabayashi MShimada TSawamoto KMiyamoto KKitagawa H, et al. (2015) Saturable hepatic extraction of gemcitabine involves biphasic uptake mediated by nucleoside transporters equilibrative nucleoside transporter 1 and 2. J Pharm Sci 104:3162–3169. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yoshisue K, Chiba M, Nakanishi T, Tamai I (2018) Contribution of equilibrative nucleoside transporter(s) to intestinal basolateral and apical transports of anticancer trifluridine. Biopharm Drug Dispos 39:38–46. [DOI] [PubMed] [Google Scholar]
- Ueda K, Hosokawa M, Iwakawa S (2015) Cellular uptake of decitabine by equilibrative nucleoside transporters in HCT116 cells. Biol Pharm Bull 38:1113–1119. [DOI] [PubMed] [Google Scholar]
- Ueda K, Nakamura T, Tanaka S, Hosokawa M, Iwakawa S, Ogawara K-I (2020) Numerical analysis of apparent decitabine uptake in HCT116 cells: incorporation of a bidirectional first-order kinetic parameter for ENT1 transport and Michaelis-Menten parameters for subsequent phosphorylation. Drug Metab Pharmacokinet 35:124–130. [DOI] [PubMed] [Google Scholar]
- Vlachodimou A, Konstantinopoulou K, IJzerman AP, Heitman LH (2020) Affinity, binding kinetics and functional characterization of draflazine analogues for human equilibrative nucleoside transporter 1 (SLC29A1). Biochem Pharmacol 172:113747. [DOI] [PubMed] [Google Scholar]
- Ward JL, Sherali A, Mo ZP, Tse CM (2000) Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J Biol Chem 275:8375–8381. [DOI] [PubMed] [Google Scholar]
- White JC, Rathmell JP, Capizzi RL (1987) Membrane transport influences the rate of accumulation of cytosine arabinoside in human leukemia cells. J Clin Invest 79:380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley JS, Jones SP, Sawyer WH (1983) Cytosine arabinoside transport by human leukaemic cells. Eur J Cancer Clin Oncol 19:1067–1074. [DOI] [PubMed] [Google Scholar]
- Wright NJ, Lee S-Y (2019) Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat Struct Mol Biol 26:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao SYM, Ng AML, Cass CE, Baldwin SA, Young JD (2011) Nucleobase transport by human equilibrative nucleoside transporter 1 (hENT1). J Biol Chem 286:32552–32562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JD, Yao SYM, Baldwin JM, Cass CE, Baldwin SA (2013) The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med 34:529–547. [DOI] [PubMed] [Google Scholar]
- Young JD, Yao SYM, Sun L, Cass CE, Baldwin SA (2008) Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 38:995–1021. [DOI] [PubMed] [Google Scholar]
- Zdrazil B, Hellsberg E, Viereck M, Ecker GF (2016) From linked open data to molecular interaction: studying selectivity trends for ligands of the human serotonin and dopamine transporter. MedChemComm 7:1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]