

Abstract
Severe acute respiratory syndrome has relapsed recently as novel coronavirus causing a life threat to the entire world in the absence of an effective therapy. To hamper the replication of the deadly SARS CoV-2 inside the host cells, systematic in silico virtual screening of total 267,324 ligands from Asinex EliteSynergy and BioDesign libraries has been performed using AutoDock Vina against RdRp. The molecular modeling studies revealed the identification of twenty-one macrocyclic hits (2–22) with better binding energy than remdesivir (1), marketed SARS CoV-2 inhibitor. Further, the analysis using rules for drug-likeness and their ADMET profile revealed the candidature of these hits due to superior oral bioavailability and druggability. Further, the MD simulation studies of top two hits (2 and 3) performed using GROMACS 2020.1 for 10 ns revealed their stability into the docked complexes. These results provide an important breakthrough in the design of macrocyclic hits as SARS CoV-2 RNA replicase inhibitor.
Keywords: COVID-19, SARS CoV-2, Molecular docking, MD simulations, RdRp, ADMET assay
Abbreviations: ACE2, angiotensin converting enzyme 2; ADMET, absorption, distribution, metabolism, excretion and toxicity; BBB, blood-brain barrier; BOILED, brain or intestinal estimated permeation method; COVID-19, corona virus disease 2019; E, envelope protein; FDA, food and drugs administration; HBA, hydrogen bond acceptor; HBD, hydrogen bond donor; HERG, human ether-a-go-go-related gene; LOAEL, oral rat chronic toxicity; M, membrane protein; MD, molecular dynamics; N, nucleocapsid protein; NSPs, non-structural proteins; pp1a/b, polyproteins; RdRp, RNA dependent RNA polymerase; S, spike glycoprotein; SARS CoV-2, severe acute respiratory syndrome 2; ssRNA, single stranded ribonucleic acid; UTR, untranslated region; WHO, world health organization
Graphical abstract
1. Introduction
Novel coronavirus (CoV) has been recognized as a deadly respiratory pathogen and causative agent for severe acute respiratory syndrome coronavirus-2 (SARS CoV-2) or coronavirus disease 2019 (COVID-19) [1] since its outbreak in the Hubei province of Wuhan, China in 2019 [2,3]. It has caused an unprecedented pandemic worldwide with the higher fatality rate than the previously known SARS CoV. The background of the virus traces back nearly two decades to a closely related previously emerged pathogen having genomic similarity of 75–80% [4]. The epidemiological study on the virus brings in to notice the first case to be reported on December 2019 in China, which was then observed with the aggressive or violent form by January 2019 with increasing number of cases transmitting in the other parts of the world [5,6]. By January 2019, approximately eighteen countries were reported to be infected with the virus emerged in China led to the declaration as “Public Health Emergency of International Concern” by World Health Organization (WHO) on Mar 11, 2020 [7]. As on Jul 9, 2021 there have been reported 185,291,530 confirmed cases with 4,010,834 fatali cases worldwide [8]. The entire world has to suffer from the huge numbers of challenges due to this global pandemic as all the vital sectors like health, education, economic, social, pharmaceutical, etc. have been widely affected. Unemployment, events cancellation, prohibited public places, declined foreign and domestic trades during the pandemic caused a huge fall in the global economy [9,10].
The novel SARS CoV-2 viral pathogens responsible for the global pandemic have been derived from the previously emerged strain reported in 2002–03 which are suspected to be transmitted into humans through several animal hosts such as bats, civets, pangolins and camels [11]. The clinical symptoms in the patients infected with coronaviruses may vary from moderate symptomatic to severe asymptomatic characteristics including headache, cough, cold, tiredness, aches, fever, loss of taste or smell, diarrhoea, discoloration of finger or toes, breathing difficulties to dysfunction of organs. The viruses such as SARS and MERS are transmitted into humans through the direct or indirect exposure of the respiratory secretions [12,13].
The large genome of the coronaviruses including 30 kb long nucleotide sequence has been comprised within the single stranded positive sense enveloped RNAs [14]. The crown like structural morphology of these viruses observed under electron microscope are due to the club-like glycoproteins [13]. The CoV belongs to the β-coronaviruses among the four major genuses of the large family Nidovirales [15]. The critical insights into the viral pathogenesis and replication may shine a light towards the comprehensive understanding regarding the virus as presented in Fig. 1 . The pathogenesis of corona virus begins with the fusion and attachment to the host receptor angiotensin converting enzyme-2 (ACE2) through its spike glycoprotein. Upon invasion and after releasing its genomic material inside the host cell, the genome gets translated into several polypeptides followed by the translation into sixteen non-structural polyproteins (NSPs). The 3’ end of the sub-genomic RNA gets transcribed into several structural proteins which are assembled on the endoplasmic reticulum (ER) and golgi body to release the viral progenies outside the plasma membrane [16].
Fig. 1.

Life cycle of corona virus inside the host cell. ACE2:Angiotensin Converting Enzyme 2; 3’UTR:Untranslated region; pp1a/pp1ab: polyproteins; S: spike glycoprotein; M: membrane protein; E: envelope protein; N: nucleocapsid protein.
Despite of current antiviral strategies including the anti-viral agents and vaccines to combat the deadly coronavirus, these could not prove themselves effective against the virulent disease. In order to generate the scopes for effective inhibitors of SARS CoV-2, several drugs have been repurposed and implemented to combat this ailment, though they have not shown any reliable results necessitating the demand for some potential inhibitors [17,18]. In view of this fact, several vaccines containing live attenuated, RNA-DNA derived, viral vectored, protein contained and inactivated vaccines have been developed with positive results against the catastrophic virus, SARS CoV-2 [19]. In contrary, the vaccines under phase III trials have also been noted with several side effects after the vaccination such as headache, fatigue, chills, diarrhoea, arthralgia, myalgia, redness, itching, swelling, etc. [20] To exemplify, a nucleotide analog, remdesivir was declared under emergency use authorization by United States food and drug administration (USFDA) and was evaluated under in vivo and in vitro studies for the inhibition of viral replication inside the host body [21]. Despite of its efficacy, it has not been found effective solution to tackle the race for the inhibition of the deadly virus due to its impotency in patients with immunodeficiency and heart failure, hypotension, respiratory dysfunction, as well as renal impairment [22].
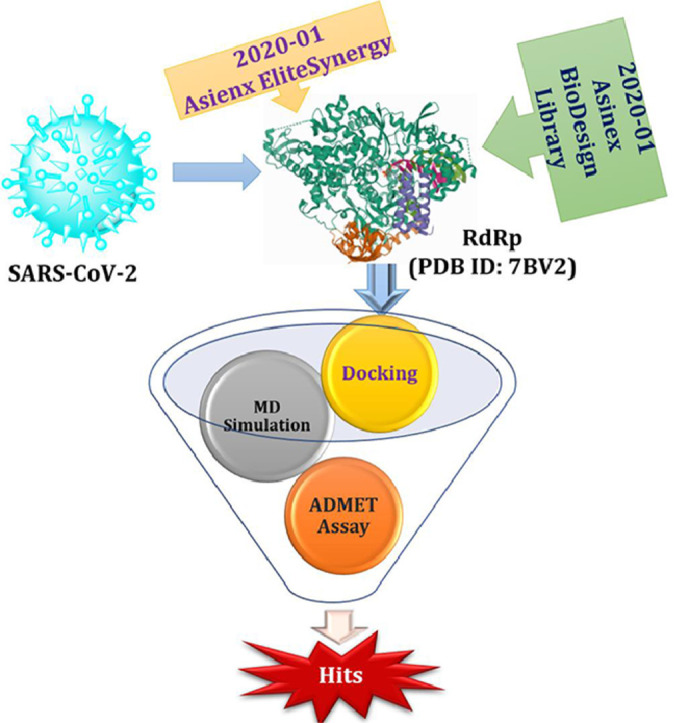
The conventional approach to the drug design and development may take plenty of resources such as cost, time and manpower with no guarantee to afford the desired candidates with efficacy and potency. Therefore, the computer assisted drug design (CADD) have been preferred over several years above the traditional approaches reducing the cost-burden by benefiting the feasibility and improved results after removing the gap between chemical and biological science [23]. Among these CADD tools, the use of molecular modeling should not be undermined as they provide significant interactions of identified hits against their biological targets to understand their mode of action [24], [25], [26], [27], [28]. Recently, we have performed in silico screening of significant phytochemicals against RNA-dependent RNA Polymerase (RdRp) and main protease (Mpro) using molecular docking followed by their druggability using ADMET assay and stability of their complexes using molecular dynamics (MD) simulations in search of effective SARS CoV-2 inhibitors [29]. Motivated by the potential of computational chemistry and in search of potent inhibitors of SARS CoV-2, herein we have performed the virtual screening of total 267,324 ligands from 2020–01 Asinex EiteSynergy (91,473) and BioDesign (175,851) libraries using AutoDock Vina against RdRp using molecular docking followed by their further assessment using rules for druggability, ADMET assay and MD simulations.
2. Results and discussion
2.1. Analysis of protein structure
Xu et al. have reported the cryo-EM structure of the RdRp from SARS-CoV-2, co-crystallized with remdesivir having the resolution of 2.8 Å (PDB ID: 7BV2) [30,31]. In this complex, partial double-stranded RNA template was implanted into the central channel of the RdRp where remdesivir was covalently fused into the primer strand at the first replicated base pair and terminated the chain elongation. The core component of the viral replication complex is the non-structural proteins (nsp12) of the RdRp [32,33].
2.2. Molecular docking
The molecular docking has been performed against the selected protein (PDB ID: 7BV2) [30] using AutoDock Vina [34] to evaluate the binding mode of ligand and interactions in the active site. The required 267,324 ligands have been obtained from 2020–01 Asinex EiteSynergy (91,473) and BioDesign (175,851) library from the Asinex online database, a part of PubChem database consists of small molecules, which can be easily synthesized in organic chemistry labs [35]. The preprocessed protein and ligands in PDBQT format have been used for the molecular docking process. The binding energy of the hit molecules (2–22) have been compared with the co-crystallized ligand remdesivir (1, Table 1 , Entry 1). Further, these hits (2–22) have been found with better binding energy as compared to other promising RdRp inhibitors such as ribavirin (23), penciclovir (24), favipravir (25), [36] molnupiravir (26) and sofosbuvir (27) [37]. Total twenty-one identified macrocyclic hits have been found to possess binding energy lesser than -12.5 Kcal/mol and presented in Table 1 and their 2D-possess with the formed interactions have been presented in Figs. S1–S4 (see supporting informartion).
Table 1.
The identified hits (2–22) having more binding energy than clinically used remdesivir (1) and other RdRp inhibitors (23–27).
| Compound No. | Dataset ID | 2D-Structure | Binding energy (Kcal/mol) |
|---|---|---|---|
| 1 | Remdesivir | 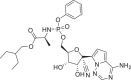 |
-9.2 |
| 2 | LAS 51620435 | 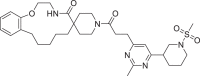 |
-13.1 |
| 3 | LAS 51620429 | 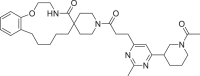 |
-13 |
| 4 | LAS 51609341 | 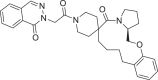 |
-13 |
| 5 | LAS 51620411 | 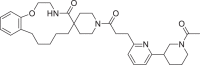 |
-12.9 |
| 6 | LAS 51609377 | 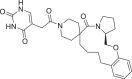 |
-12.8 |
| 7 | LAS 51620382 | 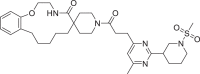 |
-12.8 |
| 8 | LAS 51624260 | 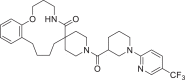 |
-12.8 |
| 9 | LAS 51624264 | 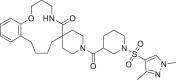 |
-12.8 |
| 10 | LAS 51624295 | 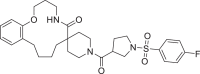 |
-12.8 |
| 11 | LAS 52131039 | 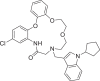 |
-12.8 |
| 12 | LAS 51609335 | 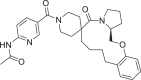 |
-12.7 |
| 13 | LAS 51614073 | 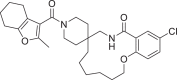 |
-12.7 |
| 14 | LAS 51609336 | 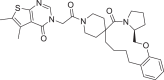 |
-12.6 |
| 15 | LAS 51620402 | 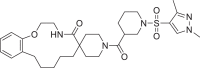 |
-12.6 |
| 16 | LAS 51620430 | 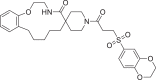 |
-12.6 |
| 17 | LAS 51620433 | 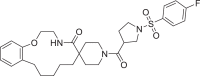 |
-12.6 |
| 18 | LAS 51624245 | 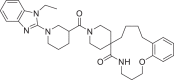 |
-12.6 |
| 19 | LAS 51609330 | 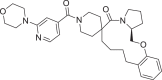 |
-12.5 |
| 20 | LAS 51620424 | 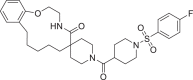 |
-12.5 |
| 21 | LAS 51624284 | 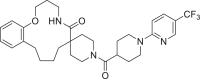 |
-12.5 |
| 22 | LAS 51624301 | 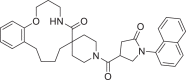 |
-12.5 |
| 23 | Ribavirin | 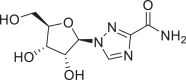 |
-7.4 |
| 24 | Penciclovir | 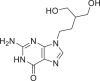 |
-7.2 |
| 25 | Favipravir | 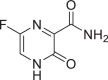 |
-6.7 |
| 26 | Molnupiravir | 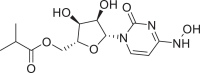 |
-10 |
| 27 | Sofosbuvir | 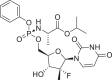 |
-8.7 |
The 3D-poses of identified ligands 2–7 bound at the active site of RdRp have been generated using Biovia Discovery Studio 2020 [38] and presented in Fig. 2 a–f. Compound 2 formed conventional hydrogen bonds (HB) with the amino acid Asn496 from chain A and nucleotides Ade11 and Ade13 of chain P, Ade14, Ade15 and Ade19 of chain T. Further, it formed van der Waals interactions with Ile548 and Ade14 of chain P, π-anion interaction with Urd18 (chain P), π-alkyl interaction with Ala547, Ade13 and Ade14 (chain T, Fig. 2a). Additionally, the presence of sulfonyl group in the structure of 2 could lead the formation of π-sulfur interaction with Ade15 of chain P. Pyrimidine derivative 3 made conventional HB with Ade14 of chain P and Ade11 of chain T, C-H bond with Lys500 and Urd12 of chain T, π-π bond with Phe441, π-alkyl bond with Ala547 and Ile548 along with π-anion bond with Urd17 of chain P (Fig. 2b). Compound 4 formed conventional HB with the amino acid, Asn496 and nucleotides, Ade13 and Ade14 of chain P and Ade14 and 15 of chain T. Additionally, it formed π-π bond with Urd12 of chain T (Fig. 2c). By the replacement of 2-methylpyrimidine ring of compound 3 with pyridine ring in compound 5 retained almost similar interactions including van der Waals bond (Lys500), π-π bond (Phe441, π-alkyl bond with Ala547 and Ile548), π-anion bond (Urd17 and Urd18 of chain P) and conventional HB (Ade14 of chain P and Ade11 of chain T, Fig. 2d). Compound 6 found to form conventional HB with Ade14, Gua16 and Ade19 of chain P, Ade13 of chain T, Lys500 and Asn497. It also formed C-H bond with Asn497, π-anion bond with Ade19 of chain P and π-alkyl bond with Ala547 and Urd18 of chain P (Fig. 2e). N-Methylsulfonyl piperidine substituted compound 7 interacted at the active site of RdRp by forming π-alkyl bond (Ala547, Ile548 and Ade13 of chain T), π-sulfur bond (Ade14 of chain P), π-anion bond (Urd17 and Urd18 of chain P), C-H bond (Lys500) and conventional HB (Ade11 of chain T and Ade14 and 15 of chain P, Fig. 2f). In general, these interactions with the ligands have supported our hypothesis on the tight binding of identified ligands into the active site of protein.
Fig. 2.

The 3D-interactions of ligands 2 (a), 3 (b), 4 (c), 5 (d), 6 (e) and 7 (f) complexed with RdRp. Poses have been generated using Biovia Discovery Studio 2020 [38].
2,4-Disubstituted pyridine derivative 8 has been found with binding energy of -12.8 Kcal/mol (Table 1) due to interaction with RdRp including π-alkyl bond with Ade19 of chain P and conventional HB with Ade11 of chain T, Ade14 and Ade19 of chain P (Fig. 3 a). 1,3-Dimethyl-1H-pyrazol-4-yl sulfonyl derivative compound 9 (Fig. 3b) found to bind tightly through the formation of π-anion bond with Urd13 of chain P, non-polar C-H bond with Urd18 of chain P, π-sulfur bond with Ade19 of chain P and HB with Ade11 (chain T) Ade14 and Ade19 (chain P). Interactions including C-H bond (Lys500), π-alkyl bond (Ala547), π-sulfur bond (Ade14 of chain T), π-anion bonds (Asp845 and Urd18 of chain P) and conventional HB (Ade13, Ade14 of chain T and Gua16, Ade15 of chain P) have been found with the docked complex of 1-sulfonyl pyrrolidine-3-carboxamide 10 against RdRp (Fig. 3c) with similar binding energy. Apart from the interactions including conventional HB (Ade14 of P chain, Ade13 and Ade14 of T chain), two π-π bonds (Urd12) and π-alkyl bond (Urd10 from chain T), N-cyclopenyl indole derivative 11 formed halogen interactions between chlorine atom present in it with Cyt15 and Ade14 of chain T (Fig. 3d). The replacement of pyrimidine-2,4(1H,3H)-dione-6-methyl in ligand 6 with pyridin-2-yl-acetamide in 12 has been resulted in almost similar binding energy (-12.8 and -12.7 kcal/mol, respectively) along the with formation of π-anion bond with Ade19 of chain P, π-alkyl bond with Ala547 and Urd18 of chain P, conventional HB with Ade14, Ade15 and Ade19 belonging to chain P (Fig. 3e). Several π-alkyl interactions are found in docked complex of compound 13 with amino acids Phe441, Ile548, Lys500, nucleotides Urd17, Gua16 (chain P) and Ade11, Urd12 (chain T). Further, ligand 13 formed conventional HB with Lys545, Arg836 and Ade14 of chain T and π-anion bond with Urd18 of T chain and π-σ bond with Ala547 (Fig. 3f).
Fig. 3.

3-D interactions of ligands 8 (a), 9 (b), 10 (c), 11 (d), 12 (e) and 13 (f) with RdRp (PDB ID: 7BV2). Interactive poses of hits have been generated using Biovia Discovery Studio 2020 [38].
Compound 14, with binding energy of -12.6 kcal/mol, formed conventional HB with Ade19 of chain P and Ade11, Ade14 and Cyt15 of chain T, π-anion bond with Urd13 of chain P. Additionally, nucleotide Urd18 has found with π-π bond, π-sulfur bond and π-alkyl bond with the ligand 14 (Fig. 4 a). Compound 15, a metamer of compound 9, formed completely dissimilar interactions in comparison with 9 such as C-H bond with Lys500, π-sulfur bond with Ade13 of T chain, π-alkyl bonds with Ala547 and Ile548, π-anion bonds with Urd17 and Urd18 from chain P and conventional HB with Asn496, Ade11 and Ade13 of chain T (Fig. 4b). Next, 2,3-dihydrobenzo[b][1,4]dioxin-6-sulfonyl derivative 16 found to form π-sulfur bond with Ade14 of chain T, π-alkyl bond with Ile548, π-anion bond with Urd17 and Urd18 of chain P and HBs with the amino acid Asn496 and nucleotides Ade15 and Ade19 from P chain and Ade13 and 14 from T chain (Fig. 4c). Compound 17 being N-(4-flurophenylsulfonyl)pyrrolidine derivative formed two π-alkyl bonds (Ala547), π-sulfur bond (Ade17 of chain T), C-H bonds (Lys500 and Urd12 of chain T), π-anion bonds (Asp845, Urd17 and 18 from P chain), conventional HB (Ade13, Ade14 and Cyt15 of chain T). Further, 4-fluoro on phenyl ring interacted with Asn496 through halogen interactions (Fig. 4d). N-Ethyl 2-substitued benzo[d]imidazole derivative 18 interacted with the protein through C-H bonds with Gua16 and Urd17 of chain P and π-alkyl bond with Ade15, Gua16, Ade19 and Urd20 of chain P (Fig. 4e). 2-Morpholinyl-piperdin-4-yl compound 19 has been found to interact with π-anion bond with Ade19 of P chain, π-alkyl bond with Urd18 of P chain and conventional HB with Ade13 of chain T, Ade19 of chain P and Asn497 (Fig. 4f) with binding energy of -12.5 kcal/mol.
Fig. 4.

3D interactions of ligands compound 14 (a), compound 15 (b), compound 16 (c), compound 17 (d), compound 18 (e) and compound 19 (f) with 7BV2. Poses have been generated using Biovia Discovery Studio 2020 [38].
Ligands 20–22 have been observed with similar binding energy of -12.5 kcal/mol (Table 1). Ligand 20 formed conventional HB and π-sulfur bond with Ade15 of chain P, C-H bonds with Lys500 and Ade14 of chain P, π-alkyl bonds with Ala547 and Ile548, π-anion bonds with Urd17 and 18 of chain P. One of the fluorine atom of phenyl ring interacted with Urd13 of chain P (Fig. 5 a). Compound 21, being the positional isomer of 8, made π-anion bond with Urd17 of chain P and conventional HB with Lys846, Arg858, and Ade11 of chain T and Ade14 of chain P. Additionally, two fluorine atoms of trifluoromethyl group in 21 were found to interact with Asp845 (Fig. 5b). α-Naphthyl-1H-pyrrolidonone derivative 22 formed conventional HB with Ade15 of chain P, C-H bond with Asn496, π-alkyl bonds with Ala547 and Urd18 of chain P and π-anion bonds with Arg836 and Ade19 of chain P (Fig. 5c) to accommodate on the active site of RdRp.
Fig. 5.

3D interactions of ligands 20 (a), 21 (b) and 22 (c) with 7BV2 (RdRp). Poses have been generated using Biovia Discovery Studio 2020 [38].
2.3. Analysis of physicochemical properties and ADMET parameters
The identified macrocyclic hits have been analyzed for the several physicochemical properties (Table 2 ) such as molecular weight (MW), hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), number of rotatable bonds (RB) and partition co-efficient (Log P) using SwissADME [39] and pkCSM [40,41]. All the hits have better physicochemical properties than clinically used remdesivir (1). Almost all the hits have molecular weight beyond 500 including remdesivir. No compound from the hits has found violating the criteria of hydrogen bond acceptors, hydrogen bond donors and rotatable bonds. Only six compounds (8, 11, 13, 18, 21 and 22) exceeded the maximum limit of Log P value. These exceeded properties may be tackled through novel sophisticated techniques during development of formulation with a view of improving the drug-likeness. Thus, most of the hits may have potential to be a better drug molecule in comparison with remdesivir (1, Table 2).
Table 2.
Physicochemical properties of the macrocyclic hits (2–22) and marketed drug, remdesivir (1).a
| Comp No. | MWb | HBDc | HBAd | RBe | Log Pf | No. of violationsg |
|---|---|---|---|---|---|---|
| 1 | 602.58 | 4 | 12 | 14 | 2.31 | 2 |
| 2 | 625.82 | 1 | 8 | 6 | 3.78 | 1 |
| 3 | 589.77 | 1 | 6 | 6 | 4.36 | 1 |
| 4 | 528.64 | 0 | 5 | 3 | 3.80 | 1 |
| 5 | 574.75 | 1 | 5 | 6 | 4.66 | 1 |
| 6 | 494.58 | 2 | 5 | 3 | 2.01 | 0 |
| 7 | 625.82 | 1 | 8 | 6 | 3.78 | 1 |
| 8 | 572.66 | 1 | 7 | 4 | 5.24 | 2 |
| 9 | 585.76 | 1 | 7 | 4 | 3.05 | 1 |
| 10 | 571.70 | 1 | 7 | 4 | 3.76 | 1 |
| 11 | 560.08 | 1 | 5 | 3 | 7.05 | 2 |
| 12 | 504.62 | 1 | 5 | 4 | 4.06 | 1 |
| 13 | 513.07 | 1 | 4 | 2 | 6.12 | 2 |
| 14 | 562.72 | 0 | 5 | 3 | 4.48 | 1 |
| 15 | 585.76 | 1 | 7 | 4 | 3.05 | 1 |
| 16 | 570.70 | 1 | 7 | 5 | 3.54 | 1 |
| 17 | 571.70 | 1 | 7 | 4 | 3.76 | 1 |
| 18 | 571.75 | 1 | 4 | 4 | 5.19 | 2 |
| 19 | 532.67 | 0 | 5 | 3 | 3.94 | 1 |
| 20 | 585.73 | 1 | 7 | 4 | 4.15 | 1 |
| 21 | 572.66 | 1 | 7 | 4 | 5.24 | 2 |
| 22 | 553.69 | 1 | 4 | 3 | 5.11 | 2 |
Table 3.
Predicted absorption and distribution properties of the hits (2–22) and remdesivir (1).a
| Comp No. | Ali log Sb | MRc | tPSA (Å2)d | CaCO2 permeabilitye | Human intestinal absorption (% absorbed)f | VDss (Human)g | Fraction unbound (Human)h | P-gp inhibition (yes/no)i |
|---|---|---|---|---|---|---|---|---|
| 1 | -6.01 | 150.43 | 213.36 | 0.51 | 69.44 | -0.38 | 0.03 | Yes |
| 2 | -5.89 | 182.34 | 130.18 | 0.99 | 98.37 | -0.40 | 0.00 | Yes |
| 3 | -5.57 | 178.39 | 104.73 | 1.00 | 97.33 | -0.09 | 0.02 | Yes |
| 4 | -5.37 | 157.77 | 84.74 | 1.49 | 94.72 | 0.28 | 0.00 | Yes |
| 5 | -5.56 | 175.62 | 91.84 | 0.98 | 89.36 | 0.04 | 0.01 | Yes |
| 6 | -3.95 | 143.15 | 115.57 | 1.04 | 78.89 | -0.13 | 0.21 | Yes |
| 7 | -5.89 | 182.34 | 130.18 | 0.90 | 92.22 | -0.47 | 0.00 | Yes |
| 8 | -6.56 | 161.6 | 74.77 | 0.79 | 90.74 | -0.02 | 0.00 | Yes |
| 9 | -5.05 | 168.38 | 122.22 | 0.95 | 86.35 | -0.45 | 0.00 | Yes |
| 10 | -5.75 | 161.52 | 104.4 | 0.95 | 95.03 | -0.31 | 0.00 | Yes |
| 11 | -6.79 | 165.08 | 64.96 | 1.03 | 91.72 | 0.55 | 0.13 | Yes |
| 12 | -4.89 | 149.43 | 91.84 | 0.88 | 95.13 | 0.06 | 0.06 | Yes |
| 13 | -7.47 | 149.63 | 71.78 | 0.65 | 89.23 | 0.46 | 0.00 | Yes |
| 14 | -6.77 | 165.58 | 112.98 | 0.92 | 93.39 | 0.14 | 0.02 | Yes |
| 15 | -5.24 | 168.38 | 122.22 | 1.02 | 86.64 | -0.51 | 0.00 | Yes |
| 16 | -5.78 | 158.12 | 119.62 | 1.13 | 87.27 | -0.31 | 0.00 | Yes |
| 17 | -5.94 | 161.52 | 104.4 | 1.02 | 95.32 | -0.39 | 0.00 | Yes |
| 18 | -6.66 | 178.16 | 79.7 | 0.74 | 90.82 | 0.61 | 0.02 | Yes |
| 19 | -5.15 | 160.85 | 75.21 | 1.45 | 96.05 | 0.02 | 0.00 | Yes |
| 20 | -6.31 | 166.33 | 104.4 | 1.00 | 100 | -0.39 | 0.00 | Yes |
| 21 | -6.56 | 161.6 | 74.77 | 0.77 | 90.62 | -0.19 | 0.00 | Yes |
| 22 | -6.19 | 171.7 | 78.95 | 0.76 | 93.19 | 0.48 | 0.00 | Yes |
Parameters calculated using pkCSM [40].
Aqueous solubility descriptor (≤ 0),
molar refractivity (≤ 155),
topological polar surface area (≤ 150 Å2),
Caco-2 cell permeability (log Papp in 10−6 cm/s > 0.09),
absorption (human, % > 30),
volume of distribution (human, log L/kg) (low if < − 0.15 and high if > 0.45),
fraction unbound, and
ability to inhibit the P-glycoprotein.
Rules for the drug-likeliness such as Lipinski's rule of 5 (Ro5), Ghose rule, Veber's rule, Egan's rule and Muegge's rule have been also studied to ensure the drug-likeliness of 1–22 and number of violations have presented graphically in Fig. 6 . According to the rule of 5, the molecule can serve as better drug candidate with the suitable properties including MW (≤ 500 Da), HBD (≤ 5), HBA (≤ 10), LogP (≤ 5) and RB (≤ 5) [42]. Not more than one violation of these parameters will reduce the druggability of the molecule. Most of the hits (except 8, 11, 13, 18, 21 and 22) have found to meet the desired properties suggesting their candidature for the anti-viral drug. As per the Veber's rule, compound should have the polar surface area less than 140 Å2 to have the better oral bioavailability [43]. None of hits violated Veber's rule suggesting their good oral bioavailability. All hits except 11 qualified the criteria for oral bioavailability laid down by the Egan rule with desired tPSA (0–132 Å2) and log P (-1–6) [44]. Muegge's filter includes several parameters including MW (200–600), lipophilicity (XLOGP3, -2–5), tPSA (≤ 150), cyclic rings (≤ 7), carbon atoms (> 4), heteroatoms (> 1), RB (≤ 15), HBA (≤ 10), and HBD (≤ 5) [45] and only few compounds (2, 7, 8, 11, 13, 18 and 21) violated with note more than one parameter to qualify it. Ghose rule indicated the desired requirements of MW (160–480 Da), Log P (-0.4–5.6), MR (40–130) and atoms (20–70) to be the good drug candidate [46] and all the hits meet the suggested properties. However, as compared to Remdesivir, the identified hits may have potential to act as a good drug candidate in the future as they obey the rules for druggability and drug-likeness.
Fig. 6.

Result of the hits against different violations for the rules of druggability and drug-likeness.
Next, we have studied the various ADMET (absorption, distribution, metabolism, excretion and toxicity) parameters like water solubility (Ali log S), molar refractivity (MR), topological polar surface area (tPSA), CaCO2 cell permeability, intestinal absorption, volume of distribution (Vd), unbound fraction of drug and ability to inhibit the P-glycoprotein substrate for the identified hits. All the molecules have satisfied the criteria for the Ali log S, tPSA and CaCO2 cell permeability. Most of the hits with sufficient human intestinal absorption indicated their better oral bioavailability. All the hits also qualified the criteria for Vd along with the inhibition of the P-glycoprotein substrates. Further, we analyzed the additional parameters to predict the metabolism, excretion and toxicity profile of remdesivir and these hits (Table 4 ). Most of the molecules inhibited CYP2D6 but not CYP3A4 along with medium renal clearance except compound 16 and 22 (with low clearance). All the hits except hits 4, 5, 18 were found as the substrates for renal uptake transporter in proximal convoluted tubule (OCT2). All the hits were found non-cytotoxic (hERG cell line), non-mutagenic (AMES toxicity), non-toxic to dermis (skin sensitization) and found safe through prediction of oral rat acute and (LD50) chronic toxicity (LOAEL). Thus, these macrocyclic hits (2–22) might be good and successful drug candidates in the future.
Table 4.
Metabolism, excretion and safety parameters of remdesivir (1) and identified hits (2–22).a
| Comp No. | CYP2D6 inhibitorb | CYP3A4 inhibitorc | CLTd | Renal OCT2 substratee | AMES toxicityf | hERG I toxicityg | LD50h | LOAELi | Skin sensitizationj |
|---|---|---|---|---|---|---|---|---|---|
| 1 | No | No | 0.16 | No | No | No | 2.25 | 2.27 | No |
| 2 | No | Yes | 0.56 | No | No | No | 3.09 | 1.54 | No |
| 3 | No | Yes | 0.50 | No | No | No | 3.20 | 1.33 | No |
| 4 | No | Yes | 0.27 | Yes | No | No | 2.88 | -0.08 | No |
| 5 | No | Yes | 0.49 | Yes | No | No | 3.34 | -0.45 | No |
| 6 | No | Yes | 0.42 | No | No | No | 2.40 | 0.92 | No |
| 7 | No | Yes | 0.54 | No | No | No | 2.95 | 1.51 | No |
| 8 | No | Yes | 0.11 | No | No | No | 2.70 | 0.80 | No |
| 9 | No | Yes | 0.36 | No | No | No | 2.97 | 1.78 | No |
| 10 | No | Yes | 0.37 | No | No | No | 2.80 | 1.44 | No |
| 11 | No | Yes | 0.55 | No | No | No | 3.24 | -0.19 | No |
| 12 | No | Yes | 0.37 | No | No | No | 2.65 | 1.60 | No |
| 13 | No | Yes | -0.09 | No | No | No | 2.49 | 0.71 | No |
| 14 | No | Yes | 0.05 | No | No | No | 2.92 | -0.42 | No |
| 15 | No | Yes | 0.40 | No | No | No | 3.11 | 1.63 | No |
| 16 | No | Yes | 0.43 | No | No | No | 3.05 | 1.69 | No |
| 17 | No | Yes | 0.40 | No | No | No | 2.98 | 0.94 | No |
| 18 | No | Yes | 0.44 | Yes | Yes | No | 2.37 | 1.47 | No |
| 19 | No | Yes | 0.40 | No | No | No | 3.18 | -0.12 | No |
| 20 | No | Yes | 0.41 | No | No | No | 3.03 | 0.97 | No |
| 21 | No | Yes | 0.17 | No | Yes | No | 3.00 | -0.08 | No |
| 22 | No | Yes | -0.03 | No | No | No | 2.46 | 1.79 | No |
Parameters calculated using pkCSM [40],
ability to inhibit CYP2D6 enzyme,
ability to inhibit CYP3A4 enzyme,
total renal clearance; high (>1 mL/min/kg), medium (> 0.1 to < 1 mL/min/kg) or low (≤ 0.1 mL/min/kg),
ability to inhibit renal OCT2 substrate;
AMES toxicity;
hERG I toxicity;
oral rat acute toxicity (LD50);
oral rat chronic toxicity (LOAEL);
skin sensitisation.
The Brain Or IntestinaL EstimateD permeation method (BOILED-Egg) provide the in silico estimation of accessibility of molecules to gastrointestinal (GI) tract and blood-brain barrier [47]. Hence, we also become interested to predict the permeability of the identified macrocyclic hits using SwissADME [39]. The boiled egg model revealed all the hit molecules possessed satisfactory GI absorption along with inhibition of the P-glycoprotein, a protein responsible for efflux of drugs from cells (Fig. 7 ). All the compounds (except 19) were found with probability of permeation to BBB and henceforth, the least chances of neurotoxicity of CNS with the proposed compounds 2–18 and 20–22 may be approximated.
Fig. 7.

BOILED egg model of hit molecules (2–22) generated with the help of SwissADME. The yellow and colorless regions indicate BBB and GI permeability, respectively. The blue circles indicate inhibition of P-glycoprotein.
2.4. MD simulation
To study the stability of ligand into the binding site of protein, molecular dynamics (MD) simulations provide the better understanding of ligand through several statistical parameters [48,49]. Several scientific literatures support the reliability and satisfactory stability for the MD run (≤ 10 ns) through in silico endeavor [50], [51], [52], [53], [54], [55]. In a similar line of approach, the hit complexes obtained from the evaluation of ADMET and drug-likeliness properties, were subjected for MD simulations using GROMACS 2020.1 to assess the stability of the ligands (2 and 3) in the active site of docked complex at various time points up to 10 ns [56,57].
Next, we subjected the complex of compound 2 to MD simulation studies and the graphical representation of plots of statistical parameters has been presented in Fig. 8 . The ligand-receptor complex of 2 with RdRp was found with root mean square deviation (RMSD) value with an average of 0.757 nm (Fig. 8a) for the ligand and 0.267 nm (Fig. 8b) for the protein which indicate the stability of the ligand with least deviation in the active site of remdesivir. The radius of gyration (RoG) for the same complex ranging from 2.94 to 3.01 nm with an average of 2.972 nm (Fig. 8c) indicated the compactness of the complex. The plots of solvent assessable surface area (SASA) and hydrogen bonds has been presented in Fig. 8d and Fig. 8e, respectively. The surface area accessed by the solvent molecules was found within the satisfactory range from 447–461 nm2 with an average of 454.72 nm2 (Fig. 8d). From the plot of number of HB vs simulation time, maximum four HB were found between the ligand and receptor within the time period of 10 ns (Fig. 8e).
Fig. 8.

The schematic plots of RMSD-L (a), RMSD-P (b), RoG (c), SASA (d) and HB (e) for the complex of compound 2 with RdRp.
The satisfactory stability of the ligand with least deviation in the active site of complex has been evident from RMSD value with an average of 0.942 nm (ligand, Fig. 9 a) and 0.270 nm (protein, Fig. 9b) for the ligand-receptor complex of compound 3 with RdRp. The RoG for the same complex was found in the range of 2.94–3.00 nm with an average of 2.966 nm (Fig. 9c) indicating their compactness. The surface area accessed by the solvent/water molecules was found within the range from 446–460 nm2 with an average of 454.215 nm2 (Fig. 9d). From the plot of number of HB vs time, maximum two HBs were found between the ligand and receptor within the time period of 10 ns (Fig. 9e). The electrostatic (coulombic short-range, Coul-SR) and van der Waals/hydrophobic (LJ-SR) energies have been represented in Table 5 for the complexes of identified hits 2 and 3 with RdRp. These complexes have been observed to be stabilized by the van der Waals or hydrophobic interactions significantly over the electrostatic or coulombic contributions.
Fig. 9.

The schematic plots of RMSD-L (a), RMSD-P (b), RoG (c), SASA (d) and HB (e) for the complex of compound 3 with RdRp.
Table 5.
The electrostatic steric interactions (KJ/mol) for the complex of respective compounds (2 and 3) with RdRp protein.
| Compd. No. | Energy (KJ/mol) | |
|---|---|---|
| Electrostatic interaction (Coul-SR) | van der Waals/Hydrophobic interactions (LJ-SR) | |
| 2 | -48.7869 ± 4.6 | -115.263 ± 9.1 |
| 3 | -32.4944 ± 8.6 | -108.078 ± 8.1 |
3. Conclusion
In conclusion, the in silico based virtual screening of total 267,324 ligands from 2020-01 Asinex EiteSynergy (91,473) and BioDesign (175,851) libraries against RdRp using AutoDock Vina lead to discovery of twenty-one macrocyclic hits (2–22) with better binding energy than remdesivir (1), clinically used SARS CoV-2 inhibitor. The several interactions between ligand and protein helped to understand their binding mode into the active site of protein. The drug-likeliness and druggability studied using several rules for druggability and analysis of ADMET profiles of these hits (2–22) revealed the better bioavailability and safety profile than 1. MD simulation studies up to 10 ns using GROMACS 2020.1 of the top two hits (2 and 3) indicated their stability into the active site of complex. Further, the structural modifications of these molecules may have the scope with significant inhibition of SARS CoV-2 in future.
Experimental section
Collection and preparation of data
The 3-dimentional structure of the SARS-CoV-2 RdRp evaluated through cryo electron microscopy (resolution: 2.8 Å) having antiviral remdesivir as a co-crystallised inhibitor (PDB ID: 7BV2) was obtained from RCSB protein data bank [30]. The protein was prepared for further molecular modelling using AutoDock Vina [58]. All the water molecules were deleted along with addition of polar hydrogen atoms and Kollman charges to complete the protein in terms of polarity and charges, respectively. The generation of the receptor grid box around the macromolecule of co-crystallised ligand remdesivir was achieved with the size of 90.768 × 99.896 × 99.788 Å (x, y and z) and the co-ordinates of centers 70 × 70 × 70 Å (x, y, and z). The prepared protein has been kept in PDBQT format which has been further used for the molecular docking. The present docking protocol has been validated by docking of remdesivir using the same and observed with RMSD of less than 1.
The Asinex BioDesign library 2020.1 having 175,851 molecules and Asinex EliteSynergy having 91,473 molecules was downloaded from Asinex database [35]. All the ligand molecules have been optimized and converted into PDBQT format using OpenBabel and further used for molecular docking process [59].
Molecular docking
The multi-ligand molecular docking was performed using the prepared protein using AutoDock Vina to reveal the binding affinity and interactions of the ligands with RdRp protein (PDB ID: 7BV2) [30]. All the ligands (267,324) were docked on the site using the optimized grid box and ten poses per ligand were generated. The docked pose of remdesivir was compared with the co-crystallized inhibitor remdesivir to validate the present docking protocol. The docking interactions of hits were visualized and analyzed using Biovia Discovery studio 2020 [38]. The 2D interactions of docked compounds (2–22) with RdRp have been presented in supporting information (Figs S1–S4).
Analysis of physicochemical properties, drug-likeliness and ADMET parameters
Various physicochemical properties like molecular weight, number of hydrogen bond donors, number of hydrogen bond acceptors, oil to water partition co-efficient and number of rotational bonds of the selected hits (2–22) and remdesivir (1) were studied using Swiss ADME [39] and pkCSM [40,41]. Several rules such as Lipinski's rule of 5, Ghose rule, Veber's rule, Egan's rule and Muegge's rule were applied to the selected hits using the SwissADME to ensure their druggability and to identify numbers of violated parameters. The ADMET properties like water solubility, molar refractivity, topological polar surface area, CaCO2 cell permeability, intestinal absorption, volume of distribution, fraction unbound, total renal clearance, hepatotoxicity and ability to inhibit the P-glycoprotein were also studied using pkCSM.
Molecular dynamics (MD) simulation
Two molecules (2 and 3), obtained from the manual analysis of the hits found from the results of molecular docking and ADMET analysis, were selected for MD simulation using GROningen MAchine for Chemical Simulations (GROMACS) 2020.1 [56,57] software. CHARMM36 (Chemistry at Harvard Macromolecular Mechanics) as an all atom force field [60] and CHARMM General Force Field (CGenFF) server to retrieve the topology of (2 and 3) were used [61,62]. After solvation (TIP3P water model), neutralization (Na+ and Cl− ions), equilibration [canonical (NVT) and isobaric-isothermic (NPT) ensemble for 100 ps], the complexes were subjected to molecular dynamics run for 10 ns. By analyzing the plots of temperature, pressure and energy equilibria of the trajectories (Figs S5 and S6, see supporting information), we found the system satisfactorily equilibrated.
CRediT authorship contribution statement
Prinsa R. Nagar: Methodology, Data curation, Writing – original draft, Writing – review & editing. Normi D. Gajjar: Methodology, Data curation, Writing – original draft, Writing – review & editing. Tejas M. Dhameliya: Data curation, Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Declaration of Competing Interest
The authors declare no competing financial interest.
Acknowledgments
A part work of virtual screening mentioned in the present work has been assisted by Drug Discovery Hackathon 2020 (DDH-2020) through the support from Council of Scientific and Industrial Research (CSIR), All India Council of Technical Education (AICTE) and Government of India. Authors would like to acknowledge the support provided by Centre for Development of Advanced Computing (CDAC), Government of India for accessing the virtual tool room for performing molecular modelling.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2021.131190.
Appendix. Supplementary materials
References
- 1.Nicola M., Alsafi Z., Sohrabi C., Kerwan A., Al-Jabir A., Iosifidis C., Agha M., Agha R. The socio-economic implications of the coronavirus pandemic (COVID-19): a review. Int. J. Surg. 2020;78:185–193. doi: 10.1016/j.ijsu.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu B., Guo H., Zhou P., Shi Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y., Ren R., Leung K.S.M., Lau E.H.Y., Wong J.Y., Xing X., Xiang N., Wu Y., Li C., Chen Q., Li D., Liu T., Zhao J., Liu M., Tu W., Chen C., Jin L., Yang R., Wang Q., Zhou S., Wang R., Liu H., Luo Y., Liu Y., Shao G., Li H., Tao Z., Yang Y., Deng Z., Liu B., Ma Z., Zhang Y., Shi G., Lam T.T.Y., Wu J.T., Gao G.F., Cowling B.J., Yang B., Leung G.M., Feng Z. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N. Engl. J. Med. 2020;382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou P., Lou Yang X., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Di Jiang R., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai C.C., Shih T.P., Ko W.C., Tang H.J., Hsueh P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents. 2020;55 doi: 10.1016/j.ijantimicag.2020.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L., Wang Y., Ye D., Liu Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int. J. Antimicrob. Agents. 2020;55 doi: 10.1016/j.ijantimicag.2020.105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization. Coronavirus disease 2019 (COVID-19). Situation report –51, (2020). https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200311-sitrep-51-covid-19.pdf?sfvrsn=1ba62e57_10 (accessed February 2, 2021).
- 8.WHO Coronavirus Disease (COVID-19) Dashboard. https://covid19.who.int/ (accessed July 12, 2021).
- 9.Bashir M.F., Ma B., Shahzad L. A brief review of socio-economic and environmental impact of COVID-19. Air Qual. Atmos. Health. 2020;13:1403–1409. doi: 10.1007/s11869-020-00894-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiscott J., Alexandridi M., Muscolini M., Tassone E., Palermo E., Soultsioti M., Zevini A. The global impact of the coronavirus pandemic. Cytokine Growth Factor Rev. 2020;53:1–9. doi: 10.1016/j.cytogfr.2020.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau S.K.P., Woo P.C.Y., Li K.S.M., Huang Y., Tsoi H.W., Wong B.H.L., Wong S.S.Y., Leung S.Y., Chan K.H., Yuen K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U S A. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adhikari S.P., Meng S., Wu Y.J., Mao Y.P., Ye R.X., Wang Q.Z., Sun C., Sylvia S., Rozelle S., Raat H., Zhou H. Epidemiology, causes, clinical manifestation and diagnosis, prevention and control of coronavirus disease (COVID-19) during the early outbreak period: a scoping review. Infect. Dis. Poverty. 2020;9:29. doi: 10.1186/s40249-020-00646-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prasad S., Potdar V., Cherian S., Abraham P., Basu A. Transmission electron microscopy imaging of SARS-CoV-2, Indian. J. Med. Res. 2020;151:241–243. doi: 10.4103/ijmr.IJMR_577_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Astuti Y.Indwiani. Viral structure and host responses. Diabetes Metab. Res. Rev. 2020;14:407–412. doi: 10.1016/j.dsx.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drexler J.F., Corman V.M., Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir. Res. 2014;101:45–56. doi: 10.1016/j.antiviral.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavan Kumar S.P.K., Choudhary K., Thakur N., Gaurav Suresh W., Dayaramani R., Agrawal M., Alexander A. Virology, pathogenesis, diagnosis and in-line treatment of COVID-19. Eur. J. Pharmacol. 2020;883 doi: 10.1016/j.ejphar.2020.173375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohamed K., Yazdanpanah N., Saghazadeh A., Rezaei N. Computational drug discovery and repurposing for the treatment of COVID-19: a systematic review. Bioorg. Chem. 2021;106 doi: 10.1016/j.bioorg.2020.104490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de A. Santos I., Grosche V.R., Bergamini F.R.G., Sabino-Silva R., Jardim A.C.G. Antivirals against coronaviruses: candidate drugs for SARS-CoV-2 treatment? Front. Microbiol. 2020;11 doi: 10.3389/fmicb.2020.01818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kyriakidis N.C., López-Cortés A., González E.V., Grimaldos A.B., Prado E.O. SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines. 2021;6:28. doi: 10.1038/s41541-021-00292-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menni C., Klaser K., May A., Polidori L., Capdevila J., Louca P., Sudre C.H., Nguyen L.H., Drew D.A., Merino J., Hu C., Selvachandran S., Antonelli M., Murray B., Canas L.S., Molteni E., Graham M.S., Modat M., Joshi A.D., Mangino M., Hammers A., Goodman A.L., Chan A.T., Wolf J., Steves C.J., Valdes A.M., Ourselin S., Spector T.D. Vaccine side-effects and SARS-CoV-2 infection after vaccination in users of the COVID symptom study app in the UK: a prospective observational study. Lancet Infect. Dis. 2021;21:939–949. doi: 10.1016/S1473-3099(21)00224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eastman R.T., Roth J.S., Brimacombe K.R., Simeonov A., Shen M., Patnaik S., Hall M.D. Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 2020;6:672–683. doi: 10.1021/acscentsci.0c00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinot M., Jary A., Fafi-Kremer S., Leducq V., Delagreverie H., Garnier M., Pacanowski J., Mékinian A., Pirenne F., Tiberghien P., Calvez V., Humbrecht C., Marcelin A.-G., Lacombe K. Remdesivir failure with SARS-CoV-2 RNA-dependent RNA-polymerase mutation in a B-cell immunodeficient patient with protracted COVID-19. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rifaioglu A.S., Atas H., Martin M.J., Cetin-Atalay R., Atalay V., Doǧan T. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief. Bioinform. 2019;20:1878–1912. doi: 10.1093/bib/bby061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bharatam P.V. In: Drug Discovery and Development. Poduri R., editor. Springer; Singapore: 2021. Computer-aided drug design; pp. 137–210. [Google Scholar]
- 25.Shah P., Dhameliya T.M., Bansal R., Nautiyal M., Kommi D.N., Jadhavar P.S., Sridevi J.P., Yogeeswari P., Sriram D., Chakraborti A.K. N-Arylalkylbenzo[d]thiazole-2-carboxamides as anti-mycobacterial agents: design, new methods of synthesis and biological evaluation. Med. Chem. Commun. 2014;5:1489–1495. [Google Scholar]
- 26.Jadhavar P.S., Dhameliya T.M., Vaja M.D., Kumar D., Sridevi J.P., Yogeeswari P., Sriram D., Chakraborti A.K. Synthesis, biological evaluation and structure-activity relationship of 2-styrylquinazolones as anti-tubercular agents. Bioorg. Med. Chem. Lett. 2016;26:2663–2669. doi: 10.1016/j.bmcl.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 27.Dhameliya T.M., Tiwari R., Banerjee A., Pancholia S., Sriram D., Panda D., Chakraborti A.K. Benzo[d]thiazole-2-carbanilides as new anti-TB chemotypes: design, synthesis, biological evaluation, and structure-activity relationship. Eur. J. Med. Chem. 2018;155:364–380. doi: 10.1016/j.ejmech.2018.05.049. [DOI] [PubMed] [Google Scholar]
- 28.Bhakhar K.A., Gajjar N.D., Bodiwala K.B., Sureja D.K., Dhameliya T.M. Identification of anti-mycobacterial agents against mmpL3: virtual screening, ADMET analysis and MD simulations. J. Mol. Struct. 2021;1244 [Google Scholar]
- 29.Gajjar N.D., Dhameliya T.M., Shah G.B. In search of RdRp and Mpro inhibitors against SARS CoV-2: molecular docking, molecular dynamic simulations and ADMET analysis. J. Mol. Struct. 2021;1239 doi: 10.1016/j.molstruc.2021.130488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Protein data bank. https://www.rcsb.org/ (accessed August 30, 2020).
- 31.Yin W., Mao C., Luan X., Shen D.D., Shen Q., Su H., Wang X., Zhou F., Zhao W., Gao M., Chang S., Xie Y.C., Tian G., Jiang H.W., Tao S.C., Shen J., Jiang Y., Jiang H., Xu Y., Zhang S., Zhang Y., Xu H.E. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;368:1499–1504. doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.te Velthuis A.J.W., Arnold J.J., Cameron C.E., van den Worm S.H.E., Snijder E.J. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. 2010;38:203–214. doi: 10.1093/nar/gkp904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahn D.G., Choi J.K., Taylor D.R., Oh J.W. Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Arch. Virol. 2012;157:2095–2104. doi: 10.1007/s00705-012-1404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asinex. http://www.asinex.com/ (accessed August 17, 2019).
- 36.Shagufta I.Ahmad. The race to treat COVID-19: potential therapeutic agents for the prevention and treatment of SARS-CoV-2. Eur. J. Med. Chem. 2021;213 doi: 10.1016/j.ejmech.2021.113157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian L., Qiang T., Liang C., Ren X., Jia M., Zhang J., Li J., Wan M., YuWen X., Li H., Cao W., Liu H. RNA-dependent RNA polymerase (RdRp) inhibitors: the current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021;213 doi: 10.1016/j.ejmech.2021.113201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dassault Systѐmes BIOVIA, BIOVIA Workbook, Release 2021; BIOVIA DS Visualizer, Release 2021, San Diego: Dassault Systѐmes, 2021.
- 39.Daina A., Michielin O., Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.pkCSM: pharmacokinetic properties. http://biosig.unimelb.edu.au/pkcsm/prediction (accessed February 24, 2021).
- 41.Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and devlopment settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 43.Veber D.F., Johnson S.R., Cheng H.Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 44.Egan W.J., Merz K.M., Baldwin J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000;43:3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- 45.Muegge I., Heald S.L., Brittelli D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001;44:1841–1846. doi: 10.1021/jm015507e. [DOI] [PubMed] [Google Scholar]
- 46.Ghose A.K., Viswanadhan V.N., Wendoloski J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 47.Daina A., Zoete V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11:1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durrant J.D., McCammon J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:71. doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hospital A., Goñi J.R., Orozco M., Gelpí J.L. Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinform. Chem. 2015;8:37–47. doi: 10.2147/AABC.S70333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.González Torres M., Villarreal-Ramírez E., de los A. Moyaho Bernal M., Álvarez M., González-Valdez J., Gutiérrez Uribe J.A., Leyva Gómez G., Cortez J.R.C. Insights into the application of polyhydroxyalkanoates derivatives from the combination of experimental and simulation approaches. J. Mol. Struct. 2019;1175:536–541. [Google Scholar]
- 51.Ramalho T.C., França T.C.C., Cortopassi W.A., Gonçalves A.S., Da Silva A.W.S., Da Cunha E.F.F. Topology and dynamics of the interaction between 5-nitroimidazole radiosensitizers and duplex DNA studied by a combination of docking, molecular dynamic simulations and NMR spectroscopy. J. Mol. Struct. 2011;992:65–71. [Google Scholar]
- 52.Abbas S., Nasir H.H., Zaib S., Ali S., Mahmood T., Ayub K., Tahir M.N., Iqbal J. Carbonic anhydrase inhibition of Schiff base derivative of imino-methyl-naphthalen-2-ol: synthesis, structure elucidation, molecular docking, dynamic simulation and density functional theory calculations. J. Mol. Struct. 2018;1156:193–200. [Google Scholar]
- 53.Batran R.Z., Khedr M.A., Abdel Latif N.A., Abd El Aty A.A., Shehata A.N. Synthesis, homology modeling, molecular docking, dynamics, and antifungal screening of new 4-hydroxycoumarin derivatives as potential chitinase inhibitors. J. Mol. Struct. 2019;1180:260–271. [Google Scholar]
- 54.Modi P., Patel S., Chhabria M. Structure-based design, synthesis and biological evaluation of a newer series of pyrazolo[1,5-a]pyrimidine analogues as potential anti-tubercular agents. Bioorg. Chem. 2019;87:240–251. doi: 10.1016/j.bioorg.2019.02.044. [DOI] [PubMed] [Google Scholar]
- 55.Hornak V., Simmerling C. Development of softcore potential functions for overcoming steric barriers in molecular dynamics simulations. J. Mol. Graph. Model. 2004;22:405–413. doi: 10.1016/j.jmgm.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 56.M.J. Abraham, Berk Hess, E. Lindahl, D. van der Spoel, GROMACS 2020.1 (manual version 2020.1) Zenodo, (2020). 10.5281/zenodo.4054996 (accessed September 10, 2020).
- 57.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindah E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- 58.Trott O., Olson A.J. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comb. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open babel: an open chemical toolbox. J. Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., De Groot B.L., Grubmüller H., MacKerell A.D. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2016;14:71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A.D. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu W., He X., Vanommeslaeghe K., MacKerell A.D. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012;33:2451–2468. doi: 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.